

ABSTRACT
Movement of macromolecules between the cytoplasm and the nucleus occurs through the nuclear pore complex (NPC). Karyopherins comprise a family of soluble transport factors facilitating the nucleocytoplasmic translocation of proteins through the NPC. In this study, we found that karyopherin α6 (KPNA6; also known as importin α7) was required for the optimal replication of porcine reproductive and respiratory syndrome virus (PRRSV) and Zika virus (ZIKV), which are positive-sense, single-stranded RNA viruses replicating in the cytoplasm. The KPNA6 protein level in virus-infected cells was much higher than that in mock-infected controls, whereas the KPNA6 transcript remains stable. Viral infection blocked the ubiquitin-proteasomal degradation of KPNA6, which led to an extension of the KPNA6 half-life and the elevation of the KPNA6 level in comparison to mock-infected cells. PRRSV nsp12 protein induced KPNA6 stabilization. KPNA6 silencing was detrimental to the replication of PRRSV, and KPNA6 knockout impaired ZIKV replication. Moreover, KPNA6 knockout blocked the nuclear translocation of PRRSV nsp1β but had a minimal effect on two other PRRSV proteins with nuclear localization. Exogenous restitution of KPNA6 expression in the KPNA6-knockout cells results in restoration of the nuclear translocation of PRRSV nsp1β and the replication of ZIKV. These results indicate that KPNA6 is an important cellular factor for the replication of PRRSV and ZIKV.
IMPORTANCE Positive-sense, single-stranded RNA (+ssRNA) viruses replicate in the cytoplasm of infected cells. The roles of transport factors in the nucleocytoplasmic trafficking system for the replication of +ssRNA viruses are not known. In this study, we discovered that PRRSV and ZIKV viruses needed karyopherin α6 (KPNA6), one of the transport factors, to enhance the virus replication. Our data showed that viral infection induced an elevation of the KPNA6 protein level due to an extension of the KPNA6 half-life via viral interference of the ubiquitin-proteasomal degradation of KPNA6. Notably, KPNA6 silencing or knockout dramatically reduced the replication of PRRSV and ZIKV. PRRSV nsp1β depended on KPNA6 to translocate into the nucleus. In addition, exogenous restitution of KPNA6 expression in KPNA6-knockout cells led to the restoration of nsp1β nuclear translocation and ZIKV replication. These results reveal a new aspect in the virus-cell interaction and may facilitate the development of novel antiviral therapeutics.
KEYWORDS: PRRSV, ZIKV, virus replication, nucleocytoplasmic trafficking system, karyopherin α6, KPNA6, KPNA6 turnover, porcine reproductive and respiratory syndrome virus
INTRODUCTION
Transport of macromolecules into and out of the nucleus must go through nuclear pore complexes (NPCs), macromolecular structures (>60 MDa) consisting of approximately 30 proteins, on the nuclear envelope (1, 2). Molecules smaller than 40 kDa can passively diffuse through the NPCs. Transport of macromolecules across the NPCs requires a regulated energy-dependent process, which needs either direct interaction with the NPCs or association with different types of transport receptors. The largest group of transport receptors have been designated karyopherins (3–6), which mediate the nuclear import of many cytoplasmic proteins. In the classical nuclear import pathway, cytoplasmic cargoes bearing a nuclear location sequence (NLS) are first recognized by karyopherin α (KPNA), followed by association with karyopherin β1 (KPNB1) (4). The ternary importing complex karyopherin β/α/NLS-cargo translocates through the NPCs to the nucleus.
There are seven isoforms of karyopherin α (KPNA1 to KPNA7) that have been found so far (4, 7, 8). Due to their importance in nucleocytoplasmic trafficking, KPNAs play indispensable roles in signal transduction, especially in the host immune response. For instance, KPNA1 is responsible for the nuclear translocation of signal transducer and activator of transcription protein 1 (STAT1) in interferon (IFN)-stimulated gene expression (9, 10); KPNA1 and KPNA6 transport STAT3 (11) to activate the interleukin-6 signaling pathway. Therefore, KPNAs are often targeted by viruses to interfere with the immune response. For example, Ebola virus VP24 interacts with KPNA1 in the region overlapping with phosphorylated STAT1 (pSTAT1), which blocks pSTAT1 nuclear translocation (12, 13). Hepatitis C virus (HCV) induces the cleavage of KPNB1 by NS3/4A to disrupt the IRF3 and NF-κB translocation to inhibit IFN-β production (14). Hepatitis B virus (HBV) polymerase interferes with the IFN-α-stimulated signaling by inhibiting the nuclear transportation of STAT1/2 by binding to KPNA1 competitively (15). PRRSV inhibits IFN-activated JAK/STAT signaling by blocking the nuclear translocation of the STAT1/STAT2 complex by inducing KPNA1 degradation (16).
On the other hand, many viruses, especially those with a DNA genome, have been found to exploit the karyopherins for their own benefit. DNA viruses generally deliver their genetic elements into the nucleus for propagation or integration into the host genome for latent infections. Karyopherins play an essential role in this process and thus are pivotal host dependency factors. For example, parvovirus delivers its DNA into the nucleus through karyopherin α and karyopherin β (17). The replication of human papillomavirus is inhibited by antibodies against KPNA2 and KPNB1 (18). Moreover, herpesvirus and adenovirus rely on karyopherins for their nuclear entry (19). In addition, among RNA viruses, human immunodeficiency virus (HIV) (20) and influenza A virus (21) exploit karyopherins to translocate their proteins or genetic elements into the nucleus. KPNA1 and KPNA6 were found to be positive cellular factors for the efficient replication of influenza A virus (21–24).
However, most RNA viruses, including positive-sense, single-stranded RNA (+ssRNA) viruses, replicate in the cytoplasm rather than in the nucleus. Therefore, the nucleocytoplasmic trafficking machinery has not been linked to replication of +ssRNA viruses. In particular, it remains unknown whether KPNAs contribute to the replication of +ssRNA viruses.
The +ssRNA viruses, including flavivirus, coronavirus, picornavirus, and arterivirus, cause severe human or animal diseases. Porcine reproductive and respiratory syndrome virus (PRRSV) is a small enveloped RNA virus belonging to the genus Rodartevirus of the family Arteriviridae (25, 26). This virus causes a contagious disease that is characterized by reproductive failure in sows and respiratory disease of variable severity in pigs of all ages (27). PRRS causes substantial economic losses to the swine industry and remains one of the top challenges since it was first reported in 1987. Zika virus (ZIKV) is a mosquito-borne flavivirus that caused recent outbreaks accompanied by severe manifestations, including fetal microcephaly and the Guillain-Barre syndrome (28–30). ZIKV belongs to the same Flavivirus genus of the Flaviviridae family as several other global human pathogens, including dengue virus, yellow fever virus, and West Nile virus (31).
In a previous study, we discovered that PRRSV-mediated KPNA1 degradation antagonizes IFN-activated signaling (16). Unexpectedly, we noticed that the KPNA6 protein level was higher in PRRSV-infected cells than in a mock-infected control. The objective of the present study was to examine the KPNA6 level in virus-infected cells, the mechanism of KPNA6 elevation, and its functions in viral replication. Here, we show that KPNA6 is a common host proviral factor for PRRSV and ZIKV. Infections with these viruses induce KPNA6 elevation by extending its half-life and abrogating its ubiquitin-proteasomal degradation. KPNA6 knockdown in MARC-145 cells via RNA interference (RNAi) silencing inhibits the replication of PRRSV. In addition, KPNA6 knockout in Vero cells via the clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 system suppresses ZIKV replication. The nuclear translocation of PRRSV nsp1β is blocked in KPNA6-knockout cells. The exogenous restitution of KPNA6 expression in the KPNA6-knockout cells leads to the restoration of ZIKV replication and the nuclear translocation of PRRSV nsp1β. These results demonstrate that PRRSV and ZIKV harness KPNA6 for their own proliferation.
RESULTS
PRRSV induces an increase in the KPNA6 protein level.
