

Abstract
Drug resistance and toxicity are major limitations of cancer treatment and frequently occurs in melanoma therapy. Nanotechnology can decrease drug resistance by improving drug delivery, with limited toxicity. This study details the development of nanoparticles containing arachidonyl trifluoromethyl ketone (ATK), a cytosolic phospholipase A2 inhibitor, which can inhibit multiple key pathways responsible for the development of recurrent resistant disease. Free ATK is toxic, limiting its efficacy as a therapeutic agent. Hence, a novel nanoliposomal delivery system called NanoATK was developed, which loads 61.7% of the compound and was stable at 4°C for 12 weeks. The formulation decreased toxicity-enabling administration of higher doses, which was more effective at killing melanoma cells compared to free-ATK. Mechanistically, NanoATK decreased cellular proliferation and triggered apoptosis to inhibit melanoma xenograft tumor growth without affecting animal weight. Functionally, it inhibited the cPLA2, AKT, and STAT3 pathways. Our results suggest the successful preclinical development of a unique nanoliposomal formulation containing ATK for the treatment of melanoma.
Keywords: Phospholipase A2, cyclooxygenase, inflammatory cancer, lipid mediators, arachidonyl trifluoromethyl ketone and nanoliposomes
Graphical abstract
Cytosolic PLA2 represents an attractive target for cancers in which inflammation plays a key role. Arachidonyl trifluoromethyl ketone (ATK) inhibits cytosolic PLA2 but the utility of ATK as a therapeutic agent has not been investigated due to its high lipophilicity and low solubility. A novel PEGylated ATK nanoliposomal system called NanoATK was developed that delivers ATK to cancer cells. NanoATK enhances stability, increases circulation time and lowers toxicity. NanoATK retarded xenografted melanoma tumor growth by up to 60% without affecting animal weight compared to 30% reduction in tumor development when the maximum tolerated dose of free ATK was administered.

Background
Over the past decade, significant advances have occurred in the field of melanoma therapy, including the discovery of B-RAF activating mutations and development of targeted B-RAF inhibitors as well as the expansion of clinically effective, immunotherapies 1. Nevertheless, drug resistance and toxicity remain recurring issues plaguing this disease, necessitating the design of drugs that circumvent mechanisms of resistance 2. Hence drugs targeting multiple signaling cascades to limit resistance are being developed.
Arachidonic acid (AA) signaling is a critical inflammatory pathway that is upregulated in most cancers 3. In mammalian cells, under normal physiological conditions, AA is stored as a phospholipid and released by cytosolic phospholipase A2 (cPLA2) 3. AA is an important precursor of eicosanoid synthesis that includes prostaglandins, leukotrienes, and thromboxanes, which have diverse functions in signal transduction and other biological processes 3. Mediators and cellular effectors of inflammation are important constituents of the tumor microenvironment 4. Inflammation develops in response to a malignant tumor, or an oncogenic change inducing inflammation to promote tumor development 5. Mechanisms of cancer related inflammation are being unraveled, leading to the identification of new targets that could potentially improve the treatment of melanoma 5.
Cyclooxygenases (COX) are a group of enzymes responsible for the production of prostaglandins and thromboxanes from AA 3. COX inhibitors have been tested for treating cancer but are not effective alone since the NSAIDs cannot block the generation of leukotrienes from AA 6. Gene knockdown of cPLA2 led to significant reduction in cell growth and tumor development in hormone refractory prostate cancer 7. Thus, cPLA2 represents an attractive target for cancers in which inflammation plays a key role 7. cPLA2 inhibitors such as RSC-3388 and CAY10502 have been evaluated for anti-inflammatory activity 8, 9. However, no cPLA2 inhibitor has been developed or tested for anti-cancer activity since these compounds have solubility and toxicity-related issues 10.
Arachidonyl trifluoromethyl ketone (AACOCF3; ATK) is an analog of AA that functions by inhibiting the 85-kDa cytosolic form of PLA2 without affecting the secretary PLA2 (sPLA2) and has a minimal effect on calcium independent PLA2 (iPLA2) 11. cPLA2 is a crucial mediator of eicosanoid biosynthesis generating free AA 11. ATK prevents AA release by inhibiting the activity of cPLA2 11. ATK inhibits several cPLA2 downstream mediators of inflammation including IL-1β, TNFα, and PGE2 12. The utility of ATK as a therapeutic agent has not been investigated due to its high lipophilicity and low solubility 13, 14. Since lipid-based delivery can improve the solubility of many lipophilic drugs 15-17, we determined whether loading ATK into nanoliposomes could increase the dose that could be administered to improve its tumor inhibitory efficacy.
A novel PEGylated ATK nanoliposomal system called NanoATK was developed that loads and delivers ATK to cancer cells. The nanoparticles have an average size of 70 nm and are stable at 4°C for 12 weeks. NanoATK retarded xenografted melanoma tumor growth by up to 60% without affecting animal weight compared to only a negligible approximately 30% reduction in tumor development when the maximum tolerated dose (MTD) of free ATK was administered in DMSO. Mechanistically, NanoATK inhibited cPLA2, AKT and STAT3 pathways to increase cellular apoptosis and decrease proliferation. Thus, a potential therapeutic formulation has been developed for delivering ATK to decreased melanoma tumor growth.
Material and Methods
Cell lines and culture conditions
Normal human fibroblast cell line FF2441 was provided by Craig Myers, Penn State, Hershey, PA. Mutant V600E-BRAF human melanoma cell line 1205 Lu was provided by Meenhard Herlyn, Wistar Institute, Philadelphia, PA and UACC 903 was provided by Mark Nelson, University of Arizona, Tucson, AZ. Wild type BRAF melanoma cell lines C8161.Cl9 was provided by Danny Welch, University of Kansas, Kansas City, KS and MelJuSo was provided by Judith Johnson, Institute for Immunology, Germany. Cell lines were maintained in a 37°C humidified 5% CO2 atmosphere incubator and periodically monitored for phenotypic and genotypic characteristics. The tumorigenic potential was assessed annually to validate and confirm cell line identity.
