

ABSTRACT
The hepatitis A virus (HAV) cellular receptor 1 (HAVCR1), classified as CD365, was initially discovered as an HAV cellular receptor using an expression cloning strategy. Due to the lack of HAV receptor-negative replication-competent cells, it was not possible to fully prove that HAVCR1 was a functional HAV receptor. However, biochemistry, classical virology, and epidemiology studies further supported the functional role of HAVCR1 as an HAV receptor. Here, we show that an anti-HAVCR1 monoclonal antibody that protected African green monkey kidney (AGMK) cells against HAV infection only partially protected monkey Vero E6 cells and human hepatoma Huh7 cells, indicating that these two cell lines express alternative yet unidentified HAV receptors. Therefore, we focused our work on AGMK cells to further characterize the function of HAVCR1 as an HAV receptor. Advances in clustered regularly interspaced short palindromic repeat/Cas9 technology allowed us to knock out the monkey ortholog of HAVCR1 in AGMK cells. The resulting AGMK HAVCR1 knockout (KO) cells lost susceptibility to HAV infection, including HAV-free viral particles (vpHAV) and exosomes purified from HAV-infected cells (exo-HAV). Transfection of HAVCR1 cDNA into AGMK HAVCR1 KO cells restored susceptibility to vpHAV and exo-HAV infection. Furthermore, transfection of the mouse ortholog of HAVCR1, mHavcr1, also restored the susceptibility of AGMK HAVCR1 KO cells to HAV infection. Taken together, our data clearly show that HAVCR1 and mHavcr1 are functional HAV receptors that mediate HAV infection. This work paves the way for the identification of alternative HAV receptors to gain a complete understanding of their interplay with HAVCR1 in the cell entry and pathogenic processes of HAV.
IMPORTANCE HAVCR1, an HAV receptor, is expressed in different cell types, including regulatory immune cells and antigen-presenting cells. How HAV evades the immune response during a long incubation period of up to 4 weeks and the mechanism by which the subsequent necroinflammatory process clears the infection remain a puzzle that most likely involves the HAV-HAVCR1 interaction. Based on negative data, a recent paper from the S. M. Lemon and W. Maury laboratories (A. Das, A. Hirai-Yuki, O. Gonzalez-Lopez, B. Rhein, S. Moller-Tank, R. Brouillette, L. Hensley, I. Misumi, W. Lovell, J. M. Cullen, J. K. Whitmire, W. Maury, and S. M. Lemon, mBio 8:e00969-17, 2017, https://doi.org/10.1128/mBio.00969-17) suggested that HAVCR1 is not a functional HAV receptor, nor it is it required for HAV infection. However, our data, based on regain of the HAV receptor function in HAVCR1 knockout cells transfected with HAVCR1 cDNA, disagree with their findings. Our positive data show conclusively that HAVCR1 is indeed a functional HAV receptor and lays the ground for the identification of alternative HAV receptors and how they interact with HAVCR1 in cell entry and the pathogenesis of HAV.
KEYWORDS: African green monkey cells, CD365, CRISPR/Cas9, HAVCR1, human hepatoma Huh7 cells, Vero E6 cells, cellular receptor, gene editing, hepatitis A virus, mouse ortholog, TIM1, KIM1
INTRODUCTION
The monkey ortholog of HAVCR1 (CD365; also known as TIM1 or KIM1) was first identified as a hepatitis A virus (HAV) cellular receptor by an expression cloning strategy using a monoclonal antibody (mAb) that blocked infection of African green monkey kidney (AGMK) cells (1). Because receptor-negative HAV-permissive cells were not available (2), we used biochemistry and classical virology to further support the role of HAVCR1 as an HAV receptor, including neutralization of HAV by soluble forms of HAVCR1 that altered the viral particles and released the viral genome (3–5) and mutagenesis analysis that showed that HAV bound to the immunoglobulin-like (IgV) domain of HAVCR1 (6). As expected by the high degree of homology, human HAVCR1 was also identified as an HAV receptor (6). Early on, it was clear that additional HAV receptors might exist, as implied by the nomenclature used to designate the novel glycoprotein (HAV cellular receptor 1). We also showed that mouse liver cells contained an alternative HAV receptor different from the HAVCR1 mouse ortholog (7). Further studies on HAVCR1 revealed its natural function as a phosphatidylserine (PS) receptor that binds apoptotic cells and modulates immune responses (8). Furthermore, HAVCR1 has been implicated as a cell entry factor of enveloped viruses via interaction with the PS present on the viral membrane by viral apoptotic mimicry (9), a mechanism similar to that used by phagocytic cells to engulf and clear apoptotic cells to prevent disease.
HAV, a small nonenveloped RNA virus that belongs to the Picornaviridae, is the main cause of acute hepatitis in humans (for reviews, see references 10 and 11). HAV is mainly transmitted via the fecal-oral route and rarely transmitted parenterally by blood and blood products. The target organ of HAV replication is the liver, from where the virus is mainly shed by hepatocytes to the bile and released to the environment via the feces as free viral particles (vpHAV).
Cells infected with viruses produce extracellular vesicles containing viral genomes capable of transmitting infection to other cells (12). Like cells infected with many other viruses, HAV-infected cells produce exosomes, a type of extracellular vesicle that buds into intracellular multivesicular bodies (13), which are capable of infecting cells (14). Exosomes containing HAV (exo-HAV) are found in the bloodstream, but their role in pathogenesis, if any, is poorly understood and unlikely to have a significant effect in the fecal-oral route of transmission. A controlled necroinflammatory process in the liver triggered by an unknown mechanism(s) 2 to 4 weeks after infection causes the typical signs of hepatitis A (HA), including jaundice. The concomitant appearance of anti-HAV antibodies and acute liver damage generally results in viral clearance and resolution of the disease in about 2 months.
Soluble forms of HAVCR1 neutralized detergent-treated vpHAV purified by sedimentation in sucrose gradients (3, 4), indicating that HAVCR1 interacts directly with the viral particles. Because exosomes contain PS on their membranes (15), it is likely that exo-HAV also use HAVCR1 as a functional receptor to infect cells, an evolutionary trait that may enhance HAV infectivity.
