

Abstract
Malaria continues to be one of the deadliest diseases worldwide, and the emergence of drug resistance parasites is a constant threat. Plasmodium parasites utilize the methylerythritol phosphate (MEP) pathway to synthesize isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), which are essential for parasite growth. Previously, we and others identified that the Malaria Box compound MMV008138 targets the apicoplast and that parasite growth inhibition by this compound can be reversed by supplementation of IPP. Further work has revealed that MMV008138 targets the enzyme 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (IspD) in the MEP pathway, which converts MEP and cytidine triphosphate (CTP) to cytidinediphosphate methylerythritol (CDP-ME) and pyrophosphate. In this work, we sought to gain insight into the structure–activity relationships by probing the ability of MMV008138 analogs to inhibit Pf IspD recombinant enzyme. Here, we report Pf IspD inhibition data for fosmidomycin (FOS) and 19 previously disclosed analogs and report parasite growth and Pf IspD inhibition data for 27 new analogs of MMV008138. In addition, we show that MMV008138 does not target the recently characterized human IspD, reinforcing MMV008138 as a prototype of a new class of species-selective IspD-targeting antimalarial agents.
Keywords: Plasmodium, malaria, MEP pathway, MMV008138, IspD, structure–activity studies
Graphical Abstract
Human malaria is caused by five species of Plasmodium. Around 250 million cases of malaria occur every year with 95% of the infections caused by Plasmodium falciparum and Plasmodium vivax.1 Malaria continues to be one of the deadliest diseases worldwide, and the continued emergence of drug resistance is a constant threat. Therefore, efforts to identify and characterize new leads for development of antimalarial drugs with different mechanisms of action are still needed. In order to catalyze the development of new antimalarials, the Medicines for Malaria Venture (MMV) and SCYNEXIS assembled the Malaria Box,2 an open access library composed of 400 compounds originally identified by phenotypic screening of nearly 6 000 000 compounds from the research libraries of Saint Jude Children’s Research Hospital, Novartis, and GlaxoSmithKline.3 Over 290 assays had been performed to screen the Malaria Box, and substantial information about these compounds is now available.4
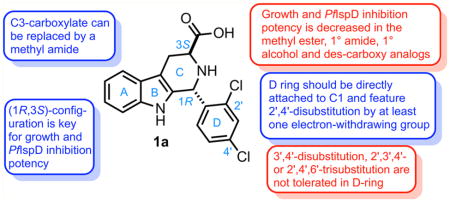
Malaria parasites contain a vestigial plastid called the apicoplast, which performs vital functions such as the biosynthesis of isoprenoid precursors, fatty acids, and part of the heme.5,6 Plasmodium parasites utilize the methylerythritol phosphate (MEP) pathway to synthesize isopentenyl pyrophosphate (IPP) and dimethylallyl pyrophosphate (DMAPP), which are essential for parasite growth.7,8 This pathway is absent in humans, who rely on the mevalonate pathway instead. Recently, it has been suggested that the MEP pathway and the biosynthesis of the isoprenoid precursors IPP and DMAPP represent the sole essential function of this organelle during asexual intraerythrocytic development of the parasites9 as well as during gametocytogenesis.10 The strongest support for this proposal stems from the observation that both loss of the apicoplast function and loss of the organelle can be chemically rescued by supplementing the growth medium with IPP. Therefore, the growth inhibitory effect of drugs that directly target biosynthesis of isoprenoid precursors or indirectly disrupt their biosynthesis by interfering with processes essential for apicoplast biogenesis, such as apicoplast DNA replication, transcription, and protein translation, may be reverted by IPP supplementation.9 As a result, the reversal of growth inhibition by IPP supplementation can be used as a phenotypic screening diagnostic to identify compounds that target the apicoplast, thus identifying their mechanism of action and narrowing their potential molecular targets.9 Previously, we identified the drug-like Malaria Box compound MMV008138 (Figure 1) using this method and proposed that the molecular target may be within the MEP pathway, especially because we noted that MMV008138 did not present a delayed death phenotype and the apicoplast was not lost in the presence of IPP, similar to fosmidomycin (FOS).11 Further analyses revealed that the active stereoisomer of this molecule is (1R,3S)-configured12,13 and that its MEP pathway target is the 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (IspD) (E.C.2.7.7.60) which converts methylerythritol phosphate (MEP) and cytidine triphosphate (CTP) to cytidine diphosphate methylerythritol (CDP-ME) and pyrophosphate.12,14
Figure 1.
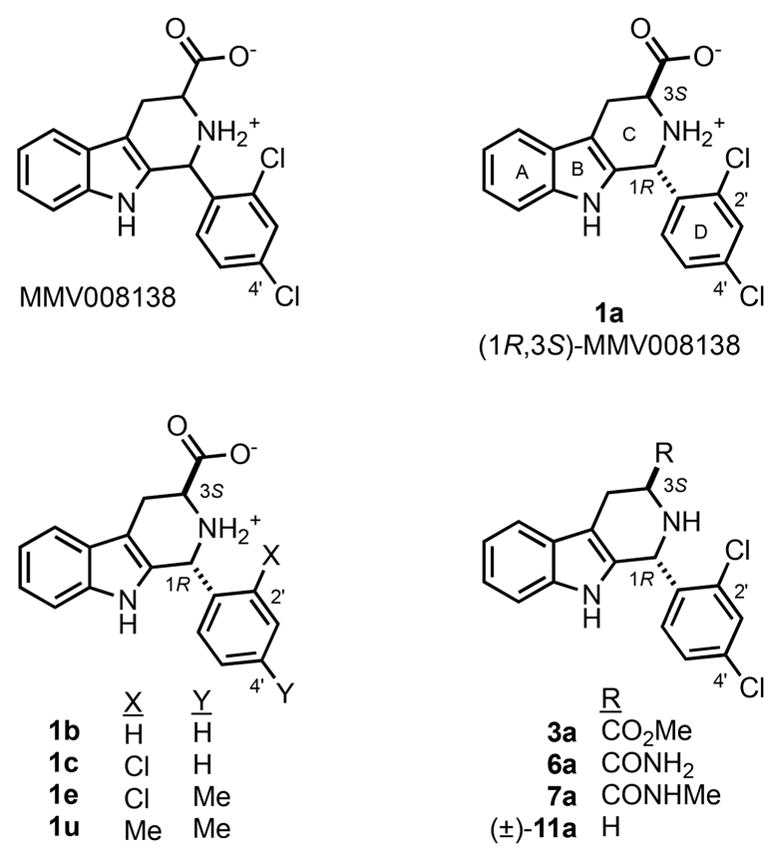
MMV008138 as originally disclosed, its active stereoisomer 1a ((1R,3S)-MMV008138), and selected analogs.
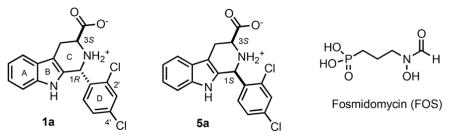
One of the most striking features of (1R,3S)-MMV008138 (henceforth 1a, Scheme 1) is its species selectivity toward the malarial IspD.12,14 Though 1a was reported to inhibit P. falciparum IspD with Ki values in the 7–13 nM range, it did not inhibit Arabidopsis thaliania, Mycobacterium tuberculosis, or Escherichia coli IspDs at concentrations up to 10 μM.12,14 This E. coli IspD (EcIspD) insensitivity is especially important since the MEP pathway is present in the human microbiome.15 Therefore, unlike antibiotics such as doxycycline (DOX) that kill malaria parasites by interfering with the apicoplast’s genome expression16 or FOS that inhibits 1-deoxy-D-xylulose 5-phosphate reductoisomerase (DXR) in the MEP pathway also affecting the gut microbiome,8,17–19 compound 1a may not affect beneficial gut bacteria. Moreover, doxycycline is used for prophylaxis in endemic areas with multidrug resistance and in combination therapies; however, it is not recommended for pregnant women and children under the age of 8.20 Therefore, antimicrobial agents that are parasite-specific are ideally suited for the development of malaria chemopreventive agents that are not toxic to pregnant women and children and do not impact the human intestinal microbiome. Indeed, we previously confirmed that 1a had no effect on E. coli growth at concentrations up to 500 μM.13
Scheme 1.