Analysis of human melanoma patient tumors
Melanoma tumor specimens from human patients were randomly selected according to the protocols approved by the Institutional Review Board at The Pennsylvania State University and the Cooperative Human Tissue Network. Informed consent was provided according to the Declaration of Helsinki. Tissue samples from 26 patients with metastatic melanoma were surgically removed, snap-frozen in liquid nitrogen, and stored at −80°C until preparation of protein lysate. Tumors were pulverized with a mortar and pestle that had been chilled in liquid nitrogen and protein lysates extracted as reported previously 18. Western blotting was used to measure levels of cPLA2 protein, normalized to ERK1/2 using ImageJ software.
cPLA2 activity assay
cPLA2 enzyme activity in melanoma cell line lysate was assayed using a commercial cPLA2 activity kit from Cayman chemicals according to manufacturer's protocol. UACC 903 cells were lysed in Hepes buffer pH 7.4 containing 1 mM EDTA. For cPLA2 specific activity in total protein lysates, residual sPLA2 was removed using a membrane filter with a molecular weight cut off of 30 kDa using an Amicon centrifuge concentrator. To avoid any interference with iPLA2 activity, cell lysate was treated with a specific inhibitor, bromoenol lactone. Cell lysates were treated with ATK or NanoATK (0.5 to 10 μmol/L). DMSO and empty nanoliposome served as the negative control for 100% activity. Reactions were initiated by addition of arachidonoyl thio-PC as the substrate for 1 hour followed by addition of 5,5′-dithio-bis-(2-Nitrobenzoic acid) (DTNB). Thio-PC released from the substrate due to cPLA2 activity reacts with DTNB to form a colorimetric reaction, which can be measured at 414 nm 12.
Preparation of NanoATK
ATK was encapsulated into a nanoliposome by combining L-α-phosphatidylcholine (ePC) and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N- [methoxy(polyethylene glycol)-2000] ammonium salt (DPPE-PEG-2000) in chloroform at 80:20 mol % for a final lipid concentration of 25 mg/mL (Avanti Polar Lipids). 5.0 mg of ATK was added to 1.0 mL of nanoliposome solution. The mixture was then dried under nitrogen gas and resuspended in 0.9% saline at 60°C. Following rehydration, the mixture was sonicated at 60°C for 10 minutes followed by extrusion at 60°C through a 100-nm polycarbonate membrane using an Avanti Mini Extruder (Avanti Polar Lipids). Particle size and charge characteristics were measured using a Malvern Zetasizer (Malvern Instruments) 17, 19, 20.
Characterization of NanoATK
(a) Drug encapsulation
Encapsulation of ATK in the liposomal formulation was estimated by UV-visible spectrophotometry (SPECTRAmax M2 plate reader) 17, 19, 20. Free drug not incorporated into the nanoliposomes was separated using a 10 kDa Centricon filter tube (Millipore). 1.0 mL of NanoATK in a 10 kDa Centricon filter tube was centrifuged at 3,750 rpm for 30 minutes. Next, 0.5 mL of purified NanoATK was combined with 0.5 mL of 1:1 ratio of chloroform to methanol to destroy the liposomal structure and release the drug into the solution. Following vortexing for 10 minutes, precipitated lipids were separated following centrifugation at 10,000 rpm for 15 minutes. The supernatant was then used to measure the amount of ATK, calculated from a standard curve from 0.01 and 0.1 mg/mL. 1:1 ratio of chloroform to methanol was used as the reference blank. Percentage drug incorporated in the liposome was calculated as the free drug/total drug × 100 17, 19, 20.
(b) Stability
Stability of NanoATK stored at 4°C was assessed weekly, comparing size and zeta potential charge using the Malvern Zetasizer. IC50 was measured on UACC 903 cells by MTS assay to assess cell killing efficacy by NanoATK 17, 19, 20.
(c) In-vitro drug-release kinetics of NanoATK
Drug release kinetics was measured for 1 mL of purified NanoATK by dialysis in 1 L of 10 mM reduced glutathione at room temperature through a molecular weight cut off 25 kDa membrane (Spectra Por) 21. NanoATK from the dialysis bag was removed at different time points and the amount of ATK released at each time point estimated using UV-visible spectrophotometry 17, 19.
(d) Hemolytic activity
Fresh human blood was drawn and placed into an EDTA test tube. Erythrocytes were separated from the plasma by centrifugation at 1,500 rpm for 10 minutes at 4°C using PBS. A 5% v/v solution of human erythrocytes in PBS was treated with ATK or NanoATK (10 - 40 μmol/L), ethanol (negative control) and empty liposome (negative control) or 1% Triton X-100 (positive control). Samples were incubated at 37°C for 60 minutes and then centrifuged at 12,000 rpm for 10 minutes. Next, supernatants were transferred to a 96-well plate, and absorption at 540 nm was measured. Amount of hemoglobin released in the presence of 1% Triton X-100 was set at 100% lysis and % hemolysis was calculated as absorbance of the samples at 540 nm/absorbance of the positive control X100 17.
Cell viability studies
Viability studies for melanoma and normal cell lines following treatment with NanoATK were measured by MTS assay 18, 22-29. 5×103 cells were seeded per well into a 96-well plate in 100 μL of media and after 48 hours, cells were treated with 10-100 μmol/L of NanoATK for 72 hours for melanoma cell lines (UACC 903, 1205 Lu, C8161. Cl9, and MelJuSo) or normal cell lines (FOM103 and FF2441). IC50 killing efficacy of NanoATK (in μmol/L) for each respective cell line was measured from 3 independent experiments using GraphPad Prism version 4.01.
Cellular proliferation and apoptosis rates
5×103 UACC 903 or 1205 Lu melanoma cells were seeded in 96-well plates, followed by treatment with 10 to 60 μmol/L of NanoATK for 72 hours. Percentage proliferating or apoptotic cells were quantified by a colorimetric cell proliferation ELISA BrdU kit (Roche Applied Sciences, Indianapolis, IN) or fluorimetric Apo-ONE Homogenous caspase-3/7 assay kit as previously reported (Promega, Madison, WI) 18, 22-29.
Western blot analysis
Melanoma cell lysates were harvested by addition of RIPA lysis buffer and samples processed as described previously 17, 23-26. 0.75×106 melanoma cells were plated in 100 mm culture dishes and 48 hours later, treated with NanoATK (20 - 50 μmol/L) for 24 hours. Protein lysates were collected for Western blotting. Blots were probed with antibodies according to each supplier's recommendations: antibodies to pAKT (S473), AKT (pan) (11E7), pSTAT3 (Y705), STAT3, cPLA2, and phospho cPLA2 (S505) from Cell Signaling Technology; CYCLIN D1, p21, p27, COX-2 and secondary antibodies from Santa Cruz Biotechnology. Immunoblots were visualized using the enhanced chemiluminescence detection system or Super Signal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific).
