

Abstract
Rationale
Cardiac fibrosis plays a critical role in the pathogenesis of heart failure (HF). Excessive accumulation of extracellular matrix (ECM) resulting from cardiac fibrosis impairs cardiac contractile function and increases arrhythmogenicity. Current treatment options for cardiac fibrosis, however, are limited and there is a clear need to identify novel mediators of cardiac fibrosis to facilitate the development of better therapeutics. Exploiting co-expression gene network analysis on RNA sequencing data from failing human heart, we identified thioredoxin domain containing 5 (TXNDC5), a cardiac fibroblast (CF)-enriched endoplasmic reticulum (ER) protein, as a potential novel mediator of cardiac fibrosis and we completed experiments to test this hypothesis directly.
Objective
To determine the functional role of TXNDC5 in the pathogenesis of cardiac fibrosis.
Methods and Results
RNASeq and Western blot analyses revealed that TXNDC5 mRNA and protein were highly upregulated in failing human left ventricles (LV) and in hypertrophied/failing mouse LV. In addition, cardiac TXNDC5 mRNA expression levels were positively correlated with those of transcripts encoding transforming growth factor β1 (TGFβ1) and ECM proteins in vivo. TXNDC5 mRNA and protein were increased in human CF (hCF) under TGFβ1 stimulation in vitro. Knockdown of TXNDC5 attenuated TGFβ1-induced hCF activation and ECM protein upregulation independent of SMAD3, whereas increasing expression of TXNDC5 triggered hCF activation and proliferation and increased ECM protein production. Further experiments showed that TXNDC5, a protein disulfide isomerase, facilitated ECM protein folding and that depletion of TXNDC5 led to ECM protein misfolding and degradation in CF. In addition, TXNDC5 promotes hCF activation and proliferation by enhancing JNK activity via increased reactive oxygen species, derived from NAD(P)H oxidase 4. TGFβ1-induced TXNDC5 upregulation in hCF was dependent on ER stress and activating transcription factor 6-mediated transcriptional control. Targeted disruption of Txndc5 in mice (Txndc5-/-) revealed protective effects against isoproterenol (ISO)-induced cardiac hypertrophy, reduced fibrosis (by ∼70%) and markedly improved LV function; post-ISO LV ejection fraction was 59.1±1.5 vs 40.1±2.5 (P<0.001) in Txndc5-/- vs wild-type mice, respectively.
Conclusions
The ER protein TXNDC5 promotes cardiac fibrosis by facilitating ECM protein folding and CF activation via redox-sensitive JNK signaling. Loss of TXNDC5 protects against β agonist-induced cardiac fibrosis and contractile dysfunction. Targeting TXNDC5, therefore, could be a powerful new therapeutic approach to mitigate excessive cardiac fibrosis, thereby improving cardiac function and outcomes in HF patients.
Keywords: TXNDC5, cardiac fibrosis, heart failure, ER, JNK, oxidative stress. Subject Terms: Basic Science Research, Fibrosis, Heart Failure, Mechanisms
Introduction
Heart failure (HF) is a major and growing public health problem, afflicting more than 23 million individuals worldwide.1 While advances in care and treatment options over the past few decades have reduced dramatically the mortality rates associated with cardiovascular diseases such as hypertensive heart disease, acute coronary syndrome, congenital and valvular heart diseases,2, 3 the incidence of HF and mortality rates in HF patients have improved only marginally.4-6 The prevalence of HF is on the rise, and the 5-year mortality remains at ∼50% for symptomatic HF, worse than that for many cancers.7 A complex blend of molecular, structural and neurohumoral alterations may account for the progressive and recalcitrant nature of HF,8, 9 and there is a clear and immediate need for novel therapies to improve the outcomes of HF patients.
In addition to cardiac muscle abnormalities, cardiac fibrosis is a key contributor to structural and functional remodeling in HF.10, 11 Excessive accumulation of extracellular matrix (ECM) resulting from cardiac fibrosis can increase wall stiffness, leading to diastolic dysfunction.12 Replacement of diseased/dying cardiac muscle cells with fibrotic tissue also contributes to worsened ventricular systolic function.10, 11 Beyond mechanical dysfunction, cardiac fibrosis also contributes to electrical abnormalities by slowing conduction through the myocardium.13 In addition, increased cardiac fibrosis promotes enhanced automaticity and early after depolarizations causing triggered activity.14 These electrical abnormalities associated with cardiac fibrosis can foster life-threatening arrhythmias. Cardiac fibrosis, therefore, represents an important therapeutic target for the management of contractile dysfunction and arrhythmias in HF.
The pathogenesis of cardiac fibrosis involves alterations in the cellular and neurohumoral environments that lead to changes in fibroblast activity and ECM turnover.15-17 The expression levels of local and circulating hormones of the renin-angiotensin-aldosterone system (RAAS), endothelin 1 and transforming growth factor-β1 (TGFβ1) increase in response to mechanical and metabolic stresses, triggering increased activation and proliferation of fibroblasts.15-17 Activated fibroblasts transition into α-smooth muscle actin (α-SMA)-expressing myofibroblasts, which are responsible for producing an excessive amount of ECM that characterizes fibrotic tissue.18 Myofibroblasts also modify ECM turnover by modulating the balance between matrix metalloproteinases (MMPs) and their natural inhibitors (tissue inhibitor of MMP; TIMPs) to promote fibrosis.19 The changes in the ECM also alter the signals that cardiomyocytes receive from their scaffolding environment, triggering the activation of gene programs that are associated with cardiac hypertrophy and contractile dysfunction.17
Currently, there are no clinical therapies directly targeting cardiac fibrosis. Antagonists of RAAS, such as angiotensin converting enzyme inhibitor, angiotensin II receptor and aldosterone receptor blockers, have been shown to improve ventricular function and slow progression of myocardial fibrosis.20-22 The use of these treatments, however, is limited by their hypotensive effects and their inability to stop fibrosis progression. Broad inhibitors of TGFβ such as pirfenidone and tranilast, on the other hand, have been shown to attenuate fibrosis by inhibiting fibroblast activation and ECM deposition without affecting blood pressure in experimental models.23-25 The widespread clinical use of these agents, however, is limited due to undesirable side effects, including liver toxicity.26 More recently, several groups have made significant progress in preclinical studies to mitigate cardiac fibrosis by targeting various fibrogenic molecules including TGFβ-activated kinase 1,27 p38,28 endothelin receptors,29, G protein-coupled receptor kinase 2,30 and miR-21.31 The clinical efficacies of these anti-fibrotic strategies, however, remain to be demonstrated. There is, therefore, a clear and pressing need to identify additional novel mediators of cardiac fibrosis presentation and progression in response to pathological stimuli to facilitate the development of alternative therapeutic strategies targeting cardiac fibrosis.
In the studies presented here, we identified thioredoxin domain containing 5 (TXNDC5), a cardiac fibroblast (CF)-enriched endoplasmic reticulum (ER) protein, as a novel mediator of cardiac fibrosis. TXNDC5 is upregulated in failing human and mouse hearts and TXNDC5 and fibrogenic protein expression levels are strongly correlated. Results presented here suggest that TXNDC5 promotes cardiac fibrosis by redox-dependent CF activation, as well as by enhancing ECM production by facilitating ECM protein folding. In addition, we show here that targeted deletion of Txndc5 protects against β agonist-induced cardiac fibrosis and LV dysfunction in mice. Taken together, these results indicate that modulating TXNDC5 could be a powerful new approach to reduce fibrosis and ECM accumulation, thereby improving cardiac function and outcomes in individuals suffering from HF.
Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
A detailed Methods section can be found in the Online Data Supplement.
Results
RNA sequencing and gene co-expression network analysis links TXNDC5 to cardiac fibrosis
To identify novel mediators of cardiac fibrosis, we performed gene co-expression network analysis on an RNA sequencing (RNASeq) dataset, obtained from human failing (HF, n=16) and non-failing (NF, n=8) left ventricles (LV) reported by our group previously (GSE46224).32 In line with advanced HF status, the expression levels of markers of HF, including atrial natriuretic factor (ANF/NPPA) and brain-type natriuretic peptide (BNP/NPPB), were significantly increased in the HF, compared to the NF, LV samples (Figure 1A). The expression levels of genes encoding ECM proteins, such as type I collagens α1 (COL1A1) and α2 (COL1A2), type III collagen α1 (COL3A1), elastin (ELN), and fibronectin (FN1), as well as CCN family member 2 (CCN2), profibrotic cytokines (TGFβ1/TGFB1, TGFβ2/TGFB2) and transcription factors (SMAD2 and SMAD7), were also significantly (P<0.05) upregulated in the HF LV samples (Figure 1A), consistent with the presence of extensive myocardial fibrosis in failing human hearts.
Figure 1. RNASeq and co-expression gene network analyses identified TXNDC5 as a potential novel mediator of cardiac fibrosis.