Advances in the genetic manipulation of cells using RNA-guided clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 genome-editing technology allows the knockout (KO) of genes in culture cells, providing novel tools to interrogate gene function. Here, we used the CRISPR/Cas9 technology to knock out the monkey ortholog of HAVCR1 (mkHAVCR1) in HAV-permissive cells and study the function of HAVCR1 as an HAV receptor. A similar approach was recently undertaken by the Stanley M. Lemon and Wendy Maury (Lemon/Maury) labs and suggested that HAVCR1 was not a functional HAV receptor (16). However, this study from the Lemon/Maury labs contains significant flaws, including the HAVCR1 knockout strategy, which left soluble forms of HAVCR1 containing the IgV binding domain, and the use of cell lines expressing multiple HAV receptors. In our study, we used a clean knockout strategy that introduced deletions in the mkHAVCR1 IgV binding domain of 100 bp in one copy and 101 nucleotides in the other copy of the mkHAVCR1 gene that are critical for HAV binding. In addition to the mkHAVCR1 IgV deletions of 100 to 101 bp, our knockout strategy resulted in reading frame shifts that prevented expression of the other domains of mkHAVCR1. We also used a protective anti-HAVCR1 mAb to screen for cell lines that mainly use HAVCR1 as an HAV receptor. The anti-HAVCR1 mAb protected AGMK cells against HAV infection but only partially protected monkey Vero E6 and human hepatoma Huh7 cells, indicating that these cell lines express alternative HAV receptors. Therefore, we knocked out the mkHAVCR1 gene in AGMK cells. The resulting mutant cells, termed AGMK HAVCR1 KO cells, did not express mkHAVCR1 and were resistant to HAV infection. Transfection of the human HAVCR1 cDNA into AGMK HAVCR1 KO cells restored the susceptibility to HAV infection. Transfection of the cDNA of the mouse ortholog of HAVCR1, mHavcr1, into AGMK HAVCR1 KO cells also restored vpHAV and exo-HAV infection. Our data clearly show that HAVCR1 and mHavcr1 are indeed functional HAV receptors that mediate infection of vpHAV and exo-HAV. We concluded that HAVCR1 is required for HAV infection of AGMK cells and plays a significant role in HAV infection of other cell lines, including Vero E6 and Huh7 cells.
RESULTS
Cell lines express multiple HAV receptors.
Our previous work suggested that some cells express HAV cellular receptors unrelated to HAVCR1 (7). We further studied the expression of alternative HAV cellular receptors in HAV-susceptible cells using protective anti-HAVCR1 mAb 1D12 to block HAV infection (17). To do so, we developed a CFU assay based on the selection of cells that gained resistance to the antibiotic blasticidin (Bsd) upon infection with a recombinant HAV containing a Bsd resistance gene (HAV-Bsd). We used attenuated HAV-Bsd (18) to infect monkey AGMK and Vero E6 cells and pathogenic HAV-Bsd (19–21) to infect human hepatoma Huh7 cells due to their optimal viral growth in the corresponding cell lines. Because HAV does not cause a cytopathic effect, cells infected with HAV-Bsd grew in the presence of Bsd, forming colonies, whereas uninfected cells died due to the antibiotic selection. Cells were infected with vpHAV or exo-HAV purified from HAV-Bsd-infected cells (Fig. 1A and B). Before performing the protection assay, we determined that the AGMK, Vero E6, and Huh7 cells expressed the monkey or human orthologs of HAVCR1 at the cell surface (Fig. 1C to E), indicating that the 1D12 epitope was conserved in monkeys and humans. Pretreatment with anti-HAVCR1 mAb 1D12 significantly reduced the number of Bsd-resistant CFU in AGMK cells (Fig. 1F) infected with exo-HAV (72%, P < 0.05) or vpHAV (96%, P < 0.01) (Fig. 1I), indicating that cells were protected against HAV infection and suggesting that HAVCR1 is the main HAV receptor in these cells. A smaller reduction in the number of CFU was observed in mAb 1D12-treated Vero E6 cells (Fig. 1G), indicating that these cells express alternative HAV receptors that resulted in partial protection against exo-HAV (45%, P < 0.05) and vpHAV (37%, P < 0.05) infection (Fig. 1J). Interestingly, mAb 1D12 partially protected Huh7 cells against exo-HAV (40%, P < 0.01) (Fig. 1H) but not vpHAV (Fig. 1F), indicating that HAVCR1 functions as a receptor for exo-HAV in these cells. Because mAb 1D12 did not protect Huh7 cells against vpHAV infection, we concluded that, in addition to HAVCR1, the Bsd-resistant Huh7 cells express an alternative vpHAV receptor(s). Taken together, these experiments show that HAVCR1 functions as the main HAV receptor in AGMK cells and that Vero E6 and Huh7 cells express an alternative HAV receptor(s), in addition to HAVCR1.
FIG 1.

HAVCR1 mediates HAV infection in cells expressing HAVCR1 and other alternative receptors. (A and B) Purification of exo-HAV and vpHAV from supernatants of AGMK cells infected with attenuated HAV-Bsd (A) or Huh7 cells infected with pathogenic HAV-Bsd (B) by isopycnic ultracentrifugation in iodixanol gradients. The HAV RNA in gradient fractions collected after isopycnic ultracentrifugation was quantified by RT-qPCR. Fractions 11 and 12, containing exo-HAV (density, 1.10 to 1.11 g cm−3), and fraction 19 or 20, containing vpHAV (density, 1.25 to 1.28 g cm−3), were collected and used for further experimentation. Arrows mark the exo-HAV and vpHAV peaks. GE, genome equivalents. (C to E) Expression of mkHAVCR1 at the cell surface of AGMK (C) and Vero E6 (D), and human HAVCR1 at the cell surface of Huh7 (E) cells stained with protective anti-HAVCR1 mAb 1D12 and analyzed by flow cytometry (black lines). Cells were also stained with an isotype mAb as a negative control (gray filled curves). (F to H) Protection of AGMK (F), Vero E6 (G), and Huh7 (H) cells against HAV infection by treatment with anti-HAVCR1 mAb 1D12. Cells pretreated with 50 μg/ml of mAb 1D12 (+) or medium (−) for 30 min at room temperature were infected with HAV-Bsd for 24 h, trypsinized, and selected with the antibiotic Bsd. At 12 days postinfection, cell colonies were fixed and stained with crystal violet (dark spots), and 6-well plates were imaged using a flatbed scanner. (I to K) The number of Bsd-resistant CFU was quantitated and graphed as the mean colony numbers from duplicate plates. Bars represent SDs for duplicate wells. Differences between control and mAb-treated cells were analyzed by Student's unpaired t test. *, P < 0.05; **, P < 0.01. Results are representative of those from two independent experiments.