In our study of the PRRSV virus-host interaction, we discovered that PRRSV infection of MARC-145 cells led to a higher expression level of KPNA6, whereas the KPNA2 level remained unchanged (Fig. 1A). To confirm the KPNA6 elevation, we infected MARC-145 cells with two PRRSV strains, VR-2385 and VR-2332, at a multiplicity of infection (MOI) of 1 and then harvested the cells at 24 h postinfection (hpi). We found that the virus-infected cells exhibited a 2-fold increase in KPNA6 protein level compared to mock-infected cells (Fig. 1B). MARC-145 cells are derived from the kidney of an African Green monkey, which is not the natural host of PRRSV (32). Pigs are the only host animals for PRRSV infection, and the main target cells for PRRSV infection are pulmonary alveolar macrophages (PAMs) (27). Thus, we examined KPNA6 level in PAMs after PRRSV infection. The result showed that PRRSV VR-2385 infection also induced KPNA6 elevation (Fig. 1C).
FIG 1.
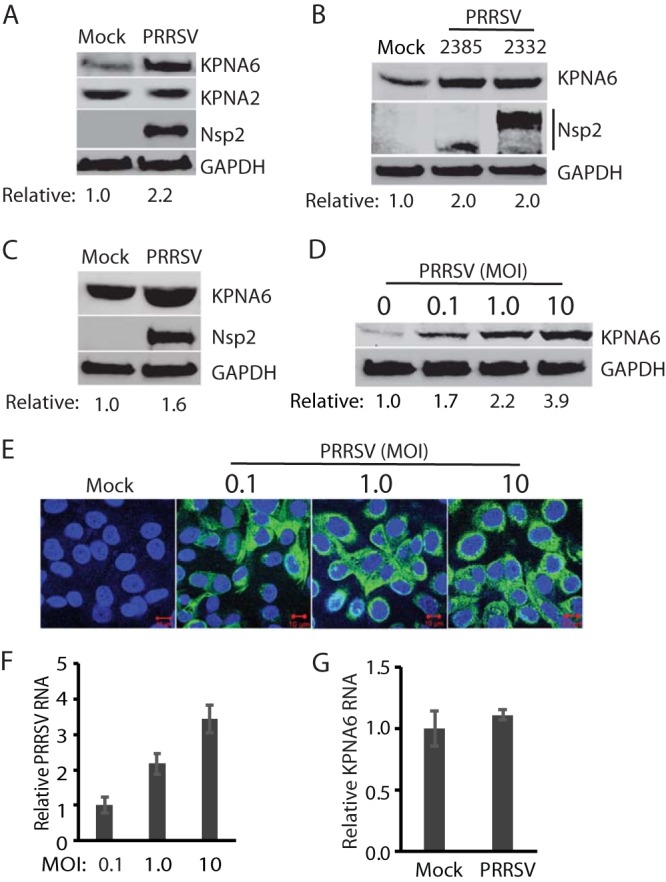
PRRSV infection induces elevation of the KPNA6 protein level but has a minimal effect on the KPNA6 transcript level. (A) KPNA6 protein in PRRSV-infected MARC-145 cells is higher than in a mock-infected control, whereas there is minimal change in the KPNA2 level. The cells were infected with PRRSV strain VR-2385 at an MOI of 1 and harvested at 24 hpi. WB with antibodies against KPNA6, KPNA2, PRRSV nsp2, and GAPDH was performed. The relative levels of KPNA6 after normalization with GAPDH are shown below the images. (B) Elevation of KPNA6 level in MARC-145 cells infected with different PRRSV strains. The cells were infected with VR-2385 and VR-2332 at an MOI of 1, incubated for 24 h, and harvested for WB with antibodies against KPNA6 and GAPDH, and pig anti-PRRSV serum. (C) The KPNA6 level increased in primary PAM cells infected with PRRSV strain VR-2385 at an MOI of 1. (D) Dose-dependent increase of KPNA6 protein level by PRRSV. MARC-145 cells were infected with the incremental MOI of PRRSV VR-2385 and harvested at 24 hpi. The relative levels of KPNA6 are shown below the images. (E) IFA of PRRSV-infected cells. The panel of overlay IFA images shows PRRSV-nsp2 in green and nuclear DNA staining with DAPI in blue. The scale bars in the low right of the images denote 10 μm. (F) Relative PRRSV RNA levels in MARC-145 cells infected with incremental MOIs. The cells were harvested at 24 hpi for RNA isolation and RT-qPCR. Error bars represent the standard errors of three repeated experiments. (G) The transcript level of KPNA6 remains stable in PRRSV-infected cells at 24 hpi as detected by RT-qPCR.
To determine whether the virus-induced KPNA6 elevation is a dose-dependent response, we infected MARC-145 cells with PRRSV VR-2385 at MOIs of 0.1, 1, and 10 and assessed the KPNA6 level by Western blotting. The result showed that KPNA6 protein levels increased in the PRRSV-infected cells in a dose-dependent manner (Fig. 1D). Compared to the mock-infected cells, PRRSV infection increased KPNA6 levels by 1.7-, 2.2-, and 3.9-fold in cells inoculated with PRRSV at MOIs of 0.1, 1, and 10, respectively, at 24 hpi. Immunofluorescence assay (IFA) results showed that almost all of the cells inoculated at MOIs of 1 and 10 were infected, whereas most of the cells inoculated at an MOI of 0.1 were positive (Fig. 1E). The increase in the PRRSV RNA level in infected cells was positively correlated to the increase in the virus inoculum (Fig. 1F).
Multiple factors, such as enhanced mRNA transcription or translation or less protein degradation, can contribute to elevated KPNA6 expression in virus-infected cells. First, we detected the KPNA6 transcript level in PRRSV-infected cells. Reverse transcription and real-time PCR (RT-qPCR) analysis suggested that the KPNA6 mRNA levels were similar between PRRSV-infected and control cells (Fig. 1G). This result indicates that the elevation of KPNA6 protein level was not due to alteration at the transcriptional level.
The KPNA6 half-life is extended, whereas its polyubiquitination level is reduced in the PRRSV-infected cells.
Since the KPNA6 increase in PRRSV-infected cells was not due to a change in its transcriptional level and PRRSV infection stabilized KPNA6, we wondered whether the virus infection might extend the KPNA6 half-life. To address this question, we infected MARC-145 cells with PRRSV and treated the cells with cycloheximide, a protein translation inhibitor, at 24 hpi. The KPNA6 half-life in PRRSV-infected cells was 32 h compared to 12 h in mock-infected cells (Fig. 2A). The results suggest that an extended KPNA6 half-life may account for its higher levels in viral infections.
FIG 2.
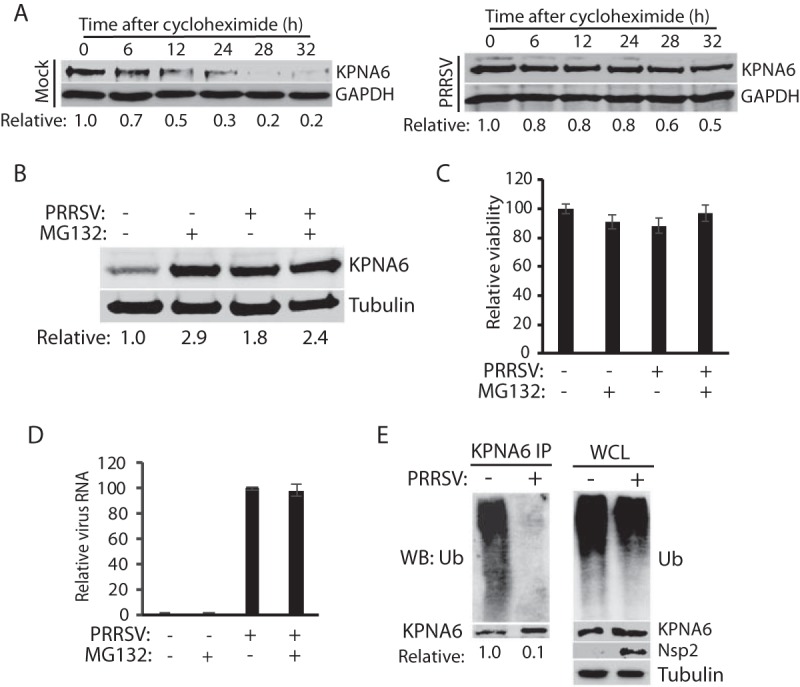
PRRSV extends KPNA6 half-life and reduces KPNA6 polyubiquitination. (A) PRRSV infection extends KPNA6 half-life. MARC-145 cells were infected with VR-2385 at an MOI of 1. The cells were treated with cycloheximide at 24 hpi and harvested at the indicated time (h) for WB. Mock-infected cells at the corresponding time points were included as controls. The relative levels of KPNA6 are given below the images. (B) MG132 treatment blocks KPNA6 turnover in MARC-145 cells. The cells were infected with VR-2385 at an MOI of 1. At 18 hpi, the cells were treated with MG132 and, 6 h later, harvested for WB with antibodies against KPNA6 and tubulin. The MG132-negative wells were treated with the solvent, DMSO. Mock-infected cells were included as controls. The relative levels of KPNA6 are shown below the images after normalization with tubulin. (C) Cell viability assay of MARC-145 cells treated with MG132 or DMSO and/or infected with PRRSV. (D) PRRSV viral RNA level in cells treated with MG132 or DMSO. (E) PRRSV infection reduces KPNA6 polyubiquitination. MARC-145 cells were infected with PRRSV VR-2385 at an MOI of 1 and harvested for IP with KPNA6 antibody at 24 hpi, followed by WB with the ubiquitin (Ub) antibody. WB of the whole-cell lysate (WCL) with the antibodies against ubiquitin, KPNA6, PRRSV nsp2, and tubulin was conducted. The relative levels of Ub after normalization with KPNA6 are shown below the images.