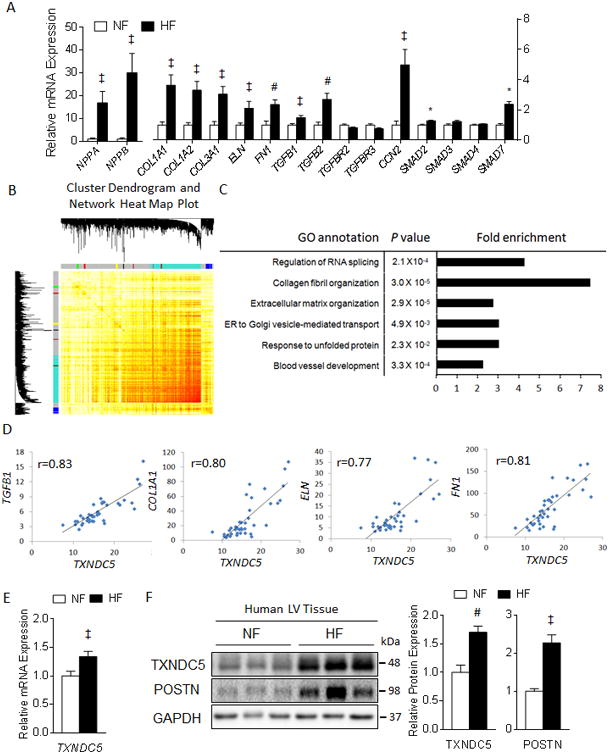
(A) RNASeq analysis revealed upregulation of fibrogenic genes (including ECM protein genes COL1A1, COL1A2, COL3A1, ELN and FN1, matricellular protein CCN2 and genes involved in TGFβ signaling such as TGFB1, TGFB2, SMAD2 and SMAD7) and HF markers (ANF/NPPA and BNP/NPPB) in human HF, compared to NF, LV. (B) Cluster dendrogram and network heat map plot of the 15 gene modules identified by Weighted Gene Co-expression Network Analysis (WGCNA) on the human LV RNASeq data. (C) Gene ontology analysis revealed that module Turquoise was enriched in genes that are involved in the pathogenesis of cardiac fibrosis. (D) TXNDC5 expression in human LV showed strong positive correlations with TGFβ1 and ECM genes including COL1A1, ELN and FN1. The transcript (E) and protein (F) expression levels of TXNDC5 were significantly upregulated in human HF, compared to NF, LV samples (‡P<0.05, #P<0.01, *P<0.001).
Pairwise correlations encompassing all detectable cardiac mRNA species in human LV samples were computed and a gene co-expression network was constructed using the Weighted Gene Co-expression Network Analysis (WGCNA).33 A total of 15 gene co-expression modules were identified and color-coded (Figure 1B). Gene ontology analysis revealed that module Turquoise, a clustered gene module containing 1071 genes and assigned with the turquoise color label (Supplemental Table I), was significantly enriched in genes that are involved in collagen fibril organization (7.5-fold enrichment, P=3.0 × 10-5) and extracellular matrix organization (2.8-fold enrichment, P=2.9 × 10-5) (Figure 1C), two biological processes that are critical for the development of cardiac fibrosis. Among the genes in module Turquoise, the expression level of the gene encoding the ER protein, thioredoxin domain containing 5 (TXNDC5), showed a strong positive correlation with those of genes encoding TGFβ1 (r=0.83) and various ECM proteins, including COL1A1 (r=0.80), ELN (r=0.77) and FN1 (r=0.81) (Figure 1D). In addition, TXNDC5 transcripts were significantly (P<0.05, Figure 1E) upregulated in human HF, compared with NF, LV samples. The protein expression levels of TXNDC5, as well as of periostin (POSTN), a matricellular protein secreted by activated CF,34, 35were also strongly upregulated in failing human LV (Figure 1F). Analysis of an RNASeq dataset (GSE35350) obtained from a mouse model of cardiac hypertrophy, induced by transverse aortic constriction (TAC),36 also revealed significant (P<0.01) upregulation of Txndc5 with cardiac hypertrophy and a strong positive correlation between the transcript expression levels of Txndc5 and fibrogenic factors, including Tgfb1, Postn, Acta2 (gene encoding α-SMA) and of genes encoding various ECM proteins, such as Col1a1, Fn1 and Eln (Supplemental Figure I). Taken together, the observed upregulation of TXNDC5/Txndc5 expression in fibrotic human and mouse hearts and the strong positive correlations with genes involved in cardiac fibrosis suggest a potential role for TXNDC5 in the pathogenesis of myocardial fibrosis during cardiac hypertrophy and heart failure.
TXNDC5 is enriched in cardiac fibroblasts and upregulated in response to TGFβ1
Expression analysis demonstrated that TXNDC5 was selectively enriched in primary human cardiac fibroblasts (hCF): mRNA and protein expression levels of TXNDC5 in hCF were significantly higher than those in human LV cardiomyocytes (hCM) by 5.9-and 11.1-fold, respectively (Figure 2A, B). Consistent with the observed enrichment of TXNDC5 in hCF, immunohistochemical staining of sections from a mouse model of isoproterenol (ISO)-induced HF and fibrosis (see below) revealed strong expression of TXNDC5 in myocardial interstitium, but not in myocytes, a pattern similar to that observed for other fibrogenic proteins, such as COL1A1 (Figure 2C), CCN2 and POSTN (Supplemental Figure IIA). Using a Col1a1-GFPTg (GFP driven by Col1a1 enhancer/promoter) transgenic mouse line that allows visualizing active cardiac fibroblasts with GFP,37 immunofluorescence staining showed that TXNDC5 staining is highly co-localized with GFP-positive, collagen-producing CF (Supplemental Figure IIB) in fibrotic mouse myocardium induced by ISO treatment.
Figure 2. TXNDC5 is highly enriched in cardiac fibroblasts and upregulated in response to TGFβ1 stimulation Transcript.
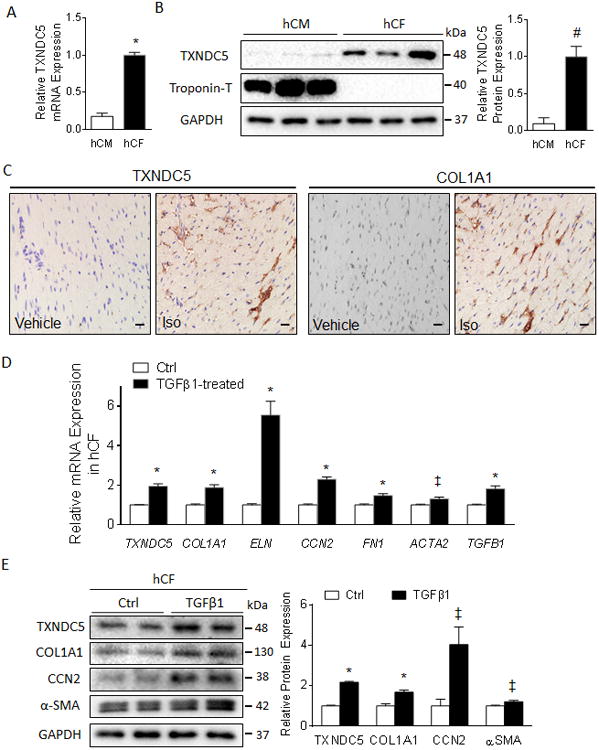
(A) and protein (B) expression analyses in isolated human cardiac fibroblasts (hCF) and cardiomyocytes (hCM) revealed strong enrichment of TXNDC5 in hCF compared to hCM. (C) Immunohistochemical staining of cardiac sections from a mouse model of isoproterenol (ISO)-induced heart failure showed strong staining of TXNDC5 in the myocardial intersitium but not in myocytes (left), similar to the distribution pattern of type 1 collagen (right) (Scale bar=20 μm). TGFβ1 treatment (4 ng/ml for 48 hours) in hCF induced significant upregulation of TXNDC5 mRNA (D) and protein (E) expression, as well as of various fibrogenic proteins including COL1A1, CCN2 and ACTA2/α-SMA, compared to vehicle-treated control cells (Ctrl) (‡P<0.05, #P<0.01, *P<0.001).
TGFβ1 expression is strongly upregulated in failing myocardium and contributes to fibrosis by promoting CF activation, proliferation and ECM production.38 As the expression levels of TXNDC5 and TGFβ1 are highly correlated in the myocardium, we hypothesized that TXNDC5 expression in hCF could be regulated by TGFβ signaling. Consistent with this hypothesis, TGFβ1 treatment (4 ng/ml for 48 hours) of hCF induced significant upregulation of the TXNDC5 mRNA (2.0-fold, P<0.001, Figure 2D) and protein (2.2-fold, P<0.001, Figure 2E) expression levels, compared to vehicle-treated control cells (Ctrl). TGFβ1-induced upregulation of TXNDC5 in hCF was accompanied by increased expression of several fibrogenic gene transcripts and proteins including COL1A1, ELN, CCN2 and ACTA2/α-SMA (Figure 2D,E). In addition, immunocytochemical staining revealed that TXNDC5 is strongly expressed in TGFβ1-activated/α-SMA expressing, but not in quiescent/α-SMA non-expressing, hCF (Supplemental Figure IIC).
TXNDC5 depletion attenuates TGFβ1-induced CF activation and proliferation, and the upregulation of ECM proteins
To determine whether TXNDC5 is requisite in the biological functions of CF, lentiviral vectors carrying TXNDC5/Txndc5-targeted shRNAs were utilized to knockdown TXNDC5/Txndc5 in human (hCF) and mouse (mCF) cardiac fibroblasts. Consistent with the pro-fibrotic effects of TGFβ signaling, TGFβ1 treatment increased the proliferative activity in non-targeted shRNA (shScr)-treated hCF (Figure 3A), as well as the transcript (Supplemental Figure IIIA) and protein expression levels of the ECM proteins COL1A1 and CCN2 and the myofibroblast marker αSMA (Figure 3B). Knockdown of TXNDC5 in hCF, however, eliminated TGFβ1-induced fibroblast activation and transformation into myofibroblasts, as evidenced by inhibited cellular proliferation (Figure 3A) and reduced αSMA expression (Figure 3B) in spite of TGFβ1 stimulation. Knockdown of TXNDC5 in hCF reduced expression of the ECM proteins COL1A1 and CCN2 at baseline and in response to TGFβ1 stimulation (Figure 3B), but had no effect on COL1A1 and CCN2 transcript expression levels (Supplemental Figure IIIA). Similar to the results in hCF, reduced ECM (COL1A1, ELN, CCN2) and α-SMA protein expression levels, as well as impaired responsiveness to TGFβ1 stimulation, were observed in mCF (Figure 3C) and in NIH-3T3 mouse fibroblasts (Supplemental Figure IIIB, C) with Txndc5 knockdown.
Figure 3. Knockdown of TXNDC5 prevented TGFβ1-induced CF activation and ECM protein upregulation.
