

ABSTRACT
WD repeat-containing protein 5 (WDR5) is essential for assembling the VISA-associated complex to induce a type I interferon antiviral response to Sendai virus infection. However, the roles of WDR5 in DNA virus infections are not well described. Here, we report that human cytomegalovirus exploits WDR5 to facilitate capsid nuclear egress. Overexpression of WDR5 in fibroblasts slightly enhanced the infectious virus yield. However, WDR5 knockdown dramatically reduced infectious virus titers with only a small decrease in viral genome replication or gene expression. Further investigation of late steps of viral replication found that WDR5 knockdown significantly impaired formation of the viral nuclear egress complex and induced substantially fewer infoldings of the inner nuclear membrane. In addition, fewer capsids were associated with these infoldings, and there were fewer capsids in the cytoplasm. Restoration of WDR5 partially reversed these effects. These results suggest that WDR5 knockdown impairs the nuclear egress of capsids, which in turn decreases virus titers. These findings reveal an important role for a host factor whose function(s) is usurped by a viral pathogen to promote efficient replication. Thus, WDR5 represents an interesting regulatory mechanism and a potential antiviral target.
IMPORTANCE Human cytomegalovirus (HCMV) has a large (∼235-kb) genome with over 170 open reading frames and exploits numerous cellular factors to facilitate its replication. HCMV infection increases protein levels of WD repeat-containing protein 5 (WDR5) during infection, overexpression of WDR5 enhances viral replication, and knockdown of WDR5 dramatically attenuates viral replication. Our results indicate that WDR5 promotes the nuclear egress of viral capsids, the depletion of WDR5 resulting in a significant decrease in production of infectious virions. This is the first report that WDR5 favors HCMV, a DNA virus, replication and highlights a novel target for antiviral therapy.
KEYWORDS: human cytomegalovirus, WDR5, IINMs, capsid, nuclear egress
INTRODUCTION
The Herpesviridae family encompasses eight human-pathogenic members which are classified into three subfamilies (the Alpha-, Beta-, and Gammaherpesvirinae subfamilies) on the basis of their host range, cell tropism and site of latency, model of replication, and sequence similarity (1, 2). Human herpesvirus 5, a ubiquitous opportunistic pathogen also known as human cytomegalovirus (HCMV), is responsible for congenital infection in developed countries (0.6% to 0.7%) (3) and in developing countries (1% to 5%) (4), and approximately 50 to 90% of adults globally have been infected with HCMV (5).
The life cycle of HCMV proceeds within the nucleus and cytoplasm. After virus entry, capsid formation as well as double-stranded DNA synthesis and encapsidation occurs in an enlarged host cell nucleus (6). HCMV replicates and packages its double-stranded viral genome within or at the periphery of nuclear replication compartments (NRCs) in the nucleus (7–9). HCMV capsids have a diameter of about 85 nm, which prevents their direct transport into the cytoplasm through intact nuclear pores (which have a diameter of about 39 nm) (6, 10–12). Therefore, HCMV nuclear egress occurs in several steps: (i) capsids move from NRCs toward the periphery of the nucleus via F-actin filaments (13), which may help capsids gain contact with the inner nuclear membrane (INM). (ii) Viral nuclear egress complexes (NEC), encompassing viral proteins, such as pUL50, pUL53, and RASCAL, recruit viral kinase pUL97 and cellular proteins, including p32/gC1qR, emerin, protein kinase C, etc., to phosphorylate nuclear lamins. This in turn disrupts the nuclear lamina barrier to permit infoldings of the inner nuclear membrane (IINMs) so that capsids can undergo primary envelopment, budding into the perinuclear space. (iii) Enveloped capsids in the perinuclear space then fuse with the outer nuclear membrane (ONM) and undergo deenvelopment to be released into the cytoplasm (14–22). In the cytoplasm, viral tegument proteins, including pp28, pp65, pp71, pp150, and pUL48, sequentially surround the capsids. Viral envelope glycoproteins, including gB, gH, gL, gM, gN, and gO, are present in the Golgi apparatus-derived secretory vesicles within small transport vacuoles (23). The tegumented capsids are thought to be enveloped via budding into glycoprotein-containing vacuoles in the cytoplasm (23–25). Finally, virions exit the host cells by utilizing the cellular transport machinery and complete the viral life cycle (6).
WD repeat-containing protein 5 (WDR5), a member of the WD-40 repeat protein family, is found in several multisubunit complexes, such as histone 3 at lysine 4 (H3K4) methyltransferases of the SET1 family (SET1A, SET1B, MLL1, MLL2, MLL3, and MLL4) (26–30). Methylation of H3K4 is usually associated with transcriptionally active promoters (31). Hence, previous studies of WDR5 have focused on epigenetic modulation through H3K4 trimethylation (32–35). It is also documented that WDR5 plays an essential role in reprogramming and self-renewing embryonic stem cells and maintaining their pluripotency (36, 37) and promoting cancer cell proliferation and tumorigenesis in leukemia (32, 38, 39). Moreover, WDR5 has been shown to play an antiviral role in Sendai virus (SeV) infection by a mechanism involving viral RNA-triggered type I interferon (IFN) signaling (40). However, it remains to be determined whether WDR5 plays a similar role in HCMV (DNA virus) infection.
In this study, we found that WDR5 expression increases during HCMV infection and this serves to enhance HCMV replication. Furthermore, knockdown of WDR5 resulted in defects in capsid nuclear egress and reduced production of infectious virions. Our findings suggest that WDR5 contributes to HCMV replication.
RESULTS
HCMV infection upregulates WDR5.
Cellular protein WDR5 has been reported to be recruited to the VISA-associated complex (also known as MAVS, IPS-1, and Cardif) upon infection by Sendai virus (SeV) and to play an essential role in activating IFN regulatory factor 3 (IRF3) and NF-κB signaling (40). To investigate whether there was a corresponding role for WDR5 in HCMV infection, WDR5 and viral proteins were quantified in infected human embryonic lung fibroblasts (HELs) during HCMV infection. As expected, the levels of representative viral proteins, including immediate early (IE; IE1 and IE2), early (pUL44), and late (gB) proteins, increased as the virus infection progressed. However, the protein levels of WDR5 increased dramatically (Fig. 1A, right), a finding which is distinct from that seen during SeV infection, in which no change in the WDR5 protein level was noted (40). In contrast, herpes simplex virus 1 (HSV-1), a member of the Alphaherpesvirinae subfamily, caused a decrease in WDR5 protein levels (Fig. 1A, left). These data suggest that the increase in WDR5 may be a unique cellular response to HCMV infection.
FIG 1.

HCMV infection upregulates WDR5. HELs were mock infected (M) or infected with HSV-1 or HCMV (V) at an MOI of 3 and then harvested at 36 hpi (HSV-1) or the times indicated (HCMV). (A) Relative protein levels were determined from IBs of cell lysates that were probed for the indicated proteins; actin is a loading control. Protein levels relative to those in mock-infected controls were determined by densitometry and are indicated below each WDR5 blot. Representative images from three independent experiments are shown. (B) Total RNA was isolated, and the relative levels of WDR5 transcripts were determined by qRT-PCR; GAPDH is an internal control. Data were normalized to the levels in mock-infected cells to provide fold changes after HCMV infection. Data were analyzed by one-way ANOVA. *, P < 0.05; ***, P < 0.001. (C) HCMV-infected HELs were treated with cycloheximide (CHX; 100 μg/ml) at 72 hpi, and the levels of WDR5 were determined by IB at 4-h intervals after CHX addition and normalized to the levels measured at 0 h. Representative images from three independent experiments are shown (top); relative levels are presented as means ± SDs (bottom). Data were analyzed by one-way ANOVA. ***, P < 0.001. (D) At 72 hpi, HCMV-infected HELs were treated with DMSO (−) or MG132 (6 μM) (+) and then harvested 8 h later and analyzed to determine the extent of WDR5 ubiquitination by immunoprecipitation (IP) with WDR5 antibody followed by IB with antiubiquitin antibody (right). The levels of ubiquitinated proteins or specific proteins were determined by probing the IBs of input samples with antiubiquitin (Ub) antibody or antibodies to the indicated proteins (left).
The protein quantity measured at a steady state is usually the combined outcome of gene expression, translation, and protein degradation. Therefore, quantitative reverse transcription-PCR (qRT-PCR) was performed to examine WDR5 mRNA levels. As shown in Fig. 1B, compared to the levels in the mock-infected controls at the same time points, the mRNA level of WDR5 increased with progression of HCMV infection. Upregulation occurred at 12 h postinfection (hpi), and the mRNA levels continuously increased to about 6-fold of those for the mock-infected controls at 96 hpi. The stability of WDR5 was also examined in mock- and HCMV-infected HELs. To inhibit de novo protein synthesis, cells were treated with 100 μM cycloheximide (CHX) starting at 72 hpi and then harvested at 4-h intervals up to 84 hpi. Following CHX treatment, the WDR5 protein level rapidly decreased in mock-infected cells but was maintained at a constant level in HCMV-infected HELs (Fig. 1C). These data suggest that HCMV infection stabilizes WDR5.
