

Abstract
Drug resistance has been reported for every antimalarial in use highlighting the need for new strategies to protect the efficacy of therapeutics in development. We have previously shown that resistance can be suppressed with a population biology trap: by identifying situations where resistance to one compound confers hypersensitivity to another (collateral sensitivity), we can design combination therapies that not only kill the parasite but also guide its evolution away from resistance. We applied this concept to the Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) enzyme, a well validated antimalarial target with inhibitors in the development pipeline. Here, we report a high-throughput screen to identify compounds specifically active against PfDHODH resistant mutants. We additionally perform extensive cross-resistance profiling allowing us to identify compound pairs demonstrating the potential for mutually incompatible resistance. These combinations represent promising starting points for exploiting collateral sensitivity to extend the useful lifespan of new antimalarial therapeutics.
Keywords: DHODH, malaria, collateral sensitivity, drug resistance, drug combinations
Over the last 15 years, renewed efforts to control malaria disease and transmission have led to a 37% reduction in incidence and a 60% reduction in mortality worldwide.1 Effective treatment is a cornerstone of malaria eradication efforts. However, the emergence of drug resistance threatens these fragile gains. Resistance has been reported for every antimalarial that has been in clinical use,2 and there is an urgent need to develop not only new antimalarial drugs but also strategies to combat resistance and prolong the useful lifespan of these therapies.
The problem of resistance is not limited to antimalarial drugs but is a widespread observation in the treatment of all infectious agents and many cancers.3,4 The strong evolutionary pressure exerted by drug treatment results in the selection of resistant organisms or cells. The current strategy to prevent the emergence of drug resistance is to combine two drugs with different mechanisms of action. Combining therapies with different modes of action can help delay resistance, but this must be balanced with possible toxic or counterproductive effects.5 The concept is that resistance is far less likely to emerge to both of the drugs simultaneously. However, in practice, there are many examples of this strategy failing in part because of different pharmacological properties of the paired drugs.6−10
We have previously demonstrated that an alternate approach based on evolutionary principles could provide a viable path toward suppressing resistance.11 The underlying hypothesis is that a mutation that leads to resistance to a particular drug also has consequences for the fitness of that organism, creating new vulnerabilities which could potentially be exploited. One such potential consequence is collateral sensitivity, in which resistance to one drug causes an increase in sensitivity to another chemical agent.12 In practical terms, once an enzyme carries a mutation that confers drug resistance, that enzyme has increased sensitivity to other small molecules that preferentially recognize the altered or mutant form. The concept is then to combine a wild-type specific drug with a mutant specific drug in order to block resistance from emerging. We found two such instances in our original work: a molecule that was specific for chloroquine-resistant parasites and was inactive against sensitive parasites.13 A second combination targeted dihydroorotate dehydrogenase (DHODH), one of the newly identified targets for antimalarial drug development.11,13 Subsequently, other examples have been published including inhibitors of PfATPase4,14 antibacterials,15,16 and cancer therapeutics.17,18
The purpose of the work described here was to further investigate the strategy of designing drug combinations based on collateral evolutionary forces. We focused on the enzyme DHODH in part because we had demonstrated the feasibility of this approach previously and because it is one of the drug targets currently being targeted for development under MMV sponsorship. The goal was to identify potential compounds that could be combined to target DHODH wild-type and mutant forms.
This project is a collaboration between the GlaxoSmithKline (GSK) Tres Cantos Open Lab and Harvard University and was conducted at both institutions. We sought to more comprehensively probe the extent of collateral sensitivity for the Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH) drug target. To do so, we performed a high throughput screen of wild-type and mutant DHODH enzyme in order to identify chemotypes that were preferentially active against resistant forms. Further validation of these molecules against a larger panel of PfDHODH mutant parasites allowed us to understand the networks of cross-resistance and collateral sensitivity for this target and to identify promising compound combinations designed to suppress the emergence of resistance.
Results and Discussion
To identify PfDHODH inhibitors, and in particular compounds active against the mutant form of the enzyme, we performed a high throughput screen (HTS) of select GSK chemical libraries. We screened the E182D mutant enzyme as it had independently arisen in selections using diverse chemical scaffolds, and our previous studies suggested it could represent an optimal fitness-resistance compromise for the enzyme.13 To screen the mutant and wild-type enzymes, we utilized a previously optimized in vitro colorimetric assay that measures enzyme activity by coupling the oxidation of the dihydroorotate (DHO) substrate with the reduction of 2,6-dichloroindophenol (DCIP).19,20 The mutant (E182D) enzyme was recombinantly expressed and tested against select libraries at GSK, amounting to a total of 130 887 small molecules assessed. Data for the inhibition of the wild-type (WT) enzyme was previously obtained by GSK (1.1% hit rate, personal communication) and used as a comparator for the mutant data. Compounds were first tested at a single dose of 5 μM, and hits were defined as those demonstrating at least 50% inhibitory activity when compared to vehicle control wells. These 458 hit compounds (0.35% overall hit rate) were cherry-picked and run in full dose–response against both the wild-type and mutant enzymes to determine the half-maximal inhibitory concentration (IC50). This resulted in 118 primary hits with potent IC50 values. Comparison of the mutant IC50 relative to wild-type allowed us to classify compounds as being WT-active (ratio >2), E182D-active (ratio <0.5), and equally potent (ratio between 0.5 and 2) (Figures 1a and S1, Table S1). Of particular interest for additional study are the 18 mutant-active and 21 equipotent molecules as they represent promising starting points to test our targeting resistance concept.
