

ABSTRACT
Pathogenic human immunodeficiency virus (HIV)/simian immunodeficiency virus (SIV) infection of humans and rhesus macaques (RMs) induces persistently high production of type I interferon (IFN-I), which is thought to contribute to disease progression. To elucidate the specific role of interferon alpha (IFN-α) in SIV pathogenesis, 12 RMs were treated prior to intravenous (i.v.) SIVmac239 infection with a high or a low dose of an antibody (AGS-009) that neutralizes most IFN-α subtypes and were compared with six mock-infused, SIV-infected controls. Plasma viremia was measured postinfection to assess the effect of IFN-α blockade on virus replication, and peripheral blood and lymphoid tissue samples were analyzed by immunophenotypic staining. Consistent with the known antiviral effect of IFN-I, high-dose AGS-009 treatment induced a modest increase in acute-phase viral loads versus controls. Four out of 6 RMs receiving a high dose of AGS-009 also experienced an early decline in CD4+ T cell counts that was associated with progression to AIDS. Interestingly, 50% of the animals treated with AGS-009 (6/12) developed AIDS within 1 year of infection compared with 17% (1/6) of untreated controls. Finally, blockade of IFN-α decreased the levels of activated CD4+ and CD8+ T cells, as well as B cells, as measured by PD-1 and/or Ki67 expression. The lower levels of activated lymphocytes in IFN-α-blockaded animals supports the hypothesis that IFN-α signaling contributes to lymphocyte activation during SIV infection and suggests that this signaling pathway is involved in controlling virus replication during acute infection. The potential anti-inflammatory effect of IFN-α blockade should be explored as a strategy to reduce immune activation in HIV-infected individuals.
IMPORTANCE Interferon alpha (IFN-α) is a member of a family of molecules (type I interferons) that prevent or limit virus infections in mammals. However, IFN-α production may contribute to the chronic immune activation that is thought to be the primary cause of immune decline and AIDS in HIV-infected patients. The study presented here attempts to understand the contribution of IFN-α to the natural history and progression of SIV infection of rhesus macaques, the primary nonhuman primate model system for testing hypotheses about HIV infection in humans. Here, we show that blockade of IFN-α action promotes lower chronic immune activation but higher early viral loads, with a trend toward faster disease progression. This study has significant implications for new treatments designed to impact the type I interferon system.
KEYWORDS: HIV, HIV therapy, SIV, immune activation, interferons, rhesus macaque, type I interferon
INTRODUCTION
Simian immunodeficiency virus (SIV) infection of rhesus macaques (RMs) is typically characterized by high levels of virus replication, failure of the host immune response to control the virus, chronic high levels of immune activation, and, ultimately, progression to a severe immune deficiency that is very similar to what is observed in human immunodeficiency virus (HIV)-infected humans (1). The SIV-associated chronic immune activation is characterized by uncontrolled lymphocyte proliferation, aberrant production of proinflammatory cytokines (i.e., tumor necrosis factor [TNF] and interferon gamma [IFN-γ]), high levels of lymphocyte apoptosis, and irreparable damage to the architecture of lymphoid tissues (2). While HIV-associated immune activation and inflammation are decreased when patients are placed on long-term antiretroviral therapy (ART), the overall levels of immune activation remain higher than those observed in non-HIV-infected age-matched individuals (3–6). While the causes of HIV/SIV-associated immune activation are complex and not fully understood, comparative studies of pathogenic SIV infection of macaques and nonpathogenic SIV infection of natural hosts (e.g., sooty mangabeys [7] and African green monkeys [8]) point to chronic activation of the type I interferon (IFN-I) pathway as a potential major contributor to the chronic immune activation that leads to AIDS progression.
The IFN-I pathway is a crucial component of the early innate immune response to many virus infections. The term IFN-I covers an array of several different types of interferon homologs in primates, all of which bind to the IFN receptor: 13 IFN-α subtypes, in addition to IFN-β, IFN-ε, IFN-κ, and IFN-ω (9). Many cell types, upon sensing a conserved viral motif via innate immune receptors (e.g., IFI-16, cGAS, and RIG-I), upregulate the expression of IFN-I, which in turn induces the expression of a cascade of interferon-stimulated genes (ISGs), which include a large number of viral restriction factors that have evolved to specifically target viral functions through binding to specific molecular motifs (9). Human and rhesus macaque ISGs that can restrict HIV and SIV replication include TRIM5α, APOBEC-3G, BST-2/tetherin, SAMHD1, and GAS genes (9, 10). In addition to these well-characterized antiviral restriction factor genes, IFN-I induces the expression of numerous ISGs whose functions are still unclear. Some of these genes may be evolutionary relics that helped to limit past virus infections, while others could potentially be responsible for modulating adaptive immune responses to intracellular antigens.