WDR5 can be degraded through the proteasome-dependent pathway (41), and the ubiquitin-proteasome system is the main pathway to regulate proteolysis of target proteins (42). Thus, the increased stability of WDR5 during HCMV infection is presumably a consequence of reduced ubiquitin-proteasomal degradation or reduced WDR5 ubiquitination. The levels of ubiquitinated WDR5 were examined in mock- and HCMV-infected HELs. A substantial amount of ubiquitinated WDR5 was detected in mock-infected HELs, and the amount was increased by addition of the proteasome inhibitor MG132 (Fig. 1D, right). This finding was consistent with a previous report (41). Notably, in HCMV-infected HELs the levels of ubiquitinated WDR5 were dramatically reduced, even in the presence of MG132 (Fig. 1D, right). Taken together these results indicate that HCMV infection upregulates WDR5 protein levels by increasing WDR5 mRNA and suppressing ubiquitin-proteasomal degradation of WDR5.
WDR5 affects HCMV replication.
HCMV infection not only increased the expression of WDR5 but also inhibited its degradation. Thus, the increased expression of WDR5 during HCMV infection could have an impact on HCMV replication. To aid in our investigations, human telomerase reverse transcriptase (hTERT)-immortalized HEL cells (HELFs) (43) were employed to construct lentivirus-transduced cell lines that overexpressed WDR5 or in which WDR5 was knocked down. Overexpression of WDR5 had no significant impact on HCMV viral genome replication (Fig. 2A) or viral protein expression throughout infection (Fig. 2B) or caused only a minimal difference in infectious virus yields (Fig. 2C, left and middle). In contrast, HSV-1 replication was clearly suppressed by WDR5 overexpression (Fig. 2C right). These data suggest that the levels of WDR5 that are induced following HCMV infection are sufficient to support HCMV replication.
FIG 2.
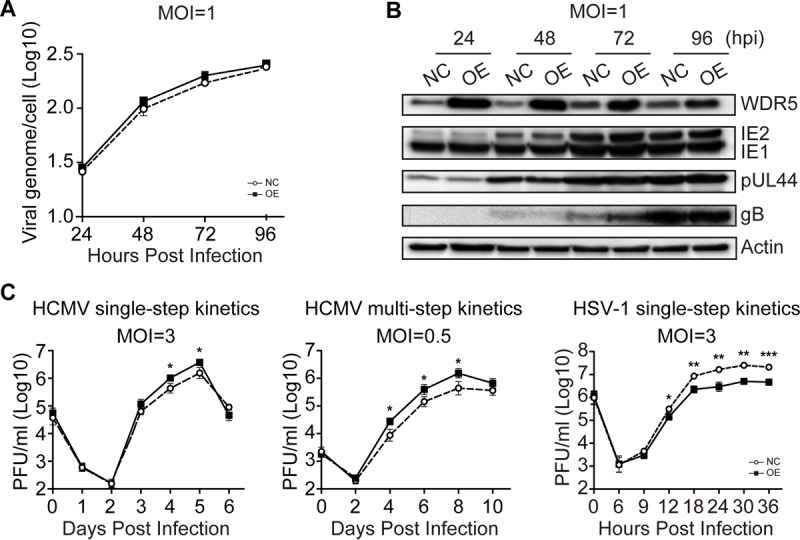
WDR5 overexpression enhances HCMV genome replication, viral gene expression, and infectious virus yield. HELFs were transduced with lentiviruses designed to overexpress WDR5 (OE) or the empty vector control (NC) and then infected with HCMV or HSV-1 at the indicated MOIs. Infected cells and culture supernatants were collected at the times indicated and analyzed to determine viral genome copy number, viral protein level, and infectious virus titers. (A) Viral genome replication. The HCMV genome copy number was determined by qPCR and normalized to the amount of cellular DNA (GAPDH). Results are shown as means ± SDs. (B) IB was used to determine relative WDR5 or viral protein levels in cell lysates. Representative images from three independent experiments are shown. (C) Growth kinetics of viral infection. NC and OE cells were infected with HCMV at an MOI of 3 (left) or 0.5 (middle) or HSV-1 at an MOI of 3 (right). Supernatant samples were collected at the indicated times postinfection, and infectious virus was titrated by a plaque formation assay. The average results from three independent ones are shown. Significance was analyzed by one-way ANOVA. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
WDR5 expression was then knocked down using short hairpin RNAs (shRNAs). Lentiviruses expressing scrambled shRNA (sh-Scram) or three WDR5-specific shRNAs (sh-W1, sh-W2, and sh-W3) were transduced into HELFs. Knockdown efficiencies are shown in Fig. 3A. All three WDR5-specific shRNAs decreased WDR5 protein levels, with sh-W3 being the most effective. Clonal cell lines isolated from HELFs that were transduced with sh-W3 were screened for WDR5 expression, and one, designated KD, was identified as expressing low levels of WDR5. A control cell line, designated Ctl, was isolated in parallel from sh-Scram-transduced HELFs. Lastly, KD cells were transduced with a lentivirus expressing WDR5, and clonal cell lines were screened to identify a cell line, designated Rec, in which normal WDR5 expression was restored. As seen in Fig. 3B, WDR5 expression in KD cells was significantly decreased, while Rec cells expressed WDR5 at levels that were comparable to those for Ctl cells. The viability of KD and Rec cells was slightly lower than that of Ctl cells, but the differences were not statistically significant (Fig. 3C).
FIG 3.

Knockdown of WDR5 suppresses HCMV replication. (A) Efficiency of WDR5 knockdown. HELFs were transduced with lentiviruses expressing scrambled shRNA (sh-Scram) or three WDR5-specific shRNAs (sh-W1, sh-W2, sh-W3), and WDR5 levels were examined by IB at 48 h postransduction. WDR5 levels relative to those in sh-Scram-transduced cells are listed below each blot. (B) WDR5 protein level in the established cell lines. WDR5 levels in stable cell lines derived from HELFs transduced with sh-Scram (Ctl cells) or sh-W3 (KD cells) or KD cells transduced with WDR5-expressing lentivirus (Rec cells) were examined by IB. WDR5 levels relative to those in Ctl cells (after normalization to the actin level) are listed below the corresponding blots. (C) Cell viability. The viability of cells of the Ctl, KD, and Rec cell lines were determined by MTT assay. Data were from three independent experiments and were analyzed by one-way ANOVA followed by the Bonferroni post hoc test. Results are presented as means ± SDs. NS, not significant. (D) Growth kinetics of viruses. Ctl, KD, or Rec cells were infected with HCMV or HSV-1 at the indicated MOIs. Culture supernatants were collected at the indicated times postinfection, and infectious virus titers were determined by a plaque formation assay. Data were collected from three independent experiments, performed in triplicate, and analyzed by one-way ANOVA (right). Results are presented as means ± SDs. **, P < 0.01; ***, P < 0.001. When a comparison among experimental groups was needed (left and middle), the Bonferroni post hoc test was conducted for multiple-test corrections. **, P < 0.0033; ***, P < 0.0003.
The Ctl, KD, and Rec cells were then infected with HCMV at a multiplicity of infection (MOI) of 3 or 0.5, and infectious virus titers in the cell culture were determined at different days postinfection (dpi) to generate single- and multistep growth curves. As expected, similar viral growth kinetics were observed in Ctl and Rec cells; however, the infectious yield was significantly decreased in KD cells, with the most significant effect being observed at an MOI of 0.5. The virus titers produced by KD cells were 8.9-fold lower than those produced by Ctl cells at 5 dpi at an MOI of 3 and 22-fold lower at 6 dpi at an MOI of 0.5. This suppression was maintained to 10 dpi at the low MOI (Fig. 3D, left and middle). In contrast, HSV-1 replication was significantly enhanced by knockdown of WDR5 (Fig. 3D, right). These results suggest that HCMV promotes WDR5 expression to enhance its replication efficiency.
Knockdown of WDR5 does not affect viral entry but slightly attenuates viral genome replication and viral gene expression.
The replication of HCMV can be affected at different stages of the virus life cycle, such as viral entry, viral genome replication, viral gene expression, capsid assembly, viral genome encapsidation, nuclear egress, tegumentation, envelopment, and release of mature virions (20). To determine which of these steps are affected by WDR5, viral replication at each step was compared in Ctl, KD, and Rec cells.
Initially, viral entry was compared among Ctl, KD, and Rec cells. Following viral entry and uncoating, the viral tegument protein pp65 rapidly translocates to the nucleus. Thus, cell-associated pp65 is commonly used as an indicator for viral attachment or entry (44–46). To measure pp65 translocation, cells were infected at an MOI of 5 and incubated at 4°C for 1 h to allow virus to attach. Cells were then incubated for 1 h at 37°C to allow virus entry, washed to remove unattached virus, trypsinized, and analyzed by immunoblotting (IB) to determine the levels of cell-associated pp65. Similar pp65 amounts were observed with Ctl, KD, or Rec cells (Fig. 4A). IE1 gene expression is another commonly used marker of HCMV entry and also indicates initiation of viral replication. IE1 protein was examined by immunofluorescence analysis (IFA) at 12 h following infection at an MOI of 1. Similar percentages of IE1-positive cells were observed for Ctl, KD, and Rec cells (Fig. 4B). These data indicate that viral entry is not affected by knockdown of WDR5.
FIG 4.