Synthesis of 1a and Its D-Ring Analogs
Subsequent to our discovery of 1a as a MEP-pathway inhibitor,11 we prepared 34 close structural analogs designed to probe P. falciparum growth inhibition structure–activity relationships (SAR).13 We determined that, in addition to (1R,3S)-configuration, potent growth inhibition required 2′,4′-disubstitution of the D-ring, featuring at least one electron-withdrawing substituent. For example, whereas 1a inhibited P. falciparum (Dd2 strain) growth with a half-maximum inhibitory concentration (IC50) value of 250 ± 50 nM, neither 1b (bearing an unsubstituted phenyl ring) nor 1u (bearing 2′,4′-dimethyl substitution) inhibited P. falciparum growth at 10 000 nM (Figure 1). However, 2′-chloro substituted analog 1c showed weak growth inhibition, and 1e (2′-chloro-4′-methyl substituted) nearly recapitulated the potency of 1a. The carboxy substituent at C3 of the C-ring also proved to be essential; replacement with CO2Me (3a) or H ((±)-11a) abrogated growth inhibition potency. However, weak growth inhibition was restored with the first amide analog 6a, and methyl amide derivative 7a proved equipotent to 1a.
In the present work, we sought to gain insight into these structure–activity relationships by probing the ability of these compounds and new analogs to inhibit Pf IspD recombinant enzyme, the proposed parasite target of 1a. In this paper, we report Pf IspD inhibition data for FOS and 19 previously disclosed analogs and report growth inhibition and Pf IspD inhibition data for 27 new analogs of 1a. As we will show below, these analogs show an excellent correlation between P. falciparum Dd2 strain growth inhibition and Pf IspD inhibition, confirming the proposed antimalarial mode of action. In addition, we expanded the biological studies to assess cytotoxicity and stage specificity profile of 1a.
RESULTS AND DISCUSSION
Structure–Activity Relationships
We first measured the kinetic parameters of recombinant IspD (KmMEP = 12.0 ± 2.5 μM, kcatMEP = 7.6 ± 0.6 s−1, KmCTP = 9.3 ± 2.5 μM, kcatCTP = 11.7 ± 1.2 s−1) using a PhosphoWorks Fluorimetric Pyrophosphate Assay Kit (AAT Bioquest, Inc.) which directly measures pyrophosphate (PPi) released from the IspD-catalyzed reaction (MEP + CTP → CDP-ME + PPi,) (see Figure S2). Previously reported kinetic parameters of the substrate CTP for this construct were KmCTP = 59 μM and kcatCTP = 0.43 s−1.14 In addition, Wu et al. reported kinetic parameters of the substrate MEP of KmMEP = 61 μM and kcatMEP = 0.16 s−1 using a histidine- and maltose binding protein-tagged Pf IspD protein.12 However, in both cases, different detection assays (EnzChek Phosphate and Pyrophosphate Assay Kits) were used to indirectly measure release of pyrophosphate through its subsequent conversion to phosphate. After the kinetic parameters were determined, we first studied the inhibitory properties of 1a, its three diastereomers 5a, ent-1a, and ent-5a, and the antimalarial FOS (Table 1). As was first reported by Wu et al.,12 we confirmed that, among the 4 stereoisomers of MMV008138, (1R,3S)-configured 1a is the most potent inhibitor of Pf IspD (IC50 = 44 ± 15 nM). Thus, the stereochemical dependence of P. falciparum growth inhibition potency correlates very well with Pf IspD target engagement. Furthermore, as expected, the IspC (DXR)-targeting antimalarial FOS is a poor inhibitor of Pf IspD (~4% inhibition at 10 μM). We would also note that the toxicological selectivity of 1a is much higher than that of FOS: whereas 1a has no effect on E. coli growth at 2000-fold of its P. falciparum growth inhibition IC50 value, FOS shows an E. coli MIC only 14-fold higher than its P. falciparum growth inhibition IC50 value. This toxicological selectivity is expected, since 1a does not engage EcIspD.12,14 Interestingly, while (1S,3R)-configured ent-1a and (1S,3S)-configured 5a have no effect on E. coli growth at 250 and 125 μM, (1R,3R)-configured ent-5a inhibited 18 ± 6% of E. coli growth at 250 μM.
Table 1.
Pf IspD Inhibition Potency of MMV008138 Stereoisomers and FOS
| |||||
|---|---|---|---|---|---|
| compound | configuration | P. falciparum growth inhibition IC50 (nM)a | % recovery (200 μM IPP)c | PfIspD IC50 (nM)d | % E. coli growth inhibition (18 h)e |
| 1a | (1R,3S) | 250 ± 70b | 100 @ 2.5 μMb | 44 ± 15 | NI @ 500 μMb |
| 5a | (1S,3S) | >10 000b | NDb | NI @ 1 μM | NI @ 125 μM |
| ent-1a | (1S,3R) | >10 000b | NDb | NI @ 1 μM | NI @ 250 μM |
| ent-5a | (1R,3R) | 3000 ± 200b | 60 ± 5 @ 10 μMb | 27 ± 5% inh. @ 1 μM | 18 ± 6 @ 250 μM |
| FOS | NA | 880 ± 70b | 100b | 4 ± 2% inh. @ 10 μM | 11.99 ± 0.02 μM (MIC) |
MRA-150, chloroquine-resistant (intermediate), pyrimethamine-resistant, mefloquine-resistant.
Data reported previously.13
Drug at indicated concentration.
Recombinant P. falciparum IspD IC50 values measured at 60 nM Pf IspD.
Percent growth inhibition at the indicated concentration or the minimum inhibitory concentration (MIC). Values represent averages of at least two independent assays ± SEM. NA signifies “not applicable”. ND signifies “not determined”. NI signifies “no inhibition”.
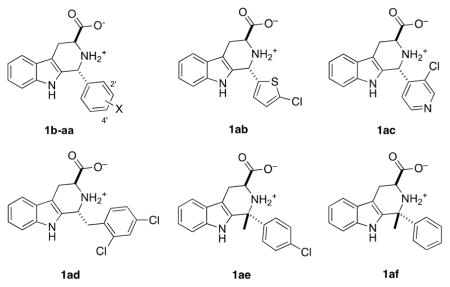
As alluded to above, previously, we observed a very tight D-ring SAR for analogs of 1a; only analogs bearing at least one sterically small, electron-withdrawing substituent at 2′ and/or 4′ were potent P. falciparum growth inhibitors (e.g., 1e–g).13 To further test this hypothesis, 17 new acid analogs of 1a were prepared and compared to previously synthesized analogs (Table 2). As can be seen, P. falciparum growth inhibition potency is retained in compounds 1h–l that feature single replacement of the Cl atoms of 1a with F (1h,i), Br (1j,k), or both Cl atoms of 1a with Br (1l). Within experimental error, compounds 1j and 1k are equipotent to 1a, but compounds featuring 2′-I,4′-F, 2′-F,4′-I, or 2′-Br,4′-I substitution (1m–o) were less potent growth inhibitors. Nevertheless, compounds 1e–n all showed full rescue upon addition of 200 μM IPP, confirming their action as MEP pathway inhibitors. However, replacement of one of the Cl atoms of 1a with OMe, OH, or CO2H (1p–s) significantly reduces growth inhibition potency. Compounds featuring very large electron-withdrawing groups (1t) or electron-releasing substituents at 2′,4′ (1u, 1v) are also poor P. falciparum growth inhibitors. A few compounds featuring 3′,4′-disubstitution (1w, 1x), trisubstitution (1y, 1z, 1aa), and heterocyclic modification (1ab, 1ac) were also explored and found to be poor P. falciparum growth inhibitors. Two modifications of the C–D-ring junction of 1a were explored. Compound 1ad features the insertion of a CH2 unit between the C- and D-rings of 1a, and this modification abrogated growth inhibition. Second, preparation of a derivative of 1a that featured replacement of the C1–H with methyl was attempted via a ketone Pictet-Spengler reaction with 2′,4′-dichlorophenyl methyl ketone (vide infra). Although the expected product of this reaction could not be isolated, compounds 1ae and 1af were successfully prepared by ketone Pictet-Spengler reactions with 4-chlorophenyl methyl ketone and acetophenone. However, these compounds also did not inhibit growth of the parasite below 5 μM. Thus, the new compounds synthesized reinforce our earlier conclusion that potent growth inhibition by analogs of 1a requires 2′,4′-disubstitution of the D-ring, featuring at least one electron-withdrawing substituent.
Table 2.