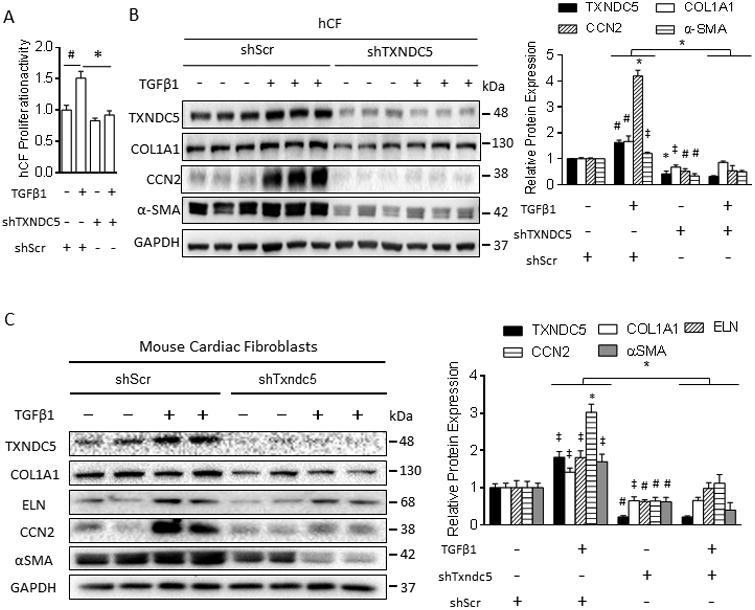
(A) TGFβ1 treatment increased the proliferation rate of control hCF (transduced with shScr); knockdown of TXNDC5 (shTXNDC5) abolished TGFβ1-induced cellular proliferation in hCF. (B) Knockdown of TXNDC5 in hCF prevented TGFβ1-induced upregulation of fibrogenic proteins including αSMA, COL1A1 and CCN2 (All significant symbols indicate comparisons to baseline shScr group without TGFβ1 treatment, except the symbol above the bars, which indicates the significant level of differences between groups of TGFβ1+shScr and TGFβ1+shTXNDC5). (C) Knockdown of Txndc5 in mouse cardiac fibroblasts (mCF) also diminished TGFβ1-induced upregulation of αSMA and ECM proteins COL1A1, ELN and CCN2 (n=6 in each group, ‡P<0.05, #P<0.01, *P<0.001).
TXNDC5 is essential for maintaining ECM protein stability in CF
To determine how TXNDC5 contributes to the regulation of ECM protein expression in CF, a cycloheximide protein chase assay was conducted to evaluate the stability of ECM proteins in hCF with and without TXNDC5 knockdown. As shown in Figure 4A and Supplemental Figure IVA, TXNDC5 knockdown in hCF led to accelerated degradation of ECM proteins COL1A1, ELN and CCN2, compared to shScr-treated cells. This observation suggests a critical role for TXNDC5 in maintaining the stability of ECM proteins in CF.
Figure 4. Knockdown of TXNDC5 in hCF led to accelerated ECM protein degradation owing to ECM protein misfolding and subsequent removal through ER-associated protein degradation (ERAD).
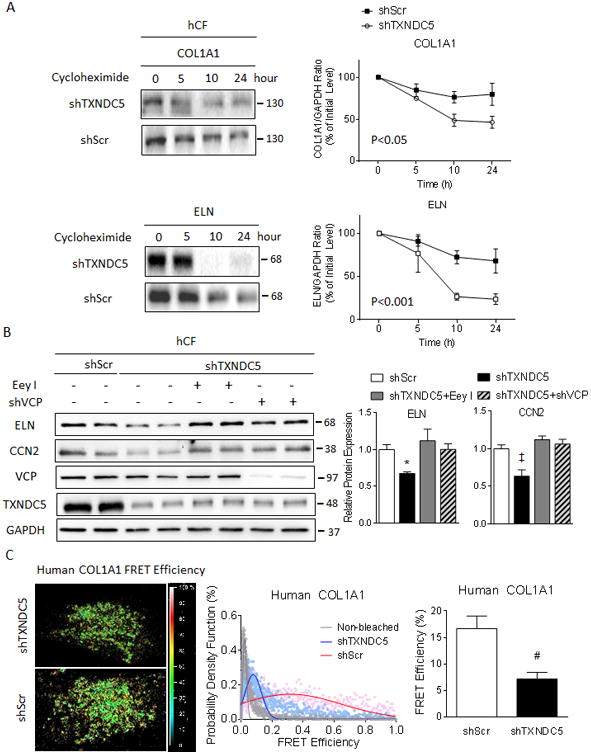
(A) A cycloheximide protein chase assay revealed accelerated degradation of COL1A1 and ELN proteins in hCF with TXNDC5 knockdown (shTXNDC5-transduced), compared to control (shScr-transduced) cells. (B) ERAD inhibitor Eey I or shVCP treatment in hCF reversed the reduction in ELN and CCN2 protein expression resulting from knockdown of TXNDC5. (C) A fluorescence resonance energy transfer (FRET)-based protein folding assay using a dual fluorescence-labeled COL1A1 construct in hCF showed significantly reduced COL1A1 FRET efficiency in cells with knockdown of TXNDC5 (shTXNDC5, n=10), compared to scrambled control (shScr, n=10), indicating reduced COL1A1 folding with TXNDC5 depletion (‡P<0.05, #P<0.01, *P<0.001).
TXNDC5 is an ER resident protein with thioredoxin activity known to catalyze protein disulfide bond isomeration,39 suggesting that TXNDC5 might facilitate protein folding in the ER. It has previously been demonstrated that nascent polypeptides destined for the secretory pathway fold in the ER, facilitated by ER chaperons and protein disulfideisomerases,40 whereas misfolded proteins are subjected to ER-associated protein degradation (ERAD) via the ubiquitin-proteasome system.41 We hypothesized that TXNDC5 functions to facilitate the folding of ECM proteins in the ER, allowing ECM proteins to mature and subsequently travel through the secretory pathway. Depletion of TXDNC5, therefore, may cause ECM protein misfolding and subsequent degradation by ERAD. To test this hypothesis directly, we first treated hCF with an ERAD inhibitor eeyarestatin I (Eey I, which blocks ERAD by inhibiting valosin containing protein [VCP], an essential component of the protein complex mediating the retrotranslocation of misfolded substrates from ER to cytosol, 0.5 μmol/L) or shRNA targeting VCP (shVCP) to block ERAD. As shown in Figure 4B, both Eey I treatment and VCP knockdown by shRNA (Supplemental Figure IVB) reversed the reduction of ELN and CCN2 protein caused by TXNDC5 depletion in hCF. These results suggest that ECM protein degradation resulting from TXNDC5 depletion in hCF is mediated through ERAD.
Additional co-immunoprecipitation experiments demonstrated that TXNDC5 associates with several ECM proteins, including COL1A1, CCN2 and ELN in hCF (Supplemental Figure IVC). In addition, the extent of ECM protein folding in control and TXNDC5-depleted hCF was examined using a fluorescence resonance energy transfer (FRET)-based protein folding assay that has been shown to detect protein folding and unfolding in cells successfuly.42 Human COL1A1 fused with cyan fluorescent protein (CFP) at the N terminus and yellow fluorescent protein (YFP) at the C terminus (CFP-COL1A1-YFP) was expressed in control and TXNDC5-depleted hCF; efficient FRET (from CFP to YFP) would only be detected when the COL1A1 ternary fusion proteins fold properly and bring CFP and YFP into close proximity (illustrated in Supplemental Figure IVD, bottom). Application of a standard acceptor photobleaching FRET protocol in hCF expressing CFP-COL1A1-YFP revealed that TXNDC5 knockdown significantly (P<0.01) reduced FRET efficiency (Figure 4C), suggesting decreased COL1A1 protein folding in hCF with TXNDC5 depletion. A similar reduction in the folding of mouse ELN protein was observed in NIH-3T3 mouse fibroblasts with TXNDC5 depletion (Supplemental Figure V). Taken together, these results suggest that TXNDC5 functions to maintain the proper folding of ECM proteins, such as collagen and elastin, in CF. When TXNDC5 expression is reduced, ECM protein unfolding and breakdown are increased via an ER-associated degradation pathway.
TXNDC5 activates fibroblasts through SMAD-independent, redox-sensitive regulation of JNK signaling
The observations that TXNDC5 depletion in mCF and hCF attenuated αSMA expression and cellular proliferation in response to TGFβ1 stimulation suggest that TXNDC5 is required for TGFβ1-induced CF activation and transdifferentiation into myofibroblasts. To determine whether elevated TXNDC5 expression is sufficient to trigger CF activation, proliferation and ECM production, we overexpressed TXNDC5 in hCF. These experiments revealed that ectopic overexpression of TXNDC5 resulted in increased protein expression levels of COL1A1, αSMA and POSTN (Figure 5A), secretion of type 1 collagen (Figure 5B), as well as cellular proliferation in hCF (Figure 6B).
Figure 5. TXNDC5-dependent activation of hCF is associated with increased JNK, but not SMAD3 or ERK, activity.