Since the KPNA6 half-life was extended by viral infections, we speculated that there might be a delay in the degradation of KPNA6. Since the ubiquitin-proteasome system is involved in the turnover of most cellular proteins, we hypothesized that the treatment of cells with proteasome inhibitors, such as MG132, would block KPNA6 degradation. As expected, MG132-treated MARC-145 cells exhibited a higher level of KPNA6 than did mock-treated cells (Fig. 2B). PRRSV infection did not result in an additive effect to MG132 treatment in the elevation of KPNA6 or vice versa. Due to its short treatment time (6 h), MG132 did not cause apparent cytotoxicity (Fig. 2C) and had a minimal effect on PRRSV replication (Fig. 2D). These results indicated that the proteasome blocking stabilized KPNA6 to a level similar to that of the PRRSV infection.
Based on these results, KPNA6 appeared to be degraded via the ubiquitin-proteasomal pathway. The KPNA6 polyubiquitination level was expected to be downregulated in PRRSV-infected cells. To confirm this, we conducted an immunoprecipitation (IP) assay of KPNA6 from MARC-145 cells in the presence or absence of PRRSV infection, followed by immunoblotting with a ubiquitin antibody. The result showed that the KPNA6 ubiquitination level in the PRRSV-infected cells was 10-fold lower than that of the mock-infected control, whereas there was minimal difference in the total ubiquitination level of whole-cell lysates between infected and control cells (Fig. 2E). Collectively, the extension of KPNA6 half-life through reduction of its ubiquitination may lead to virus-mediated KPNA6 upregulation.
PRRSV nsp12 protein upregulates the KPNA6 protein level in cells.
We speculated that some PRRSV proteins were responsible for the KPNA6 elevation in the virus-infected cells. To identify the viral proteins, we transfected HEK293 cells with plasmids encoding the PRRSV nonstructural proteins (nsp) and assessed their effects on the KPNA6 level. Western blotting (WB) results showed that PRRSV nsp1β, nsp7α, nsp7β, and nsp12 led to a higher KPNA6 level, compared to the empty vector, whereas the other PRRSV proteins had less effect (Fig. 3A). Since nsp12 induced the highest elevation of KPNA6, we selected it to further confirm its effect by using a stable HEK293 cell line expressing nsp12 that was established earlier in the laboratory. WB result showed there was a 2.1-fold increase in KPNA6 protein level in the nsp12-stable cells compared to the control HEK293 cells (Fig. 3B). In addition, an IP assay showed that PRRSV nsp12 also reduced KPNA6 ubiquitination (Fig. 3C). This suggests that nsp12 interferes with KPNA6 turnover in cells. However, no physical interaction between KPNA6 and nsp12 was detected in the IP (Fig. 3C), suggesting that mechanisms other than direct protein binding might mediate the effect.
FIG 3.
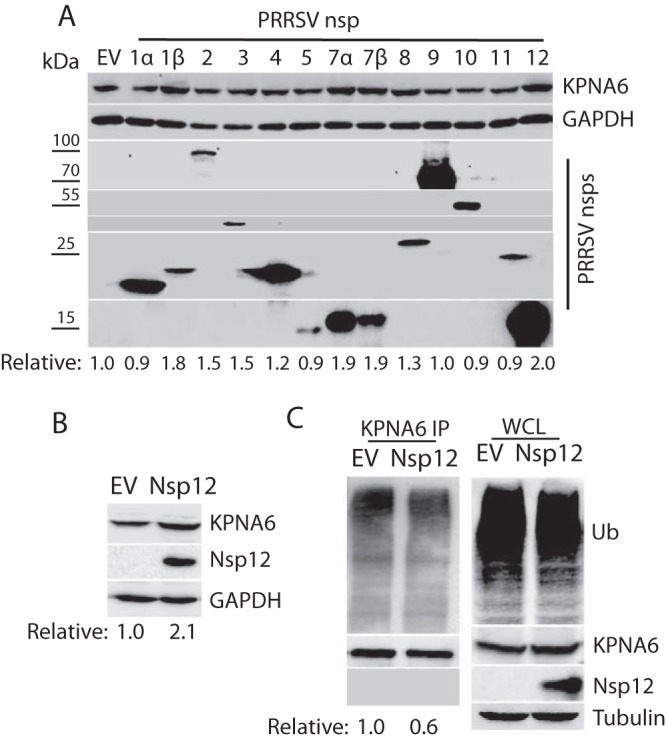
PRRSV nsp12 induces KPNA6 elevation. (A) Screening of PRRSV nsp to identify the viral protein that is responsible for the elevation of KPNA6. Empty vector (EV) was included as a control. At 36 h posttransfection, HEK293 cells were harvested for WB. The lower panel of images show the bands of PRRSV nsp. The relative levels of KPNA6 protein are shown below the images. Molecular mass markers are denoted on left of the images in kilodaltons. (B) Higher KPNA6 level in the nsp12-stable HEK293 cells than in the control. The relative levels of KPNA6 are shown below the images. (C) Lower KPNA6 polyubiquitination in the HEK293 cells transfected with nsp12 plasmid. The MG132 treatment was done before the cell harvesting. KPNA6 IP, followed by WB with an antibody against ubiquitin (Ub), was performed. The relative levels of Ub are shown below the images. WB of WCL with the antibodies against Ub, KPNA6, PRRSV nsp12, and tubulin was conducted.
KPNA6 silencing impairs PRRSV replication.
To determine whether KPNA6, stabilized in the virus-infected cells, could play a positive role in viral replication, we generated a KPNA6 shRNA construct for knockdown of KPNA6 expression in MARC-145 cells via RNAi silencing. A random shRNA was included as a negative control. Cells were transfected with shRNA plasmids, and stable cell lines were generated under the selection pressure of the antibiotic G418. WB results showed KPNA6 was reduced to less than one third in the cells with stable expression of KPNA6 shRNA, whereas the KPNA1 level showed only a minimal change (Fig. 4A). The KPNA6 knockdown had no detectable effect on cell viability (Fig. 4B). To investigate the effect of KPNA6 knockdown on PRRSV replication, we infected the KPNA6-silenced cells with VR-2385. A viral titration assay showed that the KPNA6-silenced cells produced a significantly lower virus yield (a fraction of 1/750) compared to control cells (Fig. 4C).
FIG 4.