HAVCR1 knockout blocks HAV infection.
Because Vero E6 and Huh7 cells express alternative yet unidentified HAV receptors, we focused our studies on AGMK cells, in which mkHAVCR1 functions as the main HAV receptor (Fig. 1). To knock out the mkHAVCR1 gene in AGMK cells, two single guide RNAs (sgRNA) were designed to introduce a deletion in mkHAVCR1 exon 2 (Fig. 2A), which codes for the IgV domain that interacts with HAV (6). An AGMK cell clone that contained deletions of 100 bp in one copy and 101 bp in the other copy of the mkHAVCR1 gene was identified and termed AGMK HAVCR1 KO cells. Nucleotide sequence analysis corroborated that the mkHAVCR1 gene in AGMK HAVCR1 KO cells contained the 100- and 101-bp deletions in the IgV domain, resulting in the knockout of mkHAVCR1 and reading frame shifts that prevented the expression of all the other domains of mkHAVCR1 (Fig. 2B). As expected, AGMK HAVCR1 KO cells did not express mkHAVCR1 at the cell surface, whereas parental AGMK cells expressed high levels of the receptor (Fig. 2C). Immunofluorescence (IF) analysis of cells infected with a virus preparation containing both exo-HAV and vpHAV (Fig. 1A) showed that AGMK HAVCR1 KO cells were resistant to HAV infection (Fig. 2D). These results indicated that HAVCR1 is required for HAV infection of AGMK cells.
FIG 2.

Knockout of mkHAVCR1 in AGMK cells prevents HAV infection. (A) Two single guide RNAs (sgRNA) in exon 2 of the mkHAVCR1 gene, which codes for the IgV binding domain, were designed to knock out the gene in AGMK cells using the CRISPR/Cas9 gene editing technology. (B) Nucleotide sequence alignment of AGMK parental (wild type [WT]) and AGMK HAVCR1 knockout (AGMK HAVCR1 KO) cells shows a 100-nucleotide deletion in exon 2 resulting in a frameshift mutation. (C) Analysis of the expression of mkHAVCR1 in AGMK HAVCR1 KO cells. mkHAVCR1 expression at the cell surface of AGMK parental (black line) and AGMK HAVCR1 KO (red line) cells was determined by flow cytometry analysis using anti-HAVCR1 mAb 1D12. Parental AGMK cells were also stained with an isotype control (filled gray curve). (D) IF analysis of HAV-infected cells. AGMK parental and AGMK HAVCR1 KO cells were infected with HAV-Bsd for 4 days, and the cells were fixed and permeabilized, stained with anti-HAV neutralizing mAbs (green fluorescence), counterstained with DAPI nuclear dye (blue fluorescence), and observed under a fluorescence microscope. Fluorescence and phase-contrast micrographs from representative fields were taken at a magnification of ×400. The experiments whose results are shown in panels D and E were repeated at least 3 times with similar results.
mkHAVCR1 is required for infection of both exo-HAV and vpHAV.
IF analysis suggested that mkHAVCR1 was required for the infection of both exo-HAV and vpHAV (Fig. 2D). Therefore, we analyzed infection of AGMK HAVCR1 KO and parental cells with purified exo-HAV or vpHAV. A growth kinetics analysis of cells infected with exo-HAV or vpHAV using reverse transcription-quantitative PCR (RT-qPCR) showed a marked increase in the number of HAV genomes in parental AGMK cells (P < 0.01) but not in AGMK HAVCR1 KO cells (Fig. 3A). These RT-qPCR results correlated with the increase in the number of Bsd-resistant CFU in the parental cells compared to the low background of Bsd-resistant CFU in AGMK HAVCR1 KO cells (Fig. 3B). At long absorption times, we observed the appearance of a very low background level of Bsd-resistant CFU in AGMK HAVCR1 KO cells (compare the number of colonies at 12 versus 72 h of absorption), suggesting that HAV is capable of entering cells inefficiently in cell culture by, for instance, (i) binding of partially uncoated vpHAV to the cell surface and release of viral RNA into the cells, (ii) using an alternative HAV receptor(s) expressed in only a few cells or at a low density, or (iii) binding of exo-HAV to the cell surface and release of HAV genomes contained in the exo-HAV. Alternatively, this very low background level may also represent an artifact of the CFU assay with no relevance to in vivo infection. The significant decrease in the number of Bsd-resistant CFU in AGMK HAVCR1 KO cells clearly indicated that HAVCR1 is needed for the efficient infection of HAV. Taken together, our results confirmed that both exo-HAV and vpHAV require mkHAVCR1 for infection.
FIG 3.

HAVCR1 is required for infection of exo-HAV and vpHAV. Growth of exo-HAV and vpHAV in AGMK parental and AGMK HAVCR1 KO cells analyzed by RT-qPCR (A) and Bsd-resistant CFU assay (B). Infected cells were trypsinized at 12 to 72 h postinfection, 50% of the cells were used for RT-qPCR, and the remaining cells were seeded in 6-well plates for the Bsd-resistant CFU assay. RT-qPCR results are the mean ± SD for duplicate wells. Differences between means were analyzed by t test. **, P < 0.01. Dark dots in the Bsd-resistant CFU assay derive from HAV-infected cell colonies, and clear surfaces are free from cells. At 72 h postinfection, cell colonies formed an almost confluent monolayer. Data are representative of those from two experiments.
Transfection of HAVCR1 cDNA restores susceptibility of AGMK HAVCR1 KO cells to HAV infection.