In addition to its direct antiviral role, IFN-I is involved in the generation and maintenance of antiviral cellular immune responses, as indicated by a series of mouse studies in which the IFN-I pathway was knocked out (11–13). In addition, it has been shown that plasmacytoid dendritic cells (pDCs), which are the main producers of IFN-I in vivo, accumulate at sites where CD8+ T cells are undergoing activation and therefore help to create a microenvironment conducive to effective antiviral and antitumor immunity (14). Nevertheless, the specific role of IFN-I in the generation of antiviral CD8 responses remains incompletely understood. Several recent studies employing strategies to block IFN-I signaling in mouse models of chronic viral infection have provided some clues. Blockade of IFN-I both prior to and during chronic infection with lymphocytic choriomeningitis virus (LCMV) clone 13 reduces immune activation and expression of coinhibitory molecules (e.g., PD-1) (15, 16). These immunomodulatory changes promoted the more effective clearance of virus-infected cells, thereby allowing the CD8+ T cell-mediated clearance of LCMV from these animals. Further studies have shown that IFN-β is the primary IFN-I inducing the immune activation and downstream inhibition of CD8 antiviral responses (17). More recently, two groups have extended these findings to the model of HIV infection of humanized mice and showed that blockade of IFN-I signaling reduces immune activation and expression of coinhibitory markers on CD8+ T cells (18, 19). The reinvigorated antiviral cellular immune response reduced HIV viremia, as well as the levels of both total cell-associated HIV-DNA and replication-competent virus.
One recent in vivo study of the effects of IFN-I blockade in SIV-infected rhesus macaques has shown that IFN-I does indeed have a significant impact on the natural history and replication of SIV (20). In that study, the authors utilized an IFN receptor antagonist to block signaling of all IFN-I subtypes just prior to SIV infection. They found that blockade of IFN-I during the early stages of infection resulted in significantly higher viral loads and more rapid CD4+ T cell decline during the chronic phase of infection, which was associated with faster progression to AIDS in the IFN-I-blockaded animals despite a decrease in activation markers on lymphocytes. However, the authors were unable to determine the contributions of the blockade of the various IFN-I subtypes on the outcome of SIV infection, since the IFN antagonist blocks all IFN-I subtypes from interactions with their receptors.
Despite the antiviral activities of IFN-I, several lines of evidence suggest that persistently high levels of IFN-I production correlate with long-term immune activation during chronic HIV/SIV infection (9). For example, downmodulation of IFN-I production and ISG upregulation during the chronic phase of infection are key features of nonpathogenic SIV infection of the natural hosts, sooty mangabeys and African green monkeys (7, 8). Additionally, exogenous administration of IFN-α (as in treatment of hepatitis C virus [HCV] infection) has an antiproliferative effect on lymphocytes (21), which suggests that IFN-I may have a detrimental effect on T cell homeostasis in the context of a chronic, persistent virus infection, like that of HIV (22).
In this study, we attempted to characterize the roles of the different IFN-I subtypes during pathogenic SIV infection of rhesus macaques by blocking the effects of IFN-α (but not other type I interferons) through administration, just prior to SIVmac239 infection, of an antibody that neutralizes 11 of the 13 subtypes of rhesus macaque IFN-α. IFN-α blockade resulted in a trend toward higher viral loads in treated animals at day 7 postinfection. Subsequently, 6 out of 12 IFN-α-blockaded animals developed AIDS-related complications during the year of follow-up compared to only 1 of 6 control animals. While the treatment had little effect on the numbers of circulating CD4+ and CD8+ T cells, treated animals exhibited lower levels of PD-1+ Ki67+ CD4+ T cells and PD-1+ CD8+ T cells and significantly lower levels of B cell proliferation during the chronic phase of infection. Furthermore, plasma cytokine levels were reduced in treated animals at 3 months postinfection. The lower levels of activated lymphocytes in IFN-α-blockaded animals supports the hypothesis that IFN-α signaling contributes to lymphocyte activation during SIV infection. Furthermore, blockade of IFN-α in chronically HIV-infected, ART-treated humans may help to prevent chronic immune activation and the resultant inflammation-mediated morbidities associated with long-term treatment of HIV infection.