Effect of WDR5 knockdown on viral entry, genome replication, and viral gene expression. Ctl, KD, or Rec cells were mock infected (M) or infected with HCMV (V) at the indicated MOIs. Cells were harvested or fixed at the indicated times postinfection and processed for determination of viral entry, viral genome replication, or viral gene expression. (A) Viral entry determined by input pp65 levels. Cells were harvested at 1 hpi and analyzed by IB to detect pp65. Protein levels were quantified by densitometry, normalized to the amount of actin, and compared to the corresponding levels in Ctl cells. WDR5 and pp65 protein levels relative to those in Ctl cells are listed below the corresponding blots. (B) Viral entry determined by IE1 expression. Ctl, KD, or Rec cells on coverslips were infected with HCMV at an MOI of 1 and fixed at 12 hpi. HCMV IE1 protein (red) was detected by IFA, and nuclei were counterstained with DAPI (blue). Total cells and IE1-positive cells were counted in 20 random fields selected from each experiment. Data were analyzed by one-way ANOVA followed by the Bonferroni post hoc test. Representative images (left) and quantifications (right) are shown. Results are medians ± SDs. NS, not significant. (C) Viral genome level. HCMV-infected Ctl, KD, and Rec cells were assessed for the HCMV genome copy number at the indicated times postinfection by qPCR. Data were analyzed by one-way ANOVA followed by the Bonferroni post hoc test. Results are shown as means ± SDs. *, P < 0.0167; ***, P < 0.0003. (D) Viral protein level. HCMV-infected Ctl, KD, and Rec cells were collected at the indicated times postinfection, and cell lysates were subjected to IB to detect the indicated proteins. Representative images from three independent experiments are shown.
Viral genome replication and viral gene expression were then compared in Ctl, KD, and Rec cells. Cells were infected with HCMV at an MOI of 1 and harvested at different times postinfection, and viral genome copy numbers were determined by quantitative PCR (qPCR). At 96 hpi, viral genome copy numbers in infected KD cells were reduced 1.7-fold compared with those in Ctl cells and 1.5-fold compared with those in Rec cells (Fig. 4C). The levels of representative viral gene products, including viral immediate early (IE; IE1/IE2), early (pUL44), and late (gB) proteins, were examined by IB. The protein levels of IE1/IE2 and pUL44 were similar or only mildly decreased in KD cells compared to Ctl or Rec cells. However, the level of the late protein gB was lower in KD cells than in Ctl or Rec cells (Fig. 4D). These data suggest that WDR5 knockdown affects the later events of the viral life cycle, rather than viral entry, genome replication, or immediate early and early viral gene expression.
Knockdown of WDR5 does not affect capsid formation.
The lack of an observed difference in viral entry along with only a modest decrease in viral genome replication and viral gene expression appeared to be inconsistent with the ∼22-fold reduction in infectious virus titers produced by KD cells (Fig. 3D, middle). Therefore, we focused our studies on the potential impact on late events of the HCMV life cycle, including virus assembly, egress, and release. Transmission electron microscopy (TEM) was initially used to investigate which steps are affected by WDR5 knockdown. First, the numbers of capsids were compared among infected Ctl, KD, and Rec cells at 96 hpi (MOI = 0.5). In the replication cycle of HCMV, viral capsids assemble in the nucleus before egress into the cytoplasm for further tegumentation, envelopment, and release (20, 47, 48). Representative micrographs showed that HCMV capsids were produced in all the three cell lines (Fig. 5A to F). Ctl, KD, and Rec cells on average contained about 221.0, 151.2, and 168.5 capsids per cell, respectively. Thus, the number of capsids in KD cells was about 1.5-fold and 1.1-fold lower than that in Ctl and Rec cells, respectively, but these differences were not statistically significant (Fig. 5G). In infected cell nuclei, HCMV genome packaging results in three types of capsids: A capsids lack DNA and appear empty, B capsids also lack DNA but contain scaffold proteins that produce a wagon wheel appearance, and C capsids contain electron-dense DNA and are therefore capable of maturing into infectious virions (6, 20, 49–51). The ratios of A, B, and C capsids were not significantly different between infected Ctl, KD, and Rec cells (Fig. 5H). These results indicate that knockdown of WDR5 does not significantly affect the efficiency of viral capsid assembly or genome packaging.
FIG 5.

Knockdown of WDR5 does not affect capsid formation. Ctl cells (A and B), KD cells (C and D), or Rec cells (E and F) were infected with HCMV at an MOI of 0.5 and fixed at 96 hpi for analysis by TEM. To evaluate the formation of capsids in the nuclei, capsid quantification and analysis were performed. Cell sections were captured at a magnification of ×1,700 (A, C, and E), and the boxed regions were enlarged to a magnification of ×9,600 (B, D, and F) to show the capsids. Cyto, cytoplasm; Nuc, nucleus. (G) For each experiment, the total numbers of A, B, or C capsids per cell were quantitated. (H) The prevalence of A, B, and C capsids in each cell type was calculated as the percentage of the total number of capsids per cell. (I) Capsids that localized outside of NRCs (B, D, and F, white arrows) were counted in 12 nuclei of each cell type. Data were collected from three independent experiments and analyzed by the Kruskal-Wallis test. The medians from each experiment are indicated, and their values are shown to the right of each plot. The means from the three experiments are shown above the data for each cell line. NS, not significant.
Most capsids form in NRCs, which can be identified by TEM as electron-dense, darkly staining inclusions in the nuclei, and are then transported from NRCs to the INM for nuclear egress (51). The numbers of capsids located in the periphery of the nucleus and outside NRCs (Fig. 5B, D, and F, white arrows) were quantified and compared in infected cells (Fig. 5I). There were, on average, 12.8, 9.6, and 11.2 capsids per nucleus that localized outside NRCs in infected Ctl, KD, and Rec cells, respectively, and again, the difference in the number of capsids per nucleus among the cells was not significant. This suggests that the migration of capsids from NRCs toward the nuclear periphery is not affected by WDR5 knockdown.
Knockdown of WDR5 impairs capsid nuclear egress.
Capsid nuclear egress refers to the translocation of capsids through the nuclear membrane (52). IINMs are the primary site for nuclear egress of HCMV capsids (21, 53), and viral proteins pUL50 and pUL53 are crucial for the proper formation of IINMs by contributing to the assembly of NEC. pUL50 interacts with pUL53 and forms a rim around the nucleus of the infected cell; thus, pUL53 can serve as a marker of NEC (17, 22, 54). To evaluate the effects of WDR5 knockdown on capsid nuclear egress, cells were costained with pUL53 and the HCMV major capsid protein (MCP). MCP-positive cells represented the cells that expressed capsid proteins or had capsids assembled. Among MCP-positive cells, the percentage of pUL53-positive cells increased and pUL53 formed a continuous nuclear rim as the infection progressed in all three cell types. By 48 hpi, a low percentage of cells had a continuous nuclear rim of pUL53 and pUL53 almost exclusively localized to the nucleus, with minor localization occurring at the perinuclear region. As the infection progressed, the percentage of cells with a continuous nuclear rim stained with anti-pUL53 antibodies continued to increase. As shown in Fig. 6A, the MCP-positive rates were not significantly different among Ctl, KD, or Rec cells, which was consistent with the similar capsid numbers per cell observed by TEM (Fig. 5G). However, the fraction of KD cells with a pUL53 rim was significantly decreased relative to that of Ctl and Rec cells during the entire infection time course. Moreover, some of the nuclear rims in KD cells exhibited discontinuous pUL53 staining and presented as dashed rims (Fig. 6A and B, white arrows). By 72 hpi, pUL53 gradually formed punctate structures that have been shown to coincide with IINMs (54). At 120 hpi, there were significantly more punctate pUL53 structures per pUL53-positive Ctl cell (12.2) or Rec cell (11.2) than per pUL53-positive KD cell (5.7) (Fig. 6C).
FIG 6.

Knockdown of WDR5 affects capsid nuclear egress by impairing NEC formation. Ctl, KD, or Rec cells were infected with HCMV at an MOI of 1, and the cells were fixed at the indicated times for IFA. (A) MCP (cyan) and pUL53 (red) were determined by IFA, and nuclei were counterstained with DAPI (blue). Representative images from three independent experiments are shown. (B) Cells staining positive for MCP or pUL53 were counted in 20 random fields. The percentages of pUL53-positive cells among MCP-positive populations during the course of HCMV infection are shown. Data from three independent experiments were analyzed by one-way ANOVA followed by the Bonferroni post hoc test. The results are presented as the means ± SDs. NS, not significant; *, P < 0.0167; **, P < 0.0033; ***, P < 0.0003. (C) Average numbers of pUL53 puncta per nuclear rim at 120 hpi in Ctl, KD, or Rec cells. Data were collected from three independent experiments and analyzed by the Kruskal-Wallis test followed by post hoc Dunn's multiple-comparison test. The medians from each experiment are indicated to the right of each plot, and the means from the three experiments for each cell line are shown above the data. NS, not significant; ***, P < 0.001.
pUL50 and pUL53 are crucial for NEC formation. To further determine whether WDR5 participates in NEC formation, pUL50 and pUL53 were transiently expressed in Ctl, KD, and Rec cells. When expressed individually, pUL50 mainly localized to the perinuclear rim and cytoplasm, while pUL53 principally localized in the nucleoplasm. Consistent with previous reports (21, 53, 54), when coexpressed, both pUL50 and pUL53 localized to the nuclear rim. In KD cells, colocalization of pUL50 and pUL53 formed discontinuous dashed-rim structures (Fig. 7). These data suggest that WDR5 depletion alters NEC formation.
FIG 7.