To further confirm that HAVCR1 is a functional receptor required for HAV infection, we transfected cells with human HAVCR1 cDNA or empty vector and infected the cell transfectants with a preparation of HAV that contained both exo-HAV and vpHAV, as shown in Fig. 1A. AGMK HAVCR1 KO cell transfectants expressed HAVCR1 at the cell surface (Fig. 4A) and gained susceptibility to HAV infection, as determined by RT-qPCR (P < 0.05) (Fig. 4B) and the number of Bsd-resistant CFU (P < 0.01) (Fig. 4C), whereas vector-transfected cells showed background levels of Bsd-resistant CFU (Fig. 4C, middle). We performed a similar experiment, using HAV without the Bsd resistance marker, using RT-qPCR analysis, which showed that the Bsd resistance marker did not have an effect in the lack of susceptibility of the AGMK HAVCR1 KO cells to HAV infection and the gain of susceptibility after HAVCR1 cDNA transfection (data not shown). These data further confirm that HAVCR1 is a functional HAV receptor because HAVCR1 KO cells regained susceptibility to HAV infections upon transfection of HAVCR1 cDNA.
FIG 4.
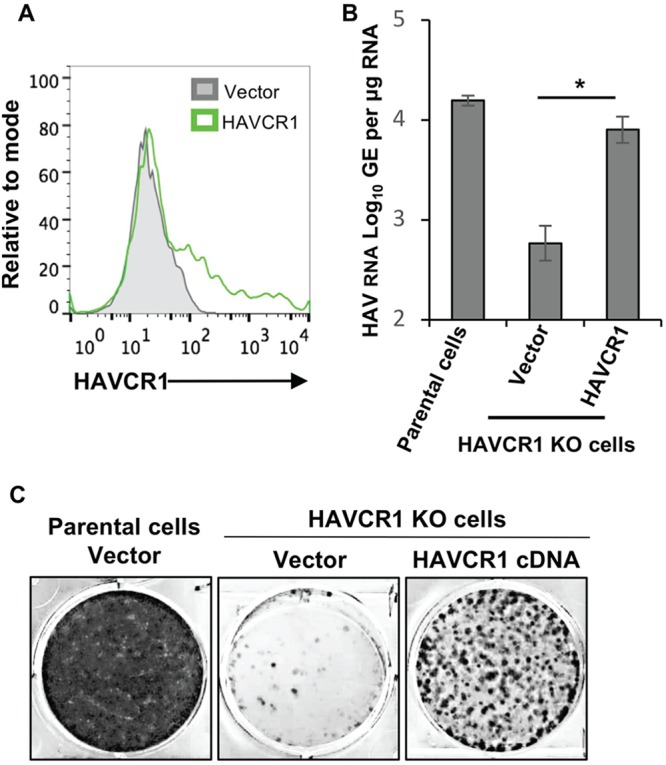
Transfection of HAVCR1 cDNA restores susceptibility to HAV infection. (A) Expression of HAVCR1 at the cell surface of vector-transfected (gray filled histogram) or HAVCR1-transfected (green line) AGMK HAVCR1 KO cells analyzed by flow cytometry. Transfectants were infected with an HAV preparation containing exo-HAV and vpHAV for 72 h and trypsinized. (B and C) Half of the cells were used for extraction of intracellular RNA for HAV-specific RT-qPCR (B), and the remaining cells were used for Bsd-resistant CFU analysis (C). RT-qPCR results are the mean ± SD for duplicate wells. Differences between means were analyzed by t test. *, P < 0.05. Dark dots in the Bsd-resistant CFU assay derive from HAV-infected cell colonies, and clear surfaces are free from cells. Data are representative of those from three experiments.
The mouse ortholog of HAVCR1 is also an HAV functional receptor.
The IgV binding domain of HAVCR1 and its mouse ortholog, mHavcr1, share approximately 60% homology (22) and are structurally similar (8), suggesting that mHavcr1 may also function as an HAV receptor. In a previous work, we showed that mHavcr1 bound less HAV than HAVCR1 expressed at the cell surface of HEK293 cells (17), suggesting that HAV had a lower affinity for mHavcr1 than for HAVCR1. The lack of a receptor-minus replication-permissive cell line at that time precluded us from testing whether mHavcr1 could also work as a functional HAV receptor. The development and characterization of AGMK HAVCR1 KO cells herein as a receptor-negative replication-permissive cell line for HAV allowed us to test whether mHavcr1 could indeed function as an HAV receptor. To do so, we transfected AGMK HAVCR1 KO cells with the cDNA of mHavcr1 or control HAVCR1. The mHavcr1 and HAVCR1 cell transfectants expressed similar levels of these receptors at the cell surface (Fig. 5A). RT-qPCR analysis showed a significant increase of HAV genomes in the AGMK HAVCR1 KO cells transfected with the mHavcr1 cDNA compared to vector-transfected cells upon infection with exo-HAV (P < 0.01) or vpHAV (P < 0.001) (Fig. 5B), which was similar to the increase observed in the AGMK HAVCR1 KO cells transfected with human HAVCR1 cDNA (Fig. 4B). The Bsd-resistant CFU analysis was consistent with the RT-qPCR analysis, showing a significant increase in the number of colonies of the mHavcr1 and HAVCR1 transfectants (Fig. 5C) compared to vector-transfected cells (P < 0.05). To rule out the possibility that residual membranes associated with vpHAV could mediate infection of vpHAV via binding of PS to the HAVCR phospholipid-binding pocket, we treated purified vpHAV with detergent (1% Sarkosyl) and repurified it in an iodixanol gradient (Fig. 5D). Sarkosyl-treated and untreated vpHAV migrated at the same density in the iodixanol gradients, indicating that there were no lipids associated with the vpHAV particles. Furthermore, there were no significant differences in the infectivity of Sarkosyl-treated or untreated vpHAV in AGMK HAVCR1 KO cell transfectants expressing mHavcr1 or HAVCR1 (Fig. 5E), further confirming that these two receptors mediate infection of vpHAV. Finaly, IF analysis using anti-HAV neutralizing monoclonal antibodies (mAbs) revealed the typical punctuated cytoplasmic fluorescence of HAV-infected cells in the mHavcr1 transfectants infected with exo-HAV or vpHAV, whereas vector-transfected cells showed the background level of fluorescence (Fig. 5F), confirming that the mHavcr1 cell transfectants regained susceptibility to HAV infection. Taken together, these data indicate that mHavcr1 is a functional HAV receptor that mediates infection of exo-HAV and vpHAV.