Tumorigenicity assessments
Experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee at The Pennsylvania State University College of Medicine and ensured the humane care of the animals. Tumor kinetics were measured by subcutaneous injection of 1×106 UACC 903 or 1205 Lu cells in 0.2 mL of DMEM supplemented with 10% FBS above both left and right rib cages of 3 to 4 week-old female athymic-foxn1nu nude mice (Harlan). Eight days later, when a fully vascularized 50-75 mm3 tumor had formed, mice were randomly divided into vehicle control and experimental groups (4 mice/group; 2 tumors/mice) and treated intravenously with nanoliposomes or intraperitoneally injected with free drug daily for 3-4 weeks. Experiments were undertaken independently twice. Body weight and dimensions of developing tumors were measured on alternate days 30-32.
Statistical Analysis
Statistical significance was determined using the One-way / Two-way ANOVA GraphPad PRISM Version 4.01 software. Results represent at least two to three independent experiments and are shown as averages ± S.E.M. Number of asterisks in the figures indicates the level of statistical significance as follows: *, p < 0.05; **, p < 0.01 and ***, p < 0.001.
Results
cPLA2 expression is elevated in melanoma patient tumors and cell lines
Elevated cPLA2 protein expression levels occur in various cancers including those of the colon 33, blood 34, prostate 7 and breast 35. cPLA2 expression was measured by Western blotting in a panel of melanoma patient tumors and melanoma cell lines representing radial growth phase (WM35), vertical growth phase (WM115, WM278, WM3211) and metastatic stage tumors (1205 Lu, A375M, UACC 903, WM793, WM164). Ninety-six percent of melanoma patient tumors had elevated cPLA2 expression compared to normal melanocytes (FOM103) indicating a potentially significant role of cPLA2 in this disease (Fig. 1A). Similarly, all melanoma cell lines except for A375M had higher levels of cPLA2 expression compared to either normal control melanocytes (FOM103) or fibroblasts (FF2441) (Fig. 1B). There was no significant correlation with the expression levels of cPLA2 with the stage of cancer or BRAF mutational status suggesting a broad potential to target cPLA2 in melanoma.
Figure 1. cPLA2 expression is elevated in melanoma.

1A. Elevated expression levels of cPLA2 in tumors from metastatic melanoma patients. Western blot analysis of melanoma patient tumors suggests increased expression of cPLA2 protein. ERK1/2 served as a protein loading control against which to normalize cPLA2 expression. 1B. cPLA2 expression increased in all melanoma cell lines isolated at different stages in melanoma progression except for A375M. ERK1/2 was used as a control for equal protein loading.
To establish the importance of cPLA2 signaling in melanoma, approaches to target cPLA2 were explored and ATK (Fig. 2A) was identified as a specific inhibitor that could kill melanoma cells 13, 36, 37. Various in-vitro studies found ATK to be a slow inhibitor of cPLA2 with 500-fold more potency in inhibiting cPLA2 than sPLA2 and iPLA2 13, 37. ATK dissolved in DMSO on average killed melanoma cells 3-fold more effectively than average normal cells (Fig. 2B). The average IC50 of ATK for killing normal FOM103 and FF2441 was 60 μmol/L compared with 22 μmol/L for the average of four melanoma cells. The specificity of ATK for inhibiting cPLA2 was measured in melanoma cell lysates with IC50 found to be 1.85 μmol/L (Fig. 2C). In order to evaluate the efficacy in tumor models, a 14-day repeated dose study to identify the MTD for the free form of ATK was measured from 5 to 30 mg/kg body weight. Animal mortality occurred at 20 and 30 mg/kg body weight when administered i.p. daily (Table 1). Weight loss of 14.5% occurred with 15 mg/kg of free ATK. Hence, the MTD of free ATK was established as 10 mg/kg and the efficacy was evaluated in a UACC 903 xenograft nude mouse model. Vascularized tumors formed by day 8 and mice were treated daily via intraperitoneal injections of ATK at 5 and 10 mg/kg in DMSO or vehicle control for 3-4 weeks. ATK led to an approximately 30% reduction in tumor volume (Fig. 2D). To enhance the efficacy and reduce potential toxicity, ATK was encapsulated into a nanoparticle 14.
Figure 2. Inhibition of cPLA2 by ATK led to a minimal decrease in tumor development.

2A. Structure of arachidonyl trifluoromethyl ketone (ATK). 2B. Efficacy of ATK for killing normal versus melanoma cells. ATK was 3-fold more effective at killing melanoma cell lines than normal cells. 2C. cPLA2 inhibitory activity of ATK. Activity of cPLA2 following treatment with ATK was evaluated in UACC 903 cells using a commercial cPLA2 activity kit. 2D. Effect of ATK on melanoma tumor growth. Nude mice with pre-existing tumors were treated with i.p. injections of ATK dissolved in DMSO at a daily dose of 5 and 10 mg/kg body weight. DMSO was used as the vehicle control. ATK treated animals did not show any significant changes in body weight (2D; inset).
Table 1. Maximum tolerated dose (MTD) and in-vivo tumor inhibitory efficacy for ATK.
Repeated 14-day dosing of ATK administered to Swiss Webster mice and body weights monitored every two days to establish the MTD. Similarly, nude mice with pre-existing tumors were treated with i.p. injections of free ATK for in-vivo tumor inhibitory efficacy assessment (3 mice/group; 2 tumors/mice). Experiments were undertaken independently twice. Body weight and dimensions of developing tumors in nude mice were measured on alternate days.
| Drug dose (mg/kg body weight) | ATK administered in DMSO | |
|---|---|---|
| % weight loss at day 14 in Swiss Webster mice | % in-vivo tumor inhibition at day 21 in Nude mice | |
| 5 | No weight loss | 12.9 ± 6.30 |
| 10 | 3.1 ± 3.52 | 32.5 ± 5.73 |
| 15 | 14.5 ± 0.95 | - |
| 20 | Death in 10 days | - |
| 30 | Death in 8 days | - |
Development of NanoATK
To identify the optimal lipid formulation to enable loading of sufficient quantity of ATK into nanoliposomes, several formulations were evaluated and results for size, zeta potential, membrane fluidity and surface hydration were assessed. The liposomal formulation selected included the PEGylated nanoliposomal formulation made of 80:20 mol % ePC: DPPE PEG-2000 called NanoATK, which is diagrammatically illustrated in Figure 3A. NanoATK was not predicted to have altered cPLA2 inhibitory activity when formulated as nanoliposomes compared to free drug, and as predicted, it retained cPLA2 inhibitory activity similar to that of free ATK dissolved in ethanol (Fig. 3B). Size of the nanoliposomes in saline ranged between 67.58 ± 5.87 nm with a zeta potential of -0.35 ± 0.04 mV (Fig. 3C).