Growth Inhibition (P. falciparum, E. coli) and Pf IspD Inhibition of D-Ring Variants of 1a
| |||||
|---|---|---|---|---|---|
| compounda | X | P. falciparum growth inhibition IC50 (nM)c | % recovery (200 μM IPP)e | PfIspD IC50 (nM)f | % E. coli growth inhibition (18 h)h |
| 1b | H | >10 000d | ND | >5000g | NI @ 250 μM |
| 1c | 2′-Cl | 3280 ± 990d | 60 ± 5 @ 10 μM | ~1000 | NI @ 250 μM |
| 1d | 4′-Cl | 1170 ± 60d | 50 ± 7 @ 10 μM | 510 ± 90 | NI @ 250 μM |
| 1e | 2′-Cl,4′-Me | 410 ± 40d | 100 @ 2.5 μM | 82 ± 10 | NI @ 500 μMc |
| 1f | 2′-Me,4′-Cl | 700 ± 90d | 100 @ 2.5 μM | 260 ± 50 | NI @ 500 μMc |
| 1g | 2′,4′-F2 | 780 ± 175d | 100 @ 5 μM | 230 ± 10 | NI @ 500 μMc |
| 1h | 2′-F,4′-Cl | 860 ± 80 | 100 @ 5 μM | 140 ± 30 | NI @ 250 μM |
| 1i | 2′-Cl,4′-F | 433 ± 55 | 100 @ 10 μM | 100 ± 10 | NI @ 250 μM |
| 1j | 2′-Cl,4′-Br | 320 ± 60 | 100 @ 5 μM | 34 ± 11 | NI @ 250 μM |
| 1k | 2′-Br,4′-Cl | 360 ± 40 | 100 @ 5 μM | 31 ± 4 | NI @ 125 μM |
| 1l | 2′,4′-Br2 | 590 ± 20 | 100 @ 10 μM | 84 ± 14 | NI @ 125 μM |
| 1m | 2′-I,4′-F | 970 ± 180 | 100 @ 10 μM | 140 ± 70 | NI @ 250 μM |
| 1n | 2′-F,4′-I | 3343 ± 496 | 100 @ 10 μM | 130 ± 20 | NI @ 125 μM |
| 1o | 2′-Br,4′-I | 1500 ± 200 | 80 ± 5 @ 10 μM | ND | ND |
| 1p | 2′-Cl,4′-OMe | >5000 | ND | ND | ND |
| 1q | 2′-OMe,4′-Cl | 2500 ± 600 | ND | ND | ND |
| 1rb | 2′-Cl,4′-OH | >5000 | ND | ND | ND |
| 1s | 2′-Cl,4′-CO2H | NI @ 10 000 | ND | ND | ND |
| 1t | 2′,4′-(CF3)2 | >10 000d | ND | >5000g | NI @ 250 μM |
| 1u | 2′,4′-Me2 | 70% inh. @ 10 000d | ND | ~1000 | NI @ 250 μM |
| 1v | 2′,4′-(OMe)2 | >20 000d | ND | >5000g | NI @ 250 μM |
| 1w | 3′,4′-Cl2 | >10 000 | ND | NI @ 500 nM | NI @ 250 μM |
| 1x | 3′,4′-(OMe)2 | >20 000d | ND | ND | ND |
| 1y | 2′,6′-F2,4′-Cl | 1800 ± 150 | 80 ± 8 @ 10 μM | ND | ND |
| 1z | 2′,3′,4′-F3 | ~20 000 | ND | ND | ND |
| 1aa | 2′-Br,4′-F,5′-OMe | >10 000 | ND | ND | ND |
| 1ab | NA | >10 000 | ND | ND | ND |
| 1ac | NA | >5000 | ND | ND | ND |
| 1ad | NA | >10 000 | ND | ND | ND |
| 1ae | NA | >10 000 | ND | ND | ND |
| 1af | NA | >5000 | ND | ND | ND |
All compounds are trans-configured and derived from (S)-Trp-OMe; all are (1R,3S) except for 1ac, which is (1S,3S) due to a Cahn-Ingold-Prelog priority switch.
This compound was tested as an approximate 1:1 mixture of the (1R,3S) and (1S,3S)-stereoisomers.
MRA-150, chloroquine-resistant (intermediate), pyrimethamine-resistant, mefloquine-resistant.
Data reported previously.13
Drug at indicated concentration.
Recombinant P. falciparum IspD IC50 values measured at 60 nM Pf IspD.
Approximately 10% inhibition at 1000 nM.
NA signifies “not applicable”. NI signifies “no inhibition”. ND signifies “not determined”. Values represent averages of at least two independent assays ± SEM.
The Pf IspD inhibition potencies of these compounds largely follow the same trend (Tables 1 and 2). Unsubstituted or monosubstituted D-ring derivatives (1b–d, IC50 = 510 to >5000 nM) are weaker inhibitors than 1a (IC50 = 44 ± 15 nM). Close analogs of 1a featuring 2′,4′-disubstitution with at least one small electron-withdrawing group (1e–l) feature Pf IspD IC50 values ranging from 31 to 260 nM, consistent with their potent growth inhibition (IC50 = 320–860 nM). Note that several potent compounds (1a, 1e, 1i–l) have Pf IspD IC50 values close to the nominal enzyme concentration in the assay (60 nM), and thus, these values represent our current best estimates. This nominal enzyme concentration represents a practical current lower limit, on the basis of the sensitivity of the assay (0.1 μM of PPi); if assay at lower nominal enzyme concentration could be performed, it is possible that the measured IC50 values of these compounds would be lower. Therefore, differences in the measured Pf IspD IC50 values of 1a, 1e, and 1i–l (31–100 nM) may not be reflective of actual differences in target-site affinity of these compounds. Finally, as was seen for growth inhibition, replacement of the C2′ and C4′ Cl groups of 1a with CF3, Me, or OMe (1t–v) or 3′,4′-Cl2 substitution (1w) significantly reduced Pf IspD inhibition. Thus, the antimalarial activity of D-ring analogs of 1a appears very well correlated to the extent of their engagement with Pf IspD. This tight SAR suggests that the D-ring projects into a very well-defined cavity of Pf IspD and is not solvent-exposed. The requirement that at least one of the substituents at C2′ or C4′ must be a halogen (cf. 1a vs 1e/1f vs 1u) is curious and could be a consequence of a number of phenomena. It is possible that halogen-bonding21 to one or more Pf IspD backbone carbonyl groups contributes to affinity, as was characterized crystallographically for the 1,3-dihalobenzene moieties present in the pseudillin inhibitors of A. thaliana IspD.22
We then examined several of the carboxylic acid replacement analogs of 1a disclosed in our earlier publication (3a, 6a, 7a, 7e, 11a), and six new ones (7i–k, 8a, 9a, 10a, Table 3). As mentioned earlier, methyl ester (3a) was a poor P. falciparum growth inhibitor, and as expected, this compound does not measurably inhibit Pf IspD at 1000 nM. The 1° amide 6a does inhibit growth of P. falciparum in the 1 μM range, and its potency to inhibit Pf IspD is similar. Methyl amides (7a, 7e, 7i–k) proved to be excellent replacements for the carboxylic acid group, both for growth inhibition (IC50 = 190–506 nM) and for Pf IspD inhibition (IC50 = 21–360 nM). With respect to growth inhibition, 7j and 7k are equipotent to 1a (within error), and 7a, 7e, and 7i–k all show 100% rescue upon supplementation with 200 μM IPP. The methyl amides also show very weak inhibition of E. coli growth at 250–500 μM. Finally, the ethyl amide (8a), N,N-dimethyl amide (9a), 1° alcohol analog (10a), and racemic des-carboxy derivative 11a showed poor growth and Pf IspD inhibition. Thus, within this limited series, antimalarial activity is also well-correlated to Pf IspD engagement. These data and those in Table 1 show that the carboxylate group of 1a plays a critical role in interacting with Pf IspD; however, the nature of this interaction is not clear. A purely electrostatic interaction of the carboxylate of 1a with Pf IspD is ruled out by the potency of methyl amide 7a. Yet, the carboxylate cannot simply serve as a hydrogen-bond acceptor, since the isosteric 1° amide 6a does not potently engage Pf IspD. In any event, the carboxylate and methylamide groups of 1a and 7a must project into a rather constricted pocket, since ethyl amide 8a and N,N-dimethyl amide 9a dramatically lose potency relative to methyl amide 7a.
Table 3.