RESULTS
Study design.
The role of IFN-I in pathogenic HIV and SIV infections of humans and RMs is not completely understood. While IFN-I is a primary mediator of innate antiviral immunity, it is unclear to what extent the IFN-α subtypes versus IFN-β contribute to this antiviral activity early during SIV infection. In order to characterize the balance between IFN-α and IFN-β in both limiting viral replication and inducing chronic immune activation, we used an antibody that neutralizes 11 of the 13 IFN-α subtypes (AGS-009; Argos Therapeutics). Eighteen RMs were split into three treatment groups of six animals each and administered AGS-009 at different concentrations (group A, 100 mg/kg of body weight; group B, 10 mg/kg; group C, 0 mg/kg) the day prior to intravenous (i.v.) infection with 3,000 50% tissue culture infective doses (TCID50) of SIVmac239 (Fig. 1). During the year-long follow-up observation, blood, rectal biopsy specimens (RB), lymph nodes (LN), and PAXGene tubes (Preanalytix) were taken at regular intervals for immunophenotyping, viral load determination, and gene expression analysis. A second dose of AGS-009 was administered to 3 animals from the treated groups (group A, 50 mg/kg; group B, 10 mg/kg) at day 182 postinfection.
FIG 1.

IFN-α blockade study design. Three groups of six animals were given either a high dose (group A; 100 mg/kg), low dose (group B; 10 mg/kg), or mock infusion (group C) of AGS-009 1 day prior to SIVmac239 i.v. infection. The animals were monitored for approximately 1 year. Peripheral blood, lymph nodes, and rectal biopsy specimens were collected for virological and immunological analyses.
IFN-α blockade was associated with a nonsignificant trend toward higher viral loads in SIV-infected RMs.
To assess the effect of IFN-α blockade on viral replication, the viral load in the plasma of infected animals was measured at regular intervals after treatment with AGS-009 and high-dose i.v. infection with SIVmac239 (Fig. 2A and B). While differences in plasma viral loads between study groups were not statistically significant, we observed that plasma viremia at day 7 postinfection was on average 0.5 log unit higher in group A (i.e., receiving a high dose of AGS-009) than in controls (Fig. 2A). Setpoint viral loads were similar between groups for the duration of the study. Interestingly, one animal (RYh13) controlled SIVmac239 infection to below 1,000 copies per milliliter despite having been treated with 100 mg/kg AGS-009, suggesting that, in some cases, IFN-α is not required to achieve early viral control (Fig. 2B).
FIG 2.

SIVmac239 plasma viral loads, cell-associated DNA loads, and survival. (A) Geometric means of SIVmac239 plasma viral load by treatment group. (B) Plasma viral loads of individual animals. The dashed lines represent animals sacrificed due to simian AIDS. (C) SIVmac239 DNA copies per 10,000 CD4+ cells were measured by qPCR on microbead-sorted CD4+ cells. Cell equivalents were determined assuming 2 copies of albumin per cell. Means and standard errors are shown. (D and E) Kaplan-Meier curves showing the percentage of animals surviving in (D) all three treatment groups and (E) treated versus control animals.
In order to assess the impact of IFN-α blockade on the level of SIV-infected circulating CD4+ T cells, we longitudinally assessed (i.e., days 14, 28, 42, and 70 postinfection) the number of cell-associated SIV DNA copies relative to albumin in CD4+ T cells purified by magnetic bead separation from peripheral blood mononuclear cells. As shown in Fig. 2C, the level of cell-associated viral DNA declined between days 14 and 70 with similar rates in all three experimental groups. Taken together, these data indicate that blockade of IFN-α with AGS-009 resulted in only a modest increase of virus replication in SIV-infected RMs.
IFN-α blockade induced a trend toward decreased survival in SIV-infected RMs.