pUL50 and pUL53 colocalize in transfected Ctl, KD, or Rec cells. Uninfected Ctl, KD, or Rec cells were transiently transfected with plasmids carrying UL50-DsRed or hemagglutinin (HA)-UL53. Cells were fixed at 48 h posttransfection and stained with a mouse antihemagglutinin primary antibody, followed by Alexa Fluor 488-conjugated goat anti-mouse immunoglobulin secondary antibody (green). Nuclei were counterstained with DAPI (blue). Cells in the upper two rows were transfected with each plasmid independently, while cells in the next three rows were cotransfected with both plasmids simultaneously. Areas magnified in the bottom panels are framed by boxes in the panels above. Bars, 10 μm.
IINMs are the primary location for HCMV capsid nuclear egress (6, 10, 18). Using TEM, IINMs appeared as tubular infoldings of the inner nuclear membrane with their lumens adjacent to the nucleoplasm. Whether WDR5 knockdown alters IINMs was examined. The structure of IINMs was examined by TEM at 120 hpi and compared among infected Ctl, KD, and Rec cells. Representative images of IINMs (red dashed lines) were obtained in all of the infected Ctl, KD, or Rec cells (Fig. 8A to I). Quantifying the number of cells with IINM structures revealed that 38.6% of Ctl cells and 34.4% of Rec cells contained IINMs, while the corresponding fraction in KD cells was significantly lower (15.8%), as shown in Fig. 8J. The numbers of IINMs per IINM-containing nucleus were also quantified. On average, there were 2.5, 1.2, and 1.3 IINMs in each IINM-containing Ctl, KD, and Rec cell, respectively. The number of IINMs in KD cells was significantly lower than the number in Ctl cells (Fig. 8K). Due to the limitations of TEM, IINMs were observed in only less than half of the infected cells. The numbers of capsids associated with IINMs were further quantified in IINM-containing cells. Among IINM-containing cells, each KD cell contained an average of 4.5 capsids associated with IINM, whereas each Ctl and Rec cell contained 25.4 or 20.8 IINM-associated capsids. Thus, KD cells contained 5.7-fold and 4.5-fold fewer IINM-associated capsids than Ctl and Rec cells (Fig. 8L).
FIG 8.

Knockdown of WDR5 impairs formation of IINM. Ctl (A to C), KD (D to F), or Rec (G to I) cells were infected with HCMV at an MOI of 0.5 and then fixed at 120 hpi for TEM. Micrographs were captured at a magnification of ×1,700 (A, D, and G), while the boxed regions are enlarged to a magnification of ×3,500 (E) or ×6,800 (B, C, F, H, and I). IINMs are delineated by dotted red lines. (J) For each experiment, 12 cells with identifiable nuclei were randomly selected to calculate the percentages of cells containing IINMs. (K and L) The numbers of IINMs per cell (K) and the numbers of IINM-associated capsids per cell (L) were determined for each cell type. Data were collected from three independent experiments and analyzed by the Kruskal-Wallis test followed by post hoc Dunn's multiple-comparison test. The medians for each experiment are indicated to the right of each plot, and the means from the three experiments for each cell line are shown above the data. NS, not significant; *, P < 0.05; ***, P < 0.001.
In summary, the fraction of cells that contained IINMs, the number of IINMs per cell, and the number of capsids associated with IINMs and presumably undergoing nuclear egress were significantly reduced in KD cells. Normalizing these data to the fraction of KD cells forming IINMs indicated that there were 2.4-fold fewer IINM-containing cells in the KD cell line than in the Ctl cell line (38.6% Ctl cells/15.8% KD cells) and 2.2-fold fewer IINM-containing cells in the KD cell line than in the Rec cell line (34.4% Rec cells/15.8% KD cells). Furthermore, when the fold decrease in IINMs was then normalized to the respective change in the number of capsids associated with IINMs, KD cells had approximately 13.7-fold fewer IINM-associated capsids than Ctl cells (2.4 × 5.7) and 9.9-fold fewer IINM-associated capsids than Rec cells (2.2 × 4.5). Taken together, these results indicate that WDR5 knockdown impairs capsid nuclear egress by altering NEC formation, impairing IINM formation, and reducing capsid recruitment to IINMs.
Knockdown of WDR5 decreases viral particles in cytoplasm.
To assess the impact of WDR5 knockdown on production of viral particles in the cytoplasm, cells were infected and the numbers of viral particles in the cytoplasm were quantified. TEM images revealed a large number of viral particles, including virions, in the cytoplasm of Ctl cells (Fig. 9A to C) and Rec cells (Fig. 9G to I) but very few in KD cells (Fig. 9D to F). The median values for the number of cytoplasmic viral particles per cell in Ctl, Rec, and KD cells were 47.6, 34.0, and 4.5, respectively. Thus, the number of cytoplasmic viral particles in KD cells was 10.6- and 7.6-fold less than the number in Ctl and Rec cells, respectively, and these differences were statistically significant (Fig. 9J). The 10.6-fold difference in the numbers of cytoplasmic viral particles was consistent with the 13.7-fold decrease in IINM-associated capsids. Finally, restoration of WDR5 almost completely rescued the production of cytoplasmic viral particles (Fig. 9G to I). These results indicate that WDR5 knockdown impairs nuclear egress of capsids, which in turn impairs viral particles in the cytoplasm and decreases infectious virus yield.
FIG 9.

Knockdown of WDR5 decreases the number of cytoplasmic virions. Ctl (A to C), KD (D to F), or Rec (G to I) cells were infected with HCMV at an MOI of 0.5 and fixed at 120 hpi for analysis by TEM. Sections were captured at a magnification of ×1,700 (A, D, and G), and cytoplasmic compartments containing virus particles were captured at magnifications of ×3,500 (B, E, and H) and ×6,800 (C, F, and I). For each experiment, cytoplasmic virus particles located within virus assembly compartments were quantified in 10 cells. Representative images (A to I) and quantitations (J) are shown. The medians from each experiment are indicated to the right of each plot, and the medians from the three experiments for each cell line are shown above the data. Data were collected from three independent experiments and analyzed by the Kruskal-Wallis test followed by post hoc Dunn's multiple-comparison test. NS, not significant; ***, P < 0.001.
DISCUSSION
HCMV utilizes sophisticated strategies to exploit host factors in order to optimize viral replication. Upon HCMV infection, the virus initiates cell cycle arrest at the G1/S and G2/M phases to create an optimal cellular environment for viral replication by modulating multiple cell cycle regulators (44, 55). At the very immediate early stage, pp71 disrupts Daxx, IE1 decreases Sp100A to inhibit the intrinsic cellular immunity (56–58), and IE2 interacts with HDAC2 to relieve its repressive effect on the viral polymerase promoter (59). Upon HCMV nuclear egress, viral proteins pUL50 and pUL53 form a complex and recruit the cellular proteins p32/gC1qR, emerin, and protein kinase C to alter the structure of the nuclear membrane to facilitate the nuclear egress of capsids (21). During HCMV cytoplasmic virion assembly, the Golgi apparatus, the trans-Golgi network, and early endosomes are driven to form a viral assembly compartment to facilitate virion maturation (60).
In the present study, we found that HCMV infection increased the expression of and stabilized the cellular protein WDR5. Overexpression of WDR5 slightly enhanced HCMV replication, while knockdown significantly attenuated HCMV replication. To identify the steps in which WDR5 is involved, the replicative cycle of HCMV replication was investigated in WDR5-depleted cells. Our data indicate that formation of the NEC and IINMs was significantly impaired, suggesting that the nuclear egress of HCMV capsids is a key step in which WDR5 is involved.
Among the numerous cellular factors exploited by HCMV, proteins encoded by IFN-stimulated genes directly exhibit restrictive effects on microbial infections (61). However, HCMV has evolved mechanisms to allow or enhance expression of certain IFN-stimulated genes and repurposes some of them to facilitate virus production. This is distinct from the mechanisms adopted by other viruses, especially RNA viruses (62). These host factors are as follows: (i) gamma interferon (IFN-γ)-inducible protein 16 (IFI16) senses double-stranded DNA, activates IRF3 to initiate the innate immune response (63), and restricts HCMV replication by downregulating expression of viral early and late genes (64). In return, pp65 (pUL83) blocks the nuclear DNA sensing of IFI16 by hijacking IFI16 to activate HCMV major immediate early promoter (MIEP) (65). In late stages of HCMV infection, pUL97 mislocalizes IFI16 into the virus assembly compartment and entraps it in mature virions to compromise its restriction activity (66). (ii) Bone marrow stromal cell antigen 2 (BST2) is also present in HCMV virions and enhances viral entry. In addition, ectopic expression of BST2 in HCMV naturally nonpermissive HEK293M cells could allow virus entry (45). (iii) IFN-induced transmembrane proteins (IFITMs) are exploited by HCMV to promote the formation of the virus assembly compartment to facilitate the virus assembly process (67). (iv) Viperin interacts with viral protein vMIA (the viral protein of UL37 exon 1), resulting in its relocation from the endoplasmic reticulum to the mitochondria and mediating the generation of ATP, enhancing HCMV infectivity (68).
Other cellular factors not associated with innate immunity are also exploited by HCMV. For example, p53, a key regulator of the cell cycle and apoptosis, is activated and stabilized by HCMV infection in fibroblasts. The activated p53 is sequestered into the viral replication center, so that cell apoptosis is avoided (69). vMIA plays an antiapoptosis role via sequestering Bax in mitochondria and blocking Bax-mediated apoptosis (70). These factors work together to keep the infected cell alive and facilitate virus replication.