Growth Inhibition (P. falciparum, E. coli) and Pf IspD Inhibition of C3-Carboxy Replacement Variants of 1a
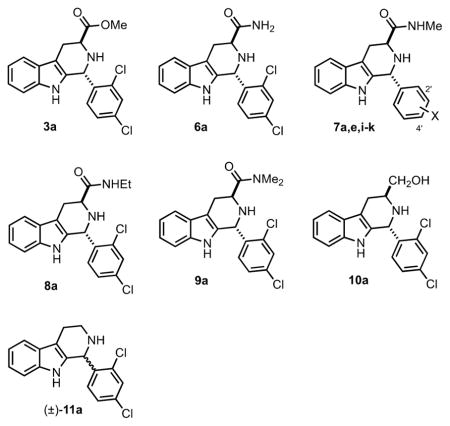
| |||||
|---|---|---|---|---|---|
| compounda | X | P. falciparum growth inhibition IC50 (nM)b | % recovery (200 μM IPP)d | PfIspD IC50 (nM)e | % E. coli growth inhibition (18 h)f |
| 3a | 2′,4′-Cl2 | 6800 ± 1400c | 20 ± 10 @ 20 μM | NI @ 1000 | NI @ 250 μM |
| 6a | 2′,4′-Cl2 | 1200 ± 100c | 50 ± 7 @ 10 μM | ~1000 | NI @ 250 μM |
| 7a | 2′,4′-Cl2 | 190 ± 30c | 100 @ 2.5 μM | 57 ± 10 | NI @ 500 μMc |
| 7e | 2′-Cl,4′-Me | 340 ± 50c | 100 @ 2.5 μM | 360 ± 40 | NI @ 500 μMc |
| 7i | 2′-Cl,4′-F | 506 ± 45 | 100 @ 2.5 μM | 278 ± 27 | 21 ± 3 @ 250 μM |
| 7j | 2′-Cl,4′-Br | 300 ± 40 | 100 @ 5 μM | 21 ± 6 | NI @ 250 μM |
| 7k | 2′-Br,4′-Cl | 340 ± 60 | 100 @ 5 μM | 31 ± 4 | NI @ 250 μM |
| 8a | 2′,4′-Cl2 | ~5000 | ND | ~1000 | NI @ 250 μM |
| 9a | 2′,4′-Cl2 | >20 000 | ND | ~5% inh. @ 1000 | NI @ 250 μM |
| 10a | 2′,4′-Cl2 | >10 000 | ND | ND | ND |
| 11a | 2′,4′-Cl2 | 10 000 ± 1600c | NI @ 20 μM | 7% inh. @ 1000 | 45 ± 10 @ 250 μM |
Except for (±)-11a, all compounds are trans- and are (1R,3S)-configured.
MRA-150, chloroquine-resistant (intermediate), pyrimethamine-resistant, mefloquine-resistant.
Data reported previously.13
Drug at indicated concentration.
Recombinant P. falciparum IspD IC50 values measured at 60 nM Pf IspD.
Percent growth inhibition at the indicated concentration. NI signifies “no inhibition”; ND signifies “not determined”. Values represent averages of at least two independent assays ± SEM.
Pf IspD Inhibitor 1a Is Cytocidal in Late Trophozoite/Early Schizont Stages but Not in Ring Stage
In our previous study, we demonstrated that 1a (MMV008138) targets the apicoplast, where it inhibits elongation and disturbs the mitochondrial membrane; these effects are reversed upon IPP supplementation.11 However, the parasite’s development was arrested at the early schizont stage, since DNA replication (assessed by Hoechst staining) was still observed after 40 h of treatment with 2.5 μM of 1a.11 Therefore, to better assess if 1a is cytocidal and parasite stage specific, we determined the half-maximum lethal concentration (LC50) at 72 h using highly synchronous cultures starting at ring stage and bolus incubation times ranging from 6 to 24 h. At each indicated time, 1a was washed out and parasites were returned to culture to complete 72 h of growth, at which point growth was measured by the SYBR Green assay (Figure 2). In parallel, cultures were smeared and stained with Giemsa for stage assessment of 1a-treated and control parasites at the time that 1a was washed out. The SYBR Green assay at 72 h confirmed that parasites were able to grow similarly to untreated controls following 6 and 12 h incubation with 1a; therefore, the LC50 values could not be determined. However, when incubation with 1a was extended to the late trophozoite/early schizont stage (18 h), the LC50 value was 1.20 ± 0.08 μM. When the drug incubation was extended to 24 h, the measured LC50 value further decreased to 0.46 ± 0.02 μM, which was very close to the measured IC50 value at 72 h performed simultaneously with these assays (0.35 ± 0.03 μM). The cytocidal action of 1a, therefore, manifests at 18–24 h, which is the time needed for parasites to progress from ring to late trophozoite/early schizont stage. This transition marks the period of highest metabolic activity of the MEP pathway, as discussed below (Figure 3). The absence of growth inhibition when cultures were treated for only 6 or 12 h is interesting (Figure 2A), given that the MEP pathway is known to be active through the entire asexual intraerythrocytic cycle.18,23 Analysis of MEP pathway intermediates at ring (12 h postinvasion, hpi), trophozoite (19 hpi), and schizont (33 hpi) showed that IPP concentrations increase markedly between 19 and 33 hpi (Figure 3), which is in agreement with our cytocidal analysis showing that the measured LC50 value in cultures exposed for only 24 h was very close to the IC50 value measured at 72 h. Cassera et al. have previously shown that transcript abundance of genes involved in the MEP pathway and the isoprenoid products dolichol and ubiquinone all peak at the schizont stage.23 Therefore, one potential explanation for the lack of growth inhibition observed at 72 h when 1a was present for only 6 or 12 h is that isoprenoid products were present at a sufficient level at the time that 1a treatment started to allow parasites to progress through ring and early trophozoite stages until 1a was washed out.
Figure 2.

LC50 value of 1a decreases with increasing drug exposure time. (A) Parasite growth was measured at 72 h by the SYBR Green assay in cultures where 1a was washed out at 6 or 12 h, and parasites were returned to culture to complete 72 h. A representative growth inhibition curve at 72 h from parasites continuously exposed to 1a is also shown. (B) 1a becomes cytocidal at 18 h drug bolus time when parasites reach late trophozoite/early schizont stages. The reported values represent averages and SEM of at least three independent assays.
Figure 3.

Highly synchronous parasites were recovered at different stages of P. falciparum intraerythrocytic cycle (12 hpi: rings; 19 hpi: trophozoites; 33 hpi: schizonts), and the presence of the MEP pathway intermediates was assessed by quantitative LC-MS/MS where only CDP-ME, MEcPP, and IPP were detected. The levels of all three detected intermediates increased markedly between trophozoite (19 hpi) and schizont (33 hpi) stages. The response of each detected metabolite is expressed as the mean and SEM of two biological replicates. 1: 1-Deoxy-D-xylulose-5-phosphate (DOXP) reductoisomerase; 2: IspD; 3: CDP-ME kinase; 4: 2C-methyl-D-erythritol-2,4-cyclodiphosphate (MEcPP) synthase; 5: 4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMBPP) synthase; 6: HMBPP reductase.
Methylamide Derivative 7a Inhibits CDP-ME Formation in Vivo Better than the Carboxylic Acid 1a
In 2015, we reported that the methylamide derivative 7a was equipotent to 1a for P. falciparum growth inhibition,13 and in this study, we determined that its Pf IspD inhibition values are identical within experimental error (44 ± 15 and 57 ± 10 nM, respectively, Tables 1 and 3). As a further means to compare these compounds, we assessed in vivo IspD inhibition in P. falciparum at schizont stage by quantitative liquid chromatography–mass spectrometry (LC-MS/MS, Figure 4) in highly synchronous schizont stage parasites treated for 2 and 4 h with 1 μM 1a or 7a (~4 times the IC50 value). FOS was used as positive control at 10 μM (~10 times the IC50 value). After 2 h of drug exposure, 7a caused 93 ± 5% decrease in the cellular levels of the Pf IspD product CDP-ME, while 1a caused 67 ± 2% decrease. The 27% greater reduction caused by 7a was statistically significant by the Benjamini-Hochberg significance test. After 4 h of treatments, 1a caused 84 ± 8% decrease in the levels of CDP-ME while the reduction caused by 7a remained the same. Interestingly, FOS treatment did not cause significant reduction of the CDP-ME levels after 2 or 4 h of drug exposure. As mentioned above, FOS inhibits MEP synthesis and does not inhibit the enzymatic activity of recombinant Pf IspD (Table 1). Previously, Zhang et al. reported that parasites treated with 5 μM FOS for 10 h starting at ring stage presented increased levels of MEP and an unknown metabolite, presumptively identified as 2-C-methylerythrose 4-phosphate, and reduced levels of CDP-ME.24 Accumulation of MEP and the potential 2-C-methylerythrose 4-phosphate was unexpected, and the authors hypothesized that FOS may be indirectly inhibiting Pf IspD through the intermediate 2-C-methylerythrose 4-phosphate that accumulates following FOS treatment or through other control mechanisms that remain to be identified. Because our experiments involved shorter treatments at a different P. falciparum stage, the results cannot be directly compared; however, in both cases, FOS induced unexpected levels of metabolites downstream of its enzyme target DXR, suggesting that FOS is triggering other control mechanisms that regulate the MEP pathway in the malaria parasite.