Figure 1.
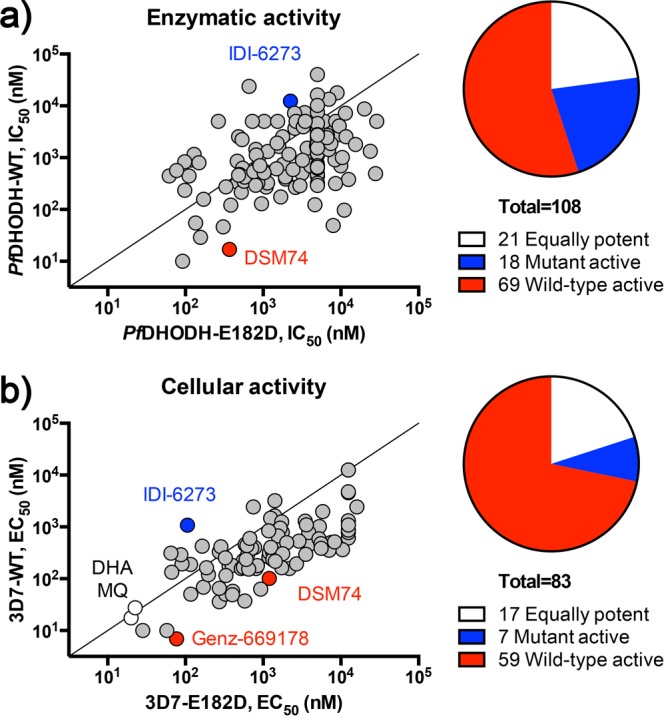
Identification of 3D7-E182D mutant active, equally potent, and wild-type active DHODH inhibitors. (a) A high-throughput screen of select GSK libraries using wild-type and E182D recombinant PfDHODH identified 118 primary hits. On the basis of the IC50 ratio of E182D/WT, compounds were classified as E182D mutant active (n = 18), equally potent (n = 21), or wild-type active (n = 69). Control compounds are indicated on the plot: IDI-6273 (blue), mutant active control; DSM74 (red), wild-type active control. (b) Cell-based validation of 85 active compounds. Compounds were classified into three groups based on the EC50 ratio of E182D/WT: equally potent (n = 17), mutant active (n = 7), or wild-type active (n = 59). Control compounds are indicated on the plot: IDI-6273 (blue), mutant active control; DSM74 and Genz-669178 (red), wild-type active controls; dihydroartemisinin (DHA) and mefloquine (MQ) (white), non-DHODH inhibitor controls.
To further validate their cellular mode of action, we counter-screened the 118 hit compounds identified from the enzymatic screen for activity against the 3D7-WT and 3D7-DHODH:E182D mutant parasite lines in a whole-cell dose–response assay. Despite having established inhibitory activity against the PfDHODH enzyme, it is possible that the cell-based activity of our compounds could fail to permeate cells or involve additional pathways other than PfDHODH, thus confounding our results. To help rule out off-target activity in the cell-based assay, we also included a Dd2-ScDHODH transgenic parasite along with its wild-type parental line, Dd2-attB, in our secondary screen. The transgenic Dd2-ScDHODH strain expresses the cytosolic type 1 DHODH from S. cerevisiae (ScDHODH) and is resistant to P. falciparum electron transport chain (ETC) inhibitors. Expression of the yeast enzyme bypasses the parasite’s dependency on ubiquinone for DHODH activity in the pyrimidine biosynthesis pathway.21 Ablation of compound activity in this cell line relative to its parent functionally validates its cellular mechanism of action as inhibition of DHODH or downstream effectors in the ETC. Compounds were first assessed for potency against each of the four strains. Among the primary hits, 29 compounds showed poor potency (<40% inhibition at 20 μM) to both 3D7-WT and 3D7-E182D and were removed from further study. An additional 12 compounds were discarded as they showed greater than 40% inhibition against the Dd2-ScDHODH strain at the lowest dose of 0.2 μM, suggesting off-target activity (Table S1).
The remaining 85 compounds (secondary hits) were then tested in full dose–response (12-point titrations in triplicate), and 50% effective concentrations (EC50 values) were determined for both the 3D7-WT and 3D7-DHODH:E182D strains. On the basis of their EC50 ratio (E182D/WT), compounds were classified as WT-active (ratio >2), E182D-active (ratio <0.5), and equally potent (ratio between 0.5 and 2) (Figure 1b). Using these parameters, we identified 59 WT-active, 7 E182D-active, and 17 equally potent compounds. On the basis of overall cellular potency and compound availability, 23 molecules representing ten distinct chemotypes were selected for further characterization (Figure S2). Of note are compounds 10 (TCMDC-124417), 9 (TCMDC-124402), 8 (TCMDC-123826), and 15 (TCMDC-125162), which share structural similarity to the triazolopyrimidine clinical candidate, DSM265.32,33 We also detected a cluster of molecules (compounds 2, 4, 6, 7, 11, and 19) with structural similarity to GSK3, an inhibitor identified in our previous study as demonstrating increased potency against mutant parasites.