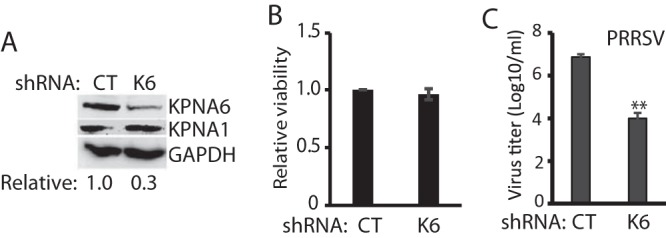
KPNA6 silencing leads to lower replication of PRRSV. (A) KPNA6 protein level in the MARC-145 cells stably transfected with KPNA6-shRNA. CT, control shRNA; K6, shRNA against KPNA6. The WB analyses of KPNA6, KPNA1, and GAPDH were conducted. (B) Relative viability of shRNA-transfected MARC-145 cells. (C) Lower PRRSV yield in cells with KPNA6 silencing than in control MARC-145 cells (**, P < 0.01). MARC-145 cells were infected with PRRSV VR-2385 at an MOI of 0.1. The virus was harvested at 48 hpi for titration.
PRRSV nsp1β relies on KPNA6 to translocate into the nucleus.
Although PRRSV replicates in the cytoplasm, some viral proteins, including nsp1α, nsp1β, and nucleocapsid protein (N), translocate into the nucleus during infection. The nsp1β protein has been known to restrain host mRNA in the nucleus to inhibit host protein synthesis (33). The nsp1α protein may inhibit IFN promoter activation in the nucleus (34). N protein localizes in the nucleus and affects host nuclear processes (35).
To determine whether KPNA6 is responsible for the translocation of these viral proteins with nuclear localization, we established KPNA6-knockout HeLa cells via the CRISPR/Cas9 system. The empty vector containing Cas9, but no target guide RNA (gRNA), was also used to establish stable HeLa cells as controls. We confirmed the deletion of KPNA6 in the KPNA6−/− cells (Fig. 5A). The KPNA6 knockout had no detectable effect on the cell viability (Fig. 5B). We then transfected the KPNA6−/− and control HeLa cells with plasmids expressing PRRSV nsp1α, nsp1β, and N proteins for IFA to determine their subcellular locations. Confocal microscopy results showed that KPNA6 knockout blocked the nuclear translocation of PRRSV nsp1β, while there was minimal change in subcellular location of the other two proteins tested (Fig. 5C). This indicates that KPNA6 facilitates nsp1β nuclear translocation and may contribute to optimal virus replication.
FIG 5.

KPNA6 knockout blocks nuclear translocation of PRRSV nsp1β, whereas exogenous KPNA6 expression restores its nuclear translocation. (A) WB of KPNA6 protein in the KPNA6-knockout HeLa cells. CTRL, control cells stably transfected with empty vector with Cas9; KPNA6−/−, KPNA6-knockout HeLa cells. WB analyses of KPNA6, KPNA2, and GAPDH were conducted. (B) Relative cell viabilities of the KPNA6−/− and CTRL HeLa cells. (C) Confocal microscopy of the KPNA6−/− and CTRL HeLa cells with transient expression of the PRRSV proteins. The cells were transfected with plasmids encoding HA-nsp1α, HA-nsp1β, and N-YFP proteins. IFA with antibody against HA was done. The overlay IFA images show HA-nsp1α and HA-nsp1β in red fluorescence, N-YFP in green fluorescence, and nuclear DNA staining with DAPI in blue. The scale bars in the lower right portion of each image denote 10 μm. (D) Transient exogenous expression of KPNA6 in KPNA6−/− HeLa cells restores the nuclear translocation of PRRSV nsp1β. The cells were transfected with Myc-KPNA6 and HA-nsp1β plasmids. Empty vector (EV) was included as a control. IFA with antibodies against Myc and HA was done. The overlay images of the green and blue channels (“Overlay (G+B)”) are shown. Nuclear DNA was stained with DAPI in blue. The scale bars in the low right portion of each image denote 10 μm.
To exclude the possibility of an off-target effect by small guide RNA (sgRNA), we transfected KPNA6 and nsp1β plasmids into KPNA6-knockout HeLa cells to determine whether the exogenous restitution of KPNA6 would restore the nuclear localization of nsp1β. The results of confocal microscopy showed that restitution of KPNA6 restores the nuclear translocation of nsp1β completely (Fig. 5D). This result confirms that KPNA6 is required for nsp1β nuclear translocation.
ZIKV infection induces KPNA6 elevation and requires KPNA6 for optimal replication.
PRRSV, a member of the +ssRNA virus, replicates in the cytoplasm of infected cells. Nevertheless, KPNA6, a karyopherin, is responsible for the nucleocytoplasmic transportation of cellular proteins. To further test whether KPNA6 elevation is specific for PRRSV infection, we used another +ssRNA virus, ZIKV, and determined the KPNA6 protein level. ZIKV also induced an elevation in the KPNA6 protein level compared to mock-infected cells (Fig. 6A). ZIKV infection was confirmed by IFA detection of E protein using antibody against the flavivirus envelope protein (E) (Fig. 6A). Thus, KPNA6 increase seems to be a common feature of the two +ssRNA viruses tested.
FIG 6.
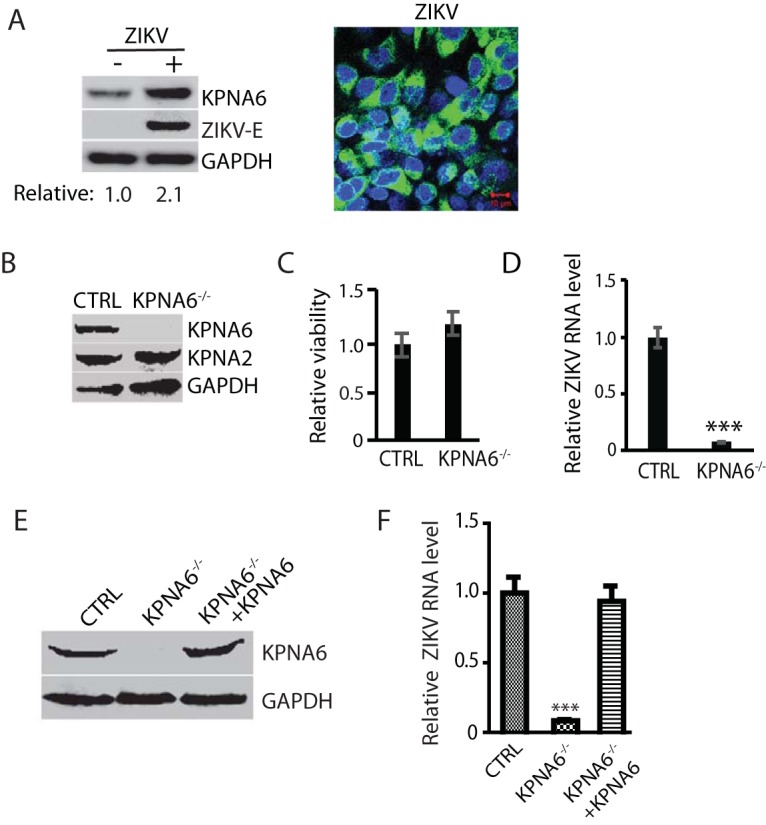
ZIKV infection also increases KPNA6 protein level and requires KPNA6 for efficient replication. (A) KPNA6 increase in ZIKV-infected Vero cells. The cells were infected with ZIKV PRVABC59 strain at an MOI of 10 and harvested at 24 hpi. WB with antibodies against KPNA6, ZIKV E protein, and GAPDH was conducted. The relative levels of KPNA6 are shown below the images. IFA with antibody against E protein was conducted. The image on the right shows an overlay of E protein in green and nuclear DNA staining with DAPI in blue. The scale bar in the lower right portion of the image denotes 10 μm. (B) KPNA6 protein level in KPNA6-knockout Vero cells. CTRL, control cells stably transfected with the empty vector; KPNA6−/−, KPNA6-knockout Vero cells via the CRISPR/Cas9 system. WB of KPNA6, KPNA2, and GAPDH was done. (C) Relative cell viabilities of CTRL and KPNA6−/− Vero cells. (D) ZIKV replication in KPNA6−/− Vero cells is significantly lower than that in CTRL cells as detected by RT-qPCR (***, P < 0.001). CTRL and KPNA6−/− Vero cells were infected with ZIKV PRVABC59 strain at an MOI of 0.1. RNA was isolated at 24 hpi, and RT-qPCR was conducted to detect the ZIKV RNA level. (E) Stable expression of exogenous KPNA6 in KPNA6−/− Vero cells restores the KPNA6 protein level. (F) Stable expression of exogenous KPNA6 in KPNA6−/− Vero cells restores the ZIKV replication level. The Vero cells were infected with ZIKV PRVABC59 strain at an MOI of 0.1. RNA was isolated at 24 hpi, and RT-qPCR was conducted.