Figure 3. Development of NanoATK, a nanoliposomal formulation of ATK.

3A. Schematic representation of NanoATK with a phospholipid bilayer and the ATK drug embedded in the lipid layers. 3B. cPLA2 inhibitory activity of NanoATK. The activity of cPLA2 following treatment with NanoATK compared to the free drug was evaluated in UACC 903 cells using a commercial cPLA2 activity kit. 3C. Size and charge of NanoATK. Size of NanoATK in saline was approximately 67 nm with an approximately neutral charge. The particle size and charge characteristics were established using a Malvern Zetasizer. 3D. Loading efficiency of NanoATK. NanoATK was centrifuged in 10 kDa Centricon filter columns, nanoliposomal integrity was destroyed by adding a 1:1 ratio of chloroform, methanol and amount of drug retained was evaluated spectrophotometrically. 3E. Stability of NanoATK. ATK nanoliposomes were stored in saline at 4°C for 12 weeks. NanoATK maintained cell killing IC50 of approximately 30 μmol/L with no significant change in size or charge of the nanoliposomes. 3F. ATK release from the NanoATK liposomal formulation. ATK nanoliposomes were suspended in a dialysis bag with a cut off pore size of 25 kDa in 1L of 10 mM Glutathione and the drug release measured at the indicated time points. 3G. Hemolytic activity of NanoATK was compared with free ATK in DMSO. Free ATK in DMSO-induced 2-3% hemolysis compared with 0.5% for nanoliposomal ATK. Hemoglobin released following erythrocyte lysis in TritonX-100 served as a control for 100% lysis. Percentage of hemolysis was calculated as absorbance of the samples at 540 nm/absorbance compared to the positive control Triton X-100.
Physiochemical and stability characterization of ATK nanoliposomes
Free ATK was solubilized during nanoliposome manufacture but a certain amount predicted to be loosely bound to the exterior lipids of the nanoparticle. To measure the proportion encapsulated in the lipid shell or free loosely bound drug, the free drug was removed by Centricon Ultra centrifugation followed by lysis of the nanoliposomes. The amount of each drug in the lipid shell was then estimated using the absorption maxima of the compound extrapolated from a standard curve of ATK ranging from 0.01 and 0.1 mg/mL. Encapsulation efficiency for ATK in nanoliposomes was found to be 62% (Fig. 3D).
The stability of NanoATK was evaluated for a period of 12 weeks. Size and charge of the NanoATK along with its IC50 for inhibiting survival of UACC 903 cells was estimated once every two weeks. Size, charge as well as the IC50 of NanoATK did not significantly vary over 12 weeks, indicating stability of the formulation (Fig. 3E). Drug release kinetics of ATK was measured by dialyzing NanoATK in 10 mM glutathione over 72 hours, at which point the nanoliposomes were lysed and remaining drug estimated by extrapolation off of a standard curve (Fig. 3F). ATK release occurred at a steady rate over 72 hours with approximately 70% release at 48-72 hours (Fig. 3F). The nanoliposomal formulation reduced hemolysis occurring with the free drug as measured by the hemolytic assay 17. A hemolytic assay was performed to examine whether NanoATK caused the same level of red blood cell lysis as ATK in ethanol. ATK at 40 μmol/L in ethanol induced 2.9% hemolysis, which was significantly reduced to 0.55% by NanoATK (Fig. 3G).
NanoATK kills cancer cells more effectively than the normal cells
Efficacy of free ATK versus NanoATK for killing metastatic melanoma cells was compared to normal cells (Table 2). There was no significant difference between ATK and NanoATK treatments in normal control cells versus melanoma cells indicating that the activity of ATK is not affected by nanoliposomal formulation. On average NanoATK was 2-fold less toxic to normal control cells compared to melanoma cell lines (Table 2). NanoATK concentration of 58 to 63 μmol/L killed 50% of normal cells following 72 hours exposure compared with 20 to 28 μmol/L for cell lines derived from advanced stage melanomas (Table 2). NanoATK inhibited the growth of melanoma cell lines at an average IC50 of 20 μmol/L irrespective of B-RAF mutational status.
Table 2. NanoATK killed melanoma more effectively than normal cells.
Normal and melanoma cells were seeded into a 96-well plate and, after 48 hours, treated with increasing concentrations of free ATK (control) or NanoATK for 72 hours. Number of viable cells was measured using MTS and percentage decrease in viability calculated. IC50 values in μmol/L for each respective cell line was measured from 3 independent experiments using GraphPad Prism version 4.01 (GraphPad Software, La Jolla, CA).
| Normal cell lines | Melanoma cell lines | |||||
|---|---|---|---|---|---|---|
| FF2441 | FOM103 | UACC 903 | 1205 Lu | C8161.Cl9 | MelJuSo | |
| ATK | 63.33 ± 10.3 | 57.93 ±10.4 | 26.32 ± 2.1 | 26.12 ± 4.4 | 18.55 ± 3.1 | 22.76 ± 4.8 |
| NanoATK | 58.32 ± 9.3 | 63.54 ±12.8 | 28.01 ± 3.3 | 25.32 ± 3.7 | 19.67 ± 2.2 | 20.85 ± 7.1 |
| Mutational status | ||||||
| B-RAF | --- | --- | V600E | V600E | WT | WT |
| PTEN | --- | --- | Deleted | Deleted | WT | WT |
| NRAS | --- | --- | WT | WT | WT | Q61L |
NanoATK reduced melanoma tumor growth with negligible toxicity
In order to determine whether the MTD improved with NanoATK compared to free ATK, a 14-day repeated dose study was measured at 5-40 mg/kg (Supplementary Table 1). There was no weight loss in animals with treatment up to 40 mg/kg of NanoATK, indicating an increase in the MTD with the nanoliposomal formulation. Higher doses of NanoATK could not be evaluated, since this was the maximal dose that could be loaded into a nanoliposome. NanoATK dose dependently reduced melanoma tumor growth from 5-40 mg/kg with maximum efficacy at 30 and 40 mg/kg in the initial evaluation on pre-existing xenografted tumors following subcutaneous injection of cells in nude mice (Supplementary Table 1). Based on the initial evaluation, we selected the doses of 30 and 40 mg/kg body weight of NanoATK for daily intravenous administration. NanoATK at 30-40 mg/kg led to significantly reduced tumor volume by 58% and 55% for UACC 903 (Fig. 4A) and 1205 Lu (Fig. 4B) cell lines, respectively, compared to control nanoliposomes lacking ATK. Tumor growth and body weight measured at 2-day intervals for 3 to 4 weeks showed no significant differences between NanoATK or nanoliposomes lacking ATK, suggesting negligible toxicity (Figs. 4A & 4B; inset).