Figure 4.

In vivo IspD inhibition assessed by quantitative LC-MS/MS. Highly synchronous schizont stage parasites were treated for 2 and 4 h with 1 μM 1a or 7a. FOS was used as positive control at 10 μM. After 2 h of drug exposure, 7a caused a greater reduction on the cellular levels of the Pf IspD product CDP-ME than 1a. The detected difference was statistically significant by the Benjamini-Hochberg significance test. Reduction of the cellular levels of the MEcPP and the final MEP pathway product, IPP, did not show significant differences among the three treatments after 2 and 4 h of exposure to 1a, 7a, and FOS as assessed by the Benjamini-Hochberg significance test. Results are expressed as a percentage of untreated control ± SEM of two biological replicates.
On the other hand, the reduction of the cellular levels of the methylerythritol cyclodiphosphate (MEcPP) and the final MEP pathway product, IPP, did not show significant differences among the three treatments after 2 and 4 h of exposure to 1a, 7a, and FOS as assessed by the Benjamini-Hochberg significance test. However, the overall reduction of the MEcPP and IPP levels was greater after 4 h of treatment with the three inhibitors where >90% reduction of the IPP levels was achieved (Figure 4). Overall, these results are in agreement with the results described above showing that 1a is cytocidal in late trophozoite/early schizont stages where there is a higher demand of isoprenoid products such as dolichol and ubiquinone.23
Human IspD Is Not Targeted by 1a
Isoprenoid synthase domain-containing (IspD-like) proteins belong to a large family of glycosyltransferases conserved from bacteria to mammals, and defective function of a IspD-like protein disrupts glycosylation of α-dystroglycan causing Walker–Warburg syndrome, a congenital muscular dystrophy accompanied by a variety of brain and eye malformations.25,26 Recently, functional studies revealed that human IspD is a ribitol-5-phosphate cytidyltransferase.27–29 Because of the possibility that drugs directed against IspD enzymes from pathogenic bacteria and parasites could also inhibit the human IspD enzyme causing undesired side effects, the validity of IspD as drug target has been questioned.29,30 In order to assess if 1a inhibits human IspD activity, its enzymatic activity was measured by LC-MS/MS to monitor CDP-ribitol formation in the presence of 20 and 200 μM of 1a. The CDP-ribitol formation was not inhibited by the presence of 1a at either concentration (Figure 5). This result was expected since 1a has been shown to be active against P. falciparum and P. vivax but not bacterial or plant IspD homologues.12,14 Therefore, our results support the validity of Pf IspD as drug target and the further development of this chemotype for malaria prophylaxis and pharmacotherapy. As other Pf IspD inhibitor chemotypes emerge, testing against hIspD will help elucidate the structural basis of this important selectivity.
Figure 5.
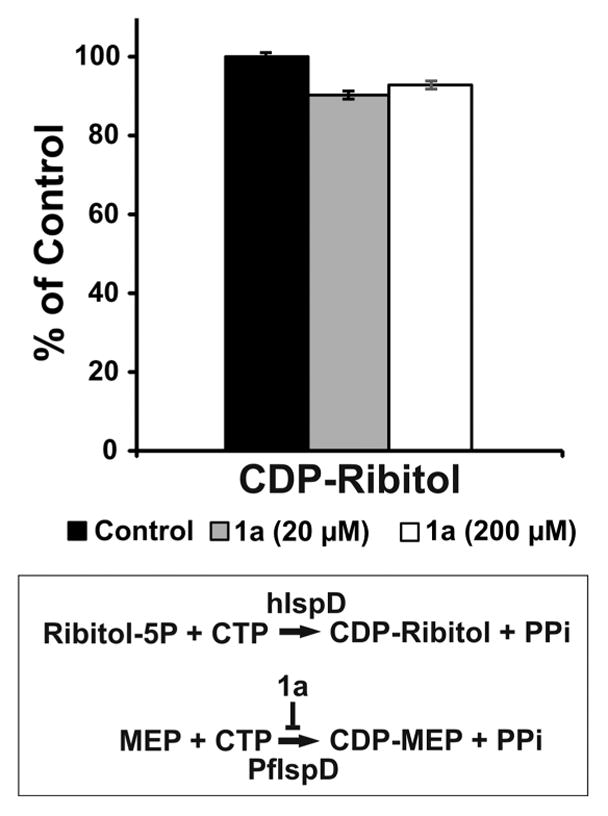
Human IspD is not targeted by 1a. Human IspD enzymatic activity in the absence or presence of 20 and 200 μM of 1a was measured by LC-MS/MS to monitor CDP-ribitol formation. The CDP-ribitol formation was not inhibited by the presence of 1a at either concentration. The small reductions observed were not statistically significant by the Benjamini-Hochberg significance test. Mean ± SEM values were calculated from two independent assays.
METHODS
Synthesis of Analogs of 1a
Compound 1a and its D-ring variants 1b–ae were prepared by Pictet-Spengler reaction of the requisite aldehydes with (S)-Trp-OMe as described in Scheme 1. The trans-configured methyl ester intermediates 3a–ac were separated from their cis-isomers by column chromatography on silica gel (using dichloromethane–ethyl acetate–hexanes mixtures to elute, as described in the Supporting Information). The relative configuration of each isomer was determined by 13C NMR according to the method of Cook and co-workers,31 and in every case examined thus far, the cis-isomer eluted before the trans-isomer. Compounds 1h–s, 1w, and 1y–af have not been previously described. C1-methyl analogs 1ae and 1af were prepared by ketone Pictet-Spengler reaction32 with (S)-Trp-OMe (Scheme 2). Note that application of this protocol to 2,4-dichlorophenyl methyl ketone did not give the expected C1-methyl analog of 1a. Methyl ester 3a, 1° amide 6a, and methyl amides 7a, 7e, and (±)-11a were prepared as described previously.13 New methyl amides 7i–k were prepared by treating the corresponding methyl esters 3i–k with methylamine in ethanol. Ethyl amide 8a and N,N-dimethyl amide 9a were prepared by HATU coupling of 1a with ethylamine and dimethylamine, respectively. The 1° alcohol analog 10a was prepared by LiAlH4 reduction of 1a.
Scheme 2.
Synthesis of C1-Methyl Analogs of 1a
P. falciparum Culture
Parasite Dd2 (MRA-150) strain was maintained in O+ human erythrocytes at 4% hematocrit in RPMI 1640 media supplemented with 2 g/L glucose (Sigma-Aldrich), 2.3 g/L sodium bicarbonate (Sigma-Aldrich), 50 mg/L hypoxanthine (Sigma-Aldrich), 5.94 g/L HEPES, 20 mg/L gentamycin (GIBCO Life Technologies), and 5 g/L Albumax I (GIBCO Life Technologies). Parasites were kept at 37 °C under reduced oxygen conditions (5% CO2, 5% O2, and 90% N2). Synchronous cultures in ring stage (>95%) were obtained by two cycles of 5% sorbitol treatment.