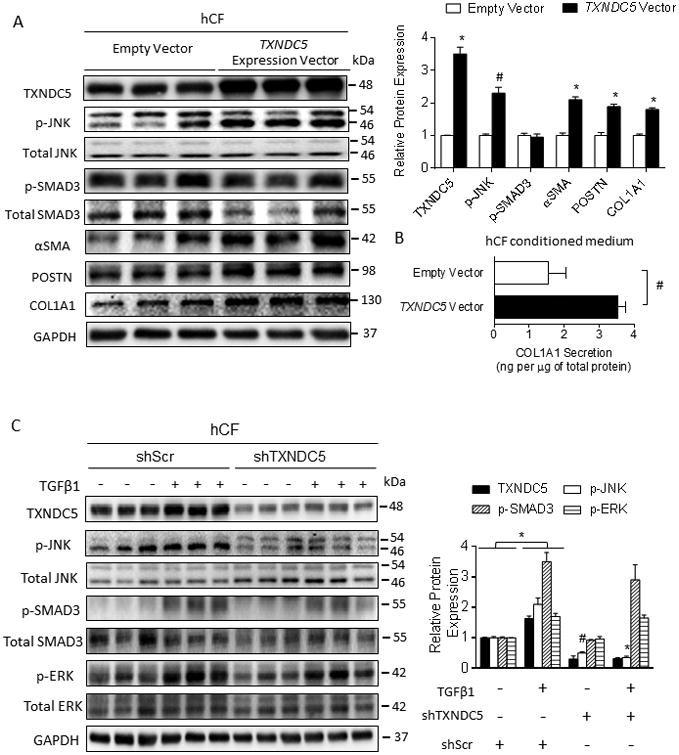
(A) Overexpression of TXNDC5 in hCF was sufficient to trigger hCF activation (as reflected in increased αSMA and POSTN protein levels), and COL1A1 production. Phosphorylation of SMAD3 was not affected by ectopic TXNDC5 expression. (B) Forced expression of TXNDC5 led to significantly increased secretion of type 1 collagen (COL1A1) from hCF. (C) Knockdown of TXNDC5 in hCF abrogated TGFβ1-induced phosphorylation of JNK, but not of SMAD3 or ERK. Phosphorylated JNK, SMAD3 and ERK were expressed relative to total JNK, SMAD3 and ERK, respectively (#P<0.01, *P<0.001).
Figure 6. TXNDC5-mediated activation and proliferation of hCF require JNK activity triggered by NOX-derived ROS.
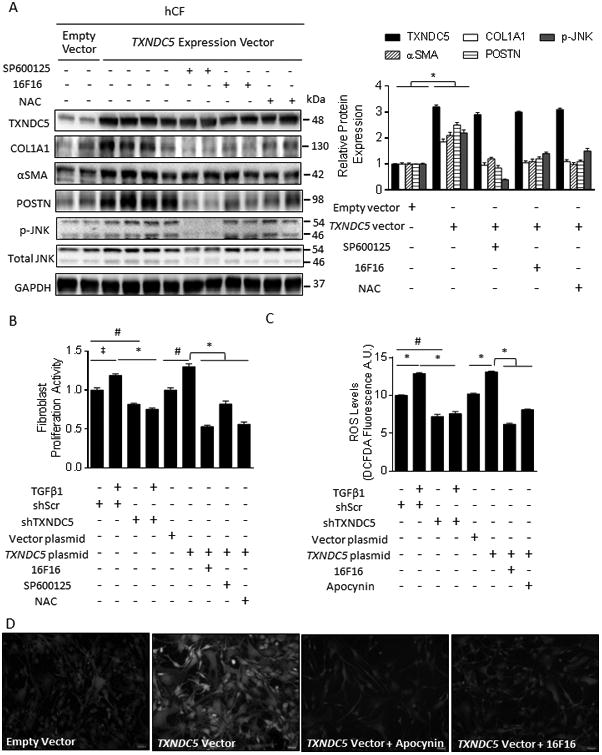
(A) Treatment with a JNK inhibitor (SP600125, 10 μmol/L), PDI inhibitor (16F16, 20 μmol/L) or ROS scavenger N-acetylcysteine (NAC, 15 mmol/L) abolished upregulation of αSMA and POSTN (markers for hCF activation and myofibroblast formation) and COL1A1 induced by ectopic TXNDC5 expression in hCF. Note that 16F16 and NAC treatment also reduced JNK phosphorylation resulting from TXNDC5 overexpression. (B) Knockdown of TXNDC5 abolished TGFβ1-induced hCF proliferation, whereas overexpression of TXNDC5 was sufficient to induce hCF proliferation. Pharmacological inhibition of JNK (with SP600125), PDI (with 16F16) or ROS (with NAC) abrogated hCF proliferation induced by TXNDC5 overexpression. (C) Knockdown of TXNDC5 abrogated TGFβ1-induced ROS elevation in hCF, whereas TXNDC5 overexpression was sufficient to increase ROS levels, which could be abolished by the PDI inhibitor, 16F16, or the NOX inhibitor, apocynin (2 mmol/L). (D) Representative photomicrographs illustrating increased ROS levels (measured using DCFDA fluorescence signal intensity) in hCF with ectopic TXNDC5 expression, which is diminished by the NOX inhibitor apocynin and by the PDI inhibitor, 16F16 (‡P<0.05, #P<0.01, *P<0.001).
Because TXNDC5 is required for TGFβ1-induced CF activation (Figure 3), we hypothesized that TXNDC5 may regulate CF activity through effectors downstream of the TGFβ signaling pathway. Consistent with this hypothesis, we observed that knockdown of TXNDC5 in hCF reduced TGFβ1-induced phosphorylation/activation of c-Jun N-terminal kinase (JNK, Figure 5C), a critical TGFβ downstream mediator that promotes fibroblast activity.43, 44 In contrast, TGFβ1-induced phosphorylation of SMAD3 and ERK was not affected by knockdown of TXNDC5 (Figure 5C). Also consistent with the results of TXNDC5 knockdown, TXNDC5 overexpression in hCF was sufficient to trigger the activation of JNK, but not SMAD3 (Figure 5A). Moreover, pharmacological inhibition of JNK with SP600125 (10 μmol/L) abolished the upregulation of COL1A1, αSMA and POSTN (Figure 6A), as well as hCF proliferation (Figure 6B), observed with ectopic TXNDC5 expression. These data collectively demonstrate that TXNDC5 promotes the activity and proliferation of hCF through JNK signaling.
As JNK is redox-sensitive and TGFβ1 is known to promote ROS formation in fibroblasts through increased NAD(P)H oxidase (NOX) activity,45 we hypothesized that TXNDC5 modulates JNK activity by regulating intracellular ROS levels. Consistent with this hypothesis, knockdown of TXNDC5 reduced TGFβ1-induced ROS production, whereas overexpression of TXNDC5 increased ROS levels in hCF (Figure 6C, D). Notably, the increased JNK activity, α-SMA/POSTN expression (Figure 6A) and cellular proliferation (Figure 6B) in hCF following ectopic TXNDC5 expression were abolished by the treatment with ROS scavenger N-acetylcysteine (NAC, 15 mmol/L). In addition, ROS production induced by TXNDC5 overexpression was suppressed by the NOX inhibitor apocynin (2 mmol/L, Figure 6C, D), demonstrating that TXNDC5-induced ROS production is derived from NOX. Because NOX4 and 5 are the predominant NOX enzymes in CFs and only NOX4 is upregulated by TGFβ1,45 we hypothesized that NOX4 is responsible for TXNDC5-induced ROS production in CFs. Consistent with this hypothesis, treatment with NOX1/4 inhibitor GSK137831 (5μmol/L) or siRNA targeting NOX4 (siNOX4) abolished TXNDC5 overexpression-induced ROS production, cellular proliferation (Supplemental Figure VI A-C) and ECM protein upregulation in hCF (Supplemental Figure VID). Taken together, these data suggest that NOX4-derived ROS productionn is essential for TXNDC5-induced JNK activation, cellular proliferation and ECM expression in hCF.
The activity of NOX is known to be modulated by protein disulfide isomerases (PDIs),46, 47 suggesting that TXNDC5 may induce NOX4-derived ROS generation via its PDI activity. In agreement with this hypothesis, treatment of hCF with a PDI inhibitor 16F16 (20 μmol/L) prevented the increase in ROS produced by TXNDC5 overexpression (Figure 6C, D). Activation of hCF resulting from TXNDC5 overexpression was also significantly reduced by 16F16, as shown by reduced αSMA and POSTN protein expression (Figure 6A) and decreased proliferation (Figure 6B). To determine the requirement for the PDI activity of TXNDC5 in its pro-oxidant and fibrogenic functions directly, we generated a TXNDC5 mutant that lacks PDI activity by introducing cysteine-to-alanine mutations in both ends (i.e. CGHC to AGHA) of each of its 3 thioredoxin domains, which are required for its PDI enzyme activity (TXNDC5 AAA mutant; the mutation scheme is illustrated in Supplemental Figure VIIA). In contrast to WT TXNDC5, the TXNDC5 AAA mutant failed to induce ROS production, cell proliferation (Supplemental Figure VIA, B), fibroblast activation (reflected by αSMA and POSTN expression levels) and/or ECM protein expression (Supplemental Figure VIIB) in hCF, suggesting that the PDI activity of TXNDC5 mediates the observed pro-oxidant and fibrogenic effects.
TGFβ1-induced TXNDC5 upregulation in CFs Is dependent on increased ER stress and activating transcription factor 6 (ATF6)
Additional experiments were focused on determining how TXNDC5 is regulated by TGFβ signaling. Because TXNDC5 mRNA was upregulated in response to TGFβ1 stimulation, we hypothesized that the promoter activity of TXNDC5 is regulated by the transcription factor(s) downstream of the TGFβ signaling pathway. Although SMAD proteins play critical roles in controlling the transcription of various TGFβ downstream genes, neither the human nor the mouse TXNDC5/Txndc5 promoter harbors a SMAD binding element, indicating that TXNDC5/Txndc5 could either be regulated indirectly by SMADs or by a SMAD-independent TGFβ pathway. TGFβ, for example, is known to trigger increased ER stress and unfolded protein response (UPR),48, 49 which are mediated by multiple transcription factors including X-box binding protein 1 (XBP1), activating transcription factor 4 and 6 (ATF6). In addition, TXNDC5 is located in the ER and facilitates protein folding (Figure 4 and Supplemental Figure V), suggesting that TXNDC5 may function downstream of ER stress pathways. Consistent with this hypothesis, both the human and the mouse TXNDC5/Txndc5 promoters contain putative ATF6 and XBP1 binding sites.39 In addition, increased ER stress level was observed in human failing heart and in fibrotic mouse myocardium, induced by ISO-treatment (Supplemental Figure VIIIA-C).