Figure 4. NanoATK inhibits melanoma tumor growth.

4A & 4B. Effect of NanoATK on melanoma tumor growth. Nude mice with pre-existing tumors were treated with i.v. injections of NanoATK at a daily dose of 30 or 40 mg/kg body weight. Empty nanoliposomes were used as controls. NanoATK significantly decreased tumor volume by 58% and 55% for UACC 903 (4A) and 1205 Lu (4B) cells respectively, compared to vehicle control treated animals. Tumor growth and body weight were measured at 2-day intervals for 3 to 4 weeks. NanoATK treated animals did not show any significant changes in body weight, suggesting negligible toxicity (4A & 4B; inset).
NanoATK decreased cellular proliferation and triggered apoptosis of melanoma cells
To identify the mechanism through which NanoATK kills cancer cells, cellular proliferation and apoptosis rates following treatment were measured 17. Increasing concentrations of NanoATK from 10 to 60 μmol/L decreased cellular proliferative potential of UACC 903 and 1205 Lu cells, which was measured by bromodeoxyuridine (BrdU) incorporation. For both cell lines, 40 μmol/L NanoATK reduced BrdU incorporation by more than 50% (Fig. 5A). Cellular apoptosis was measured in UACC 903 and 1205 Lu cells by a caspase-3/7 enzymatic activity. Dose dependent increase in caspase-3/7 activity was observed in both these cell lines upon NanoATK treatment (Fig. 5B). Furthermore, the levels of proliferation and apoptosis markers were also evaluated by Western blot analysis (Figs. 5C & 5D). NanoATK led to decreased levels of CYCLIN D1 and increased levels of p21 and p27, which are key indicators of proliferation in melanoma cells 17 (Fig. 5C). Moreover, NanoATK also led to a dose dependent increase in the levels of cleaved PARP and LC3B indicators of apoptosis and autophagy respectively (Fig. 5D).
Figure 5. NanoATK decreased melanoma cell survival and triggered apoptosis.
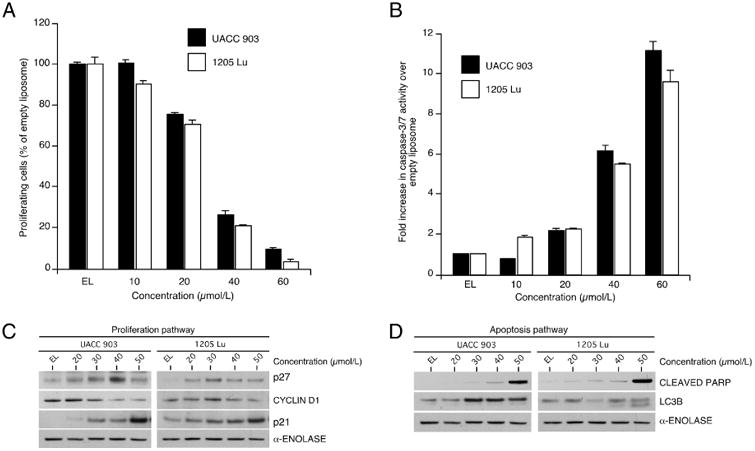
5A & 5B. Cell proliferation (5A) and apoptosis rates (5B) following treatment of cultured melanoma cells with NanoATK. Cellular proliferation and apoptosis rates were measured for UACC 903 and 1205 Lu melanoma cells following treatment with 10 to 60 μmol/L of NanoATK compared to control empty nanoliposomes for 72 hours. 5C & 5D. Westernblotting analysis evaluated levels of proliferation and apoptosis markers. Proliferation (5C) and apoptosis/autophagy (5D) markers following treatment of cultured melanoma cells with NanoATK.
Inhibition of cPLA2 by NanoATK caused a shutdown of COX-2/AKT/STAT3 pathways
Melanoma cell lines have elevated cPLA2 activity to promote cellular proliferation while decreasing apoptosis rates 10. Treatment with 20 to 50 μmol/L NanoATK decreased expression of cPLA2 activity, leading to an increase of COX-2 protein levels, which in turn increased COX-2 signaling (Fig. 6A). Increase in COX-2 protein expression has been used to indicate effective inhibition of COX-2 activity and is a compensatory response to inhibition 19, 38. The functions of COX-2 in metabolizing free AA into various lipid mediators responsible for tumor growth occurs through activation of multiple key pathways like PI3K-AKT, and STAT3 signaling in various cancer types 19, 38. Consistent decreases in the members of PI3K-AKT and STAT3 signaling pathways were observed following NanoATK treatment, which are important signaling cascades promoting melanoma development 39. Decreased levels of pAKT (Fig. 6B) and pSTAT3 (Fig. 6C), key effectors of PI3K-AKT and STAT3 signaling, were observed upon treatment with 20-50 μmol/L of NanoATK. Therefore, the mechanism leading to inhibition of proliferation and induction of apoptosis pathways occur primarily through reduction of cPLA2/AKT/STAT3 signaling pathways.
Figure 6. NanoATK inhibits key signaling pathways regulating melanoma progression. 6A, 6B & 6C.