An inhibitor with dual P. falciparum Cytochrome b (PfCytb) and PfDHODH activities was recently reported,22 prompting us to assess whether some of our hits also demonstrate the ability to inhibit both enzymes. A compound with dual activity could be incorrectly scored as a PfDHODH mutant selective compound due to the confounding effects of its PfCytb activity. To test this and rule out any such false-positives in our data set, we assayed our set of 23 secondary hits in the Dd2-ScDHODH parasite line in both the presence and absence of 1 μM proguanil (Table S2). The small molecule proguanil works synergistically with Cytochrome b (Cytb) inhibitors in P. falciparum and can rescue the apparent resistance observed in the ScDHODH parasite line.21,22 In contrast, proguanil has no effect on the activity of PfDHODH inhibitors when added to assay conditions.22,23 As summarized in Table S2, all tested compounds were at least 10-fold less active against Dd2-ScDHODH compared to the parental Dd2-attB, and the addition of proguanil largely had no effect on EC50. This is comparable to the control PfDHODH inhibitor, Genz669178. However, proguanil dramatically improved the cell-based activity of compound 21 (TCMDC-136129) similar to the effect seen for the control PfCytb inhibitor atovaquone. On the basis of these results, we hypothesize that 21 targets both PfDHODH and PfCytb.
To further validate the dual cellular activity of 21, we utilized extracellular flux analysis to directly test the ability of the molecule to inhibit specific steps of the electron transport chain in cells. Using methodology developed in our group, the oxygen consumption rate (OCR) of saponin-freed schizonts can be directly measured as an output of Complexes I–IV of the electron transport chain.24 Inhibitors of any of the enzymes along the chain result in a reduction of OCR; by varying the input energy source provided to the cells, the system allows for the functional identification of the specific target of the compound.24 When schizonts were assayed in RPMI medium, which contains fuel molecules such as glucose and glutamine, all dehydrogenases function normally and provide electrons to the downstream Cytbc1 complex (Complex III). Inhibition of Cytbc1 results in a reduction of the OCR (Figure 2c). However, in these media conditions, DHODH inhibition does not significantly alter the OCR, presumably because electron flow from other dehydrogenases mask the effect of DHODH inhibition. As shown in Figure 2c, Genz669178 and compound 1 (N16306-26-3), inhibitors that only target PfDHODH, had no effect on the OCR, while 21 and the control Cytb inhibitor antimycin A were able to dramatically decrease OCR, exhibiting their direct inhibition of Cytb. This finding was further demonstrated in minimal media assay conditions when the only fuel source provided was glycerol 3-phosphate (G3P) (Figure 2e). In contrast, when the assay is conducted in minimal media conditions in which DHO is the only substrate provided (and only DHODH is activated), OCR reduction by DHODH inhibitors can be observed. As expected, 21, 1, and the control Genz669178 decreased OCR after DHO was added to schizonts (Figure 2d).
Figure 2.

Extracellular flux analysis confirms the inhibitory activity of compound 21 against both PfCytbc1 and PfDHODH. (a) A schematic representation of target identification using extracellular flux analysis. When G3P is provided as a fuel source, Cytbc1 inhibitors such as antimycin A and compound 21 disrupt the electron flow of the ETC resulting in a reduction in the OCR. When DHO is the only fuel source provided, thus activating DHODH, electron flow and the OCR are reduced by DHODH inhibitors, such as Genz669178 and compounds 1 and 21. (b) Structures of compounds tested in the extracellular flux assay. Genz669178 is a control DHODH inhibitor and antimycin A is a control Cytbc1 inhibitor. (c) Antimycin A and compound 21 decreased OCR in RPMI media, indicating Cytb inhibition. Compound 1 and the DHODH inhibitor control Genz669178 do not have an effect on the OCR, similar to media-only controls. All data represent means ± SD (n = 3). (d) Compound 1, compound 21, and Genz669178 reduced the DHO-induced OCR, indicating their DHODH activity, while the Cytb inhibitor, antimycin A, did not. All data represent means ± SD (n = 3). (e) As observed in RPMI media conditions, only compound 21 and antimycin A reduced the OCR when G3P was the sole substrate. All data represent means ± SD (n = 3).
We additionally tested the direct inhibition of the PfCytbc1 complex using an in vitro enzymatic assay. Mitochondria were isolated from saponin-released parasites and cytochrome c reductase activity was measured by the method of Fry and Pudney.25 Addition of compound 21 reduced enzymatic activity in a dose-dependent manner resulting in an IC50 of 40 nM (Table S3). The PfDHODH specific inhibitor, DSM1, did not have an effect on activity. Taken together, these results provide evidence supporting our hypothesis that 21 directly inhibits both PfDHODH and Complex III.
The overall aim of our study was to identify inhibitors that target mutant forms of the PfDHODH enzyme. To extend the findings of our screening efforts, which focused on the E182D mutant, we assayed 17 of our cherry-picked compounds against a broader panel of five DHODH mutant parasite lines. These parasites were generated from in vitro selections with DHODH inhibitors of varying chemical classes (Table S4).11,13 All resistant cell lines have point mutations in the PfDHODH locus resulting in amino acid changes in residues lining the inhibitor binding pocket of the enzyme (Figure 3a).