To further confirm the role of KPNA6 in ZIKV replication, we established KPNA6-knockout Vero cells via the CRISPR/Cas9 system. The empty vector containing Cas9, but no target guide RNA (gRNA), was also used to establish stable Vero cells as controls. WB result showed significant reduction of KPNA6 in the stable KPNA6−/− Vero cells (Fig. 6B). The KPNA6 knockout had no detectable negative effect on cell viability (Fig. 6C). To test the effect of the KPNA6 knockout on virus replication, we infected the KPNA6−/− Vero and control Vero cells with ZIKV PRVABC59 strain. The KPNA6−/− Vero produced significantly lower ZIKV replication than control cells (Fig. 6D). These results demonstrate that KPNA6 is needed for ZIKV replication.
Furthermore, we transfected KPNA6−/− Vero cells with KPNA6 plasmid and established stable cells for exogenous restitution of KPNA6 expression to exclude the off-target effect of CRISPR/Cas9 system on ZIKV replication. The WB result showed the successful restitution of KPNA6 in the KPNA6−/− Vero cells (Fig. 6E). To determine whether the exogenous KPNA6 restitution could restore ZIKV replication in the KPNA6−/− Vero cells, we infected the cells with ZIKV PRVABC59 strain and harvested the cells at 24 hpi. The parental KPNA6−/− Vero and control cells were included as controls. The result of the RT-qPCR analysis showed that the expression of exogenous KPNA6 in KPNA6−/− Vero cells could restore ZIKV replication (Fig. 6F). This result substantiates that KPNA6 is required for ZIKV replication.
DISCUSSION
Karyopherins are critical transport factors in the canonical nucleocytoplasmic trafficking system, which is essential for the signal transduction of the host immune response. Accordingly, karyopherins have been found to be targeted by many viruses for an evasion of the host antiviral response, including Ebola virus (12, 13), HCV (14), HBV (15), poliovirus (36), and PRRSV (16). On the other hand, viruses, including DNA viruses (17), HIV (20), and influenza A virus (21), that replicate in the nucleus exploit karyopherins to translocate their proteins or genetic elements into the nucleus.
In this study, we discovered that KPNA6 is required by two cytoplasmic +ssRNA viruses, PRRSV and ZIKV, to facilitate their replication. The viruses protected KPNA6 from the ubiquitin-proteasome degradation. In addition, KPNA6 knockdown or knockout impairs viral replication. KPNA6 is involved in the nuclear translocation of PRRSV nsp1β. Notably, exogenous restitution of KPNA6 expression in KPNA6-knockout cells restores the nuclear translocation of PRRSV nsp1β and ZIKV replication. These data indicate that KPNA6 is a positive cellular factor for both PRRSV and ZIKV.
Our data showed that both PRRSV and ZIKV led to the elevation of the KPNA6 protein level. The KPNA6 transcript level underwent minimal change in the PRRSV-infected cells. The KPNA6 elevation was dose dependent on the PRRSV inoculum amount. The half-life of KPNA6 was extended from 12 h in the mock-infected cells to 32 h in the PRRSV-infected cells, a finding that is consistent with the time kinetics of KPNA6 in infected cells. These results suggest that the virus infection stabilizes the KPNA6 protein by protecting it from the ubiquitin-proteasome degradation. Consistent with this postulation, the MG132 treatment increased the KPNA6 level. The KPNA6 polyubiquitination level in the virus-infected cells was significantly lower than in mock-infected cells. The MG132 treatment lasted 6 h in this experiment and did not affect the PRRSV replication. However, ubiquitin-proteasome system plays a critical role in flavivirus replication, and MG132, added before or soon after virus inoculation, was shown to strongly repress flavivirus RNA translation and replication (37).
Among the PRRSV nsp tested, nsp1β, nsp7α, nsp7β, and nsp12 were found to induce the KPNA6 elevation when individually overexpressed. It is not known whether they work synergistically in PRRSV-infected cells to exert the effect on KPNA6 elevation. However, none of them is known to have a direct effect on the ubiquitin-proteasome pathway. The nsp1β protein antagonizes the host IFN induction and downstream JAK/STAT signaling (38). The nsp1β protein induces the degradation of KPNA1 to block STAT1 nuclear translocation. Interestingly, nsp1β can induce the elevation of KPNA6, a member of the same subfamily as KPNA1. The nsp1β dependence on KPNA6 for its nuclear translocation may account for the KPNA6 stabilization. nsp7 is highly conserved and further cleaved into nsp7α and nsp7β, which are known to induce a strong humoral immune response in PRRSV-infected pigs (39). nsp12 has been reported to interact with many cellular proteins with high probability in a proteomic study (40). Among the cellular proteins, chaperone HSP70 has been verified to interact with nsp12, and inhibition of HSP70 negatively affects PRRSV replication. nsp12 induces serine 727 phosphorylation of STAT1 to activate expression of inflammatory cytokines while having no effect on tyrosine 701 phosphorylation that is activated by IFNs (41). The induction of S727 phosphorylation depends on the p38 mitogen-activated protein kinase pathway. Our results confirmed that nsp12 induced the KPNA6 elevation in nsp12-stable HEK293 cells. nsp12 might indirectly exert the effect, since no direct interaction between nsp12 and KPNA6 was detected using co-IP. Further study is needed to delineate detail mechanisms on the viral interference with the KPNA6 turnover.
KPNA6 silencing or knockout dramatically inhibits PRRSV and ZIKV replication, demonstrating that KPNA6 is required for the viral replication. The expression of exogenous KPNA6 in KPNA6-knockout cells restores ZIKV replication, which substantiates the requirement of KPNA6 in the optimal viral proliferation. Since we did not restore KPNA6 in KPNA6-silenced MARC-145 cells, we cannot completely exclude the possibility that the reduced PRRSV replication in KPNA6 knockdown cells could be in part due to off-target effects. Nevertheless, the restoration of ZIKV replication in KPNA6-knockout cells with the expression of exogenous KPNA6 clearly demonstrates that KPNA6 is required for the viral replication. PRRSV infection leads to heavy economic losses to the swine industry. ZIKV is a significant global public health problem. However, prevention and therapeutic strategies against these viral pathogens are either limited or unavailable. Our finding may facilitate the development of antiviral strategies by modulating KPNA6 or blocking the interaction of KPNA6 with the viral proteins.
Even though +ssRNA viruses replicate in the cytoplasm, some viral proteins localize to the nucleus. Therefore, we speculated that KPNA6 might take charge of the nuclear transport of some of the viral proteins. Indeed, PRRSV nsp1β was found to depend on KPNA6 for the nuclear translocation. This result was substantiated by the restoration of nsp1β nuclear translocation in KPNA6-knockout HeLa cells with expression of exogenous KPNA6. Our data suggest that nsp1β nuclear localization is important for PRRSV replication. Yet, we did not determine the nsp1β subcellular location in PRRSV-infected MARC-145 cells with KPNA6 silencing because we lacked antibody that detects nsp1β of VR-2385. On the other hand, our result does not exclude other potential mechanisms of KPNA6 to enhance viral replication, since with ZIKV infection, the nuclear translocation of ZIKV proteins, including C and NS5, was not affected by KPNA6 knockout (data not shown). Further studies to examine the mechanisms of KPNA6 in enhancing the replication of these +ssRNA viruses are warranted.
In addition, elevation of KPNA6 protein level was also found in cells infected by other +ssRNA viruses, such as HCV and porcine epidemic diarrhea virus (PEDV) (data not shown). HCV, a member of the Hepacivirus genus of the Flaviviridae family (42), can cause liver disease upon infection, which could lead to cirrhosis and hepatocellular carcinoma. PEDV, a member of the genus Alphacoronavirus of the family Coronaviridae (43), causes an acute and highly contagious enteric disease in nursery piglets with a high mortality rate, which has led to tremendous economic losses in swine industry (44). Our data suggest that that KPNA6 might be a potential broad proviral protein for +ssRNA viruses.