To test this hypothesis directly, we first examined the interrelationship between TGFβ signaling, ER stress and TXNDC5 expression in hCF. As shown in Supplemental Figure IX, TGFβ1 treatment markedly increased ER stress signaling activity, especially in the branches of ATF6 and inositol-requiring enzyme 1 α (IRE1α)-XBP1, and significantly upregulated the protein expression levels of TXNDC5 in hCF. Treating hCF with broad ER stress inhibitors 4-phenylbutyrate (4-PBA, 0.5 mmol/L) or tauroursodeoxycholic acid (TUDCA, 0.5 mmol/L) suppressed TGFβ1-induced ER stress, as evidenced by reduced expression level of activated ATF6 (ATF-p50) (Figure 7A), and abolished TGFβ1-induced upregulation of TXNDC5 protein (Figure 7A). These results suggest that TGFβ1-induced upregulation of TXNDC5 in hCF is mediated by ER stress.
Figure 7. TGFβ1 induces TXNDC5 expression in CF through TGFβ1-ER stress-ATF6 signaling axis.
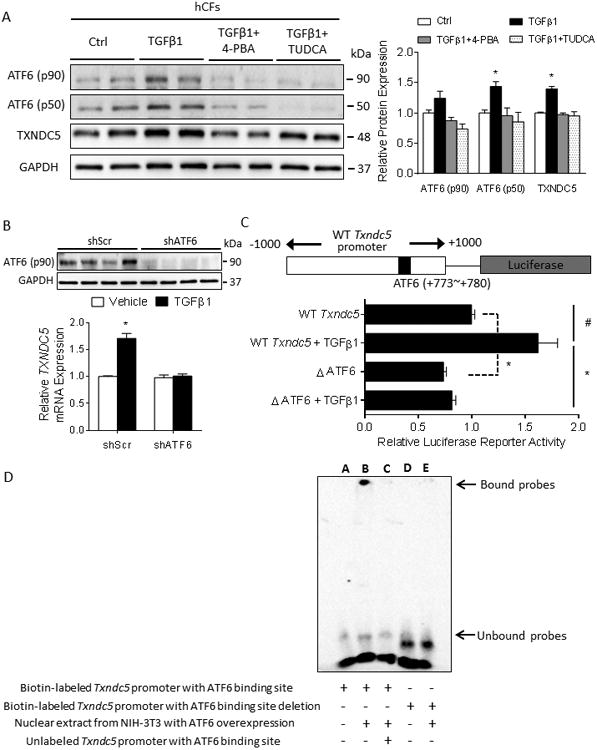
(A) TGFβ1 treatment in hCF increased ER stress (as evidenced by the upregulated ER stress markers ATF6 p50 and p90) and TXNDC5 protein expression levels, which could be abrogated by the treatment with general ER stress inhibitors 4-phenylbutyrate (4-PBA, 0.5 mmol/L) or tauroursodeoxycholic acid (TUDCA, 0.5 mmol/L) (B) Knockdown of ATF6 in hCF prevented the upregulation of TXNDC5 mRNA in response to TGFβ1 stimulation. (C) Schematic illustration of the mouse Txndc5 promoter luciferase reporter construct, which contains an ATF6 binding site (TGACGTGG, +773∼+780). Deletion of the ATF6 binding site significantly reduced TGFβ1-induced transcriptional activity of the Txndc5 promoter in the absence or presence of TGFβ1. (D) Electrophoretic mobility shift assay showed biotin-labeled Txndc5 promoter probe containing ATF6 binding site was shifted (lane B) when treated with nuclear extract from NIH-3T3 fibroblasts with ectopic ATF6 expression. Unlabeled Txndc5 promoter DNA was used as competitor (lane C) and revealed the specificity of ATF6 binding to Txndc5 promoter. Biotin-labeled Txndc5 promoter probe with ATF6 binding site deletion failed to interact with ATF6 (lane D, E).(#P<0.01, *P<0.001).
To determine ifTGFβ signaling regulates TXNDC5 expression through ATF6 and/or XBP1, two ER stress pathway transcription factors with putative binding sites in the TXNDC5/Txndc5 promoter, hCF with ATF6 or XBP1 knockdown were treated with TGFβ1, followed by assays to quantify TXNDC5 mRNA expression. These experiments showed that knockdown of ATF6 (Figure 7B), but not XBP1 (Supplemental Figure XA), prevented the upregulation of TXNDC5 mRNA with TGFβ1 stimulation. In further experiments, wild-type (WT Txndc5) and ATF6 binding site (TGACGTGG, +773∼+780)-deleted (ΔATF6) mouse Txndc5 promoter luciferase constructs (Figure 7C) were transfected into NIH-3T3 fibroblasts. Although TGFβ1 treatment significantly increased WT Txndc5 promoter activity, deletion of the ATF6 binding site significantly reduced Txndc5 promoter activity at baseline and in response to TGFβ1 stimulation (Figure 7C). A physical interaction between ATF6 and the Txndc5 promoter was confirmed using an electrophoretic mobility shift assay (Figure 7D and Supplemental Figure XB). Taken together, these data demonstrate that TGFβ1-induced TXNDC5 upregulation in CF is dependent on increased ER stress and ATF6-mediated transcriptional regulation.
In vivo targeted deletion of Txndc5 protects against β agonist-induced cardiac fibrosis and myocardial dysfunction
To explore directly the physiological role of TXNDC5 in cardiac functioning in vivo, we generated a mouse line with targeted deletion of Txndc5 (Txndc5-/-) using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) genome editing technology (details provided in the Supplemental Methods).50 Txndc5-/- mice, which harbor a 6.8 kb deletion encompassing exons 2 & 3 of Txndc5 (Supplemental Figure XIA), were born at the expected Mendelian frequency and, at baseline, showed no detectable developmental defects or structural anomalies. There were no discernable differences in Txndc5-/- and WT mouse heart sizes or contractile function at baseline (Figure 8 A,B and Supplemental Figure XIB).
Figure 8. Targeted deletion of Txndc5 protects against isoproterenol-induced cardiac hypertrophy, fibrosis and contractile dysfunction.
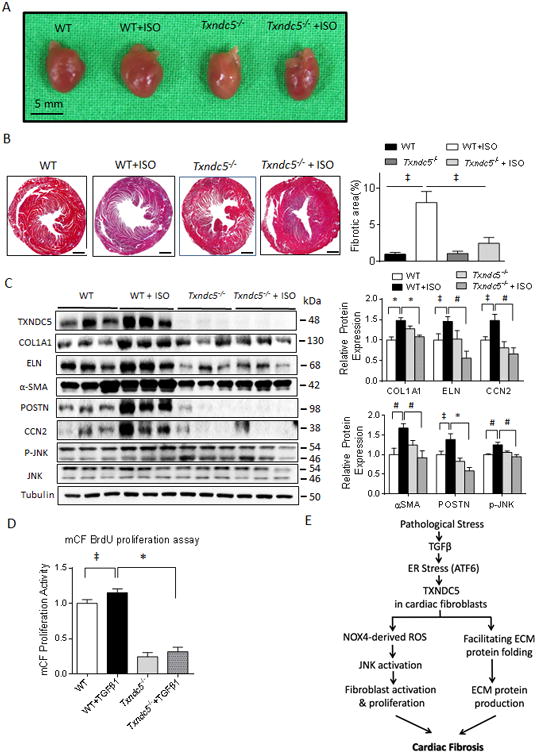
(A) Isoproterenol (ISO, 30 mg/kg/day subcutaneously for 10 days) injection led to marked increase in HW/BW ratio, an indicator of cardiac hypertrophy, in WT, but not in Txndc5-/-, mice. (B) Knockout of Txndc5 attenuated the extent of myocardial fibrosis induced by ISO injection. (C) ISO treatment led to significantly increased fibrogenic proteins (including COL1A1, ELN, CCN2, αSMA, POSTN) and p-JNK expression in WT, but not in Txndc5-/-, mouse LV. (D) Loss of Txndc5 also significantly reduced the proliferation capacity of mCF at baseline and in response to TGFβ1 stimulation. (E) Schematic summary of the proposed profibrotic mechanisms by which TXNDC5 contributes to cardiac fibrosis.(‡P<0.05, #P<0.001, *P<0.001).
To investigate the in vivo contribution of TXNDC5 to cardiac fibrosis, WT and Txndc5-/-mice were injected with isoproterenol (ISO, 30mg/kg/day subcutaneously for 10 days), a treatment paradigm shown previously to produce cardiac fibrosis, contractile dysfunction and heart failure.51, 52 As shown in Figure 8A, ISO-treatment induced significant increases in the mean ± SEM cardiac size and heart weight/body weight (HW/BW) ratio in WT mice (4.70±0.08 vs 5.75±0.07 mg/g in WT vs WT+ISO, respectively, P<0.001), but not in Txndc5-/- mice (4.60±0.31 vs 4.41±0.22 mg/g in Txndc5-/- vs Txndc5-/- +ISO, respectively, Figure 8A). In addition, Masson's trichrome staining of paraffin sections (Figure 8B) revealed that ISO-treatment was associated with extensive fibrosis in WT hearts (1.0±0.3% vs 8.1±1.5% of LV cross-sectional area in WT vs WT+ISO, respectively, P<0.05), whereas minimal fibrotic changes were observed in Txndc5-/- hearts in response to ISO (1.1±0.3% vs 2.5±0.8% of LV cross-sectional area in Txndc5-/- vs Txndc5-/- +ISO, respectively). Echocardiographic analyses (Supplemental Figure XIB) revealed significantly reduced LV contractility in post-ISO WT mice (LV ejection fraction [LVEF] 59.1±1.5 vs 40.1±2.5 % in WT vs WT+ISO, respectively, P<0.001), whereas LV contractile function was preserved in Txndc5-/- mice treated with ISO (LVEF 57.6±5.2 vs 59.0±4.1 % in Txndc5-/- vs Txndc5-/- +ISO, respectively). In addition, immunoblot analyses showed that ISO treatment significantly increased the expression levels of ECM proteins (COL1A1 and ELN), matricellular proteins CCN1 and CCN2 (but not CCN3),53 markers for CF activation (αSMA and POSTN), as well as p-JNK in WT, but not in Txndc5-/-, LV (Figure 8C and Supplemental Figure XII). Consistent with the notion that TXNDC5 plays a critical role in regulating CF activity, mCF isolated from Txndc5-/- mouse ventricles showed reduced proliferative activity compared with WT mCF, both at baseline and in response to TGFβ1 stimulation (Figure 8D). These results suggest that, in addition to preventing TGFβ1-induced CF activation and proliferation, loss of TXNDC5 protects the heart against ISO-induced cardiac hypertrophy, myocardial fibrosis and contractile dysfunction in vivo.