Figure 3.
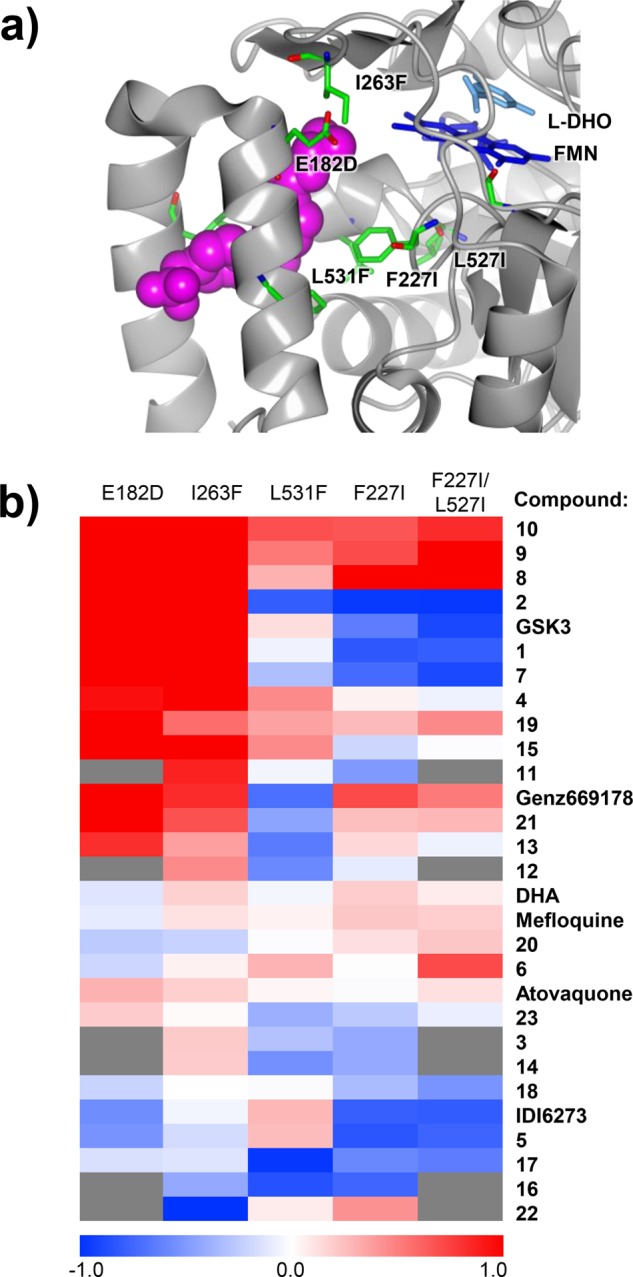
Cross-resistance profiling of multiple selected parasite lines reveals patterns of collateral sensitivity. (a) Structure of PfDHODH displaying the resistance mutations in the parasite lines profiled in this study (see Table S3). The image was generated in CCP4 mg31 using coordinates from the crystal structure of PfDHODH bound to the Genz669178 inhibitor, PDB ID: 3o8a.11 (b) Heatmap based on fold-change in EC50 of each compound across every resistant cell line tested. Both cell lines and compounds are clustered on the basis of log10-transformed activity profiles. Shades of red indicate cross-resistance, while shades of blue indicate collateral sensitivity. White and light shades indicate no significant fold-change between mutant and wild-type lines. Gray boxes indicate that the compound was not tested against the given mutant line due to limited compound availability.
We determined EC50 values for each compound assayed against the five DHODH mutants, as well as their 3D7 and Dd2 parental lines (Table S5). The compounds all demonstrate activity against the wild-type parents (EC50 range from 1.8 to 600 nM) and show decreased activity (cross-resistance), equal potency, or increased activity (collateral sensitivity) in the mutant cell lines (Table S5). To further explore patterns of cross-resistance and collateral sensitivity, the EC50 fold-change for each mutant relative to its parental line was calculated and then the log10-transformed values were visualized on a heatmap. This visualization showed that certain mutants have very similar cross-resistance profiles (e.g., I263F and E182D or F227I and the double mutant F227I/L527I) while the L531F mutant had a unique phenotypic profile. On the basis of this clustering, we tested an additional set of six secondary hits using only the I263F, F227I, and L531F mutants, as compound availability was limited (Figure 3b). Overall, we observed both patterns of cross-resistance (shades of red) and collateral sensitivity (shades of blue) in our data set (Figure 3b). For example, compounds 10, 9, 8, and 15, which are structurally related to DSM265 (see Figure S2), demonstrate significant cross-resistance in all of the cell lines tested. In contrast, we observe collateral sensitivity to compounds 17 (TCMDC-125334) and 16 (TCMDC-125331). Compound 22 (TCMDC-136379) was also of note as it demonstrated potency against the I263F mutant. This mutant was the most “pan-resistant” showing cross-resistance to most chemotypes.