In conclusion, KPNA6 plays an important role in the replication of PRRSV and ZIKV. The virus infection protects KPNA6 from degradation via inhibition of KPNA6 polyubiquitination. RNAi-mediated KPNA6 silencing or CRISPR/Cas9 knockout of KPNA6 leads to a significant reduction in PRRSV and ZIKV replication. KPNA6 is required for the nuclear translocation of PRRSV nsp1β. Notably, the expression of exogenous KPNA6 in the KPNA6-knockout cells restores nsp1β nuclear translocation and ZIKV replication. These results suggest that KPNA6 is required for PRRSV and ZIKV, which modulate the KPNA6 turnover to facilitate virus replication. This finding provides new insights on virus-cell interactions and may facilitate the development of antiviral therapeutics by blocking the viral manipulation of the cellular transport factor.
MATERIALS AND METHODS
Cells, viruses, and chemicals.
HEK293 (ATCC CRL-1573), MARC-145 (ATCC CRL12231) (32), HeLa (ATCC CCL-2), and Vero (ATCC CCL81) cells were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS) at 37°C. Aedes albopictus clone C6/36 (ATCC CRL-1660) cells were cultured in minimum essential medium with 10% FBS at 28°C.
Primary PAMs were revived from frozen stock that was previously collected from bronchoalveolar lavage fluid of 4- to 5-week-old PRRSV-negative piglets (45) and maintained in RPMI 1640 medium supplemented with 10% FBS at 37°C as previously described (45).
The PRRSV VR-2385 (46) and VR-2332 (47) strains used in this study were propagated in MARC-145 cells. ZIKV PRVABC59 strain (48) was reproduced in C6/36 cells. Virus titers were determined in cultured cells by 10-fold serial dilutions and shown as the median tissue culture infectious dose (49).
FuGeneHD (Promega, Madison, WI) was used to transfect plasmid DNA into the cells according to the manufacturer's instructions. Cycloheximide (Sigma-Aldrich, St. Louis, MO), an inhibitor of protein synthesis, was used to treat cells at a final concentration of 50 μM to test the half-life of KPNA6 (16, 50). MG132 (Sigma-Aldrich), a proteasome inhibitor, was used at a final concentration of 10 μM to treat cells for 6 h prior to harvesting cells (16, 50).
MARC-145 cells stably expressing KPNA6 shRNA were established by the transfection of cells with pSIREN-RetroQ-ZsGreen-KPNA6-shRNA and pTK-Hyg plasmids. The cells were then cultured under antibiotic hygromycin at a concentration of 50 μg/ml. The surviving cells were subjected to single-cell cloning by limited dilution. KPNA6-knockout Vero and HeLa cells by the CRISPR/Cas9 system (51, 52) were established by flow cytometry sorting of green fluorescent protein (GFP)-positive cells that were transfected with the CRISPR plasmid PX461-sgKPNA6 containing a gRNA and a CRISPR-associated endonuclease (Cas9). The empty vector containing Cas9, but no target gRNA, was also used to establish stable Vero and HeLa cells as controls. Restitution of KPNA6 expression in KPNA6-knockout Vero cells was established by the transfection of KPNA6−/− Vero cells with pCAGEN-KPNA6 and pTK-Hyg plasmids, followed by selection under the pressure of hygromycin.
Plasmids.
KPNA6 (GenBank accession number NM_012316) was cloned into pCAGEN (Addgene, plasmid 11160) (53) with a Myc tag at the N terminus as previously reported (16). Plasmids of PRRSV nsp from strain VR-2385 (GenBank accession number JX044140) were as described previously (16, 50). KPNA6 shRNAs were designed and cloned into plasmid pSIREN-RetroQ-ZsGreen according to the instructions of the manufacturer (TaKaRa Bio USA, Inc., Mountain View, CA). Plasmid pTK-Hyg (GenBank accession number U40398) was purchased from TaKaRa Bio USA. To establish KPNA6-knockout Vero cells by CRISPR/Cas9 system, a gRNA was designed and cloned into the vector pSpCas9n(BB)-2A-GFP (PX461) (a gift from Feng Zhang (Addgene plasmid # 48140)) (51). All primers used for the plasmid construction are listed (Table 1). All plasmids constructed in-house were subjected to DNA sequencing to verify the inserts.
TABLE 1.
Primers used in this study
| Primera | Sequence (5′–3′)b | Target gene/use |
|---|---|---|
| KPNA6F1 | CCGAATTCGAGACCATGGCGAGCCCAGGG | KPNA6 cloning |
| KPNA6R1 | GCCTCGAGTTATAGCTGGAAGCCCTCC | KPNA6 cloning |
| R-KPNA6-F1 | GGAAGCCTATGGCTTGGATA | KPNA6 real-time |
| R-KPNA6-R1 | CTGTTGCGTTTCATCGACTT | KPNA6 real-time |
| P7RF1 | ATCGCTCACCAAAACCAGTCC | PRRSV real-time |
| P7RR1 | CTTCAGTCGCTAGAGGGAAATGG | PRRSV real-time |
| KPNA6_S1F | GATCCAGCCTATGGCTTGGATAAATTCAAGAGATTTATCCAAGCCATAGGCTTTTTTTACGCGTG | KPNA6 shRNA |
| KPNA6_S1R | AATTCACGCGTAAAAAAAGCCTATGGCTTGGATAAATCTCTTGAATTTATCCAAGCCATAGGCTG | KPNA6 shRNA |
| KPNA6-gRNA-p2tF | CACCGAAGAGTGGTGGATCGGTTCG | KPNA6 sgRNA |
| KPNA6-gRNA-p2tR | AAACCGAACCGATCCACCACTCTTC | KPNA6 sgRNA |
| KPNA6-gRNA-p2bF | CACCGTGTTGATAACTTCATCTAT | KPNA6 sgRNA |
| KPNA6-gRNA-p2bR | AAACATAGATGAAGTTATCAACAC | KPNA6 sgRNA |
F, forward primer; R, reverse primer.
Italicized letters indicate restriction enzyme cleavage sites for cloning.
Western blot analysis.
Whole-cell lysate in Laemmli sample buffer was subjected to SDS-PAGE and Western blotting as previously described (54, 55). Antibodies against KPNA6 (Santa Cruz Biotechnology, Inc., Dallas, TX), KPNA1 (Santa Cruz), KPNA2 (Proscim, Inc., Poway, CA), PRRSV nsp2 protein (56, 57), glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz), flavivirus E protein (58), hemagglutinin (HA) tag (Thermo Fisher Scientific, Waltham, MA), cMyc tag (Thermo Fisher Scientific), ubiquitin (Santa Cruz), β-tubulin (Sigma-Aldrich), and FLAG tag (Sigma-Aldrich) were used in the blotting. Pig anti-PRRSV serum (59) was used to detect nsp2 of PRRSV strain VR-2332 since the monoclonal antibody against nsp2 (56, 57) did not work with this strain. The secondary antibodies used in this study were horseradish peroxidase-conjugated rabbit anti-goat IgG (Rockland Immunochemicals, Inc., Gilbertsville, PA), goat anti-mouse IgG (Rockland), and goat anti-swine IgG (SeraCare Life Sciences, Gaithersburg, MD). The specific reactions were detected with chemiluminescence substrate, and the signal was recorded digitally using a ChemiDoc XRS imaging system using the QuantityOne Program, version 4.6 (Bio-Rad Laboratories, Hercules, CA). All experiments were repeated at least three times to ensure reproducibility.
Reverse transcription and real-time PCR.
Total RNA was isolated from cells with the TRIzol reagent (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. Reverse transcription and real-time PCR with SYBR green detection (Thermo Fisher Scientific) were performed as described previously (50, 60). Transcript of RPL32 (ribosomal protein L32) from the same samples was detected as an internal control (61). Real-time PCR primers for KPNA6 and PRRSV are listed in Table 1. The primers for ZIKV were described previously (62). All experiments were repeated at least three times, with each experiment performed in triplicate.
IFA.