Discussion
Regulation of myocardial fibrosis by TXNDC5
The results presented here demonstrate that TXNDC5, a fibroblast-enriched ER protein, is upregulated in failing human LV and mouse heart with pressure overload-induced pathological cardiac hypertrophy, in parallel with advanced fibrosis and increased TGFβ signaling activity. The upregulation of TXNDC5 promotes cardiac fibrosis by facilitating ECM protein folding in CF, and by triggering CF activation and proliferation. TXNDC5-induced CF activation is mediated by SMAD3-independent activation of JNK via NOX4-derived ROS that requires the protein disulfide isomerase activity of TXNDC5. In addition, TGFβ1-induced upregulation of TXNDC5 in fibroblasts requires increased ER stress and ATF6-mediated transcriptional control. Consistent with the in vivo unbiased profiling results and in vitro mechanistic experiments, targeted deletion of Txndc5 mitigated β-agonist-induced cardiac fibrosis, LV remodeling and contractile dysfunction in mice. Taken together, the results presented elucidate a novel pro-fibrotic mechanism mediated by the CF-enriched ER protein TXNDC5. A schematic illustrating the regulation and function of TXNDC5 in modulating cardiac fibrosis is presented in Figure 8E.
TXNDC5 as a unique fibroblast-enriched PDI that regulates ECM turnover and CF activation
TXNDC5 is a recently discovered member of the PDI family, which facilitates the formation of disulfide bonds and correct folding of nascent polypeptides.54, 55 Among the 21 members of human PDI family genes, 16 were identified by RNASeq in human LV and 3 of these were dysregulated in HF (Supplemental Figure XIIIA).56 TXNDC5 was the only one that is highly enriched in CF among these 3 HF-associated PDI genes (Supplemental Figure XIIIB), suggesting its fibroblast-specific function that distinguishes TXNDC5 from other human PDIs. TXNDC5 contains an N-terminal signal sequence, followed by 3 thioredoxin (TRX) domains and a C-terminal ER retention signal (KDEL). It has been reported that the molecular architecture of TXNDC5 is radically different from other PDIs, such that the three TRX domains of TXNDC5 are separated, act independently and engage in rapid but promiscuous disulfide bond formation during early oxidative protein folding.57 The significance of such unique molecular properties of TXNDC5 in regulating fibroblast function, however, remains to be determined.
The observation that TXNDC5 could affect ECM protein expression through modulating protein folding suggests a previously unrecognized intracellular, post-translational mechanism that regulates ECM protein turnover, which is distinct from that of extracellular ECM regulators like MMPs, TIMPs and lysyl oxidase. As TXNDC5 expression is increased in CF with ER stress, a stimulus known to promote tissue fibrosis,48,58 it is likely that TXNDC5 functions as a component of the ER stress/UPR pathway and contributes to the profibrotic effects of elevated ER stress. The absence of TXNDC5 did not lead to developmental defects or pathological changes that are linked to excessive ER stress, which could be explained by the functional redundancy among the PDI family genes and the fact that TXNDC5 expression is restricted to certain cell types such as fibroblasts and endothelial cells. Consistent with this suggestion, TXNDC5 depletion in hCF did not lead to increased cellular apoptosis (Supplemental Figure XIV).
In addition to the canonical function of TXNDC5 as a PDI to facilitate ECM protein folding, the results presented here suggests that TXNDC5 also promotes the activation of CF, triggering fibroblast-to-myofibroblast differentiation and proliferation by increasing NOX4-derived ROS and activation of redox-sensitive JNK signaling. PDI has previously been demonstrated to promote ROS production in vascular smooth muscle cells (VSMC) by interacting with NOX enzymes including NOX1, 2, and 4, as well as by inducing the transcription of NOX1 and 4.59, 60 In contrast to VSMC, our results suggest that this non-canonical, ROS producing effect of TXNDC5 in CF is mediated through NOX4. While NOX1 and NOX2 are localized predominantly in the plasma membrane and endosomes,61 NOX4 has been identified in various intracellular compartments including mitochondria62, focal adhesions63 and ER.64, 65 It is possible that increased TXNDC5 in the ER promotes NOX4 activity directly by stabilizing NOX4 conformation or by facilitating the intracellular trafficking of NOX4 via its function as a PDI.66 Alternatively, TXNDC5 could raise NOX4-derived ROS levels indirectly by increasing NOX4 expression levels in the CF. ER ROS levels, therefore, are increased with overexpression of TXNDC5, which can further lead to elevated cytosolic ROS levels by triggering ROS production from mitochondria and/or depletion of cytosolic antioxidant glutathione,67 thereby resulting in the activation of redox-sensitive JNK signaling in the cytosol. Although the results presented cannot exclude the possibility that TXNDC5 could also promote ROS production via NOX1 or NOX2, the extremely low expression levels of NOX1/2 observed in CF45 and the fact that NOX1/2 are absent in the ER61 make this alternative hypothesis much less likely.
Targeted deletion of Txndc5 ameliorates β agonist-induced myocardial fibrosis
The results presented also demonstrate that knockout of Txndc5 has protective effects against β agonist-induced myocardial fibrosis, hypertrophy and contractile dysfunction. As a global, rather than fibroblast-specific, targeted deletion strategy was used, we cannot exclude the possibility that deletion of Txndc5 in cardiac cell types other than CF could have contributed to such cardiac protective effects. The protein expression levels of TXNDC5 in hCM, however, is less than 10% of that in hCF (Figure 2B), suggesting a minor, if any, role of TXNDC5 in modulating cardiac myocyte function directly. Although endocardial, epicardial and bone marrow-derived cells have been suggested as sources of activated cardiac myofibroblasts to varying degrees in mice subjected to cardiac injuries, pre-existing resident CF lineages are considered to be the predominant source of cardiac myofibroblasts that undergo proliferation and ECM production in the diseased heart.68, 69 We would expect, therefore, to observe similar protective effects against agonist-induced myocardial fibrosis if a fibroblast-specific Txndc5 targeted deletion mouse line were to be used.
Conclusions
In summary, the present study demonstrates a critical role for the CF-enriched ER protein TXNDC5 in promoting cardiac fibrosis by facilitating ECM protein folding as well as by triggering CF activation and proliferation via JNK signaling that is dependent on NOX4-derived ROS. Cardiac TXNDC5 expression increases in hypertrophic and failing hearts, likely under the control of a TGFβ-ER stress-ATF6 signaling axis, contributing to excessive accumulation of myofibroblasts and ECM proteins that lead to cardiac fibrosis. Our findings reveal a novel SMAD-independent mechanism that mediates a TGFβ-induced fibrogenic response in the heart. These results also suggest that targeting TXNDC5 could be a powerful novel approach to mitigate cardiac fibrosis and dysfunction.
Supplementary Material
Novelty and Significance.
What Is Known?
Cardiac fibrosis, a process that results from the activation and proliferation of cardiac fibroblasts (CF) and the secretion of excessive amounts of extracellular matrix (ECM) proteins, including type I collagen and elastin, plays a critical role in the pathogenesis of heart failure (HF).
Current therapeutic options for controlling cardiac fibrosis are limited and there is a clear and urgent need to identify novel mechanisms contributing to cardiac fibrosis to facilitate the development of potent, new therapeutics.
What New Information Does This Article Contribute?
Using co-expression gene network analysis on RNA sequencing data obtained from failing human heart, we identified thioredoxin domain containing 5 (TXNDC5), an endoplasmic reticulum (ER) protein enriched in CF, as a potential novel mediator of cardiac fibrosis.
In vitro experiments revealed that TXNDC5, downstream of TGFβ1 signaling and ER stress pathway, promotes cardiac fibrosis by facilitating the folding of ECM proteins, as well as by triggering the activation and proliferation of CF through enhancing JNK signaling via increased reactive oxygen species.
Targeted deletion of Txndc5 in mice showed protective effects against isoproterenol-induced cardiac fibrosis, hypertrophy and contractile dysfunction.
Cardiac fibrosis contributes significantly to the pathogenesis of HF by increasing the activation/proliferation of CF, resulting in excessive production/deposition of ECM proteins. Currently, there are no approved clinical therapies directly targeting cardiac fibrosis. It is essential to identify novel mediators of cardiac fibrosis to develop new therapeutic strategies targeting cardiac fibrosis. Exploiting co-expression gene network analysis on RNA sequencing data from failing human heart, we identified TXNDC5, a CF-enriched ER protein with the enzymatic activity of a protein disulfide isomerase (PDI), as a previously unrecognized critical mediator of cardiac fibrosis. Using multiple genetic, molecular and cellular methods in vitro and in vivo, we have demonstrated that TXNDC5 is a central player in a novel profibrotic pathway involving increased ECM protein folding and CF activation triggered by redox-sensitive JNK signaling, downstream of TGFβ1-induced ER stress and ATF6-mediated transcriptional control. We have shown here, for the first time, that TXNDC5, as a PDI, contributes to cardiac fibrosis by modulating the turnover of ECM proteins and CF activity. In addition to uncovering a novel molecular mechanism that is critical for cardiac fibrosis, the results presented here also demonstrate the potential of targeting TXNDC5 as a powerful new approach to treat cardiac fibrosis and heart failure.