Of particular interest were compounds with complementary activities across our panel of mutant parasite lines (Figure 4a). As noted above, all of the mutants tested demonstrated cross-resistance against compounds with a triazolopyrimidine scaffold, whereas they were sensitive to compound 17. Given these opposing phenotypic profiles, we hypothesize that a combination of 17 and 8 could provide a basis for suppression of resistance studies. Another example is the pairing of compound 6 (TCMDC-123647) and 7 (TCMDC-123823). Compound 6 is active against the E182D and I263F mutants and relatively inactive against the L531F, F227I, and F227I/L527I mutant cell lines whereas compound 7 has the opposite activity profile: highly potent against L531F, F227I, and F227I/L527I while relatively inactive against E182D and I263F (Figure 4b). The compounds 1 and 5 (TCMDC-123620) display a similar activity profile (Figure 4c). Further resistance studies are needed to test the hypothesis that any of these compound pairings could result in the suppression of resistance.
Figure 4.

Activity profiles of compound pairs exhibiting mutually incompatible resistance. Scatter plots display the EC50 values of the indicated compound for each of the selected parasite lines tested. The parental lines 3D7 (closed circle) and Dd2 (closed square) are in black, while the mutant cell lines are indicated in blue and red to represent distinctive cross-resistance phenotypes (E182D, red closed circle; I263F, red closed square; L531F, blue ×; F227I, open blue circle; F227I/L527I, open blue circle with ×). (a) Structures of compound pairs highlighted by our analysis. (b) Compound 8 is a wild-type active compound which is relatively inactive against all mutant lines tested. In contrast, compound 17 is an example of a mutant-active compound, as all mutant lines tested are more sensitive than the parental lines. These two compounds could be paired together as a strategy to target resistance. (c) Compound 7 targets the L531F, F227I, and F227I/L527I mutants, while compound 6 targets E182D and I263F. (d) An additional example of a compound pair with complementary activities. Compound 1 targets the L531F, F227I, and F227I/L527I mutants, while compound 5 targets E182D and I263F.
In this work, we took a unique HTS approach aimed at identifying small molecules that specifically target mutant forms of PfDHODH. Our primary screen identified 118 molecules with activity against either wild-type and/or mutant PfDHODH. Validation of these primary hits in cell-based assays resulted in the classification of 7 mutant active, 17 equally active, and 59 wild-type active small molecules. Extensive cross-resistance profiling of 23 cherry-pick compounds against a broad panel of PfDHODH mutant cell lines illuminated patterns of cross-resistance and collateral sensitivity. The molecules demonstrating collateral sensitivity against all or a subset of mutants represent promising starting points for further suppression of resistance studies. It is important to note that while we have observed strong correlation between mutant enzyme activity and mutant cell line activity in previous studies,11 we cannot rule out off-target effects in using a cell-based assay with the compounds explored in this study. Future efforts to evaluate this would be valuable.
Over the course of these efforts, we also discovered and validated a compound that targets both PfDHODH and PfCytb in the parasite. This presents an interesting possibility for what effectively amounts to combination treatment through a single molecule. However, questions remain as to how easily the parasite can evolve resistance to this dual-inhibitor through mutations in either target, rendering the molecule ineffective. Because there is not an evolutionary constraint preventing the parasite from mutational escape in either enzyme, we suspect that resistance to this inhibitor could arise relatively easily despite its ability to inhibit two enzymes in the parasite.
The challenge of drug resistance is often presented as a never-ending arms race between our ability to refill an arsenal with new drugs and the ability of a pathogen to evolve resistance. Our efforts focus on targeting the evolutionary changes that lead to resistance in the first place; we seek not only to refill the arsenal but also to develop ways in which we can exploit the fitness costs associated with resistance in order to trap the parasite in an evolutionary loop that favors a drug-sensitive population. Comprehensively exploring the relationships of cross-resistance and collateral sensitivity in a panel of PfDHODH mutants allowed us to develop a network of collateral sensitivity for this target and identify pairs of compounds demonstrating the potential for mutually incompatible resistance. Notably, we were able to identify complementary pairs of compounds that together targeted all of the resistance mutations tested in this study, suggesting that this may be a feasible strategy to close off potential pathways to resistance. Studies to fully explore resistance to each of these molecules individually and in combination are warranted. It will also be critical to determine whether particular resistance pathways are favored in vivo and match those from our in vitro studies. Future efforts aim to explore this and prioritize compounds that specifically block these in vivo-relevant mutants.
Methods
Reagents
l-Dihydroorotic acid (DHO), CoQD, DCIP, antimycin A, atovaquone (ATV), mefloquine (MQ), artemisinin, dihydroartemisinin (DHA), and proguanil were purchased from Sigma-Aldrich (St. Louis, MO). Genz-669178, was kindly provided by Genzyme, a Sanofi Company (Waltham, MA). IDI-6273 was purchased from ChemDiv. DSM1 and DSM74 were prepared following the literature procedure26 and were recrystallized from ethanol. 1H NMR spectra matched that reported, and HPLC analysis indicated >95% purity.
In Vitro Enzyme Activity Assay for HTS
Recombinant wild-type and E182D protein were expressed in E. coli and purified as previously described.11 Enzyme activity was measured using published protocols19,20 with slight modifications to the reaction conditions as follows: 500 μM DHO, 60 μM DCIP, 100 μm CoQD, and 0.125% Triton X-100 in a 50 μL reaction volume in 384-well plates. The absorbance at 600 nm was read every 5 min for 30 min, and the slopes of the lines were used to determine inhibition. HTS quality was assessed by Z-factor, with an overall value of ∼0.7.