We next assessed in our cohort of RMs the effect of IFN-α blockade with AGS-009 on the mortality associated with SIVmac239. We found that, in this study, AGS-009 treatment of RMs resulted in a trend toward decreased survival after i.v. SIVmac239 infection. Three out of six animals treated with 100 mg/kg AGS-009, three out of six RMs receiving 10 mg/kg AGS-009, and only one out of six control RMs were sacrificed prior to the study endpoint due to signs of simian AIDS (sAIDS) (Fig. 2D), which included but were not limited to extreme weight loss, intractable diarrhea, and lymphomas. In all, 50% of the RMs that received the anti-IFN-α antibody developed sAIDS compared to 17% of control animals (Fig. 2E). While this study was not sufficiently powerful to detect statistical differences in survival between AGS-009-treated and untreated RMs, we concluded that IFN-α blockade was associated with a trend toward decreased survival in SIVmac239-infected RMs. Interestingly, survival was not associated with plasma viral loads in any of the treatment groups (Fig. 2B, dashed lines).
IFN-α blockade had a mild impact on CD4+ T cell counts but was associated with significantly decreased levels of Ki67 and PD-1 on CD4+ and CD8+ T cells.
In order to assess the effect of IFN-α blockade on the size and phenotype of the T cell compartment peripheral blood, LN, and RB, CD4+ and CD8+ T cells and their naive and memory subsets were quantified by multiparametric flow cytometry. According to the current classification (23), naive T cells were defined as CD95neg and CD28int, while T cell memory subsets were characterized by expression of CCR7 and CD62L within the CD95pos population, where central memory (TCM) cells expressed both CCR7 and CD62L, transitional memory (TTM) cells expressed either CCR7 or CD62L, and effector memory (TEM) cells expressed neither CCR7 nor CD62L.
While mean CD4 T cell counts were not statistically distinguishable among the three groups of RMs included in this study, four out of six animals treated with a high dose of AGS-009 experienced an early and irreversible decline in peripheral CD4+ T cell numbers that was associated with disease progression (Fig. 3A). This early decline in CD4+ T cell counts distinguished group A and to some extent group B from the control group (group C), which experienced more variable and gradual declines in CD4 T cell counts (Fig. 3A to C). These differences in the tempo of CD4+ T cell decline did not accompany significant differences in the CD8 T cell compartment or differences in any CD4+ or CD8+ naive or memory T cell populations (data not shown).
FIG 3.

Immunophenotypic analysis of T cell markers and subsets in IFN-α-blockaded animals. (A to C) CD4 T cell counts (cells per cubic millimeter of blood) in the 100 mg/kg dose group (A), 10 mg/kg dose group (B), and mock-infused group (C). The dashed lines represent animals sacrificed due to simian AIDS. (D to F) Activation and proliferation markers on CD4+ T cells and their memory subsets with significant differences between the high and low doses and mock infusions in the mixed-effects regression analysis. Means and standard errors are shown. (D) Percent Ki67+ PD-1+ CD4 T cells. (E) Percent Ki67+ CD4 TEM cells. (F) Percent Ki67+ PD-1+ CD4 TEM cells. (G to K) Activation/exhaustion and proliferation markers on CD8+ T cells and their memory subsets with significant differences between the high dose and low dose and mock infusions in the mixed-effects regression analysis. Means and standard errors are shown. (G) Percent Ki67+ PD-1+ CD8 T cells. (H) Percent Ki67+ PD-1+ CD8 TTM cells. (I) Percent Ki67+ PD-1+ CD8 TEM cells. (J) Percent PD-1+ CD8 TCM cells. (K) Percent PD-1+ CD8 TTM cells.
To characterize the effect of IFN-α blockade on the levels of lymphocyte activation, we used multiparametric flow cytometric analysis to measure longitudinally the expression of markers of T cell activation and proliferation, and changes in the levels of these markers were assessed with mixed-effects regression analysis (Table 1). We found that the levels of circulating Ki67+ PD-1+ CD4+ T cells (Fig. 3D), Ki67+ CD4+ TEM (Fig. 3E), and Ki67+ PD-1+ CD4+ TEM (Fig. 3F) declined more rapidly in group A than in control animals in group C (see Table 1 for summary statistics). In addition, we found that the CD8+ T cell compartment of IFN-α-blockaded RMs exhibited a significant decline in Ki67+ and/or PD-1+ CD8+ T cells in all nonnaive memory compartments compared to control animals (Fig. 3G to K). Specifically, Ki67+ PD-1+ cells among CD8+ T cells (Fig. 3G), CD8 TTM cells (Fig. 3H), and CD8+ TEM cells (Fig. 3I), as well as PD-1+ cells among CD8+ TCM cells (Fig. 3J) and CD8+ TTM cells (Fig. 3K), declined more rapidly in group A than in group C (Table 1 shows summary statistics). We did not observe differences in CD4+ or CD8+ T cell levels or phenotypes in LN or RB (data not shown). Taken together, these data suggest that IFN-α blockade induced a significant decline in the level of T cell activation in SIV-infected RMs.