Acknowledgments
We thank the staff of the Biomedical Resource Core at the First Core Labs, National Taiwan University College of Medicine, for technical assistance. We also thank the technical services provided by the Transgenic Mouse Model Core Facility for Biopharmaceuticals, Ministry of Science and Technology, Taiwan, the Gene Knockout Mouse Core Laboratory of National Taiwan University Center of Genomic Medicine, the National RNAi Core Facility at Academia Sinica, Taiwan, and the Translational Cardiovascular Biobank and Repository at Washington University School of Medicine, supported by the Washington University Institute for Clinical and Translational Sciences (ICTS), recipient of a Clinical and Translational Sciences Award (UL1 RR024992) from the National Institutes of Health (NIH) National Center for Research Resources, the Barnes-Jewish Hospital Foundation and the Richard J. Wilkinson Trust. We thank Dr. David Brenner for providing the Col1a1-GFPTg mice.
Sources o Funding: This work was supported by the Taiwan Ministry of Science Technology (Grants 103-2320-B-002-068-MY2, 105-2628-B-002 -042-MY4to KCY), a Taiwan National Health Research Institute Career Development Grant (NHRI-EX104-10418SC to KCY), grants from National Taiwan University Hospital (NTUH.106-P02, 105-CGN01, UN106-026, 106-N3740 and VN106-12 to KCY) and the National Institutes of Health (R01 HL034161 to JMN, HL136765 and HL138223 to YF and UL1 RR024992 to the ICTS).
Nonstandard Abbreviations and Acronyms
- 4-PBA
4-phenylbutyrate
- ANF
atrial natriuretic factor
- α-SMA
α-smooth muscle actin
- ATF6
activating transcription factor 6
- BNP
brain-type natriuretic peptide
- CCN 1
2,3, CCN (CYR61, FISP12/CTGF, NOV) family members 1, 2 and 3
- CF
cardiac fibroblasts
- CFP
cyan fluorescent protein
- COL1A1
type I collagen α1
- COL1A2
type I collagen α2
- COL3A1
type III collagen α1
- CRISPR
clustered regularly interspaced short palindromic repeats
- ECM
extracellular matrix
- Eey I
Eeyarestatin I
- ELN
elastin
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- FN1
fibronectin
- FRET
fluorescence resonance energy transfer
- hCF
human cardiac fibroblasts
- hCM
human left ventricular cardiomyocytes
- HF
heart failure
- HW/BW
heart weight/body weight ratio
- IRE1α
inositol-requiring enzyme 1 α
- ISO
isoproterenol
- JNK
c-Jun N-terminal kinase
- LV
left ventricle
- mCF
mouse cardiac fibroblasts
- MMPs
matrix metalloproteinases
- NAC
N-acetylcysteine
- NF
non-failing
- NOX
NAD(P)H oxidase
- PDI
protein disulfide isomerase
- POSTN
periostin
- RNASeq
RNA sequencing
- shScr
non-targeted shRNA
- siScr
non-targeted siRNA
- TGFβ1
transforming growth factor β1
- TIMPs
tissue inhibitors of MMP
- TRX
thioredoxin
- TUDCA
tauroursodeoxycholic acid
- TXNDC5
thioredoxin domain containing 5
- UP
unfolded protein response
- VCP
valosin containing protein
- VSMC
vascular smooth muscle cells
- WGCNA
weighted gene co-expression network analysis
- WT
wild-type
- XBP1
X-box binding protein 1
- YFP
yellow fluorescent protein
Footnotes
Disclosures: None.
References
- 1.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J. 1998;19(P):P9–16. [PubMed] [Google Scholar]
- 2.Nabel EG, Braunwald E. A tale of coronary artery disease and myocardial infarction. N Engl J Med. 2012;366:54–63. doi: 10.1056/NEJMra1112570. [DOI] [PubMed] [Google Scholar]
- 3.Braunwald E. Shattuck lecture--cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med. 1997;337:1360–9. doi: 10.1056/NEJM199711063371906. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Normand SL, Wang Y, Krumholz HM. National and regional trends in heart failure hospitalization and mortality rates for Medicare beneficiaries, 1998-2008. JAMA. 2011;306:1669–78. doi: 10.1001/jama.2011.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy D, Kenchaiah S, Larson MG, Benjamin EJ, Kupka MJ, Ho KK, Murabito JM, Vasan RS. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347:1397–402. doi: 10.1056/NEJMoa020265. [DOI] [PubMed] [Google Scholar]
- 6.Jhund PS, Macintyre K, Simpson CR, Lewsey JD, Stewart S, Redpath A, Chalmers JW, Capewell S, McMurray JJ. Long-term trends in first hospitalization for heart failure and subsequent survival between 1986 and 2003: a population study of 5.1 million people. Circulation. 2009;119:515–23. doi: 10.1161/CIRCULATIONAHA.108.812172. [DOI] [PubMed] [Google Scholar]
- 7.Askoxylakis V, Thieke C, Pleger ST, Most P, Tanner J, Lindel K, Katus HA, Debus J, Bischof M. Long-term survival of cancer patients compared to heart failure and stroke: a systematic review. BMC Cancer. 2010;10:105. doi: 10.1186/1471-2407-10-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMurray J, Pfeffer MA. New therapeutic options in congestive heart failure: Part II. Circulation. 2002;105:2223–8. doi: 10.1161/01.cir.0000014771.38666.22. [DOI] [PubMed] [Google Scholar]
- 9.McMurray J, Pfeffer MA. New therapeutic options in congestive heart failure: Part I. Circulation. 2002;105:2099–106. doi: 10.1161/01.cir.0000014763.63528.9d. [DOI] [PubMed] [Google Scholar]
- 10.Conrad CH, Brooks WW, Hayes JA, Sen S, Robinson KG, Bing OH. Myocardial fibrosis and stiffness with hypertrophy and heart failure in the spontaneously hypertensive rat. Circulation. 1995;91:161–70. doi: 10.1161/01.cir.91.1.161. [DOI] [PubMed] [Google Scholar]
- 11.Schwarz F, Mall G, Zebe H, Blickle J, Derks H, Manthey J, Kubler W. Quantitative morphologic findings of the myocardium in idiopathic dilated cardiomyopathy. Am J Cardiol. 1983;51:501–6. doi: 10.1016/s0002-9149(83)80088-5. [DOI] [PubMed] [Google Scholar]
- 12.Moreo A, Ambrosio G, De Chiara B, Pu M, Tran T, Mauri F, Raman SV. Influence of myocardial fibrosis on left ventricular diastolic function: noninvasive assessment by cardiac magnetic resonance and echo. Circ Cardiovasc Imaging. 2009;2:437–43. doi: 10.1161/CIRCIMAGING.108.838367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spach MS, Boineau JP. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections: a major mechanism of structural heart disease arrhythmias. Pacing Clin Electrophysiol. 1997;20:397–413. doi: 10.1111/j.1540-8159.1997.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 14.Karagueuzian HS. Targeting cardiac fibrosis: a new frontier in antiarrhythmic therapy? Am J Cardiovasc Dis. 2011;1:101–9. [PMC free article] [PubMed] [Google Scholar]
- 15.Fredj S, Bescond J, Louault C, Potreau D. Interactions between cardiac cells enhance cardiomyocyte hypertrophy and increase fibroblast proliferation. J Cell Physiol. 2005;202:891–9. doi: 10.1002/jcp.20197. [DOI] [PubMed] [Google Scholar]
- 16.Lucas JA, Zhang Y, Li P, Gong K, Miller AP, Hassan E, Hage F, Xing D, Wells B, Oparil S, Chen YF. Inhibition of transforming growth factor-beta signaling induces left ventricular dilation and dysfunction in the pressure-overloaded heart. Am J Physiol Heart Circ Physiol. 2010;298:H424–32. doi: 10.1152/ajpheart.00529.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berk BC, Fujiwara K, Lehoux S. ECM remodeling in hypertensive heart disease. J Clin Invest. 2007;117:568–75. doi: 10.1172/JCI31044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases. J Pathol. 2003;200:500–3. doi: 10.1002/path.1427. [DOI] [PubMed] [Google Scholar]
- 19.Tsuruda T, Costello-Boerrigter LC, Burnett JC., Jr Matrix metalloproteinases: pathways of induction by bioactive molecules. Heart Fail Rev. 2004;9:53–61. doi: 10.1023/B:HREV.0000011394.34355.bb. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg B, Quinones MA, Koilpillai C, Limacher M, Shindler D, Benedict C, Shelton B. Effects of long-term enalapril therapy on cardiac structure and function in patients with left ventricular dysfunction. Results of the SOLVD echocardiography substudy. Circulation. 1995;91:2573–81. doi: 10.1161/01.cir.91.10.2573. [DOI] [PubMed] [Google Scholar]
- 21.Tsutamoto T, Wada A, Maeda K, Mabuchi N, Hayashi M, Tsutsui T, Ohnishi M, Sawaki M, Fujii M, Matsumoto T, Matsui T, Kinoshita M. Effect of spironolactone on plasma brain natriuretic peptide and left ventricular remodeling in patients with congestive heart failure. J Am Coll Cardiol. 2001;37:1228–33. doi: 10.1016/s0735-1097(01)01116-0. [DOI] [PubMed] [Google Scholar]
- 22.Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000;102:2700–6. doi: 10.1161/01.cir.102.22.2700. [DOI] [PubMed] [Google Scholar]
- 23.Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation. 2002;106:130–5. doi: 10.1161/01.cir.0000020689.12472.e0. [DOI] [PubMed] [Google Scholar]
- 24.Lee KW, Everett THt, Rahmutula D, Guerra JM, Wilson E, Ding C, Olgin JE. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006;114:1703–12. doi: 10.1161/CIRCULATIONAHA.106.624320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin J, Kelly DJ, Mifsud SA, Zhang Y, Cox AJ, See F, Krum H, Wilkinson-Berka J, Gilbert RE. Tranilast attenuates cardiac matrix deposition in experimental diabetes: role of transforming growth factor-beta. Cardiovasc Res. 2005;65:694–701. doi: 10.1016/j.cardiores.2004.10.041. [DOI] [PubMed] [Google Scholar]
- 26.Holmes JS, Arispe IE, Moy E. Heart disease and prevention: race and age differences in heart disease prevention, treatment, and mortality. Med Care. 2005;43:I33–41. [PubMed] [Google Scholar]