Parasite Culture
The erythrocytic stages of all P. falciparum strains used in this study were cultured by standard methods27 in solutions of 5% human O+ hematocrit in RPMI 1640 medium (Life Technologies) supplemented with 28 mM NaHCO3, 25 mM HEPES, 0.5% (w/v) AlbuMAX II (Life Technologies), 50 mg/mL hypoxanthine, and 25 μg/mL gentamycin. Human blood was supplied from Interstate Blood Bank. The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents. Cultures were maintained at 37 °C in a gas mixture of 1.1% O2, 4% CO2, and 95% N2 and regularly synchronized by 5% sorbitol treatment.28
Parasite Strains
Laboratory reference strains used were the 3D7 (MRA-151) and a Dd2 clone derived from MR4 line MRA-156 (MR4, BEI Resources). Parasite lines with point mutations in PfDHODH were generated via in vitro resistance selections as described11,13 (Table S3). The Dd2-ScDHODH transgenic line expressing the Saccharomyces cerevisiae DHODH was a gift from Jeff Dvorin.29
Whole-Cell Dose–Response Assay and EC50 Determination
Drug susceptibility was measured by a growth assay as previously reported.19,20 To initially verify cell-based activity of the primary HTS hits, three doses (0.2, 2.0, 20 μM) were tested in duplicate. Compounds with at least 40% inhibitory activity at the 2 μM dose were then tested in full 12-point dose–response in a 72 h assay with a SYBR-green readout. EC50 values were calculated using a nonlinear regression curve fit in Prism Software version 7 (GraphPad).
Analysis of Cross-Resistance Profiling
The extent of cross-resistance or collateral sensitivity was determined by dividing the EC50 of a given mutant line by the EC50 of the parent reference strain and taking the log10 of this EC50 ratio. These values were visualized on a heatmap generated by MultiExperimentViewer (MeV) version 4.9.0 with hierarchical clustering based on Euclidean Distance using average linkage.30 In cases where the EC50 value was outside the range of the highest dose tested, the minimum estimate as listed in Table S4 was used as a placeholder value to generate the heatmap.
Extracellular Flux Analysis Using XFe24 Analyzer
Extracellular flux analysis was conducted as previously reported24 using either unbuffered RPMI medium or mitochondria assay solution (MAS). MAS was composed of mannitol (220 mM), sucrose (70 mM), KH2PO4 (10 mM), MgCl2 (5 mM), HEPES (2 mM), EGTA (1 mM), and fatty acid free BSA (0.2% w/v). Both media were adjusted to pH 7.4, and digitonin (2 μM) was added freshly on the day of the assay. Schizonts were freed from red blood cells (RBCs) by 0.01% saponin and seeded in an XF24-well microplate with wells pretreated with CellTak cell and tissue adhesive (Fisher Scientific, CB-40241) at 11 million cells/well. Test compounds and DHO were injected at prescheduled timing as 10× in the corresponding assay medium, and measurements were performed with the following setting: mix time, 30 s; wait time, 1 min 30 s; measure time, 3 min.
Isolation of Parasite Mitochondria and Cytochrome c Reductase Activity Assay
Mitochondria were isolated and cytochrome c reductase activity was measured using a modification of the method developed by Fry and Pudney25 as previously described.29 In brief, saponin-released parasites were lysed by N2 cavitation, and the clarified lysate was further enriched for mitochondria by centrifugation and separation on a sucrose gradient. Isolated mitochondria were washed of sucrose and stored at −80 °C until the time of the assay. Cytochrome c reductase activity was measured as follows: mitochondria (40 μg/mL) were diluted in reaction buffer (250 mM sucrose, 50 mM KH2PO4, 0.2 mM EDTA, 1 mM NaN3, and 2.5 mM KCN) containing 50 μM cytochrome c. Reactions were started by addition of 25 μM decylubiquinol and monitored by reduction of cytochrome c at 550 nm. To ensure the linearity of the enzymatic reaction, only data from the first 60 s were collected.
Acknowledgments
This work was funded in part by the Tres Cantos Open Lab Foundation award TC109 to L.S.R., D.F.W., M.J.-L.M., and F.J.G.; National Institutes of Health grant R01 AI093716 supporting D.F.W., A.K.L., L.S.R., T.S.-K., and R.E.K.M.; Bill and Melinda Gates Foundation Grand Challenges Exploration grant OPP1132451 supporting D.F.W., A.K.L., T.S.-K., R.E.K.M., M.J.L.-M., and F.J.G.; the Harvard Malaria Initiative support from ExxonMobil Foundation to D.F.W., R.E.K.M., and L.S.R. We are grateful to Paul Hinkson for technical support. We thank Genzyme (Waltham, MA) for the kind gift of Genz-669178. We are also thankful to Jeff Dvorin for gift of the ScDHODH transgenic parasites and Rachel Daniels for valuable discussions and review of the manuscript.
Glossary
Abbreviations
- DHO
dihydroorotate
- DHODH
dihydroorotate dehydrogenase
- ETC
electron transport chain
- G3P
glycerol 3-phosphate
- MAS
mitochondria assay solution
- OCR
oxygen consumption rate
- RBCs
red blood cells
- WT
wild-type
- MT
mutant
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00217.