Thus, HCMV has developed sophisticated strategies to modify the host antiviral response and to direct antiviral molecules to facilitate its replication. In the current study, we found that WDR5, a host protein reported to play an essential role in induction of type I interferon and an antiviral response during infection of SeV (40), promotes HCMV replication. Overexpression of WDR5 enhanced HCMV replication but inhibited HSV-1 replication, whereas knockdown of WDR5 attenuated HCMV replication but promoted replication of HSV-1. We further found that the protein levels of WDR5 were upregulated during HCMV infection but downregulated by HSV-1 infection. HCMV and HSV-1 both belong to the Herpesviridae family, but HSV-1 is in the Alphaherpesvirinae subfamily, whereas HCMV is in the Betaherpesvirinae subfamily. HSV-1 has a wider host range, replicates more rapidly with an eclipse period of 18 to 20 h in Vero cells (71), and releases higher titers of cell-free virions. HCMV has strict species specificity (72), replicates in cells with a longer eclipse period of 48 to 72 h, and releases lower titers of cell-free virions. In clinical isolates, progeny virus remains highly cell associated. Therefore, it is possible that HCMV uses different mechanisms than HSV-1 to interact with WDR5 during its replication. Capsids of alphaherpesviruses move from the NRC to the nuclear rim by diffusion rather than by dependence on F actins (73–75). However, the migration of HCMV capsids is dependent on F actins (13, 75). Although the two subunits of NEC are highly conserved across the Herpesviridae family (pUL50 and pUL53 of HCMV, pUL34 and pUL31 of HSV-1 and pseudorabies virus, ORF27 and ORF29 of varicella-zoster virus, and BFRF1 and BFLF2 of Epstein-Barr virus [75]), viruses egress in different ways. For example, HSV-1 nuclear egress involves the US3 kinase, which is not conserved in HCMV (10). HCMV mainly forms tubular infolds of INM for capsid trafficking (6, 10, 76), while HSV-1 achieves capsid budding directly via remodeling INM without formation of tubular infoldings of INM (77–79). In HCMV infection, A, B, and C capsids associate with IINMs, which is consistent with a previous report (18). In HSV-1 infection, C capsids selectively bud into the INM (80). The mechanisms that are utilized by different herpesviruses for capsid nuclear egress require further investigation.
During nuclear egress of HCMV capsids, pUL50 interacts with pUL53 and forms a continuous nuclear rim structure in WDR5-competent cells (Ctl and Rec cells) but forms discontinuous dashed-rim structures in a lower fraction of WDR5-deficient cells (KD cells) under either HCMV infection or pUL50 and pUL53 cotransfection. These results support that knockdown of WDR5 impairs formation of NEC. pUL50 interacts with pUL53, but neither of them interacts with WDR5 directly, and another viral or cellular protein(s) may be involved in the formation of NEC. pUL97 phosphorylates pUL50 at S216 and pUL53 at S19, which is also required for the continuous nuclear rim of pUL50 and pUL53. In contrast, a discontinuous nuclear rim is observed in the absence of pUL97 or with mutant pUL50 (UL50-S216) and pUL53 (UL53-S19) (81). However, how formation of the NEC is affected by WDR5 knockdown requires additional study.
In summary, depletion of WDR5 significantly attenuates HCMV replication by decreasing NEC and IINM formation, thereby impairing capsid nuclear egress and, consequently, virion maturation in the cytoplasm. These results indicate that WDR5 contributes to HCMV replication via promoting nuclear egress.
MATERIALS AND METHODS
Ethics statement.
Human embryonic lung fibroblasts (HELs) were isolated from postmortem embryo lung tissue. The original source of the anonymized tissues was the Zhongnan Hospital of Wuhan University (Wuhan, China). The cell isolation procedures and research plans were approved by the Institutional Review Board (IRB; WIVH10201202) according to the Guidelines for Biomedical Research Involving Human Subjects at the Wuhan Institute of Virology, Chinese Academy of Sciences. The need for written or oral consent was waived by the IRB (44).
Cells and cell culture.
Human embryonic lung fibroblasts (HELs) were isolated and maintained in the lab as described previously (44, 82, 83). HELF, an immortalized human embryonic lung fibroblast cell line, was generated by introducing hTERT (kindly provided by Jason J. Chen at Columbia University) (43). Both HELs and HELFs were cultured in minimum essential medium (MEM; catalog number 41500-034; Life Technologies) supplemented with 10% fetal bovine serum (FBS; catalog number 10099-141; Life Technologies) and penicillin-streptomycin (100 U/ml and 100 μg/ml, respectively; catalog number 15140122; Life Technologies). Human embryonic kidney 293T (HEK293T) cells were purchased from ATCC (CRL-11268) and cultured in Dulbecco's modified Eagle medium (DMEM; catalog number 12900017; Life Technologies) supplemented with 10% FBS and penicillin-streptomycin as described above. All cell cultures were performed at 37°C in a humidified atmosphere containing 5% CO2.
Viruses and infections.
The HCMV Towne strain (ATCC VR-977) and a recombinant HSV-1 H129 strain created in the Luo laboratory (84) were used in this study. HCMV and HSV-1 were propagated and titrated as described previously (44, 82–85). Cells were synchronized by serum starvation prior to infection and then incubated with viral inocula for 2 h to allow for adsorption. The medium was then removed and replaced with fresh MEM as described previously (86). Infected cells were harvested at the times postinfection indicated above and in the figures.
Plasmid constructs and lentiviruses.
The human WDR5-coding sequence (GenBank accession number NM_017588) was amplified from total RNA isolated from HELs following reverse transcription and then cloned into pCDH-CMV-MCS-EF1-copGFP (pCDH; System Biosciences, USA) at the XbaI/EcoRI site to produce the WDR5 expression construct pCDH-WDR5. We purchased the shRNAs sh-Scram (5′-TTCTCCGAACGTGTCACGT-3′), sh-W1 (5′-TCAGAGGATAACCTTGTTT-3′), sh-W2 (5′-AACCTTATTGTCTCAGGAT-3′), and sh-W3 (5′-GTCGTCAGATTCTAACCTT-3′) cloned into lentiviral vector GV248 (hU6-MCS-ubiquitin-enhanced green fluorescent protein-internal ribosome entry site-puromycin) from Shanghai Genechem (Shanghai, China).
Lentiviruses expressing shRNAs were produced in HEK293T cells as described previously (43, 44). In brief, HEK293T cells were seeded onto 100-mm dishes and cotransfected, using calcium phosphate, with 15 μg of the lentiviral vector DNAs (described above) with helper plasmids pML-Δ8.9 (12 μg) and pVSV-G (8 μg). At 24 h posttransfection, the culture medium was replaced with DMEM with 10% FBS but without antibiotics. Lentiviruses were harvested, titrated, and stored at −80°C as described previously (44).
Establishment of KD, Ctl, and Rec cell lines.
A WDR5-overexpressing (OE) cell was obtained by transducing HELFs with a lentivirus expressing WDR5 (pCDH-WDR5). HELFs transduced with lentivirus produced from the empty vector (pCDH) served as a negative control (NC). The WDR5 knockdown cell line (KD) was generated by transducing HELFs with an sh-W3-expressing lentivirus which also encodes green fluorescent protein and puromycin resistance. When green fluorescence was observed in the majority of cells, cells were subcultured at 1 × 106 cells per 100-mm dish and incubated in MEM containing 8 μg/ml puromycin (catalog number P8833; Sigma-Aldrich) for 2 weeks. Survivors were harvested, and clonal lines were isolated by limiting dilution in 96-well plates. IB was used to screen the clonal lines for WDR5 reduced expression, and one of the lines was selected and designated KD. The Ctl cell line was similarly obtained by transducing HELFs with lentivirus expressing scrambled shRNA, while the Rec cell line was obtained by transducing KD cells with WDR5-expressing lentivirus (pCDH-WDR5) and screening clonal lines for restored WDR5 expression.
Cell viability.
Cell viability was analyzed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (catalog number M2128; Sigma-Aldrich) following the manufacturer's instructions. Briefly, cells were seeded at 5 × 103 cells/well in 96-well plates and incubated for 3, 4, or 5 days. The medium was then replaced with 200 μl fresh MEM without phenol red containing 0.5% MTT. After incubation at 37°C for 4 h, the medium was removed and 150 μl of dimethyl sulfoxide (DMSO) was added into each well. The plate was agitated, and the absorbance at 570 nm was determined with a reference wavelength of 630 nm using an Epoch microplate spectrophotometer (BioTek Instruments, USA).
Quantitation of virus replication.
Cells of the HELF-derived cell lines were synchronized by serum starvation for 48 h, trypsinized, and reseeded onto 6-well plates (8 × 105 cells/well) and then infected with HCMV at an MOI of 0.5, 1, or 3. After 2 h of incubation, the inoculum was removed, the cells were rinsed with phosphate-buffered saline, and fresh MEM was added. Supernatant samples were collected at various time points and stored at −80°C until they were assayed. Infectious virus titers were determined by a plaque assay (86). Briefly, HELs were seeded into 24-well dishes at a density of 1.5 × 106 cells per plate. On the following day, the cells were infected with 200 μl of supernatant sample after 10-fold serial dilution. After adsorption for 3 h, MEM with a 0.5% agarose overlay was added to each well. Plaques were counted at 7 to 10 dpi, and an average titer was derived from three independent experiments.
Quantitation of viral genome copy number.
Genomic DNA was extracted from infected cells using a genomic DNA extraction kit (catalog number DP304; Tiangen Biotech), followed by qPCR to quantitate viral DNAs using HCMV UL83 primers as described previously (44). Means and standard deviations (SD) from at least three independent experiments were calculated.