TABLE 1.
Mixed-model regression analysis of longitudinal trajectories of multiple lymphocyte populations reveals IFN-α blockade-induced changes
| Cell population | Treatment (mg/kg AGS-009 by day postinfection) | Slope relative to control | P value | Significancea |
|---|---|---|---|---|
| % Ki67+PD-1+ CD4+ T cells | 10 | −0.002971 | 0.638 | |
| 100 | −0.015282 | 0.0204 | * | |
| % Ki67+ CD4+ TEM cells | 10 | 0.026 | 0.2298 | |
| 100 | −0.06343 | 0.005 | ** | |
| % Ki67+PD-1+ CD4+ TEM cells | 10 | 0.02494 | 0.1446 | |
| 100 | −0.041388 | 0.0201 | * | |
| % Ki67+PD-1+ CD8+ T cells | 10 | 0.004006 | 0.6932 | |
| 100 | −0.027423 | 0.0097 | ** | |
| % PD-1+ CD8+ TCM cells | 10 | −0.026061 | <0.0001 | **** |
| 100 | −0.034942 | <0.0001 | **** | |
| % PD-1+ CD8+ TTM cells | 10 | −0.02862 | 0.0046 | ** |
| 100 | −0.04376 | <0.0001 | **** | |
| % Ki67+PD-1+ CD8+ TTM cells | 10 | 0.001089 | 0.9005 | |
| 100 | −0.020232 | 0.0256 | * | |
| % Ki67+PD-1+ CD8+ TEM cells | 10 | 0.016039 | 0.2045 | |
| 100 | −0.029861 | 0.0232 | * | |
| No. of NK cells per cubic milliliter blood | 10 | −0.33248 | 0.1589 | |
| 100 | 0.8025 | 0.0012 | ** | |
| No. of B cells per cubic milliliter blood | 10 | −1.0963 | 0.2307 | |
| 100 | −2.3118 | 0.0155 | * | |
| % Ki67+ B cells | 10 | −0.020671 | 0.0217 | * |
| 100 | −0.022517 | 0.0162 | * |
*, P < 0.05; **, P < 0.005; ****, P < 0.0001.
IFN-α blockade results in increased numbers of circulating NK cells and reduced proliferation of B cells.
To assess the impact of IFN-α blockade on the levels of circulating B cell and NK cells, we next measured the levels and activation states of CD20+ B cells and CD8+ CD16+ NK cells by multiparametric flow cytometry at various time points during the study. Perhaps surprisingly, given that NK cells are known to become activated upon stimulation with IFN-I during viral infections, we found that the numbers of NK cells in the RMs belonging to group A (i.e., IFN-α blockade with high-dose AGS-009) increased significantly compared to the levels of NK cells in control animals (Fig. 4A and Table 1). This difference in NK cell kinetics was not associated with differences in NK cell activation, and no significant differences in NK cell dynamics or activation were observed in LN and RB (data not shown). In contrast to NK cells, the levels of circulating CD20+ B cells in group A declined relative to the same cells in control animals (Fig. 4B). The higher level of circulating B cells observed in control RMs accompanied significantly higher levels of Ki67+ B cells (Fig. 4C). Taken together, these results suggest that IFN-I blockade has an antiproliferative effect on B cells. These differences are unlikely to be due to redistribution of B cells to lymphoid tissues, as we found no differences in the percentages of CD20+ B cells in LN among the three groups of RMs (data not shown).
FIG 4.