FIG 5.

The mouse ortholog of HAVCR1, mHavcr1, is a functional HAV receptor. (A) Expression of mHavcr1 (left) or HAVCR1 (right) at the cell surface of AGMK HAVCR1 KO cells transfected with the cDNA of mHavcr1 (light blue line), HAVCR1 (green line), or the vector (filled gray histogram), as determined by flow cytometry. (B and C) Transfectants were infected with exo-HAV or vpHAV for 72 h, and virus growth was determined by RT-qPCR (B) and the Bsd-resistant CFU assay (C), as described in the legend to Fig. 4. (D) Isopycnic ultracentrifugation of purified vpHAV treated (blue line) or not treated (gray line) with 1% Sarkosyl in iodixanol gradients, as described in the legend to Fig. 1A. (E) HAV growth in cell transfectants infected with detergent-treated or untreated vpHAV from the assay whose results are shown in panel D, determined by RT-qPCR at 72 h postinfection. (F) IF analysis of cell transfectants infected with exo-HAV or vpHAV as described in the legend to Fig. 2E. RT-qPCR results are the mean ± SD for duplicate wells. Differences between means were analyzed by t test. *, P < 0.05; **, P < 0.01; ns, no significant difference. Data are representative of those from at least two different experiments.
DISCUSSION
Several decades ago, we discovered mkHAVCR1 using protective mAb 190-4 and an expression cloning strategy and showed that mkHAVCR1 was an attachment receptor for HAV (1, 23). Due to the lack of an HAV receptor-negative replication-permissive cell line (2), we were unable to conclusively demonstrate that mkHAVCR1 and its human ortholog (24) were indeed functional HAV receptors. However, several lines of evidence gathered using biochemistry, molecular biology, and classical virology supported the role of HAVCR1 as an HAV receptor. We showed that HAV bound to the IgV domain of mkHAVCR1 (6, 25) and that Fc fusions of mkHAVCR1 neutralized, altered, and released the viral genome from HAV particles (3–5). The inverse association between HAV infection and the development of asthma and autoimmune diseases (26–29) linked to the interaction of HAV with HAVCR1 (30, 31) further supported the role of HAVCR1 in pathogenesis. Moreover, the influence of HAVCR1 polymorphisms in the severity of HA (32) and the inhibition of regulatory T cell function by the HAV-HAVCR1 interaction (17) suggest that HAVCR1 may be involved in the pathogenesis of HAV in humans. For many years, we unsuccessfully attempted to knock out HAVCR1 in HAV-susceptible cells using different technologies (unpublished results) to provide conclusive evidence of the functionality of HAVCR1 as an HAV receptor. The emergence of the CRISPR/Cas9 technology allowed us to finally knock out mkHAVCR1 in clone GL37 of AGMK cells, the same cell line used for the initial identification of mkHAVCR1 (1). Here, we show that the knockout of mkHAVCR1 in AGMK cells blocked infection of HAV mediated by viral particles (vpHAV) and exosomes containing HAV (exo-HAV). Transfection of HAVCR1 cDNA restored the susceptibility of AGMK HAVCR1 KO cells to HAV infection, further confirming that HAVCR1 is indeed a functional HAV receptor. Moreover, we also showed that the mouse ortholog of HAVCR1, mHavcr1, is a functional HAV receptor.
The data presented herein on mAb 1D12-mediated protection against HAV infection provide clear evidence that cells, such as monkey kidney Vero E6 and human hepatoma Huh7 cells, express alternative HAV receptors, in addition to mkHAVCR1 or HAVCR1. Therefore, we focused our work on AGMK cells, which mainly use mkHAVCR1 as an HAV receptor and do not express or express very low levels of an alternative HAV receptor(s). In a recent paper by the Stanley M. Lemon and Wendy Maury labs (16), it was suggested that HAVCR1 is not a functional HAV receptor because the knockout of mkHAVCR1 in Vero and HAVCR1 in Huh7 cells did not prevent HAV infection. However, as we showed herein, Vero and Huh7 cells express alternative HAV receptors, in addition to mkHAVCR1 or HAVCR1, which underscores the futility of assessing the function of a viral receptor in cell lines that also express alternative receptors. The same Lemon/Maury lab paper showed 10-fold more HAV genomes at day 14, but not at other times, in the liver of mHavcr1-Ifnar1 double-KO mice compared to Ifnar1 single-KO mice, indicating that mHavcr1 plays a significant role in the pathogenesis of HAV (16). Interestingly, our data indicated that mHavcr1 is indeed a functional HAV receptor, which is consistent with the findings in the paper from the Lemon/Maury labs of the effect of mHavcr1 in the pathogenesis of HAV in their mouse model (16). Clearly, the data presented herein using the AGMK HAVCR1 KO cells and the data in the paper from the Lemon/Maury labs (16) using mHavcr1/Ifnar1 double-KO mice show that mHavcr1 is a functional HAV receptor that has a significant role in the pathogenesis of HAV in the Ifnar1 KO mouse model. In this paper, we confirmed the existence of alternative HAV receptors different from HAVCR1, which paves the way for their identification to further understand the cell entry process of HAV. During the first 1 to 4 weeks after infection, HAV evades immune recognition, resulting in the peak of replication before viral clearance by a necroinflammatory process that resolves the acute infection (for reviews, see references 10 and 11). The understanding of the HAV-mediated mechanisms of immune evasion, control of the necroinflammatory process, and viral clearance may help explain why HAV can establish only acute infections, whereas hepatitis C virus (HCV), another human hepatotropic RNA virus, mainly establishes chronic infections. Further studies of the interaction of HAV with cellular receptors, including the interplay between HAVCR1 and an alternative receptor(s) in human liver cells and immune compartments, are likely to shed some light on the HAV cell entry and pathogenic processes and the still unresolved acute/chronic conundrum of viral infections.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
The GL37 clone of African green monkey kidney (AGMK) cells (1) was grown in Eagle′s minimum essential medium (EMEM) containing 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin-streptomycin. The Huh7-A-I clone of human hepatoma cells (Huh7 cells), which supports the stable growth of wild-type HAV (19), and Vero E6 cells (ATCC, CRL-1586) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 1% penicillin-streptomycin.