Quantitative reverse transcription-PCR (qRT-PCR).
HELs were infected at an MOI of 3 and harvested at the times postinfection indicated above and in the figures. A total of 1 × 106 cells were used for total RNA extraction using the RNAiso Plus reagent (catalog number 9109; TaKaRa), followed by treatment with 10 U of recombinant DNase I (catalog number 2270A; TaKaRa) to remove residual DNA. One microgram of RNA from each sample was reverse transcribed with a RevertAid H Minus first-strand cDNA synthesis kit (catalog number K1631; Fermentas) with random primers. Then, qPCR was performed on a real-time thermocycler (Bio-Rad; Connect) using SYBR green PCR master mix (catalog number 4309155; Applied Biosystems) in a 20-μl reaction mixture for 40 PCR cycles as described previously (44, 83, 85, 87). The PCR primers for WDR5 were 5′-GGTGGGAAGTGGATTGTGTC-3′ and 5′-GCAGCAGAGGCGATGATG-3′. The PCR primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were 5′-GAGTCAACGGATTTGGTCGT-3′ and 5′-GACAAGCTTCCCGTTCTCAG-3′.
Immunoblotting (IB).
Cells were harvested and lysed with cell lysis buffer (catalog number P0013; Beyotime) containing protease inhibitor cocktail (catalog number 04693159001; Roche). The protein concentrations of the cell lysates were determined by Bradford assay (catalog number 500-0205; Bio-Rad). Cell lysates with equal amounts of total protein were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to polyvinylidene difluoride membranes (catalog number ISEQ00010; Millipore). Membranes were sequentially probed with primary antibodies and appropriate peroxidase-conjugated secondary antibodies (Table 1), developed using SuperSignal West Femto chemiluminescent substrate (catalog number 34095; Life Technologies), detected using a FluorChem HD2 system (Alpha Innotech), and quantitated by densitometry using ImageJ software (National Institutes of Health).
TABLE 1.
Antibodies used in this study
| Antibodya (clone) | Species/isotype | Source/catalog number |
|---|---|---|
| Primary antibodies | ||
| Anti-WDR5 | Rabbit polyclonal | Millipore/07-706 |
| Antiactin (clone 6G3) | Mouse monoclonal/IgG1 | Sungene Biotech/KM9001 |
| Anti-ubiquitin B | Rabbit polyclonal | Abclonal/A0162 |
| Anti-IE1 (clone p63-27) | Mouse monoclonal/IgG2a | Gift from William J. Britt, University of Alabama, USA |
| Anti-IE1/2 (clone CH160) | Mouse monoclonal/IgG1 | Virusys/P1215 |
| Anti-UL44 (clone CH13) | Mouse monoclonal/IgG1 | Virusys/P1202-1 |
| Anti-gB (clone CH28) | Mouse monoclonal/IgG1 | Virusys/P1201 |
| Anti-pUL53 (clone 7H8) | Mouse monoclonal/IgG1 | Gift from Stipan Jonjic, University of Rijeka, Rijeka, Croatia |
| Anti-MCP (clone 28.4) | Mouse monoclonal/IgG2a | Gift from William J. Britt, University of Alabama, USA |
| Secondary antibodies | ||
| TRITC-anti-mouse IgG2a | Goat | Southern Biotech/1080-03 |
| AF647-anti-mouse IgG1 | Goat | Life Technologies/A21240 |
| Peroxidase-anti-mouse IgG | Goat | Jackson ImmunoResearch Laboratories/115-035-003 |
| Peroxidase-anti-rabbit IgG | Goat | Jackson ImmunoResearch Laboratories/111-035-003 |
TRITC, tetramethyl rhodamine isocyanate; AF647, ALexa Fluor 647.
Immunofluorescence analysis (IFA).
Synchronized HELF-derived cells were reseeded onto coverslips and infected with HCMV after attachment. Coverslips were fixed with 4% paraformaldehyde, and target proteins were detected by incubation with the primary antibodies and appropriate secondary antibodies (Table 1) as described previously (88). Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole; catalog number D9542; Sigma-Aldrich). Images were obtained using the Volocity (version 5.5) software package (PerkinElmer) on a PerkinElmer UltraVIEW VoX spinning disk laser confocal scanning microscope.
In vivo ubiquitination assay.
Mock- or HCMV-infected HELs were treated with DMSO or MG132 (6 μM) at 72 hpi, harvested after 6 h of incubation, and lysed with cell lysis buffer containing protease inhibitor cocktail at 4°C. Cell lysates containing 30 μg total protein were used for input controls. For immunoprecipitation, lysates containing 1 mg protein were incubated with 1.5 μg anti-WDR5 antibody at 4°C overnight with rotation and then incubated with protein A/G agarose beads (catalog number P2012; Beyotime) for 4 h at 4°C and analyzed by IB for WDR5 and ubiquitinated proteins. The antibodies used are listed in Table 1.
CHX treatment and protein stability assay.
To analyze WDR5 stability. HELFs that had been mock infected or HCMV infected (MOI = 3) for 72 h were treated with 100 μg/ml CHX (catalog number C7698; Sigma-Aldrich). Cell lysates were harvested at 4-h intervals and assayed by IB.
Transmission electron microscopy (TEM).
Cells were infected with HCMV at an MOI of 0.5 and harvested at 96 or 120 hpi. Cells were fixed with 2.5% (wt/vol) glutaraldehyde for 1 h at room temperature and then treated with 1% osmium tetroxide, dehydrated through a graded series of ethanol concentrations (from 30 to 100%), and embedded with an Embed 812 kit (Electron Microscopy Sciences, Fort Washington, PA). Ultrathin sections (60 to 80 nm) of embedded specimens were prepared, deposited onto Formvar-coated copper grids (200 mesh), stained with 2% (wt/vol) phosphotungstic acid (PTA; pH 6.8), and observed under a Tecnai transmission electron microscope (FEI) operated at 200 kV.
Statistical analyses.
Statistical analyses were performed using the SPSS software package (version 18.0; IBM), and graphs were generated with Prism GraphPad software (version 5.01; IBM). One-way analysis of variance (ANOVA) was used for comparison of two experimental groups, and a P value of <0.05 was considered statistically significant; when comparison among experimental groups was needed, the Bonferroni post hoc test was conducted for multiple testing corrections; a P value of <0.0167 was considered statistically significant. Statistical tests for TEM data were conducted using the Kruskal-Wallis test. This test is designed for the analysis of data that does not assume a normal distribution of errors. If the overall Kruskal-Wallis test result was statistically significant, post hoc Dunn's multiple-comparison tests were conducted, and a P value of <0.05 was considered statistically significant.
ACKNOWLEDGMENTS
We appreciate Stipan Jonjic (University of Rijeka, Rijeka, Croatia) for providing antibody to pUL53. We thank Ding Gao, Anna Du, and Pei Zhang (The Core Facility, Wuhan Institute of Virology, CAS) for technical support of electron microscopy and Xiulian Sun and Wei Fang (Wuhan Institute of Virology, CAS) for advice on statistical analyses.
This work was supported by grants from the National Natural Science Foundation of China (81620108021, 81427801, 81271850, and 81571355), the Key Basic Research Project from MOST (2015CB755601), the Sino-Africa Joint Research Center, Chinese Academy of Sciences (SAJC201605), and the Key Program of the Guangzhou Joint Healthcare Innovation Foundation (201704020229).