P. falciparum Growth Inhibition (IC50) and IPP Reversal of Growth Assays
The effects of 1a and analogs were evaluated against P. falciparum, Dd2 strain, by the SYBR Green assay as described previously in a 72 h assay since we previously reported that this class of apicoplast-targeting compounds does not present a delayed death phenotype.11 Studies were performed with the Dd2 strain since it is chloroquine resistant and we have previously showed that there is no significant differences between resistant and sensitive P. falciparum strains.11 Briefly, ring stage parasite cultures (100 μL per well, with 1% hematocrit and 1% parasitemia) were grown for 72 h in the presence of increasing concentrations of the inhibitor under reduced oxygen conditions (5% CO2, 5% O2, and 90% N2) at 37 °C. After 72 h in culture, parasite’s growth inhibition was determined by DNA quantitation using SYBR Green I as described previously.33 The half-maximum inhibitory concentration (IC50) values were calculated with GraphPad Prism (GraphPad Software, Inc.) using nonlinear regression curve fitting. The reported values represent averages of at least three independent experiments performed in triplicate, using 10-point serial dilutions, with standard errors of the mean (S.E.M.). The range for serial dilutions was adjusted accordingly for each analog after the first screening to set the IC50 value in the middle of the concentration range.
To establish reversal of growth inhibition by IPP, ring stage parasite cultures were grown for 72 h in the presence of increasing concentrations of drug and in the presence or absence of 200 μM IPP. The reported values represent averages of at least two independent experiments. SEM values are only indicated for mean values below 100%. The parasite’s growth inhibition and recovery was assessed by SYBR Green as described previously.11
Cytocidal (LC50) Assay
In order to determine the concentration of a bolus dose of the resynthesized 1R,3S-MMV008138 (1a) that kills 50% of parasites (LC50), P. falciparum (Dd2 strain) cultures were exposed to increasing concentrations of 1a and then the drug was washed away at 6, 12, 18, and 24 h to also probe stage specificity for the activity of 1a as described previously.34 Briefly, following the bolus dose incubation, plates were centrifuged at 700g for 3 min and 1a-containing medium was removed. Cell pellets were washed three times with 100 μL of medium using the same centrifuge settings and then resuspended in 100 μL of media without the inhibitor. Washed plates were then incubated at 37 °C to complete a total of 72 h after setting the assays, and growth of surviving parasites was assessed by SYBR Green. The half-maximum lethal concentration (LC50) values were calculated with GraphPad Prism software (GraphPad Software, Inc.) using nonlinear regression curve fitting, and the reported values represent averages and SEM of at least three independent assays, with each assay performed in triplicate. Parasites untreated or treated with 1a were smeared and stained with Giemsa before washing the inhibitor in order to assess stage development at the time that 1a was removed.
E. coli Growth Inhibition Assays
In order to investigate the effect of selected compounds against E. coli, strain BL21(DE3), an overnight culture of E. coli cultivated at 37 °C at 200 rpm agitation was diluted 100-fold into LB broth medium and incubated to an OD600 of ~0.6. The culture was then diluted 10 000-fold into LB broth medium. Then, 750 μL of this E. coli inoculum was inoculated into a culture tube containing 750 μL of the test compounds previously diluted in LB broth medium at three final concentrations (500, 250, and 125 μM). The final DMSO concentration was 5%. Cultures were incubated for 18 h at 37 °C and 200 rpm agitation. The following controls were performed: 100 μM fosmidomycin (FOS) treatment, which targets the MEP pathway in E. coli, media without inoculum, 5% DMSO (vehicle), and control with inoculum alone (untreated). After 18 h of incubation, bacteria growth was measured using a cell density meter. The percentage of growth was normalized to that of untreated control bacteria and potential inhibition of growth of 1a analogs was determined by comparison to the 5% DMSO control, which does not affect E. coli growth, as we described previously.13 The data represents the mean and SEM of two independent experiments.
P. falciparum IspD Protein Purification and IspD Enzymatic Assay Conditions
Plasmid containing wild-type Pf IspD (pBG1869 Pf IspD), as described in Imlay et al., was a gift from Dr. Audrey Odom.14 pBG1869 PfIspD was transformed into Arctic Express (DE3) RIL E. coli cells (Stratagene), followed by protein expression and purification as described previously (see Figure S1).14 The effect of 1a analogs on inhibiting Pf IspD enzymatic activity was measured using a PhosphoWorks Fluorimetric Pyrophosphate Assay Kit (AAT Bioquest, Inc.) following the manufacturer’s instructions.35–37 This fluorimetric assay directly measures pyrophosphate (PPi) with a linear range of 0.3 to 30 μM (detection limit of 0.1 μM) as described by the manufacturer. We measured PPi released in the enzymatic reaction catalyzed by Pf IspD (MEP + CTP → CDP-ME + PPi). The reactions were performed in a solid black 96-well microplate in a final volume of 50 μL (25 μL of test samples and 25 μL of assay solution) according to the manufacturer’s protocol. Briefly, various concentrations of 1a analogs were incubated for 10 min with 60 μM CTP, 60 μM MEP, 100 mM Tris-HCl (pH 7.4), and 1.6 mM MgCl2. Reactions were initiated by adding 60 nM Pf IspD. In assays with variable MEP, 100 μM CTP was used, and in assays with variable CTP, 100 μM MEP was used. Fluorescence signal was continuously monitored at 316 nm (Ex) and 456 nm (Em) using a Cytation-5 multimode plate reader (BioTek). The half-maximum inhibitory concentration (IC50) values were calculated using GraphPad Prism software (GraphPad Software, Inc.) from a 7-data points curve from at least three independent experimental replicates. The data represents the mean and SEM. A reaction mix without Pf IspD was performed in all assays and used for background subtraction. The range for serial dilutions was adjusted accordingly for each analog after the first screening to set the IC50 value in the middle of the concentration range.
Human IspD Protein Enzymatic Assay
Human IspD (hIspD) recombinant protein and its substrate ribitol-5-phosphate were a generous gift from Dr. Lance Wells and Dr. Osman Sheikh (University of Georgia).28,29 The effect of 1a on inhibiting hIspD enzymatic activity was measured by LC-MS/MS to monitor CDP-ribitol formation as described below for the detection of the MEP intermediates.10 Briefly, the enzymatic assay was performed using 2 μM hIspD, 50 mM Tris-HCl (pH 7.2), 2 mM MgCl2, 2 mM DTT, 1 mM CTP, and 1 mM ribitol-5-phosphate in the absence or presence of 1a at 20 and 200 μM. Reactions were carried out at 37 °C for 16 h. The reported mean and SEM values were calculated from two independent assays.
MEP Pathway Metabolite Profiling
Mycoplasma-free parasite cultures were tightly synchronized and grown to recover ring, trophozoite, and schizont stages for MEP pathway intermediates level assessment or early schizont stage (~27 h postinvasion) for drug treatments and MEP pathway intermediates level assessment. Cultures in early schizont stage were treated for 2 and 4 h with 0.1% DMSO (control), 1 μM 1a, 1 μM 7a, or 10 μM FOS. In all cases, parasites from infected erythrocytes were released from the host cell by lysis with 0.03% saponin in cold PBS containing 2 g/L glucose and washed three times with PBS/glucose by centrifuging 7 min at 10 000g and 4 °C. Metabolite extraction was performed by adding 250 μL per sample of ice-cold extraction solvent consisting of chloroform/methanol/acetonitrile (2:1:1, v/v/v) containing 1 μM isopentenyl S-thiolodiphosphate (ISPP) as internal standard. Samples were sonicated for 10 min in a water bath sonicator with ice. Then, 500 μL of ice-cold MS water was added, and the solution was mixed by vortexing and centrifuged at 12 000g and 4 °C for 4 min. The polar upper phase was transferred to a 10 kDa centricom tube and centrifuged at 12 000g and 4 °C for 10 min. The flow through was lyophilized, resuspended in 120 μL of ice-cold MS water, and centrifuged at 12 000g and 4 °C for 10 min. Samples were transferred to a total recovery vial (Waters), and two injections of 40 μL of each sample underwent LC-MS/MS analysis as previously described using ion-pair reversed phase ultraperformance liquid chromatography in tandem with mass spectrometry (IP-RP-UPLC-MS/MS).38 Data were acquired using MassLynx Software, v. 4.1, and processed using TargetLynx Application Manager (Waters). Relative quantification of the level of each metabolite was performed by determining the analyte-to-internal standard ratio (response) calculated by dividing the area of the analyte peak by the area of the internal standard (ISPP) peak. The response of each detected metabolite is expressed as the mean and SEM of two biological replicates.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (AI128362 (PRC), AI082581 (PRC), and AI108819 (MBC)), the Fralin Life Science Institute at Virginia Tech, and the Virginia Tech Center for Drug Discovery for financial support. The following reagent was obtained through the MR4 as part of the BEI Resources Repository, NIAID, NIH: Plasmodium falciparum Dd2 strain, MRA-150, deposited by D. Walliker. We thank Dr. Audrey Odom (Washington University) for the gift of the plasmid containing Pf IspD for protein expression. We thank Dr. Lance Wells and Dr. M. Osman Sheikh (University of Georgia) for the gift if the human IspD protein and ribitol-5-phosphate.