Table S1, screening results; Figure S1, screening cascade and correlation between enzyme and cell-based assays; Figure S2, structures of 23 secondary hits; Table S2, activities of 23 secondary hits in wild-type and scDHODH-expressing transgenic parasites; Table S3, mitochondrial Cytbc1 activity of compound 21; Table S4, mutant cell lines used in the study; Table S5, cross-resistance profiling in mutant DHODH cell lines (PDF)
Author Present Address
§ L.S.R.: Department of Microbiology and Immunology, Columbia University Medical Center, 701 W. 168th Street, New York, NY 10032.
Author Contributions
† L.S.R., M.J.L.-M., T.S.-K., and R.E.K.M. contributed equally.
Author Contributions
M.J.L.-M. and L.S.R. conducted enzyme-based HTS screening and analyzed the results. R.E.K.M. and T.S.-K. conducted cell-based experiments and analyzed the results. A.K.L., F.J.G., and D.F.W. reviewed and analyzed the data. L.S.R., M.J.L.-M., T.S.-K., R.E.K.M., A.K.L., and D.F.W. wrote the manuscript. All authors reviewed the final manuscript draft. A.K.L., D.F.W., and F.J.G. conceived the idea for the project.
The authors declare no competing financial interest.
This paper was published on the Web on January 22, 2018. PMID references for triazolopyrimidine clinical candidate DSM265 have been added to the paper, and the corrected version was reposted on January 31, 2018.
Supplementary Material
References
- World Health Organization (2016), World Malaria Report 2016, World Health Organization, Geneva, Switzerland. [Google Scholar]
- Wongsrichanalai C.; Pickard A. L.; Wernsdorfer W. H.; Meshnick S. R. (2002) Epidemiology of drug-resistant malaria. Lancet Infect. Dis. 2, 209–218. 10.1016/S1473-3099(02)00239-6. [DOI] [PubMed] [Google Scholar]
- Holohan C.; Van Schaeybroeck S.; Longley D. B.; Johnston P. G. (2013) Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726. 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Allen R. C.; Engelstadter J.; Bonhoeffer S.; McDonald B. A.; Hall A. R. (2017) Reversing resistance: different routes and common themes across pathogens. Proc. R. Soc. London, Ser. B 284, 20171619. 10.1098/rspb.2017.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegreness M.; Shoresh N.; Damian D.; Hartl D.; Kishony R. (2008) Accelerated evolution of resistance in multidrug environments. Proc. Natl. Acad. Sci. U. S. A. 105, 13977–13981. 10.1073/pnas.0805965105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noedl H.; Se Y.; Schaecher K.; Smith B. L.; Socheat D.; Fukuda M. M. (2008) and Artemisinin Resistance in Cambodia 1 Study, C. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 359, 2619–2620. 10.1056/NEJMc0805011. [DOI] [PubMed] [Google Scholar]
- Breman J. G. (2012) Resistance to artemisinin-based combination therapy. Lancet Infect. Dis. 12, 820–822. 10.1016/S1473-3099(12)70226-8. [DOI] [PubMed] [Google Scholar]
- Mita T.; Ohashi J.; Venkatesan M.; Marma A. S.; Nakamura M.; Plowe C. V.; Tanabe K. (2014) Ordered accumulation of mutations conferring resistance to sulfadoxine-pyrimethamine in the Plasmodium falciparum parasite. J. Infect. Dis. 209, 130–139. 10.1093/infdis/jit415. [DOI] [PubMed] [Google Scholar]
- Nagesha H. S.; Din S.; Casey G. J.; Susanti A. I.; Fryauff D. J.; Reeder J. C.; Cowman A. F. (2001) Mutations in the pfmdr1, dhfr and dhps genes of Plasmodium falciparum are associated with in-vivo drug resistance in West Papua, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 95, 43–49. 10.1016/S0035-9203(01)90329-3. [DOI] [PubMed] [Google Scholar]
- Fischbach M. A. (2011) Combination therapies for combating antimicrobial resistance. Curr. Opin. Microbiol. 14, 519–523. 10.1016/j.mib.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross L. S.; Gamo F. J.; Lafuente-Monasterio M. J.; Singh O. M.; Rowland P.; Wiegand R. C.; Wirth D. F. (2014) In vitro resistance selections for Plasmodium falciparum dihydroorotate dehydrogenase inhibitors give mutants with multiple point mutations in the drug-binding site and altered growth. J. Biol. Chem. 289, 17980–17995. 10.1074/jbc.M114.558353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal C.; Papp B.; Lazar V. (2015) Collateral sensitivity of antibiotic-resistant microbes. Trends Microbiol. 23, 401–407. 10.1016/j.tim.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukens A. K.; Ross L. S.; Heidebrecht R.; Javier Gamo F.; Lafuente-Monasterio M. J.; Booker M. L.; Hartl D. L.; Wiegand R. C.; Wirth D. F. (2014) Harnessing evolutionary fitness in Plasmodium falciparum for drug discovery and suppressing resistance. Proc. Natl. Acad. Sci. U. S. A. 111, 799–804. 10.1073/pnas.1320886110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; McNamara C. W.; Kim S. W.; Kato T. S.; Li F.; Teng C. H.; Gagaring K.; Manary M. J.; Barboa R.; Meister S.; Kuhen K.; Vinetz J. M.; Chatterjee A. K.; Winzeler E. A. (2015) Mutations in the P-type cation-transporter ATPase 4, PfATP4, mediate resistance to both aminopyrazole and spiroindolone antimalarials. ACS Chem. Biol. 10, 413–420. 10.1021/cb500616x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez de Evgrafov M.; Gumpert H.; Munck C.; Thomsen T. T.; Sommer M. O. (2015) Collateral Resistance and Sensitivity Modulate Evolution of High-Level Resistance to Drug Combination Treatment in Staphylococcus aureus. Mol. Biol. Evol. 32, 1175–1185. 10.1093/molbev/msv006. [DOI] [PubMed] [Google Scholar]