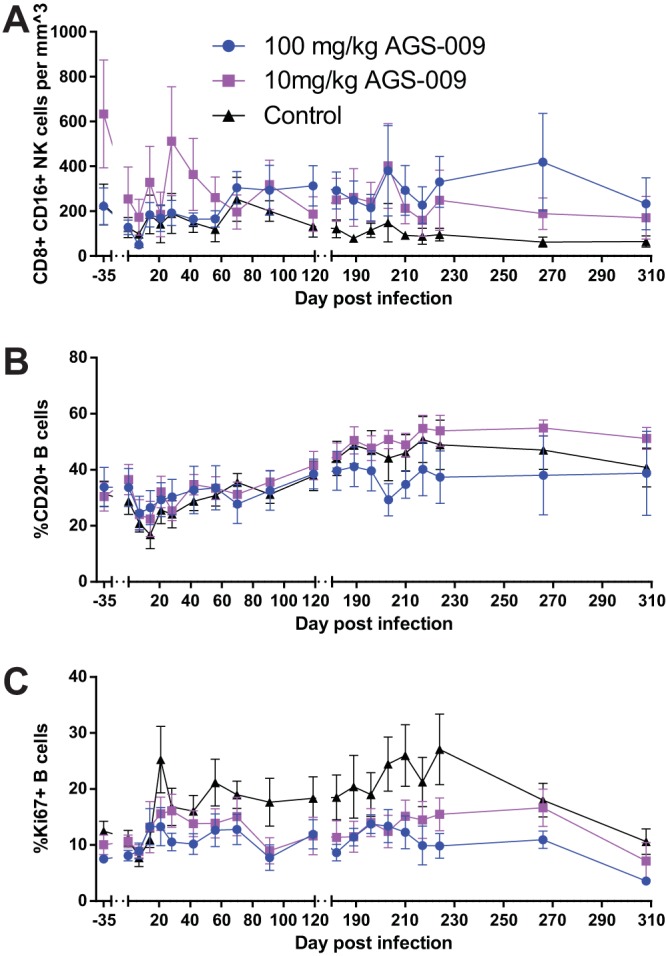
IFN-α blockade impacts levels of NK cells and proliferation of B cells in peripheral blood. (A) CD8+ CD16+ NK cells per cubic millimeter of blood. (B) Percent CD20+ B cells. (C) Percent Ki67+ B cells. Means and standard errors are shown.
IFN-α blockade is associated with a decline in plasma cytokine expression during the early chronic phase of SIVmac239 infection.
Since AGS-009 administration had a profound impact on lymphocyte activation and proliferation by flow cytometry, we hypothesized that the levels of proinflammatory cytokines would be significantly impacted, as well. We measured plasma cytokine levels using a multiplex cytometric bead array (Fig. 5). We found a significant reduction between the high-dose group and controls at day 91 in both interleukin 12 (IL-12)/23(p40) (Fig. 5A) and soluble CD40L (Fig. 5B) (Kruskal-Wallis; P > 0.05). Additionally, there were similar nonsignificant trends in a number of other cytokines, including IL-4 (Fig. 5C), TNF-α (Fig. 5D), IL-17A (Fig. 5G), IL-1β (Fig. 5H), MIP-1α (Fig. 5I), MIP-1β (Fig. 5J), IL-10 (Fig. 5K), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Fig. 5L). Thus, IFN-α blockade resulted in the impairment of downstream proinflammatory cytokine production despite the lack of a measurable effect on peripheral blood mononuclear cell (PBMC) ISG levels.
FIG 5.

IFN-α blockade results in decreases in the levels of several cytokines in plasma at day 91 postinfection. Plasma cytokine levels were measured by cytometric bead array at several time points in the first 3 months postinfection. Means and standard errors of treatment groups are shown for IL-12/23(p40) (A), soluble CD40L (B), IL-4 (C), TNF-α (D), IFN-γ (E), IL-2 (F), IL-17A (G), IL-1β (H), MIP-1α (I), MIP-1β (J), IL-10 (K), and GM-CSF (L). Differences were determined using the Kruskal-Wallis test (*, P < 0.05).
In order to more directly ascertain the consequences of the declines in lymphocyte activation and proliferation due to IFN-α blockade, we assessed the levels of downstream antiviral effector molecules in the first month of SIVmac239 infection. First, we assessed ISG expression via gene expression analysis of whole-blood total RNA hybridized to human Illumina gene chips. We chose to assess the expression levels of a panel of ISGs shown by Sandler et al. to be downregulated by IFN-I blockade (20). Despite a clear effect of AGS-009 administration on lymphocyte levels in our experiment, we did not see downregulation of this panel of ISGs during the first 4 weeks after AGS-009 administration and SIVmac239 infection (Fig. 6).
FIG 6.

IFN-α blockade did not result in significant changes to ISG expression. (A) Heat maps showing ISG expression over time. The heat maps represent log2-transformed expression values and are supervised on the abscissa (samples) and organized by unsupervised hierarchical clustering on the ordinate (transcripts). The expression values of each transcript (rows) are scaled to unity standard deviation. Unsupervised hierarchical clustering of transcripts (rows) used Euclidean distance and complete linkage methods. (B) Log2-transformed expression values of 19 ISGs over time. Means and standard errors are shown.