REFERENCES
- 1.Karlin S, Mocarski ES, Schachtel GA. 1994. Molecular evolution of herpesviruses: genomic and protein sequence comparisons. J Virol 68:1886–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davison AJ. 2002. Evolution of the herpesviruses. Vet Microbiol 86:69–88. doi: 10.1016/S0378-1135(01)00492-8. [DOI] [PubMed] [Google Scholar]
- 3.Swanson EC, Schleiss MR. 2013. Congenital cytomegalovirus infection: new prospects for prevention and therapy. Pediatr Clin North Am 60:335–349. doi: 10.1016/j.pcl.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86–102. doi: 10.1128/CMR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, Nabors LB, Cobbs CG, Britt WJ. 2002. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res 62:3347–3350. [PubMed] [Google Scholar]
- 6.Villinger C, Neusser G, Kranz C, Walther P, Mertens T. 2015. 3D analysis of HCMV induced-nuclear membrane structures by FIB/SEM tomography: insight into an unprecedented membrane morphology. Viruses 7:5686–5704. doi: 10.3390/v7112900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strang BL, Boulant S, Kirchhausen T, Coen DM. 2012. Host cell nucleolin is required to maintain the architecture of human cytomegalovirus replication compartments. mBio 3:e00301-11. doi: 10.1128/mBio.00301-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strang BL, Boulant S, Chang L, Knipe DM, Kirchhausen T, Coen DM. 2012. Human cytomegalovirus UL44 concentrates at the periphery of replication compartments, the site of viral DNA synthesis. J Virol 86:2089–2095. doi: 10.1128/JVI.06720-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bender BJ, Coen DM, Strang BL. 2014. Dynamic and nucleolin-dependent localization of human cytomegalovirus UL84 to the periphery of viral replication compartments and nucleoli. J Virol 88:11738–11747. doi: 10.1128/JVI.01889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buser C, Walther P, Mertens T, Michel D. 2007. Cytomegalovirus primary envelopment occurs at large infoldings of the inner nuclear membrane. J Virol 81:3042–3048. doi: 10.1128/JVI.01564-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pante N, Kann M. 2002. Nuclear pore complex is able to transport macromolecules with diameters of ∼39 nm. Mol Biol Cell 13:425–434. doi: 10.1091/mbc.01-06-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pogoda M, Bosse JB, Wagner FM, Schauflinger M, Walther P, Koszinowski UH, Ruzsics Z. 2012. Characterization of conserved region 2-deficient mutants of the cytomegalovirus egress protein pM53. J Virol 86:12512–12524. doi: 10.1128/JVI.00471-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkie AR, Lawler JL, Coen DM. 2016. A role for nuclear F-actin induction in human cytomegalovirus nuclear egress. mBio 7:e01254-16. doi: 10.1128/mBio.01254-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeRussy BM, Boland MT, Tandon R. 2016. Human cytomegalovirus pUL93 links nucleocapsid maturation and nuclear egress. J Virol 90:7109–7117. doi: 10.1128/JVI.00728-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mettenleiter TC. 2002. Herpesvirus assembly and egress. J Virol 76:1537–1547. doi: 10.1128/JVI.76.4.1537-1547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. 2015. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sharma M, Kamil JP, Coughlin M, Reim NI, Coen DM. 2014. Human cytomegalovirus UL50 and UL53 recruit viral protein kinase UL97, not protein kinase C, for disruption of nuclear lamina and nuclear egress in infected cells. J Virol 88:249–262. doi: 10.1128/JVI.02358-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuan MI, O'Dowd JM, Chughtai K, Hayman I, Brown CJ, Fortunato EA. 2016. Human cytomegalovirus nuclear egress and secondary envelopment are negatively affected in the absence of cellular p53. Virology 497:279–293. doi: 10.1016/j.virol.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown JC, Newcomb WW. 2011. Herpesvirus capsid assembly: insights from structural analysis. Curr Opin Virol 1:142–149. doi: 10.1016/j.coviro.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tandon R, Mocarski ES. 2012. Viral and host control of cytomegalovirus maturation. Trends Microbiol 20:392–401. doi: 10.1016/j.tim.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sonntag E, Hamilton ST, Bahsi H, Wagner S, Jonjic S, Rawlinson WD, Marschall M, Milbradt J. 2016. Cytomegalovirus pUL50 is the multi-interacting determinant of the core nuclear egress complex (NEC) that recruits cellular accessory NEC components. J Gen Virol 97:1676–1685. doi: 10.1099/jgv.0.000495. [DOI] [PubMed] [Google Scholar]
- 22.Miller MS, Furlong WE, Pennell L, Geadah M, Hertel L. 2010. RASCAL is a new human cytomegalovirus-encoded protein that localizes to the nuclear lamina and in cytoplasmic vesicles at late times postinfection. J Virol 84:6483–6496. doi: 10.1128/JVI.02462-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J Virol 77:3191–3203. doi: 10.1128/JVI.77.5.3191-3203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanarsdall AL, Howard PW, Wisner TW, Johnson DC. 2016. Human cytomegalovirus gH/gL forms a stable complex with the fusion protein gB in virions. PLoS Pathog 12:e1005564. doi: 10.1371/journal.ppat.1005564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stegmann C, Abdellatif MEA, Sampaio KL, Walther P, Sinzger C. 2016. Importance of highly conserved peptide sites of HCMV gO for the formation of the gH/gL/gO complex. J Virol 91:e01339-16. doi: 10.1128/JVI.01339-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho YW, Hong T, Hong S, Guo H, Yu H, Kim D, Guszczynski T, Dressler GR, Copeland TD, Kalkum M, Ge K. 2007. PTIP associates with MLL3- and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282:20395–20406. doi: 10.1074/jbc.M701574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song JJ, Kingston RE. 2008. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J Biol Chem 283:35258–35264. doi: 10.1074/jbc.M806900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mo R, Rao SM, Zhu YJ. 2006. Identification of the MLL2 complex as a coactivator for estrogen receptor alpha. J Biol Chem 281:15714–15720. doi: 10.1074/jbc.M513245200. [DOI] [PubMed] [Google Scholar]
- 29.Dharmarajan V, Lee JH, Patel A, Skalnik DG, Cosgrove MS. 2012. Structural basis for WDR5 interaction (Win) motif recognition in human SET1 family histone methyltransferases. J Biol Chem 287:27275–27289. doi: 10.1074/jbc.M112.364125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ernst P, Vakoc CR. 2012. WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics 11:217–226. doi: 10.1093/bfgp/els017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Xie W, Gu P, Cai Q, Wang B, Xie Y, Dong W, He W, Zhong G, Lin T, Huang J. 2015. Upregulated WDR5 promotes proliferation, self-renewal and chemoresistance in bladder cancer via mediating H3K4 trimethylation. Sci Rep 5:8293. doi: 10.1038/srep08293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. 2011. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell 43:811–822. doi: 10.1016/j.molcel.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 34.Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. 2005. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 35.Zhu ED, Demay MB, Gori F. 2008. Wdr5 is essential for osteoblast differentiation. J Biol Chem 283:7361–7367. doi: 10.1074/jbc.M703304200. [DOI] [PubMed] [Google Scholar]
- 36.Ang YS, Tsai SY, Lee DF, Monk J, Su J, Ratnakumar K, Ding J, Ge Y, Darr H, Chang B, Wang J, Rendl M, Bernstein E, Schaniel C, Lemischka IR. 2011. Wdr5 mediates self-renewal and reprogramming via the embryonic stem cell core transcriptional network. Cell 145:183–197. doi: 10.1016/j.cell.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang YW, Flynn RA, Chen Y, Qu K, Wan B, Wang KC, Lei M, Chang HY. 2014. Essential role of lncRNA binding for WDR5 maintenance of active chromatin and embryonic stem cell pluripotency. Elife 3:e02046. doi: 10.7554/eLife.02046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ge Z, Song EJ, Imamura KY, Li J, Dovat S, Song C. 2016. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 7:37740–37754. doi: 10.18632/oncotarget.9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim JY, Banerjee T, Vinckevicius A, Luo Q, Parker JB, Baker MR, Radhakrishnan I, Wei JJ, Barish GD, Chakravarti D. 2014. A role for WDR5 in integrating threonine 11 phosphorylation to lysine 4 methylation on histone H3 during androgen signaling and in prostate cancer. Mol Cell 54:613–625. doi: 10.1016/j.molcel.2014.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang YY, Liu LJ, Zhong B, Liu TT, Li Y, Yang Y, Ran Y, Li S, Tien P, Shu HB. 2010. WDR5 is essential for assembly of the VISA-associated signaling complex and virus-triggered IRF3 and NF-kappaB activation. Proc Natl Acad Sci U S A 107:815–820. doi: 10.1073/pnas.0908967107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nakagawa T, Xiong Y. 2011. X-linked mental retardation gene CUL4B targets ubiquitylation of H3K4 methyltransferase component WDR5 and regulates neuronal gene expression. Mol Cell 43:381–391. doi: 10.1016/j.molcel.2011.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hochstrasser M. 1996. Ubiquitin-dependent protein degradation. Annu Rev Genet 30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- 43.Liu XJ, Yang B, Huang SN, Wu CC, Li XJ, Cheng S, Jiang X, Hu F, Ming YZ, Nevels M, Britt WJ, Rayner S, Tang Q, Zeng WB, Zhao F, Luo MH. 2017. Human cytomegalovirus IE1 downregulates Hes1 in neural progenitor cells as a potential E3 ubiquitin ligase. PLoS Pathog 13:e1006542. doi: 10.1371/journal.ppat.1006542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu Y-R, Liu X-J, Li X-J, Shen Z-Z, Yang B, Wu C-C, Li J-F, Miao L-F, Ye H-Q, Qiao G-H. 2015. MicroRNA miR-21 attenuates human cytomegalovirus replication in neural cells by targeting Cdc25a. J Virol 89:1070–1082. doi: 10.1128/JVI.01740-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viswanathan K, Smith MS, Malouli D, Mansouri M, Nelson JA, Fruh K. 2011. BST2/tetherin enhances entry of human cytomegalovirus. PLoS Pathog 7:e1002332. doi: 10.1371/journal.ppat.1002332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev 72:249–265. doi: 10.1128/MMBR.00040-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milbradt J, Kraut A, Hutterer C, Sonntag E, Schmeiser C, Ferro M, Wagner SR, Lenac T, Claus C, Pinkert S. 2014. Proteomic analysis of the multimeric nuclear egress complex of human cytomegalovirus. Mol Cell Proteomics 13:2132–2146. doi: 10.1074/mcp.M113.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alwine J. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu X, Trang P, Shah S, Atanasov I, Kim YH, Bai Y, Zhou ZH, Liu F. 2005. Dissecting human cytomegalovirus gene function and capsid maturation by ribozyme targeting and electron cryomicroscopy. Proc Natl Acad Sci U S A 102:7103–7108. doi: 10.1073/pnas.0408826102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibson W. 1996. Structure and assembly of the virion. Intervirology 39:389–400. doi: 10.1159/000150509. [DOI] [PubMed] [Google Scholar]