Recombinant HAV containing a blasticidin (Bsd) antibiotic-selectable marker gene (HAV-Bsd) derived from pathogenic HAV (19) and attenuated HAV (18) were grown in Huh7-A-I and AGMK cells, respectively, in the presence of 5 μg/ml Bsd. Viral titers were determined by an antibiotic resistance titration assay (ARTA) (20).
Phycoerythrin (PE)-labeled anti-human CD365 (HAVCR1) 1D12 mAb (BioLegend, Inc.) and PE-labeled anti-mouse cd365 (mHavcr1) RMT1-4 mAb (BioLegend, Inc.), directed against the IgV domain of HAVCR1 and mHavcr1, respectively, and a PE-labeled mouse IgG1 isotype control (clone 15H6; Southern Biotech) or a PE-labeled rat IgG2b(κ) isotype control (clone A95-1; BD Bioscience) were used for cell surface stainings. Unlabeled mAb 1D12 was also used to protect cells against HAV infection.
Mouse anti-HAV neutralizing mAbs K2-4F2 and K3-4C8 (33) (Commonwealth Serum Laboratories, Australia) and fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin antibody (KPL Inc.) were used to detect HAV-infected cells by immunofluorescence.
Purification of exosomes and viral particles from HAV-infected cells.
Exosomes and free HAV particles were purified as previously described (16) with some modifications. Briefly, cell culture supernatants from AGMK cells infected with attenuated HAV-Bsd or Huh7-A-I cells infected with pathogenic HAV-Bsd were collected at 8 days postinfection and clarified by centrifugation. Viral particles were pelleted by ultracentrifugation at 100,000 × g for 4 h at 4°C in a SureSpin 630 rotor (Sorvall), resuspended in phosphate-buffered saline (PBS), loaded onto an 8 to 40% iodixanol step gradient, and ultracentrifuged at 140,000 × g for 18 h at 4°C in an SW55i rotor (Beckman). Approximately 20 fractions of 250 μl were collected from the top of the gradient, and the HAV RNA content of each one was quantified by RT-qPCR (see below). The density of each gradient fraction was determined by refractometry using a Refracto 30GS instrument (Mettler Toledo).
To treat vpHAV with detergent, gradient-purified attenuated vpHAV was incubated in the presence of 1% Sarkosyl (N-lauroylsarcosine sodium salt; Sigma-Aldrich) for 2 h at room temperature and repurified through iodixanol gradients as described above.
HAV genome quantification by RT-qPCR assay.
Total RNA was isolated from gradient fractions using a High Pure viral RNA kit (Roche) or from infected cell lysates using an RNeasy kit (Qiagen). HAV RNA was quantified by real-time RT-qPCR using the TaqMan Fast Virus 1-step master mix (Applied Biosystems, Inc.), HAV-specific primers, and a 6-carboxyfluorescein–MGB probe (34). Analysis was performed in a QuantStudio 6 Flex real-time PCR system (Applied Biosystems, Inc.). HAV RNA levels extracted from infected cells were normalized to the total amount of RNA present in the sample.
Flow cytometry analysis.
A total of 5 × 105 cells were trypsinized and stained to assess expression of cell surface receptors. Cells were stained with PE-labeled mouse anti-human CD365 1D12 mAb, PE-labeled rat anti-mouse cd365 RMT1-4 mAb, PE-labeled mouse isotype, or PE-labeled rat isotype. Stained cells were analyzed by flow cytometry in a Guava EasyCyte instrument (EMD Millipore Corp), and analysis was performed using FlowJo software (Tree Star, Inc., Ashland, OR).
Bsd-resistant CFU assay.
Cells were infected with HAV stocks containing exo-HAV and vpHAV or gradient-purified exo-HAV or vpHAV. For experiments using AGMK or Vero E6 cells, attenuated HAV-Bsd produced in AGMK cells was absorbed for 12 h at 37°C and washed twice with complete medium. For experiments using Huh7 cells, pathogenic HAV-Bsd produced in Huh7 cells was absorbed for 2 h and washed twice with complete medium. For the growth curve study, cells were infected at a multiplicity of infection (MOI) of 0.01 50% tissue infective dose (TCID50)/cell and incubated at 37°C under 5% CO2 for 12 to 72 h. For the transfection studies, AGMK cells were infected at an MOI of 0.1 TCID50/cell for 72 h. For anti-HAVCR1 mAb 1D12 protection studies, 90%-confluent monolayers of AGMK, Vero E6, and Huh7 cells grown in 12-well plates were treated with 50 μg/ml of 1D12 for 30 min at room temperature and infected with exo-HAV or vpHAV at an MOI of 0.1 TCID50/cell for 24 h at 37°C. After incubation, the monolayers were trypsinized and seeded in 6-well plates at 5 × 104 to 10 × 104 cells/well and grown in complete medium containing 5 μg/ml Bsd at 37°C under 5% CO2. After incubation for 10 to 12 days, cells were fixed with 80% methanol in PBS for 1 h at 4°C, stained with 0.5% crystal violet solution in 25% methanol for 20 min at room temperature, and washed extensively to remove the excess of dye. The colonies were counted, and the numbers of CFU were expressed as the mean count from duplicate wells.
Knockout of HAVCR1 gene in AGMK cells using the CRISPR/Cas9 gene editing system.
The HAVCR1 gene in AGMK cells was knocked out using the CRISPR/Cas9 technology (35) at a genome editing service (Applied StemCells, Inc.). Briefly, two single guide RNA (sgRNA) constructs specific for the monkey HAVCR1 gene were designed to introduce a deletion in the mkHAVCR1 gene exon 2 (Fig. 2A), which codes for the IgV binding domain that interacts with HAV. The dual sgRNA guided the introduction of double-stranded DNA breaks that were joined by a nonhomologous end-joining (NHEJ) DNA repair pathway. To do so, AGMK cells were transfected with an sgRNA/Cas9-2A-puromycin-coexpressing vector. Single cell clones were selected in the presence of puromycin, and 200 clones were further expanded for genotyping. A restriction fragment length polymorphism (RFLP) assay of genomic PCR products of mkHAVCR1 exon 2 was used to screen for mutated clones, and automated nucleotide sequencing was used to characterize the knocked out clones.