DISCUSSION
The effects of IFN-I on the pathogenesis of HIV and SIV infections have long been debated (9). The inability of IFN-I to uniformly prevent HIV infection combined with the observation that HIV pathogenesis is associated with chronic high levels of IFN-I and ISG expression helped fuel the idea that the antiviral effect of IFN-I was of less importance than the long-term proinflammatory effects of IFN-I that may contribute to the HIV-associated generalized immune activation and related immune decline (24). Recently, however, several studies have shown that IFN-I and downstream ISG expression may also have a beneficial role in preventing virus acquisition (20, 25). Of critical importance to the study presented here is the observation that complete IFN-I blockade via a reagent that acts as an IFN-I receptor antagonist and thus blocks all IFN-I signaling in rhesus macaques at the time of SIV infection results in significantly more rapid progression to AIDS during the early chronic phase of infection (20). IFN-I blockade was also associated with higher chronic-phase viral loads and an inability of the CD4+ T cell compartment to at least partially recover its numbers from the effects of the acute-phase virus-mediated CD4+ T cell depletion (20).
In the current study, we attempted to identify the effects of IFN-α on SIV pathogenesis by selectively blocking the interaction of IFN-α with the IFN-I receptor using an antibody (AGS-009) that binds to 11 of the 13 rhesus macaque IFN-α subtypes. Although this study was not powerful enough to see differences in survival following IFN-α blockade, we saw a trend toward faster progression to AIDS in the RMs that underwent IFN-α-blockade (6/12) than in the control animals (1/6). This higher rate of disease progression was accompanied by an average of 0.5-log-unit higher viral load at day 7 postinfection in IFN-α-blockaded animals than in control animals (Fig. 2A), and an earlier and more rapid decline of CD4+ T cells in 4/6 animals receiving the high dose of AGS-009 (Fig. 2F). While these observations were not statistically significant, they collectively suggest that blockade of IFN-α at the time of infection somewhat enhanced the ability of SIV to replicate and kill CD4+ T cells during the early stages of infection. In contrast to the results of Sandler et al. (20), we did not observe long-term differences in viral loads or CD4+ T cell counts between treated and control groups.
While this experiment did not show as dramatic an effect of IFN-α blockade as that observed by Sandler et al. (20), our study is unique in that we observed a significant reduction of the levels of PD-1+ and/or Ki67+ CD4+ and CD8+ T cell subsets, a significant reduction of proliferating peripheral CD20+ B cells, and a significant reduction in some proinflammatory cytokines. Taken together, these observations suggest that IFN-α blockade lowers the prevailing levels of lymphocyte activation and proliferation. Two non-mutually exclusive explanations could account for the IFN-α blockade-induced reduction in Ki67+ PD-1+ CD4+ T cells. One possibility is that IFN-α blockade had a relatively minor impact on virus proliferation and infection of target cells (a subset that preferentially includes Ki67+ PD-1+ CD4+ T cells), and the resultant increased virus-mediated killing of these cells could be responsible for the observed lower levels of the cells during the chronic phase of infection. On the other hand, the observed decrease in proliferating PD-1+ CD4+ T cells could be due to the impact of reduced IFN-α-mediated signaling in SIV-infected RMs undergoing IFN-α blockade. The latter possibility is supported by the concomitant observation that a number of CD8+ T cell subsets, which are not susceptible to virus-mediated killing, exhibit the same reduction in Ki67 and PD-1 levels. Similarly, the IFN-α blockade-associated increase of NK cells and decrease in proliferating peripheral CD20+ B cells may also support this hypothesis. Taking the data together, it is possible that both immunomodulatory and viral cytopathic effects have a significant impact on the dynamics of lymphocyte populations in rhesus macaques undergoing IFN-α blockade during acute SIV infection.