ABBREVIATIONS
- MEP
methylerythritol phosphate
- IPP
isopentenyl pyrophosphate
- DMAPP
dimethylallyl pyrophosphate
- CTP
cytidine triphosphate
- CDP-ME
cytidine diphosphate methylerythritol
- MEcPP
methylerythritol cyclodiphosphate
- FOS
fosmidomycin
- SAR
structure–activity relationships
- hpi
hours post-invasion
- IC50
half-maximum inhibitory concentration
- LC50
half-maximum lethal concentration
Footnotes
Author Contributions
The study and experiments were designed by M.B.C., P.R.C., D.J.S., M.M.T., M.G., E.F.M., Z.-K.Y., R.E., M.E.S., M.L.F.-M., J.H.B., M.A.C., and P.M.K. Experiments were conducted by M.G., E.F.M., Z.-K.Y., R.E., M.E.S., M.L.F.-M., J.H.B., M.A.C., and P.M.K. All authors analyzed the data. M.B.C., P.R.C., M.G., M.M.T., and D.J.S. wrote the manuscript, and all authors have read and given approval to the final version.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.7b00159.
Synthetic procedures and analytical characterization data for all the new compounds described in the paper, SDS-PAGE analysis of the Pf IspD used for enzyme inhibition studies (Figure S1), and determination of kinetic parameters for Pf IspD (Figure S2) (PDF)
References
- 1.WHO. World Malaria Report 2015. World Health Organization; Geneva: 2016. [Google Scholar]
- 2.Spangenberg T, Burrows JN, Kowalczyk P, McDonald S, Wells TNC, Willis P. The Open Access Malaria Box: A Drug Discovery Catalyst for Neglected Diseases. PLoS One. 2013;8(6):e62906. doi: 10.1371/journal.pone.0062906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guiguemde WA, Shelat AA, Garcia-Bustos JF, Diagana TT, Gamo FJ, Guy RK. Global phenotypic screening for antimalarials. Chem Biol. 2012;19(1):116–29. doi: 10.1016/j.chembiol.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Voorhis WC, Adams JH, Adelfio R, Ahyong V, Akabas MH, Alano P, Alday A, Aleman Resto Y, Alsibaee A, Alzualde A, Andrews KT, Avery SV, Avery VM, Ayong L, Baker M, Baker S, Ben Mamoun C, Bhatia S, Bickle Q, Bounaadja L, Bowling T, Bosch J, Boucher LE, Boyom FF, Brea J, Brennan M, Burton A, Caffrey CR, Camarda G, Carrasquilla M, Carter D, Belen Cassera M, Chih-Chien Cheng K, Chindaudomsate W, Chubb A, Colon BL, Colon-Lopez DD, Corbett Y, Crowther GJ, Cowan N, D’Alessandro S, Le Dang N, Delves M, DeRisi JL, Du AY, Duffy S, Abd El-Salam El-Sayed S, Ferdig MT, Fernandez Robledo JA, Fidock DA, Florent I, Fokou PV, Galstian A, Gamo FJ, Gokool S, Gold B, Golub T, Goldgof GM, Guha R, Guiguemde WA, Gural N, Guy RK, Hansen MA, Hanson KK, Hemphill A, Hooft van Huijsduijnen R, Horii T, Horrocks P, Hughes TB, Huston C, Igarashi I, Ingram-Sieber K, Itoe MA, Jadhav A, Naranuntarat Jensen A, Jensen LT, Jiang RH, Kaiser A, Keiser J, Ketas T, Kicka S, Kim S, Kirk K, Kumar VP, Kyle DE, Lafuente MJ, Landfear S, Lee N, Lee S, Lehane AM, Li F, Little D, Liu L, Llinas M, Loza MI, Lubar A, Lucantoni L, Lucet I, Maes L, Mancama D, Mansour NR, March S, McGowan S, Medina Vera I, Meister S, Mercer L, Mestres J, Mfopa AN, Misra RN, Moon S, Moore JP, Morais Rodrigues da Costa F, Muller J, Muriana A, Nakazawa Hewitt S, Nare B, Nathan C, Narraidoo N, Nawaratna S, Ojo KK, Ortiz D, Panic G, Papadatos G, Parapini S, Patra K, Pham N, Prats S, Plouffe DM, Poulsen SA, Pradhan A, Quevedo C, Quinn RJ, Rice CA, Abdo Rizk M, Ruecker A, StOnge R, Salgado Ferreira R, Samra J, Robinett NG, Schlecht U, Schmitt M, Silva Villela F, Silvestrini F, Sinden R, Smith DA, Soldati T, Spitzmuller A, Stamm SM, Sullivan DJ, Sullivan W, Suresh S, Suzuki BM, Suzuki Y, Swamidass SJ, Taramelli D, Tchokouaha LR, Theron A, Thomas D, Tonissen KF, Townson S, Tripathi AK, Trofimov V, Udenze KO, Ullah I, Vallieres C, Vigil E, Vinetz JM, Voong Vinh P, Vu H, Watanabe NA, Weatherby K, White PM, Wilks AF, Winzeler EA, Wojcik E, Wree M, Wu W, Yokoyama N, Zollo PH, Abla N, Blasco B, Burrows J, Laleu B, Leroy D, Spangenberg T, Wells T, Willis PA. Open Source Drug Discovery with the Malaria Box Compound Collection for Neglected Diseases and Beyond. PLoS Pathog. 2016;12(7):e1005763. doi: 10.1371/journal.ppat.1005763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goodman CD, McFadden GI. Targeting apicoplasts in malaria parasites. Expert Opin Ther Targets. 2013;17(2):167–77. doi: 10.1517/14728222.2013.739158. [DOI] [PubMed] [Google Scholar]
- 6.van Dooren GG, Striepen B. The algal past and parasite present of the apicoplast. Annu Rev Microbiol. 2013;67:271–89. doi: 10.1146/annurev-micro-092412-155741. [DOI] [PubMed] [Google Scholar]
- 7.Odom AR, Van Voorhis WC. Functional genetic analysis of the Plasmodium falciparum deoxyxylulose 5-phosphate reductoisomerase gene. Mol Biochem Parasitol. 2010;170(2):108–11. doi: 10.1016/j.molbiopara.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nair SC, Brooks CF, Goodman CD, Strurm A, McFadden GI, Sundriyal S, Anglin JL, Song Y, Moreno SN, Striepen B. Apicoplast isoprenoid precursor synthesis and the molecular basis of fosmidomycin resistance in Toxoplasma gondii. J Exp Med. 2011;208(7):1547–59. doi: 10.1084/jem.20110039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood-stage Plasmodium falciparum. PLoS Biol. 2011;9(8):e1001138. doi: 10.1371/journal.pbio.1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiley JD, Merino EF, Krai PM, McLean KJ, Tripathi AK, Vega-Rodriguez J, Jacobs-Lorena M, Klemba M, Cassera MB. Isoprenoid precursor biosynthesis is the essential metabolic role of the apicoplast during gametocytogenesis in Plasmodium falciparum. Eukaryotic Cell. 2015;14(2):128–39. doi: 10.1128/EC.00198-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowman JD, Merino EF, Brooks CF, Striepen B, Carlier PR, Cassera MB. Antiapicoplast and gametocytocidal screening to identify the mechanisms of action of compounds within the malaria box. Antimicrob Agents Chemother. 2014;58(2):811–9. doi: 10.1128/AAC.01500-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu W, Herrera Z, Ebert D, Baska K, Cho SH, DeRisi JL, Yeh E. A chemical rescue screen identifies a Plasmodium falciparum apicoplast inhibitor targeting MEP isoprenoid precursor biosynthesis. Antimicrob Agents Chemother. 2015;59(1):356–64. doi: 10.1128/AAC.03342-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao ZK, Krai PM, Merino EF, Simpson ME, Slebodnick C, Cassera MB, Carlier PR. Determination of the active stereoisomer of the MEP pathway-targeting antimalarial agent MMV008138, and initial structure-activity studies. Bioorg Med Chem Lett. 2015;25(7):1515–9. doi: 10.1016/j.bmcl.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imlay LS, Armstrong CM, Masters MC, Li T, Price KE, Edwards RL, Mann KM, Li LX, Stallings CL, Berry NG, O’Neill PM, Odom AR. Plasmodium IspD (2-C-Methyl-D-erythritol 4-Phosphate Cytidyltransferase), an Essential and Druggable Antimalarial Target. ACS Infect Dis. 2015;1(4):157–167. doi: 10.1021/id500047s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–9. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahl EL, Shock JL, Shenai BR, Gut J, DeRisi JL, Rosenthal PJ. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob Agents Chemother. 2006;50(9):3124–31. doi: 10.1128/AAC.00394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahl EL, Rosenthal PJ. Apicoplast translation, transcription and genome replication: targets for antimalarial antibiotics. Trends Parasitol. 2008;24(6):279–84. doi: 10.1016/j.pt.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 18.Cassera MB, Gozzo FC, D’Alexandri FL, Merino EF, del Portillo HA, Peres VJ, Almeida IC, Eberlin MN, Wunderlich G, Wiesner J, Jomaa H, Kimura EA, Katzin AM. The methylerythritol phosphate pathway is functionally active in all intraerythrocytic stages of Plasmodium falciparum. J Biol Chem. 2004;279(50):51749–59. doi: 10.1074/jbc.M408360200. [DOI] [PubMed] [Google Scholar]