Immunofluorescence assay (IFA) was carried out as reported (63) with antibodies against HA tag (Thermo Fisher Scientific), cMyc tag (Thermo Fisher Scientific), KPNA6 (Santa Cruz Biotechnology), PRRSV nsp2 protein (56, 57), and flavivirus E protein (58). The specific reactions were detected by the following conjugated secondary antibodies: goat anti-mouse IgG(H&L) Dylight 549, goat anti-mouse IgG(H&L) Dylight 488, and goat anti-rabbit IgG(H&L) Dylight 488 (Rockland Immunologicals). The cover glasses were mounted onto slides using SlowFade Gold antifade reagent containing DAPI (4′,6′-diamidino-2-phenylindole; Thermo Fisher) and observed using fluorescence microscopy.
Cell viability assay.
To test the cytotoxicity of MG132, MARC-145 cells infected with PRRSV for 24 h were treated with MG132. Dimethyl sulfoxide (DMSO)-treated and noninfected cells were included as controls. After 6 h, cell viability was detected with CellTiter-Glo (Promega, Madison, WI) according to the manufacturer's instructions. To compare the cell viability of KPNA6-knockdown and KPNA6-knockout cells to that of control cells, the same numbers of cells were plated in different wells. After 24 h, cell viability was determined with CellTiter-Glo. All experiments were repeated at least three times, with each experiment performed in triplicate.
IP.
Immunoprecipitation (IP) was conducted as described previously (50). The cell lysate was clarified and incubated with the specific antibody indicated in Results, followed by incubation with protein G-agarose (SeraCare Life Sciences). The IP complexes were subjected to Western blotting. To determine the polyubiquitination of KPNA6, we added ubiquitin aldehyde (Boston Biochem, Inc., Cambridge, MA), a specific inhibitor of ubiquitin hydrolases, to the lysis buffer at a final concentration of 2.53 μM. The IP complexes were subjected to immunoblot analysis with the antibodies against ubiquitin and KPNA6.
Statistical analysis.
Differences in indicators between treatment samples, such as the KPNA6 mRNA level between the groups in the presence of PRRSV infection and the mock-infected control, were assessed by a Student t test. A two-tailed P value of <0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Yunsheng Wang for helping us conduct flow cytometry and confocal microscopy.
Z.M., R.W., and S.L. were partially sponsored by the Chinese Scholarship Council. This study was partially funded by a seed grant from the University of Maryland, College Park, MD.
REFERENCES
- 1.Fried H, Kutay U. 2003. Nucleocytoplasmic transport: taking an inventory. Cell Mol Life Sci 60:1659–1688. doi: 10.1007/s00018-003-3070-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vasu SK, Forbes DJ. 2001. Nuclear pores and nuclear assembly. Curr Opin Cell Biol 13:363–375. doi: 10.1016/S0955-0674(00)00221-0. [DOI] [PubMed] [Google Scholar]
- 3.Mosammaparast N, Pemberton LF. 2004. Karyopherins: from nuclear-transport mediators to nuclear-function regulators. Trends Cell Biol 14:547–556. doi: 10.1016/j.tcb.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 4.Goldfarb DS, Corbett AH, Mason DA, Harreman MT, Adam SA. 2004. Importin alpha: a multipurpose nuclear-transport receptor. Trends Cell Biol 14:505–514. doi: 10.1016/j.tcb.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 5.Chook YM, Blobel G. 2001. Karyopherins and nuclear import. Curr Opin Struct Biol 11:703–715. doi: 10.1016/S0959-440X(01)00264-0. [DOI] [PubMed] [Google Scholar]
- 6.Stewart M. 2007. Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8:195–208. doi: 10.1038/nrm2114. [DOI] [PubMed] [Google Scholar]
- 7.Kelley JB, Talley AM, Spencer A, Gioeli D, Paschal BM. 2010. Karyopherin alpha7 (KPNA7), a divergent member of the importin alpha family of nuclear import receptors. BMC Cell Biol 11:63. doi: 10.1186/1471-2121-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pumroy RA, Cingolani G. 2015. Diversification of importin-alpha isoforms in cellular trafficking and disease states. Biochem J 466:13–28. doi: 10.1042/BJ20141186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBride KM, Banninger G, McDonald C, Reich NC. 2002. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J 21:1754–1763. doi: 10.1093/emboj/21.7.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reich NC, Liu L. 2006. Tracking STAT nuclear traffic. Nat Rev Immunol 6:602–612. doi: 10.1038/nri1885. [DOI] [PubMed] [Google Scholar]
- 11.Ma J, Cao X. 2006. Regulation of Stat3 nuclear import by importin alpha5 and importin alpha7 via two different functional sequence elements. Cell Signal 18:1117–1126. doi: 10.1016/j.cellsig.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Reid SP, Leung LW, Hartman AL, Martinez O, Shaw ML, Carbonnelle C, Volchkov VE, Nichol ST, Basler CF. 2006. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J Virol 80:5156–5167. doi: 10.1128/JVI.02349-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF. 2007. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 81:13469–13477. doi: 10.1128/JVI.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gagne B, Tremblay N, Park AY, Baril M, Lamarre D. 2017. Importin beta1 targeting by hepatitis C virus NS3/4A protein restricts IRF3 and NF-κB signaling of IFNB1 antiviral response. Traffic 18:362–377. doi: 10.1111/tra.12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Wu M, Zhang X, Zhang W, Zhang Z, Chen L, He J, Zheng Y, Chen C, Wang F, Hu Y, Zhou X, Wang C, Xu Y, Lu M, Yuan Z. 2013. Hepatitis B virus polymerase impairs interferon-alpha-induced STAT activation through inhibition of importin-alpha5 and protein kinase Cδ. Hepatology 57:470–482. doi: 10.1002/hep.26064. [DOI] [PubMed] [Google Scholar]
- 16.Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-alpha1 degradation. J Virol 87:5219–5228. doi: 10.1128/JVI.02643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vihinen-Ranta M, Wang D, Weichert WS, Parrish CR. 2002. The VP1 N-terminal sequence of canine parvovirus affects nuclear transport of capsids and efficient cell infection. J Virol 76:1884–1891. doi: 10.1128/JVI.76.4.1884-1891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson LM, Rose RC, LeRoux L, Lane C, Bruya K, Moroianu J. 2000. Nuclear import and DNA binding of human papillomavirus type 45 L1 capsid protein. J Cell Biochem 79:225–238. doi:. [DOI] [PubMed] [Google Scholar]
- 19.Greber UF, Fornerod M. 2005. Nuclear import in viral infections. Curr Top Microbiol Immunol 285:109–138. [DOI] [PubMed] [Google Scholar]
- 20.Zaitseva L, Cherepanov P, Leyens L, Wilson SJ, Rasaiyaah J, Fassati A. 2009. HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology 6:11. doi: 10.1186/1742-4690-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Resa-Infante P, Thieme R, Ernst T, Arck PC, Ittrich H, Reimer R, Gabriel G. 2014. Importin-alpha7 is required for enhanced influenza A virus replication in the alveolar epithelium and severe lung damage in mice. J Virol 88:8166–8179. doi: 10.1128/JVI.00270-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertram S, Thiele S, Dreier C, Resa-Infante P, Preuss A, van Riel D, Mok CK, Schwalm F, Peiris JS, Klenk HD, Gabriel G. 2017. H7N9 influenza A virus exhibits importin-alpha7-mediated replication in the mammalian respiratory tract. Am J Pathol 187:831–840. doi: 10.1016/j.ajpath.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 23.Resa-Infante P, Paterson D, Bonet J, Otte A, Oliva B, Fodor E, Gabriel G. 2015. Targeting importin-alpha7 as a therapeutic approach against pandemic influenza viruses. J Virol 89:9010–9020. doi: 10.1128/JVI.00583-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gabriel G, Herwig A, Klenk HD. 2008. Interaction of polymerase subunit PB2 and NP with importin alpha1 is a determinant of host range of influenza A virus. PLoS Pathog 4:e11. doi: 10.1371/journal.ppat.0040011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faaberg KS, Balasuriya UB, Brinton MA, Gorbalenya AE, Leung FC-C, Nauwynck H, Snijder EJ, Stadejek T, Yang H, Yoo D. 2012. Family Arteriviridae, p 796–805. In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (ed), Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, CA. [Google Scholar]
- 26.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bao Y, Ng TF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo Jle D, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O'Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, Renukaradhya GJ. 2016. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154. doi: 10.1146/annurev-animal-022114-111025. [DOI] [PubMed] [Google Scholar]