- 27.Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–63. doi: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 28.Newby LK, Marber MS, Melloni C, Sarov-Blat L, Aberle LH, Aylward PE, Cai G, de Winter RJ, Hamm CW, Heitner JF, Kim R, Lerman A, Patel MR, Tanguay JF, Lepore JJ, Al-Khalidi HR, Sprecher DL, Granger CB, Investigators S. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet. 2014;384:1187–95. doi: 10.1016/S0140-6736(14)60417-7. [DOI] [PubMed] [Google Scholar]
- 29.Anand I, McMurray J, Cohn JN, Konstam MA, Notter T, Quitzau K, Ruschitzka F, Luscher TF investigators E. Long-term effects of darusentan on left-ventricular remodelling and clinical outcomes in the EndothelinA Receptor Antagonist Trial in Heart Failure (EARTH): randomised, double-blind, placebo-controlled trial. Lancet. 2004;364:347–54. doi: 10.1016/S0140-6736(04)16723-8. [DOI] [PubMed] [Google Scholar]
- 30.Schumacher SM, Gao E, Zhu W, Chen X, Chuprun JK, Feldman AM, Tesmer JJ, Koch WJ. Paroxetine-mediated GRK2 inhibition reverses cardiac dysfunction and remodeling after myocardial infarction. Science translational medicine. 2015;7:277ra31. doi: 10.1126/scitranslmed.aaa0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 32.Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 129:1009–21. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–47. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac Fibrosis: The Fibroblast Awakens. Circ Res. 118:1021–40. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang KC, Ku YC, Lovett M, Nerbonne JM. Combined deep microRNA and mRNA sequencing identifies protective transcriptomal signature of enhanced PI3Kalpha signaling in cardiac hypertrophy. J Mol Cell Cardiol. 2012;53:101–12. doi: 10.1016/j.yjmcc.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yata Y, Scanga A, Gillan A, Yang L, Reif S, Breindl M, Brenner DA, Rippe RA. DNase I-hypersensitive sites enhance alpha1(I) collagen gene expression in hepatic stellate cells. Hepatology. 2003;37:267–76. doi: 10.1053/jhep.2003.50067. [DOI] [PubMed] [Google Scholar]
- 38.Leask A. Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res. 116:1269–76. doi: 10.1161/CIRCRESAHA.116.305381. [DOI] [PubMed] [Google Scholar]
- 39.Horna-Terron E, Pradilla-Dieste A, Sanchez-de-Diego C, Osada J. TXNDC5, a newly discovered disulfide isomerase with a key role in cell physiology and pathology. Int J Mol Sci. 2014;15:23501–18. doi: 10.3390/ijms151223501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buck TM, Wright CM, Brodsky JL. The activities and function of molecular chaperones in the endoplasmic reticulum. Semin Cell Dev Biol. 2007;18:751–61. doi: 10.1016/j.semcdb.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 42.Philipps B, Hennecke J, Glockshuber R. FRET-based in vivo screening for protein folding and increased protein stability. J Mol Biol. 2003;327:239–49. doi: 10.1016/s0022-2836(03)00077-9. [DOI] [PubMed] [Google Scholar]
- 43.Liu S, Xu SW, Kennedy L, Pala D, Chen Y, Eastwood M, Carter DE, Black CM, Abraham DJ, Leask A. FAK is required for TGFbeta-induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix-remodeling phenotype. Molecular biology of the cell. 2007;18:2169–78. doi: 10.1091/mbc.E06-12-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Javelaud D, Laboureau J, Gabison E, Verrecchia F, Mauviel A. Disruption of basal JNK activity differentially affects key fibroblast functions important for wound healing. The Journal of biological chemistry. 2003;278:24624–8. doi: 10.1074/jbc.M301942200. [DOI] [PubMed] [Google Scholar]
- 45.Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, Sorescu D. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. 2005;97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D. [DOI] [PubMed] [Google Scholar]
- 46.Trevelin SC, Lopes LR. Protein disulfide isomerase and Nox: new partners in redox signaling. Current pharmaceutical design. 2015;21:5951–63. doi: 10.2174/1381612821666151029112523. [DOI] [PubMed] [Google Scholar]
- 47.Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CX, Laurindo FR. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. The Journal of biological chemistry. 2005;280:40813–9. doi: 10.1074/jbc.M509255200. [DOI] [PubMed] [Google Scholar]
- 48.Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ. Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts. American journal of respiratory cell and molecular biology. 2012;46:731–9. doi: 10.1165/rcmb.2011-0121OC. [DOI] [PubMed] [Google Scholar]
- 49.Lenna S, Trojanowska M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr Opin Rheumatol. 2012;24:663–8. doi: 10.1097/BOR.0b013e3283588dbb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–8. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oudit GY, Crackower MA, Eriksson U, Sarao R, Kozieradzki I, Sasaki T, Irie-Sasaki J, Gidrewicz D, Rybin VO, Wada T, Steinberg SF, Backx PH, Penninger JM. Phosphoinositide 3-kinase gamma-deficient mice are protected from isoproterenol-induced heart failure. Circulation. 2003;108:2147–52. doi: 10.1161/01.CIR.0000091403.62293.2B. [DOI] [PubMed] [Google Scholar]
- 52.Wang ER, Jarrah AA, Benard L, Chen J, Schwarzkopf M, Hadri L, Tarzami ST. Deletion of CXCR4 in cardiomyocytes exacerbates cardiac dysfunction following isoproterenol administration. Gene Ther. 2014;21:496–506. doi: 10.1038/gt.2014.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10:945–63. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knoblach B, Keller BO, Groenendyk J, Aldred S, Zheng J, Lemire BD, Li L, Michalak M. ERp19 and ERp46, new members of the thioredoxin family of endoplasmic reticulum proteins. Mol Cell Proteomics. 2003;2:1104–19. doi: 10.1074/mcp.M300053-MCP200. [DOI] [PubMed] [Google Scholar]
- 55.Sullivan DC, Huminiecki L, Moore JW, Boyle JJ, Poulsom R, Creamer D, Barker J, Bicknell R. EndoPDI, a novel protein-disulfide isomerase-like protein that is preferentially expressed in endothelial cells acts as a stress survival factor. The Journal of biological chemistry. 2003;278:47079–88. doi: 10.1074/jbc.M308124200. [DOI] [PubMed] [Google Scholar]
- 56.Yang KC, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–21. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kojima R, Okumura M, Masui S, Kanemura S, Inoue M, Saiki M, Yamaguchi H, Hikima T, Suzuki M, Akiyama S, Inaba K. Radically different thioredoxin domain arrangement of ERp46, an efficient disulfide bond introducer of the mammalian PDI family. Structure. 2014;22:431–43. doi: 10.1016/j.str.2013.12.013. [DOI] [PubMed] [Google Scholar]
- 58.Matsuzaki S, Hiratsuka T, Taniguchi M, Shingaki K, Kubo T, Kiya K, Fujiwara T, Kanazawa S, Kanematsu R, Maeda T, Takamura H, Yamada K, Miyoshi K, Hosokawa K, Tohyama M, Katayama T. Physiological ER Stress Mediates the Differentiation of Fibroblasts. PloS one. 2015;10:e0123578. doi: 10.1371/journal.pone.0123578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pescatore LA, Bonatto D, Forti FL, Sadok A, Kovacic H, Laurindo FR. Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation. The Journal of biological chemistry. 2012;287:29290–300. doi: 10.1074/jbc.M112.394551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laurindo FR, Pescatore LA, Fernandes Dde C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free radical biology & medicine. 2012;52:1954–69. doi: 10.1016/j.freeradbiomed.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 61.Brown DI, Griendling KK. Nox proteins in signal transduction. Free radical biology & medicine. 2009;47:1239–53. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Graham KA, Kulawiec M, Owens KM, Li X, Desouki MM, Chandra D, Singh KK. NADPH oxidase 4 is an oncoprotein localized to mitochondria. Cancer Biol Ther. 2010;10:223–31. doi: 10.4161/cbt.10.3.12207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arteriosclerosis, thrombosis, and vascular biology. 2004;24:677–83. doi: 10.1161/01.ATV.0000112024.13727.2c. [DOI] [PubMed] [Google Scholar]
- 64.Helmcke I, Heumuller S, Tikkanen R, Schroder K, Brandes RP. Identification of structural elements in Nox1 and Nox4 controlling localization and activity. Antioxid Redox Signal. 2009;11:1279–87. doi: 10.1089/ars.2008.2383. [DOI] [PubMed] [Google Scholar]
- 65.Prior KK, Wittig I, Leisegang MS, Groenendyk J, Weissmann N, Michalak M, Jansen-Durr P, Shah AM, Brandes RP. The Endoplasmic Reticulum Chaperone Calnexin Is a NADPH Oxidase NOX4 Interacting Protein. The Journal of biological chemistry. 2016;291:7045–59. doi: 10.1074/jbc.M115.710772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laurindo FR, Fernandes DC, Amanso AM, Lopes LR, Santos CX. Novel role of protein disulfide isomerase in the regulation of NADPH oxidase activity: pathophysiological implications in vascular diseases. Antioxid Redox Signal. 2008;10:1101–13. doi: 10.1089/ars.2007.2011. [DOI] [PubMed] [Google Scholar]
- 67.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furtado MB, Costa MW, Rosenthal NA. The cardiac fibroblast: Origin, identity and role in homeostasis and disease. Differentiation; research in biological diversity. 2016;92:93–101. doi: 10.1016/j.diff.2016.06.004. [DOI] [PubMed] [Google Scholar]
- 69.Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. Journal of cellular physiology. 2010;225:631–7. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.