- Gonzales P. R.; Pesesky M. W.; Bouley R.; Ballard A.; Biddy B. A.; Suckow M. A.; Wolter W. R.; Schroeder V. A.; Burnham C. A.; Mobashery S.; Chang M.; Dantas G. (2015) Synergistic, collaterally sensitive beta-lactam combinations suppress resistance in MRSA. Nat. Chem. Biol. 11, 855–861. 10.1038/nchembio.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan A.; Nichol D.; Kinose F.; Abazeed M. E.; Marusyk A.; Haura E. B.; Scott J. G. (2017) Collateral sensitivity networks reveal evolutionary instability and novel treatment strategies in ALK mutated non-small cell lung cancer. Sci. Rep. 7, 1232. 10.1038/s41598-017-00791-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P.; Karhinen L.; Szwajda A.; Jha S. K.; Yadav B.; Aittokallio T.; Wennerberg K. (2016) Identification of selective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer 15, 34. 10.1186/s12943-016-0517-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V.; Booker M.; Kramer M.; Ross L.; Celatka C. A.; Kennedy L. M.; Dvorin J. D.; Duraisingh M. T.; Sliz P.; Wirth D. F.; Clardy J. (2008) Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 283, 35078–35085. 10.1074/jbc.M804990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin J.; Michnoff C. H.; Malmquist N. A.; White J.; Roth M. G.; Rathod P. K.; Phillips M. A. (2005) High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 280, 21847–21853. 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- Painter H. J.; Morrisey J. M.; Mather M. W.; Vaidya A. B. (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446, 88–91. 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- Phillips M. A.; Lotharius J.; Marsh K.; White J.; Dayan A.; White K. L.; et al. (2015) A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 7 (296), 296ra111. 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy J. S.; Lotharius J.; Rückle T.; Chalon S.; Phillips M. A.; Elliott S.; Sekuloski S.; Griffin P.; Ng C. L.; Fidock D. A.; Marquart L.; Williams N. S.; Gobeau N.; Bebrevska L.; Rosario M.; Marsh K.; Möhrle J. J. (2017) Safety, tolerability, pharmacokinetics, and activity of the novel long-acting antimalarial DSM265: a two-part first-in-human phase 1a/1b randomised study. Lancet Infect. Dis. 17 (6), 626–635. 10.1016/S1473-3099(17)30171-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerman B. K.; Elsworth B.; Cobbold S. A.; Nie C. Q.; McConville M. J.; Crabb B. S.; Gilson P. R. (2016) Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci. Rep. 6, 37502. 10.1038/srep37502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphemot R.; Lafuente-Monasterio M. J.; Gamo-Benito F. J.; Clardy J.; Derbyshire E. R. (2016) Discovery of Dual-Stage Malaria Inhibitors with New Targets. Antimicrob. Agents Chemother. 60, 1430–1437. 10.1128/AAC.02110-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata-Kato T.; Wirth D. F. (2016) A Novel Methodology for Bioenergetic Analysis of Plasmodium falciparum Reveals a Glucose-Regulated Metabolic Shift and Enables Mode of Action Analyses of Mitochondrial Inhibitors. ACS Infect. Dis. 2, 903–916. 10.1021/acsinfecdis.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M.; Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553. 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- Phillips M. A.; Gujjar R.; Malmquist N. A.; White J.; El Mazouni F.; Baldwin J.; Rathod P. K. (2008) Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum. J. Med. Chem. 51, 3649–3653. 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager W.; Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Lambros C.; Vanderberg J. P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420. 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- Dong C. K.; Urgaonkar S.; Cortese J. F.; Gamo F. J.; Garcia-Bustos J. F.; Lafuente M. J.; Patel V.; Ross L.; Coleman B. I.; Derbyshire E. R.; Clish C. B.; Serrano A. E.; Cromwell M.; Barker R. H. Jr.; Dvorin J. D.; Duraisingh M. T.; Wirth D. F.; Clardy J.; Mazitschek R. (2011) Identification and validation of tetracyclic benzothiazepines as Plasmodium falciparum cytochrome bc1 inhibitors. Chem. Biol. 18, 1602–1610. 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A. I.; Sharov V.; White J.; Li J.; Liang W.; Bhagabati N.; Braisted J.; Klapa M.; Currier T.; Thiagarajan M.; Sturn A.; Snuffin M.; Rezantsev A.; Popov D.; Ryltsov A.; Kostukovich E.; Borisovsky I.; Liu Z.; Vinsavich A.; Trush V.; Quackenbush J. (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378. [DOI] [PubMed] [Google Scholar]
- McNicholas S.; Potterton E.; Wilson K. S.; Noble M. E. (2011) Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 386–394. 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.