An AGMK cell clone containing a deletion of 100 bp in one copy of the mkHAVCR1 gene and a deletion of 101 bp in the other copy that introduce out-of-frame shifts in exon 2 of mkHAVCR1 was expanded, the gene deletion was verified by nucleotide sequence analysis, and the lack of mkHAVCR1 expression at the cell surface was confirmed by flow cytometry analysis staining with mAb 1D12.
Immunofluorescence (IF) analysis.
Mock- and HAV-infected AGMK parental or AGMK HAVCR1 KO cells grown in eight-well Permanox chamber slides (Nunc, Inc.) or coverslips at 37°C were fixed with 4% paraformaldehyde for 30 min at room temperature, washed, permeabilized with 0.25% Triton X-100 in PBS for 20 min at room temperature, and blocked with 2% FBS in PBS for 1 h at room temperature. Cells were treated with 0.5 μg/ml the anti-HAV K24F2 and K34C8 mAbs for 1 h at room temperature, washed, and stained with a 1:50 dilution of fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin antibody. Coverslips were mounted with ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole; Life Technologies Corp.) as a nuclear counterstain. Micrographs were taken with a Zeiss Axioscope 200 fluorescence microscope at a magnification of ×400. Mock-infected cells did not develop HAV-specific fluorescence (data not shown).
HAV-Bsd growth curve.
AGMK parental and AGMK HAVCR1 KO cells were infected with vpHAV or exo-HAV at an MOI of 0.01 TCID50/cell and incubated at 37°C under 5% CO2. At different times postinfection, cells were trypsinized and HAV infection was analyzed by RT-qPCR and Bsd-resistant CFU assays.
Transfection of cells and HAV infections.
AGMK HAVCR1 KO cells were transiently transfected with an expression plasmid containing the full-length cDNA of human HAVCR1 or the mouse ortholog, Havcr1, under the control of the EF-1alpha promoter or with an empty expression plasmid (vector). Transfections were performed in 12-well plates using the Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer′s recommendations. The expression of the receptors at the cell surface was determined by flow cytometry at 48 h after transfection. Control parental AGMK cells were transfected with empty vector.
Transiently transfected cells were infected at 48 h after transfection with HAV-Bsd harvested from infected cells containing a mixture of vpHAV and exo-HAV, purified exo-HAV or vpHAV, or detergent-treated vpHAV using an MOI of 0.1 TCID50/cell or mock infected. The susceptibility of transfected cells to HAV infection was evaluated by intracellular HAV RNA quantification by RT-qPCR, selection of Bsd-resistant CFU, and IF analyses.
Statistical analyses.
Statistics were performed using GraphPad Prism software, version 6.0 (GraphPad Software, Inc.). Significant differences were assessed by unpaired Student's t test. P values of less than 0.05 were considered significant, and those of less than 0.01 or 0.001 were considered very significant.
ACKNOWLEDGMENTS
This work was supported in part by Food and Drug Administration (FDA) intramural funding to G.K. and a program project grant from the National Institute of Allergy and Infectious Diseases (2P01-AI054456-06A1 to G.K. as collaborator). This project was supported in part by an appointment to the Research Fellowship Program at the Office of Blood Research and Review, Center for Biologics Evaluation and Research, Food and Drug Administration, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and FDA (to M.I.C).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Kaplan G, Totsuka A, Thompson P, Akatsuka T, Moritsugu Y, Feinstone SM. 1996. Identification of a surface glycoprotein on African green monkey kidney cells as a receptor for hepatitis A virus. EMBO J 15:4282–4296. [PMC free article] [PubMed] [Google Scholar]
- 2.Dotzauer A, Feinstone SM, Kaplan G. 1994. Susceptibility of nonprimate cell lines to hepatitis A virus infection. J Virol 68:6064–6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silberstein E, Dveksler G, Kaplan GG. 2001. Neutralization of hepatitis A virus (HAV) by an immunoadhesin containing the cysteine-rich region of HAV cellular receptor-1. J Virol 75:717–725. doi: 10.1128/JVI.75.2.717-725.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silberstein E, Xing L, van de Beek W, Lu J, Cheng H, Kaplan GG. 2003. Alteration of hepatitis A virus (HAV) particles by a soluble form of HAV cellular receptor 1 containing the immunoglobulin- and mucin-like regions. J Virol 77:8765–8774. doi: 10.1128/JVI.77.16.8765-8774.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silberstein E, Konduru K, Kaplan GG. 2009. The interaction of hepatitis A virus (HAV) with soluble forms of its cellular receptor 1 (HAVCR1) share the physiological requirements of infectivity in cell culture. Virol J 6:175. doi: 10.1186/1743-422X-6-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feigelstock D, Thompson P, Mattoo P, Kaplan GG. 1998. Polymorphisms of the hepatitis A virus cellular receptor 1 in African green monkey kidney cells result in antigenic variants that do not react with protective monoclonal antibody 190/4. J Virol 72:6218–6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feigelstock DA, Thompson P, Kaplan GG. 2005. Growth of hepatitis A virus in a mouse liver cell line. J Virol 79:2950–2955. doi: 10.1128/JVI.79.5.2950-2955.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. 2010. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev 235:172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amara A, Mercer J. 2015. Viral apoptotic mimicry. Nat Rev Microbiol 13:461–469. doi: 10.1038/nrmicro3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pinto R, Bosch A, Kaplan G. 2013. Hepatitis A: immune response and virus evolution, p 173–189. In Gershwin ME, Vierling JM, Manns MP (ed), Liver immunology: principles and practice, 2nd ed Springer, New York, NY. [Google Scholar]
- 11.Kaplan GG, Konduru K, Manangeeswaran M, Jacques J, Amharref N, Nakamura S. 2013. Structure, molecular virology, natural history, and experimental models, p 29–42. In Thomas HC, Zuckerman AJ, Lok ASF, Locarnini SA (ed), Viral hepatitis, 4th ed Wiley-Blackwell, Chichester, West Sussex, United Kingdom. [Google Scholar]