- 51.Tandon R, Mocarski ES, Conway JF. 2015. The A, B, Cs of herpesvirus capsids. Viruses 7:899–914. doi: 10.3390/v7030899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marschall M, Muller YA, Diewald B, Sticht H, Milbradt J. 2017. The human cytomegalovirus nuclear egress complex unites multiple functions: recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev Med Virol 27:1934. doi: 10.1002/rmv.1934. [DOI] [PubMed] [Google Scholar]
- 53.Schmeiser C, Borst E, Sticht H, Marschall M, Milbradt J. 2013. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J Gen Virol 94:2056–2069. doi: 10.1099/vir.0.052571-0. [DOI] [PubMed] [Google Scholar]
- 54.Kuan MI, O'Dowd JM, Fortunato EA. 2016. The absence of p53 during human cytomegalovirus infection leads to decreased UL53 expression, disrupting UL50 localization to the inner nuclear membrane, and thereby inhibiting capsid nuclear egress. Virology 497:262–278. doi: 10.1016/j.virol.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spector DH. 2015. Human cytomegalovirus riding the cell cycle. Med Microbiol Immunol 204:409–419. doi: 10.1007/s00430-015-0396-z. [DOI] [PubMed] [Google Scholar]
- 56.Hwang J, Kalejta RF. 2009. Human cytomegalovirus protein pp71 induces Daxx SUMOylation. J Virol 83:6591–6598. doi: 10.1128/JVI.02639-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hwang J, Kalejta RF. 2007. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral pp71 protein in human cytomegalovirus-infected cells. Virology 367:334–338. doi: 10.1016/j.virol.2007.05.037. [DOI] [PubMed] [Google Scholar]
- 58.Kim YE, Lee JH, Kim ET, Shin HJ, Gu SY, Seol HS, Ling PD, Lee CH, Ahn JH. 2011. Human cytomegalovirus infection causes degradation of Sp100 proteins that suppress viral gene expression. J Virol 85:11928–11937. doi: 10.1128/JVI.00758-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park JJ, Kim YE, Pham HT, Kim ET, Chung YH, Ahn JH. 2007. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J Gen Virol 88:3214–3223. doi: 10.1099/vir.0.83171-0. [DOI] [PubMed] [Google Scholar]
- 60.Das S, Vasanji A, Pellett PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J Virol 81:11861–11869. doi: 10.1128/JVI.01077-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rossini G, Cerboni C, Santoni A, Landini MP, Landolfo S, Gatti D, Gribaudo G, Varani S. 2012. Interplay between human cytomegalovirus and intrinsic/innate host responses: a complex bidirectional relationship. Mediators Inflamm 2012:607276. doi: 10.1155/2012/607276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Amsler L, Verweij MC, DeFilippis VR. 2013. The tiers and dimensions of evasion of the type I interferon response by human cytomegalovirus. J Mol Biol 425:4857–4871. doi: 10.1016/j.jmb.2013.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS. 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gariano GR, Dell'Oste V, Bronzini M, Gatti D, Luganini A, De Andrea M, Gribaudo G, Gariglio M, Landolfo S. 2012. The intracellular DNA sensor IFI16 gene acts as restriction factor for human cytomegalovirus replication. PLoS Pathog 8:e1002498. doi: 10.1371/journal.ppat.1002498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li T, Chen J, Cristea IM. 2013. Human cytomegalovirus tegument protein pUL83 inhibits IFI16-mediated DNA sensing for immune evasion. Cell Host Microbe 14:591–599. doi: 10.1016/j.chom.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dell'Oste V, Gatti D, Gugliesi F, De Andrea M, Bawadekar M, Cigno IL, Biolatti M, Vallino M, Marschall M, Gariglio M. 2014. Innate nuclear sensor IFI16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J Virol 88:6970–6982. doi: 10.1128/JVI.00384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xie M, Xuan B, Shan J, Pan D, Sun Y, Shan Z, Zhang J, Yu D, Li B, Qian Z. 2015. Human cytomegalovirus exploits interferon-induced transmembrane proteins to facilitate morphogenesis of the virion assembly compartment. J Virol 89:3049–3061. doi: 10.1128/JVI.03416-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Seo J, Yaneva R, Hinson ER, Cresswell P. 2011. Human cytomegalovirus directly induces the antiviral protein viperin to enhance infectivity. Science 332:1093–1097. doi: 10.1126/science.1202007. [DOI] [PubMed] [Google Scholar]
- 69.Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J Virol 80:8390–8401. doi: 10.1128/JVI.00505-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arnoult D, Bartle LM, Skaletskaya A, Poncet D, Zamzami N, Park PU, Sharpe J, Youle RJ, Goldmacher VS. 2004. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc Natl Acad Sci U S A 101:7988–7993. doi: 10.1073/pnas.0401897101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kukhanova MK, Korovina AN, Kochetkov SN. 2014. Human herpes simplex virus: life cycle and development of inhibitors. Biochemistry (Mosc) 79:1635–1652. doi: 10.1134/S0006297914130124. [DOI] [PubMed] [Google Scholar]
- 72.Burwitz BJ, Malouli D, Bimber BN, Reed JS, Ventura AB, Hancock MH, Uebelhoer LS, Bhusari A, Hammond KB, Espinosa Trethewy RG, Klug A, Legasse AW, Axthelm MK, Nelson JA, Park BS, Streblow DN, Hansen SG, Picker LJ, Früh K, Sacha JB. 2016. Cross-species rhesus cytomegalovirus infection of cynomolgus macaques. PLoS Pathog 12:e1006014. doi: 10.1371/journal.ppat.1006014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bosse JB, Enquist LW. 2016. The diffusive way out: herpesviruses remodel the host nucleus, enabling capsids to access the inner nuclear membrane. Nucleus 7:13–19. doi: 10.1080/19491034.2016.1149665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bosse JB, Virding S, Thiberge SY, Scherer J, Wodrich H, Ruzsics Z, Koszinowski UH, Enquist LW. 2014. Nuclear herpesvirus capsid motility is not dependent on F-actin. mBio 5:e01909-14. doi: 10.1128/mBio.01909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lye MF, Wilkie AR, Filman DJ, Hogle JM, Coen DM. 2017. Getting to and through the inner nuclear membrane during herpesvirus nuclear egress. Curr Opin Cell Biol 46:9–16. doi: 10.1016/j.ceb.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dal Monte P, Pignatelli S, Zini N, Maraldi NM, Perret E, Prevost MC, Landini MP. 2002. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J Gen Virol 83:1005–1012. doi: 10.1099/0022-1317-83-5-1005. [DOI] [PubMed] [Google Scholar]
- 77.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, Desalvo D. 2000. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thurlow JK, Murphy M, Stow ND, Preston VG. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J Virol 80:2118–2126. doi: 10.1128/JVI.80.5.2118-2126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sharma M, Bender BJ, Kamil JP, Lye MF, Pesola JM, Reim NI, Hogle JM, Coen DM. 2015. Human cytomegalovirus UL97 phosphorylates the viral nuclear egress complex. J Virol 89:523–534. doi: 10.1128/JVI.02426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duan YL, Ye HQ, Zavala AG, Yang CQ, Miao LF, Fu BS, Seo KS, Davrinche C, Luo MH, Fortunato EA. 2014. Maintenance of large numbers of virus genomes in human cytomegalovirus-infected T98G glioblastoma cells. J Virol 88:3861–3873. doi: 10.1128/JVI.01166-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li XJ, Liu XJ, Yang B, Fu YR, Zhao F, Shen ZZ, Miao LF, Rayner S, Chavanas S, Zhu H, Britt WJ, Tang Q, McVoy MA, Luo MH. 2015. Human cytomegalovirus infection dysregulates the localization and stability of NICD1 and Jag1 in neural progenitor cells. J Virol 89:6792–6804. doi: 10.1128/JVI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zeng WB, Jiang HF, Gang YD, Song YG, Shen ZZ, Yang H, Dong X, Tian YL, Ni RJ, Liu Y, Tang N, Li X, Jiang X, Gao D, Androulakis M, He XB, Xia HM, Ming YZ, Lu Y, Zhou JN, Zhang C, Xia XS, Shu Y, Zeng SQ, Xu F, Zhao F, Luo MH. 2017. Anterograde monosynaptic transneuronal tracers derived from herpes simplex virus 1 strain H129. Mol Neurodegener 12:38. doi: 10.1186/s13024-017-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pan X, Li X, Liu X, Yuan H, Li J, Duan Y, Ye H, Fu Y, Qiao G, Wu C, Yang B, Yang B, Tian X, Hu K, Miao L, Chen X, Zheng J, Rayner S, Rayner S, Schwartz P, Britt W, Xu J, Luo M. 2013. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J Virol 87:10968–10979. doi: 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Luo MH, Rosenke K, Czornak K, Fortunato EA. 2007. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J Virol 81:1934–1950. doi: 10.1128/JVI.01670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jiang HF, Wang W, Jiang X, Zeng WB, Shen ZZ, Song YG, Yang H, Liu XJ, Dong X, Zhou J, Sun JY, Yu FL, Guo L, Cheng T, Rayner S, Zhao F, Zhu H, Luo MH. 2017. ORF7 of varicella-zoster virus is required for viral cytoplasmic envelopment in differentiated neuronal cells. J Virol 91:e00127-17. doi: 10.1128/JVI.00127-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Duan Y, Miao L, Ye H, Yang C, Fu B, Schwartz PH, Rayner S, Fortunato EA, Luo MH. 2012. A faster immunofluorescence assay for tracking infection progress of human cytomegalovirus. Acta Biochim Biophys Sin (Shanghai) 44:597–605. doi: 10.1093/abbs/gms041. [DOI] [PMC free article] [PubMed] [Google Scholar]