- 28.Mlakar J, Korva M, Tul N, Popovic M, Poljsak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodusek V, Vizjak A, Pizem J, Petrovec M, Avsic Zupanc T. 2016. Zika virus associated with microcephaly. N Engl J Med 374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 29.Panchaud A, Stojanov M, Ammerdorffer A, Vouga M, Baud D. 2016. Emerging role of Zika virus in adverse fetal and neonatal outcomes. Clin Microbiol Rev 29:659–694. doi: 10.1128/CMR.00014-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plourde AR, Bloch EM. 2016. A literature review of Zika virus. Emerg Infect Dis 22:1185–1192. doi: 10.3201/eid2207.151990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazear HM, Diamond MS. 2016. Zika virus: new clinical syndromes and its emergence in the Western hemisphere. J Virol 90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML. 1993. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- 33.Han M, Ke H, Zhang Q, Yoo D. 2017. Nuclear imprisonment of host cellular mRNA by nsp1β protein of porcine reproductive and respiratory syndrome virus. Virology 505:42–55. doi: 10.1016/j.virol.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi X, Zhang X, Wang F, Wang L, Qiao S, Guo J, Luo C, Wan B, Deng R, Zhang G. 2013. The zinc-finger domain was essential for porcine reproductive and respiratory syndrome virus nonstructural protein-1α to inhibit the production of interferon-beta. J Interferon Cytokine Res 33:328–334. doi: 10.1089/jir.2012.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rowland RR, Yoo D. 2003. Nucleolar-cytoplasmic shuttling of PRRSV nucleocapsid protein: a simple case of molecular mimicry or the complex regulation by nuclear import, nucleolar localization and nuclear export signal sequences. Virus Res 95:23–33. doi: 10.1016/S0168-1702(03)00161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustin KE, Sarnow P. 2001. Effects of poliovirus infection on nucleocytoplasmic trafficking and nuclear pore complex composition. EMBO J 20:240–249. doi: 10.1093/emboj/20.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fernandez-Garcia MD, Meertens L, Bonazzi M, Cossart P, Arenzana-Seisdedos F, Amara A. 2011. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J Virol 85:2980–2989. doi: 10.1128/JVI.02483-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang L, Zhang YJ. 2017. Antagonizing cytokine-mediated JAK-STAT signaling by porcine reproductive and respiratory syndrome virus. Vet Microbiol 209:57–65. doi: 10.1016/j.vetmic.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J, Xu X, Tao H, Li Y, Nan H, Wang Y, Tian M, Chen H. 2017. Structural analysis of porcine reproductive and respiratory syndrome virus nonstructural protein 7alpha (NSP7α) and identification of its interaction with NSP9. Front Microbiol 8:853. doi: 10.3389/fmicb.2017.00853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong S, Liu L, Wu W, Armstrong SD, Xia D, Nan H, Hiscox JA, Chen H. 2016. Determination of the interactome of nonstructural protein12 from highly pathogenic porcine reproductive and respiratory syndrome virus with host cellular proteins using high-throughput proteomics and identification of HSP70 as a cellular factor for virus replication. J Proteomics 146:58–69. doi: 10.1016/j.jprot.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 41.Yu Y, Wang R, Nan Y, Zhang L, Zhang Y. 2013. Induction of STAT1 phosphorylation at serine 727 and expression of proinflammatory cytokines by porcine reproductive and respiratory syndrome virus. PLoS One 8:e61967. doi: 10.1371/journal.pone.0061967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manns MP, Buti M, Gane E, Pawlotsky JM, Razavi H, Terrault N, Younossi Z. 2017. Hepatitis C virus infection. Nat Rev Dis Primers 3:17006. doi: 10.1038/nrdp.2017.6. [DOI] [PubMed] [Google Scholar]
- 43.Duarte M, Gelfi J, Lambert P, Rasschaert D, Laude H. 1993. Genome organization of porcine epidemic diarrhoea virus. Adv Exp Med Biol 342:55–60. doi: 10.1007/978-1-4615-2996-5_9. [DOI] [PubMed] [Google Scholar]
- 44.Jung K, Saif LJ. 2015. Porcine epidemic diarrhea virus infection: etiology, epidemiology, pathogenesis, and immunoprophylaxis. Vet J 204:134–143. doi: 10.1016/j.tvjl.2015.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol 84:11045–11055. doi: 10.1128/JVI.00655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meng XJ, Paul PS, Halbur PG, Lum MA. 1996. Characterization of a high-virulence US isolate of porcine reproductive and respiratory syndrome virus in a continuous cell line, ATCC CRL11171. J Vet Diagn Invest 8:374–381. doi: 10.1177/104063879600800317. [DOI] [PubMed] [Google Scholar]
- 47.Benfield DA, Nelson E, Collins JE, Harris L, Goyal SM, Robison D, Christianson WT, Morrison RB, Gorcyca D, Chladek D. 1992. Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate ATCC VR-2332). J Vet Diagn Invest 4:127–133. doi: 10.1177/104063879200400202. [DOI] [PubMed] [Google Scholar]
- 48.Lanciotti RS, Lambert AJ, Holodniy M, Saavedra S, Signor Ldel C. 2016. Phylogeny of Zika virus in Western hemisphere, 2015. Emerg Infect Dis 22:933–935. doi: 10.3201/eid2205.160065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang YJ, Stein DA, Fan SM, Wang KY, Kroeker AD, Meng XJ, Iversen PL, Matson DO. 2006. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol 117:117–129. doi: 10.1016/j.vetmic.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Wang R, Ma Z, Xiao Y, Nan Y, Wang Y, Lin S, Zhang YJ. 2017. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via nsp5, which induces STAT3 degradation. J Virol 91:e02087-16. doi: 10.1128/JVI.02087-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsuda T, Cepko CL. 2004. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc Natl Acad Sci U S A 101:16–22. doi: 10.1073/pnas.2235688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang YJ, Wang KY, Stein DA, Patel D, Watkins R, Moulton HM, Iversen PL, Matson DO. 2007. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res 73:12–23. doi: 10.1016/j.antiviral.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kannan H, Fan S, Patel D, Bossis I, Zhang YJ. 2009. The hepatitis E virus open reading frame 3 product interacts with microtubules and interferes with their dynamics. J Virol 83:6375–6382. doi: 10.1128/JVI.02571-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li Y, Treffers EE, Napthine S, Tas A, Zhu L, Sun Z, Bell S, Mark BL, van Veelen PA, van Hemert MJ, Firth AE, Brierley I, Snijder EJ, Fang Y. 2014. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci U S A 111:E2172–E2181. doi: 10.1073/pnas.1321930111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo R, Katz BB, Tomich JM, Gallagher T, Fang Y. 2016. Porcine reproductive and respiratory syndrome virus utilizes nanotubes for intercellular spread. J Virol 90:5163–5175. doi: 10.1128/JVI.00036-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31:830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- 59.Wang R, Xiao Y, Opriessnig T, Ding Y, Yu Y, Nan Y, Ma Z, Halbur PG, Zhang YJ. 2013. Enhancing neutralizing antibody production by an interferon-inducing porcine reproductive and respiratory syndrome virus strain. Vaccine 31:5537–5543. doi: 10.1016/j.vaccine.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 60.Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ. 2008. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res 77:95–107. doi: 10.1016/j.antiviral.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nan Y, Wang R, Shen M, Faaberg KS, Samal SK, Zhang YJ. 2012. Induction of type I interferons by a novel porcine reproductive and respiratory syndrome virus isolate. Virology 432:261–270. doi: 10.1016/j.virol.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faye O, Faye O, Diallo D, Diallo M, Weidmann M, Sall AA. 2013. Quantitative real-time PCR detection of Zika virus and evaluation with field-caught mosquitoes. Virol J 10:311. doi: 10.1186/1743-422X-10-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Sharma RD, Paul PS. 1998. Monoclonal antibodies against conformationally dependent epitopes on porcine reproductive and respiratory syndrome virus. Vet Microbiol 63:125–136. doi: 10.1016/S0378-1135(98)00231-4. [DOI] [PubMed] [Google Scholar]