Western blot analysis of cultured cells treated with NanoATK. UACC 903 and 1205 Lu melanoma cells were treated with 20 to 50 μmol/L of NanoATK for 24 hours and cell lysates analyzed to identify changes in the expression as well as the activity of PLA2/COX-2 (6A), PI3K-AKT (6B) & STAT3 (6C). Protein quantification was performed using ImageJ. Alpha-enolase served as a control for equal protein loading.
Discussion
Development of melanoma involves both silencing and activating of multiple signaling pathways 1. Metastatic melanoma remains a challenging disease to manage using traditional chemotherapy, which is frequently accompanied by recurrent resistant disease development 40. Recent studies show promising results with selective V600E-BRAF inhibitors, vemurafenib and dabrafenib and MEK inhibitor trametinib, which were approved by the FDA for treating patients with MAPK pathway activation 1. Unfortunately, drug resistance still limits long-term efficacy and ultimately leads to the development of more aggressive disease 2. To circumvent this problem, nanoparticle based drug carriers are being developed. These include proteins, immunoglobulins, synthetic polymers, nanoliposomes and monoclonal antibodies specifically targeting melanoma cells 41, 42.
Nanotechnology-mediated drug delivery of therapeutic agents is a rapidly emerging field with potential to moderate drug resistance while improving drug delivery and reducing toxicity 43. Nanoliposomes less than 100 nm can enter tumors due to the leaky vasculature, which does not occur in normal vessels. This then provides the drug with better pharmacokinetics and bio-distribution profiles compared to the matched free agent, which is called the enhanced permeability & retention effect 44. Moreover, the development of nanotherapeutics also increases drug half-life, improves bioavailability, and can enable simultaneous delivery of multiple drugs 44. Despite rapid advances in nanotechnology, only a few clinical trials have been undertaken using this technology for melanoma treatment 15, 45. Therefore, use of nanotechnology to overcome the current challenges in the melanoma field remains at a relatively early stage in clinical development 46.
ATK can inhibit the activity of cPLA2 but not iPLA2 and sPLA2 11. PLA2 enzymes catalyze the hydrolysis of membrane glycerophospholipids to release many lipid mediators such as diacylglycerol, phosphatidic acid, lysophosphatidic acid and arachidonic acid 11. Lipid mediators generated by phospholipases, in turn, regulate intracellular signaling networks promoting a variety of cellular pathophysiological functions, including proliferation, survival, differentiation, migration, invasion, vesicle trafficking, tumorigenesis, metastasis and inflammation 11. Several studies demonstrated that PLA2 is overexpressed in carcinomas of prostate, breast, ovarian and blood, suggesting it might be a potentially important therapeutic target 7, 33-35. Unfortunately, use of ATK to target cPLA2 is limited because of its lipophilicity, which causes toxicity and reduces its bioavailability. This study suggests that a PEGylated nanoliposomal delivery system loaded with ATK, called NanoATK, can be used to enhance stability, increase circulation time, avoidance of clearance by the reticulo-endothelial system, and for lowering toxicity 47.
The mechanism leading to ATK mediated cell death has not been investigated for any cancer type. ATK contained in NanoATK seems to functions by decreasing cellular proliferation and increasing apoptosis. It inhibited the growth of pre-existing xenografted melanoma tumors by an average of 58% by reducing the activity of the cPLA2, COX-2, PI3K-AKT, and STAT3 signaling pathways. COX-2 metabolizes free arachidonic acid (AA) into various lipid mediators or eicosanoids 48. AA and its mediators promote tumor growth by activating multiple key pathways such as COX-2, PI3K-AKT, and STAT3 signaling in various cancers types 49, 50. These signaling pathways are constitutively activated in up to 50-70% of sporadic melanomas and play a prominent role in the progression and development of recurrent resistant disease 1, 2, 15, 43, 51. Simultaneous targeting of these pathways seems to cause melanoma cell death mediated by apoptosis. ATK contained in NanoATK decreased the activity of cPLA2, inhibiting downstream STAT3 and AKT signaling without affecting total protein levels in a dose dependent manner. Targeting COX-2, AKT, and STAT3 led to consistent alteration in regulatory proteins such as CYCLIN D1, p21, and p27 that are key to the functioning of the cyclin dependent kinase complex responsible for proliferation in melanoma cells 52. Similarly, NanoATK induced cleaved PARP and LC3B protein levels indicating increased cell death was occurring by apoptosis and autophagy.
In conclusion, this study demonstrates the tumor inhibitory activity of a nanoparticle based formulation of ATK called NanoATK, which is a potentially clinically viable drug containing the active ingredient ATK. NanoATK efficiently targets the major signaling pathways responsible for melanoma development by inhibiting cPLA2, with multiple effects on proteins downstream in the signaling pathway. Thus, use of this agent might more effectively treat cancer without developing possible disease resistance due to the shutdown of multiple pathways.
Supplementary Material
Acknowledgments
Grant support: NIH grants R01 CA-136667-02, RO1 CA-1138634-02, RO1 CA-127892-01A and The Foreman Foundation for Melanoma (Gavin Robertson), The Geltrude Foundation (Gavin Robertson), The Penn State Melanoma and Skin Cancer Center (Raghavendra Gowda), Gilbert Memorial Fund (Raghavendra Gowda), The James Paul Sutton Medical Research Fund (Raghavendra Gowda), The Penn State Chocolate Tour Cancer Research Fund (Raghavendra Gowda & Gavin Robertson).