- 12.van Dongen HM, Masoumi N, Witwer KW, Pegtel DM. 2016. Extracellular vesicles exploit viral entry routes for cargo delivery. Microbiol Mol Biol Rev 80:369–386. doi: 10.1128/MMBR.00063-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raposo G, Stoorvogel W. 2013. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKnight KL, Xie L, Gonzalez-Lopez O, Rivera-Serrano EE, Chen X, Lemon SM. 2017. Protein composition of the hepatitis A virus quasi-envelope. Proc Natl Acad Sci U S A 114:6587–6592. doi: 10.1073/pnas.1619519114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skotland T, Sandvig K, Llorente A. 2017. Lipids in exosomes: current knowledge and the way forward. Prog Lipid Res 66:30–41. doi: 10.1016/j.plipres.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Das A, Hirai-Yuki A, Gonzalez-Lopez O, Rhein B, Moller-Tank S, Brouillette R, Hensley L, Misumi I, Lovell W, Cullen JM, Whitmire JK, Maury W, Lemon SM. 2017. TIM1 (HAVCR1) is not essential for cellular entry of either quasi-enveloped or naked hepatitis A virions. mBio 8:e00969-17. doi: 10.1128/mBio.00969-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manangeeswaran M, Jacques J, Tami C, Konduru K, Amharref N, Perrella O, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ, Perrella A, Kaplan GG. 2012. Binding of hepatitis A virus to its cellular receptor 1 inhibits T-regulatory cell functions in humans. Gastroenterology 142:1516–1525.e3. doi: 10.1053/j.gastro.2012.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konduru K, Kaplan GG. 2010. Determinants in 3Dpol modulate the rate of growth of hepatitis A virus. J Virol 84:8342–8347. doi: 10.1128/JVI.01470-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Konduru K, Kaplan GG. 2006. Stable growth of wild-type hepatitis A virus in cell culture. J Virol 80:1352–1360. doi: 10.1128/JVI.80.3.1352-1360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Konduru K, Virata-Theimer ML, Yu MY, Kaplan GG. 2008. A simple and rapid hepatitis A virus (HAV) titration assay based on antibiotic resistance of infected cells: evaluation of the HAV neutralization potency of human immune globulin preparations. Virol J 5:155. doi: 10.1186/1743-422X-5-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tejada-Strop A, Costafreda MI, Dimitrova Z, Kaplan GG, Teo CG. 2017. Evaluation of potencies of immune globulin products against hepatitis A. JAMA Intern Med 177:430–432. doi: 10.1001/jamainternmed.2016.9057. [DOI] [PubMed] [Google Scholar]
- 22.Santiago C, Ballesteros A, Tami C, Martinez-Munoz L, Kaplan GG, Casasnovas JM. 2007. Structures of T cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity 26:299–310. doi: 10.1016/j.immuni.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Locarnini S. 1997. The attachment receptor for hepatitis A virus. Trends Microbiol 5:45–47. doi: 10.1016/S0966-842X(96)30043-7. [DOI] [PubMed] [Google Scholar]
- 24.Feigelstock D, Thompson P, Mattoo P, Zhang Y, Kaplan GG. 1998. The human homolog of HAVcr-1 codes for a hepatitis A virus cellular receptor. J Virol 72:6621–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson P, Lu J, Kaplan GG. 1998. The Cys-rich region of hepatitis A virus cellular receptor 1 is required for binding of hepatitis A virus and protective monoclonal antibody 190/4. J Virol 72:3751–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bach JF. 2005. Infections and autoimmune diseases. J Autoimmun 25(Suppl):74–80. doi: 10.1016/j.jaut.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 27.Matricardi PM, Rosmini F, Panetta V, Ferrigno L, Bonini S. 2002. Hay fever and asthma in relation to markers of infection in the United States. J Allergy Clin Immunol 110:381–387. doi: 10.1067/mai.2002.126658. [DOI] [PubMed] [Google Scholar]
- 28.Matricardi PM, Rosmini F, Ferrigno L, Nisini R, Rapicetta M, Chionne P, Stroffolini T, Pasquini P, D'Amelio R. 1997. Cross sectional retrospective study of prevalence of atopy among Italian military students with antibodies against hepatitis A virus. BMJ 314:999–1003. doi: 10.1136/bmj.314.7086.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matricardi PM, D'Amelio R, Biselli R, Rapicetta M, Napoli A, Chionne P, Stroffolini T. 1994. Incidence of hepatitis A virus infection among an Italian military population. Infection 22:51–52. doi: 10.1007/BF01780768. [DOI] [PubMed] [Google Scholar]
- 30.Umetsu DT, McIntire JJ, DeKruyff RH. 2005. TIM-1, hepatitis A virus and the hygiene theory of atopy: association of TIM-1 with atopy. J Pediatr Gastroenterol Nutr 40(Suppl 1):S43. doi: 10.1097/00005176-200504001-00026. [DOI] [PubMed] [Google Scholar]
- 31.McIntire JJ, Umetsu SE, Macaubas C, Hoyte EG, Cinnioglu C, Cavalli-Sforza LL, Barsh GS, Hallmayer JF, Underhill PA, Risch NJ, Freeman GJ, DeKruyff RH, Umetsu DT. 2003. Immunology: hepatitis A virus link to atopic disease. Nature 425:576. doi: 10.1038/425576a. [DOI] [PubMed] [Google Scholar]
- 32.Kim HY, Eyheramonho MB, Pichavant M, Gonzalez Cambaceres C, Matangkasombut P, Cervio G, Kuperman S, Moreiro R, Konduru K, Manangeeswaran M, Freeman GJ, Kaplan GG, DeKruyff RH, Umetsu DT, Rosenzweig SD. 2011. A polymorphism in TIM1 is associated with susceptibility to severe hepatitis A virus infection in humans. J Clin Invest 121:1111–1118. doi: 10.1172/JCI44182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacGregor A, Kornitschuk M, Hurrell JG, Lehmann NI, Coulepis AG, Locarnini SA, Gust ID. 1983. Monoclonal antibodies against hepatitis A virus. J Clin Microbiol 18:1237–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Costafreda MI, Bosch A, Pinto RM. 2006. Development, evaluation, and standardization of a real-time TaqMan reverse transcription-PCR assay for quantification of hepatitis A virus in clinical and shellfish samples. Appl Environ Microbiol 72:3846–3855. doi: 10.1128/AEM.02660-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]