- 19.Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Turbachova I, Eberl M, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science. 1999;285(5433):1573–1576. doi: 10.1126/science.285.5433.1573. [DOI] [PubMed] [Google Scholar]
- 20.Tan KR, Magill AJ, Parise ME, Arguin PM. Centers for Disease, C.; Prevention, Doxycycline for malaria chemoprophylaxis and treatment: report from the CDC expert meeting on malaria chemoprophylaxis. Am J Trop Med Hyg. 2011;84(4):517–531. doi: 10.4269/ajtmh.2011.10-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scholfield MR, Zanden CMV, Carter M, Ho PS. Halogen bonding (X-bonding): A biological perspective. Protein Sci. 2013;22(2):139–152. doi: 10.1002/pro.2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunfermann A, Witschel M, Illarionov B, Martin R, Rottmann M, Höffken HW, Seet M, Eisenreich W, Knölker H-J, Fischer M, Bacher A, Groll M, Diederich F. Pseudilins: Halogenated, Allosteric Inhibitors of the Non-Mevalonate Pathway Enzyme IspD. Angew Chem, Int Ed. 2014;53(8):2235–2239. doi: 10.1002/anie.201309557. [DOI] [PubMed] [Google Scholar]
- 23.Cassera MB, Merino EF, Peres VJ, Kimura EA, Wunderlich G, Katzin AM. Effect of fosmidomycin on metabolic and transcript profiles of the methylerythritol phosphate pathway in Plasmodium falciparum. Mem Inst Oswaldo Cruz. 2007;102(3):377–83. doi: 10.1590/s0074-02762007000300019. [DOI] [PubMed] [Google Scholar]
- 24.Zhang B, Watts KM, Hodge D, Kemp LM, Hunstad DA, Hicks LM, Odom AR. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry. 2011;50(17):3570–7. doi: 10.1021/bi200113y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roscioli T, Kamsteeg EJ, Buysse K, Maystadt I, van Reeuwijk J, van den Elzen C, van Beusekom E, Riemersma M, Pfundt R, Vissers LE, Schraders M, Altunoglu U, Buckley MF, Brunner HG, Grisart B, Zhou H, Veltman JA, Gilissen C, Mancini GM, Delree P, Willemsen MA, Ramadza DP, Chitayat D, Bennett C, Sheridan E, Peeters EA, Tan-Sindhunata GM, de Die-Smulders CE, Devriendt K, Kayserili H, El-Hashash OA, Stemple DL, Lefeber DJ, Lin YY, van Bokhoven H. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of alpha-dystroglycan. Nat Genet. 2012;44(5):581–5. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Willer T, Lee H, Lommel M, Yoshida-Moriguchi T, de Bernabe DB, Venzke D, Cirak S, Schachter H, Vajsar J, Voit T, Muntoni F, Loder AS, Dobyns WB, Winder TL, Strahl S, Mathews KD, Nelson SF, Moore SA, Campbell KP. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat Genet. 2012;44(5):575–80. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gerin I, Ury B, Breloy I, Bouchet-Seraphin C, Bolsee J, Halbout M, Graff J, Vertommen D, Muccioli GG, Seta N, Cuisset JM, Dabaj I, Quijano-Roy S, Grahn A, Van Schaftingen E, Bommer GT. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat Commun. 2016;7:11534. doi: 10.1038/ncomms11534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Praissman JL, Willer T, Sheikh MO, Toi A, Chitayat D, Lin YY, Lee H, Stalnaker SH, Wang S, Prabhakar PK, Nelson SF, Stemple DL, Moore SA, Moremen KW, Campbell KP, Wells L. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. eLife. 2016;5:e14473. doi: 10.7554/eLife.14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Riemersma M, Froese DS, van Tol W, Engelke UF, Kopec J, van Scherpenzeel M, Ashikov A, Krojer T, von Delft F, Tessari M, Buczkowska A, Swiezewska E, Jae LT, Brummelkamp TR, Manya H, Endo T, van Bokhoven H, Yue WW, Lefeber DJ. Human ISPD Is a Cytidyltransferase Required for Dystroglycan O-Mannosylation. Chem Biol. 2015;22(12):1643–52. doi: 10.1016/j.chembiol.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Gil J, Rodriguez-Concepcion M. Metabolic plasticity for isoprenoid biosynthesis in bacteria. Biochem J. 2013;452(1):19–25. doi: 10.1042/BJ20121899. [DOI] [PubMed] [Google Scholar]
- 31.Ungemach F, Soerens D, Weber R, DiPierro M, Campos O, Mokry P, Cook JM, Silverton JV. General method for the assignment of stereochemistry of 1,3-disubstituted 1,2,3,4-tetrahydro-β-carbolines by carbon-13 spectroscopy. J Am Chem Soc. 1980;102(23):6976–6984. [Google Scholar]
- 32.Horiguchi Y, Nakamura M, Saitoh T, Sano T. A Syntheis of Chiral 1,1,3-Trisubstituted 1,2,3,4-Tetrahydro-β-carbolines by the Pictet-Spengler Reaction of Tryptophan and Ketones: Conversion of (1R,3S)-Diastereomers into their (1S,3S)-Counterparts by Scission of the C(1)-N(2) bond. Chem Pharm Bull. 2003;51(12):1368–1373. doi: 10.1248/cpb.51.1368. [DOI] [PubMed] [Google Scholar]
- 33.Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob Agents Chemother. 2004;48(5):1803–6. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paguio MF, Bogle KL, Roepe PD. Plasmodium falciparum resistance to cytocidal versus cytostatic effects of chloroquine. Mol Biochem Parasitol. 2011;178(1–2):1–6. doi: 10.1016/j.molbiopara.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.King DT, Wasney GA, Nosella M, Fong A, Strynadka NC. Structural Insights into Inhibition of Escherichia coli Penicillin-binding Protein 1B. J Biol Chem. 2017;292(3):979–993. doi: 10.1074/jbc.M116.718403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabra A, Bessoule JJ, Atanasova-Penichon V, Noel T, Dementhon K. Host-pathogen interaction and signaling molecule secretion are modified in the dpp3 knockout mutant of Candida lusitaniae. Infect Immun. 2014;82(1):413–22. doi: 10.1128/IAI.01263-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang M, Yang X, Ren L, Li S, He X, Wu X, Liu T, Lin L, Li Y, Sun C. Biomarkers identified by urinary metabonomics for noninvasive diagnosis of nutritional rickets. J Proteome Res. 2014;13(9):4131–42. doi: 10.1021/pr500517u. [DOI] [PubMed] [Google Scholar]
- 38.Laourdakis CD, Merino EF, Neilson AP, Cassera MB. Comprehensive quantitative analysis of purines and pyrimidines in the human malaria parasite using ion-pairing ultra-performance liquid chromatography-mass spectrometry. J Chromatogr B: Anal Technol Biomed Life Sci. 2014;967:127–33. doi: 10.1016/j.jchromb.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.