Footnotes
Conflict of Interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flaherty KT. BRAF inhibitors and melanoma. Cancer J. 2011;17:505–11. doi: 10.1097/PPO.0b013e31823e5357. [DOI] [PubMed] [Google Scholar]
- 2.Dooley AJ, Gupta A. Bhattacharyya M and Middleton MR, Intermittent dosing with vemurafenib in BRAF V600E-mutant melanoma: review of a case series. Ther Adv Med Oncol. 2014;6:262–6. doi: 10.1177/1758834014548187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xing M, Post S, Ostrom RS. Samardzija M and Insel PA, Inhibition of phospholipase A2-mediated arachidonic acid release by cyclic AMP defines a negative feedback loop for P2Y receptor activation in Madin-Darby canine kidney D1 cells. J Biol Chem. 1999;274:10035–8. doi: 10.1074/jbc.274.15.10035. [DOI] [PubMed] [Google Scholar]
- 4.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 5.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chakraborti AK, Garg SK, Kumar R. Motiwala HF and Jadhavar PS, Progress in COX-2 inhibitors: a journey so far. Curr Med Chem. 2010;17:1563–93. doi: 10.2174/092986710790979980. [DOI] [PubMed] [Google Scholar]
- 7.Patel MI, Singh J, Niknami M, Kurek C, Yao M, Lu S, et al. Cytosolic phospholipase A2-alpha: a potential therapeutic target for prostate cancer. Clin Cancer Res. 2008;14:8070–9. doi: 10.1158/1078-0432.CCR-08-0566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamamoto M, Haruna T, Imura K, Hikita I, Furue Y, Higashino K, et al. Inhibitory effect of a potent and selective cytosolic phospholipase A2alpha inhibitor RSC-3388 on skin inflammation in mice. Pharmacology. 2008;81:301–11. doi: 10.1159/000117816. [DOI] [PubMed] [Google Scholar]
- 9.Cui Y, Liu X, Yang T, Mei YA, Hu C. Exposure to extremely low-frequency electromagnetic fields inhibits T-type calcium channels via AA/LTE4 signaling pathway. Cell Calcium. 2014;55:48–58. doi: 10.1016/j.ceca.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 10.Park JB, Lee CS, Jang JH, Ghim J, Kim YJ, You S, et al. Phospholipase signalling networks in cancer. Nat Rev Cancer. 2012;12:782–92. doi: 10.1038/nrc3379. [DOI] [PubMed] [Google Scholar]
- 11.Fonteh AN, Samet JM, Chilton FH. Regulation of arachidonic acid, eicosanoid, and phospholipase A2 levels in murine mast cells by recombinant stem cell factor. J Clin Invest. 1995;96:1432–9. doi: 10.1172/JCI118179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bhattacharjee A, Majumder S, Das S, Ghosh S, Biswas S, Majumdar S. Leishmania donovani-Induced Prostaglandin E2 Generation Is Critically Dependent on Host Toll-Like Receptor 2-Cytosolic Phospholipase A2 Signaling. Infect Immun. 2016;84:2963–73. doi: 10.1128/IAI.00528-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riendeau D, Guay J, Weech PK, Laliberte F, Yergey J, Li C, et al. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85-kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetraenoic acid by calcium ionophore-challenged platelets. J Biol Chem. 1994;269:15619–24. [PubMed] [Google Scholar]
- 14.Street IP, Lin HK, Laliberte F, Ghomashchi F, Wang Z, Perrier H, et al. Slow- and tight-binding inhibitors of the 85-kDa human phospholipase A2. Biochemistry. 1993;32:5935–40. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- 15.Brys AK, Gowda R, Loriaux DB, Robertson GP, Mosca PJ. Nanotechnology-based strategies for combating toxicity and resistance in melanoma therapy. Biotechnol Adv. 2016;34:565–77. doi: 10.1016/j.biotechadv.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Gowda R, Jones NR. Banerjee S and Robertson GP, Use of Nanotechnology to Develop Multi-Drug Inhibitors For Cancer Therapy. J Nanomed Nanotechnol. 2013;4:184. doi: 10.4172/2157-7439.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gowda R, Madhunapantula SV, Sharma A, Kuzu OF, Robertson GP. Nanolipolee-007, a novel nanoparticle-based drug containing leelamine for the treatment of melanoma. Mol Cancer Ther. 2014;13:2328–40. doi: 10.1158/1535-7163.MCT-14-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gowda R, Madhunapantula SV, Desai D, Amin S, Robertson GP. Simultaneous targeting of COX-2 and AKT using selenocoxib-1-GSH to inhibit melanoma. Mol Cancer Ther. 2013;12:3–15. doi: 10.1158/1535-7163.MCT-12-0492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gowda R, Kardos G, Sharma A, Singh S, Robertson GP. Nanoparticle-based Celecoxib and Plumbagin for the Synergistic Treatment of Melanoma. Mol Cancer Ther. 2017;16:440–52. doi: 10.1158/1535-7163.MCT-16-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran MA, Gowda R, Sharma A, Park EJ, Adair J, Kester M, et al. Targeting V600EB-Raf and Akt3 using nanoliposomal-small interfering RNA inhibits cutaneous melanocytic lesion development. Cancer Res. 2008;68:7638–49. doi: 10.1158/0008-5472.CAN-07-6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng S, Chang S, Lu J, Chen Z, Xie L, Nie Y, et al. Characterization of 9-nitrocamptothecin liposomes: anticancer properties and mechanisms on hepatocellular carcinoma in vitro and in vivo. PLoS One. 2011;6:e21064. doi: 10.1371/journal.pone.0021064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gowda R, Madhunapantula SV, Desai D. Amin S and Robertson GP, Selenium-containing histone deacetylase inhibitors for melanoma management. Cancer Biol Ther. 2012;13:756–65. doi: 10.4161/cbt.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gowda R, Madhunapantula SV, Kuzu OF, Sharma A, Robertson GP. Targeting multiple key signaling pathways in melanoma using leelamine. Mol Cancer Ther. 2014;13:1679–89. doi: 10.1158/1535-7163.MCT-13-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gowda R, Sharma A, Robertson GP. Synergistic inhibitory effects of Celecoxib and Plumbagin on melanoma tumor growth. Cancer Lett. 2017;385:243–50. doi: 10.1016/j.canlet.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuzu OF, Gowda R. Sharma A and Robertson GP, Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol Cancer Ther. 2014;13:1690–703. doi: 10.1158/1535-7163.MCT-13-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madhunapantula SV, Hengst J, Gowda R, Fox TE, Yun JK, Robertson GP. Targeting sphingosine kinase-1 to inhibit melanoma. Pigment Cell Melanoma Res. 2012;25:259–74. doi: 10.1111/j.1755-148X.2012.00970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madhunapantula SV, Sharma A, Gowda R, Robertson GP. Identification of glycogen synthase kinase 3alpha as a therapeutic target in melanoma. Pigment Cell Melanoma Res. 2013;26:886–99. doi: 10.1111/pcmr.12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sadashiva MP, Gowda R, Wu X, Inamdar GS, Kuzu OF, Rangappa KS, et al. A non-cytotoxic N-dehydroabietylamine derivative with potent antimalarial activity. Exp Parasitol. 2015;155:68–73. doi: 10.1016/j.exppara.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma A, Madhunapantula SV, Gowda R, Berg A, Neves RI, Robertson GP. Identification of Aurora Kinase B and WEE1 as Downstream Targets of (V600E)B-RAF in Melanoma. Am J Pathol. 2013;182:1151–62. doi: 10.1016/j.ajpath.2012.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuzu OF, Gowda R, Sharma A, Noory MA, Dinavahi SS, Kardos G, et al. Improving pharmacological targeting of AKT in melanoma. Cancer Lett. 2017;404:29–36. doi: 10.1016/j.canlet.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gowda R, Inamdar GS, Kuzu O, Dinavahi SS, Krzeminski J, Battu MB, et al. Identifying the structure-activity relationship of leelamine necessary for inhibiting intracellular cholesterol transport. Oncotarget. 2017;8:28260–77. doi: 10.18632/oncotarget.16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuzu OF, Gowda R, Noory MA, Robertson GP. Modulating cancer cell survival by targeting intracellular cholesterol transport. Br J Cancer. 2017;117:513–24. doi: 10.1038/bjc.2017.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lim SC, Cho H, Lee TB, Choi CH, Min YD, Kim SS, et al. Impacts of cytosolic phospholipase A2, 15-prostaglandin dehydrogenase, and cyclooxygenase-2 expressions on tumor progression in colorectal cancer. Yonsei Med J. 2010;51:692–9. doi: 10.3349/ymj.2010.51.5.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Runarsson G, Feltenmark S, Forsell PK, Sjoberg J, Bjorkholm M, Claesson HE. The expression of cytosolic phospholipase A2 and biosynthesis of leukotriene B4 in acute myeloid leukemia cells. Eur J Haematol. 2007;79:468–76. doi: 10.1111/j.1600-0609.2007.00967.x. [DOI] [PubMed] [Google Scholar]
- 35.Caiazza F, McCarthy NS, Young L, Hill AD, Harvey BJ, Thomas W. Cytosolic phospholipase A2-alpha expression in breast cancer is associated with EGFR expression and correlates with an adverse prognosis in luminal tumours. Br J Cancer. 2011;104:338–44. doi: 10.1038/sj.bjc.6606025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leis HJ, Windischhofer W. Inhibition of cyclooxygenases 1 and 2 by the phospholipase-blocker, arachidonyl trifluoromethyl ketone. Br J Pharmacol. 2008;155:731–7. doi: 10.1038/bjp.2008.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagase T, Uozumi N, Aoki-Nagase T, Terawaki K, Ishii S, Tomita T, et al. A potent inhibitor of cytosolic phospholipase A2, arachidonyl trifluoromethyl ketone, attenuates LPS-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2003;284:L720–6. doi: 10.1152/ajplung.00396.2002. [DOI] [PubMed] [Google Scholar]
- 38.Elrod HA, Yue P, Khuri FR, Sun SY. Celecoxib antagonizes perifosine's anticancer activity involving a cyclooxygenase-2-dependent mechanism. Mol Cancer Ther. 2009;8:2575–85. doi: 10.1158/1535-7163.MCT-09-0390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madhunapantula SV, Robertson GP. Therapeutic Implications of Targeting AKT Signaling in Melanoma. Enzyme Res. 2011;2011:327923. doi: 10.4061/2011/327923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351:998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- 41.Malas S, Harrasser M, Lacy KE, Karagiannis SN. Antibody therapies for melanoma: new and emerging opportunities to activate immunity (Review) Oncol Rep. 2014;32:875–86. doi: 10.3892/or.2014.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran MA, Watts RJ, Robertson GP. Use of liposomes as drug delivery vehicles for treatment of melanoma. Pigment Cell Melanoma Res. 2009;22:388–99. doi: 10.1111/j.1755-148X.2009.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirtane AR, Kalscheuer SM, Panyam J. Exploiting nanotechnology to overcome tumor drug resistance: Challenges and opportunities. Adv Drug Deliv Rev. 2013;65:1731–47. doi: 10.1016/j.addr.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71–9. doi: 10.1016/j.addr.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Sharma A, Madhunapantula SV, Robertson GP. Toxicological considerations when creating nanoparticle-based drugs and drug delivery systems. Expert Opin Drug Metab Toxicol. 2012;8:47–69. doi: 10.1517/17425255.2012.637916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rigon RB, Oyafuso MH, Fujimura AT, Goncalez ML, do Prado AH, Gremiao MP, et al. Nanotechnology-Based Drug Delivery Systems for Melanoma Antitumoral Therapy: A Review. Biomed Res Int. 2015;2015:841817. doi: 10.1155/2015/841817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain A, Jain SK. PEGylation: an approach for drug delivery. A review. Crit Rev Ther Drug Carrier Syst. 2008;25:403–47. doi: 10.1615/critrevtherdrugcarriersyst.v25.i5.10. [DOI] [PubMed] [Google Scholar]
- 48.Casos K, Siguero L, Fernandez-Figueras MT, Leon X, Sarda MP, Vila L, et al. Tumor cells induce COX-2 and mPGES-1 expression in microvascular endothelial cells mainly by means of IL-1 receptor activation. Microvasc Res. 2011;81:261–8. doi: 10.1016/j.mvr.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 49.Hughes-Fulford M, Li CF, Boonyaratanakornkit J, Sayyah S. Arachidonic acid activates phosphatidylinositol 3-kinase signaling and induces gene expression in prostate cancer. Cancer Res. 2006;66:1427–33. doi: 10.1158/0008-5472.CAN-05-0914. [DOI] [PubMed] [Google Scholar]
- 50.Suram S, Silveira LJ, Mahaffey S, Brown GD, Bonventre JV, Williams DL, et al. Cytosolic phospholipase A(2)alpha and eicosanoids regulate expression of genes in macrophages involved in host defense and inflammation. PLoS One. 2013;8:e69002. doi: 10.1371/journal.pone.0069002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan RJ, Flaherty KT. Resistance to BRAF-targeted therapy in melanoma. Eur J Cancer. 2013;49:1297–304. doi: 10.1016/j.ejca.2012.11.019. [DOI] [PubMed] [Google Scholar]
- 52.de Andrade BA, Leon JE, Carlos R, Delgado-Azanero W, Mosqueda-Taylor A, de Almeida OP. Immunohistochemical expression of p16, p21, p27 and cyclin D1 in oral nevi and melanoma. Head Neck Pathol. 2012;6:297–304. doi: 10.1007/s12105-012-0334-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.