The difference in the magnitudes of virological effects between IFN-α blockade (as performed in this study) and complete IFN-I blockade (20) can be instructive. One possible explanation for this difference is that IFN-α is only one component of a larger interferon response following SIV infection and that other interferons, like IFN-β, IFN-λ, and IFN-κ, may play important roles in inducing antiviral gene expression in vivo. Indeed, it has been shown that the IFN-α subtypes induced by HIV infection (α1, α2, and α5) in vitro do not significantly inhibit virus replication (26). Furthermore, a recent in vitro study of IFN-mediated effects on HIV replication has shown that IFN-β is a more potent inhibitor of HIV replication than IFN-α2 (27), the subtype of IFN used to treat HCV infection. Although rhesus macaque studies assessing the abilities of IFN-α2 (20) and IFN-β (25) to prevent SIV mucosal infection suggest that IFN-β provides more potent protection, it is difficult to compare these studies due to the confounding effects of differing IFN administration regimens and SIV infection routes and the lack of understanding of the antiviral contributions of nonclassical IFNs in these experiments. Nevertheless, we are unable to rule out the more trivial possibility that the AGS-009 reagent we used is not as effective at mediating a full IFN-α blockade as the receptor-targeted molecule used by Sandler et al. (20). However, we should note that AGS-009 was shown in humans diagnosed with systemic lupus erythematosus to broadly reduce aberrant ISG expression in a dose-dependent fashion with a maximum dose of 30 mg/kg (data not shown). In our study, we used a dose almost three times higher than that in 6 RMS and were able to observe modest changes in virus replication and survival, as well as significant decreases in the levels of Ki67 and PD-1 on CD8+ and CD4+ T cells. However, we did not observe differences in ISG expression in PBMCs, possibly due to the large and robust ISG induction associated with the contemporaneous SIV infection (Fig. 6).
Two other considerations that are pertinent to this study suggest future avenues of inquiry for type I IFN blockade experiments. First, we did not consider the effects of IFN-α blockade in the context of ART-mediated suppression of virus replication. Given that long-term IFN-I production has been hypothesized to contribute to the chronic immune activation that characterizes HIV/SIV-induced AIDS progression, even in HIV-infected individuals with ART-suppressed viremia, it will be important to elucidate the impact of IFN-α blockade on activation and virus latency in the context of ART-mediated virologic suppression. Second, it will be important to understand the impact of long-term IFN-α blockade on SIV infection-mediated immune activation and viral latency. Here, we gave only a single dose of AGS-009 just prior to SIV infection and observed modest effects on the distribution of activated lymphocyte subsets. It is unknown whether a long-term IFN-α blockade strategy would more effectively decrease lymphocyte activation without inducing SIV resistance to the antiviral effects of the ISG program. Furthermore, how IFN-α blockade affects the balance between the immune-activating effects of IFN and the antiviral effects of this critical innate immune molecule remains unclear.
The goal of this study was to determine the contribution of the early IFN-α response to the outcome of SIV infection. The results presented here support the idea that IFN-α only partially contributes to the early control of SIV infection and that other IFN-Is may be important to control acute SIV replication and thus reduce the tempo of disease progression to AIDS. However, our study suggests that IFN-α mediates significant increases in the levels of lymphocyte proliferation and activation and that blockade of IFN-α early during SIV infection significantly reduces these levels. The relative balance between these two conflicting effects of IFN-I on the outcome of HIV/SIV infection remains an open question. Future experiments directly comparing the in vivo effects of IFN-α and IFN-β (and other IFN-I) blockades during different stages (acute versus chronic) and under different conditions (e.g., under full ART suppression) of SIV infection of rhesus macaques may elucidate the innate pathways that either are most critical for control of virus replication or mediate its anti-inflammatory effect. Understanding these dual roles of IFN in HIV/SIV infection could provide clues for the development of strategies to reduce inflammation in chronically HIV-infected humans on long-term ART.
MATERIALS AND METHODS
Ethics statement.
These studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Emory University Institutional Animal Care and Use Committee (IACUC) (AWA no. A3180-01). All animals were anesthetized before the performance of any procedure, and proper steps were taken to ensure the welfare and to minimize the suffering of all the animals in the studies.
Animals, AGS-009, and study design.
Eighteen MamuB*08- and MamuB*17-negative rhesus macaques of Indian origin were selected for this study. Baseline immunophenotyping was performed approximately 35 days prior to intravenous infection with 3,000 TCID50 SIVmac239 (kindly provided by Francois Villinger). One day prior to infection, two groups of six animals were administered either 100 mg/kg or 10 mg/kg AGS-009 via saline infusion. Three animals from each treated group were then subjected to another dose of AGS-009 6 months after SIV infection. During the year-long follow-up period, PBMCs, LN biopsy specimens, and RB were collected longitudinally at various times before and after SIV infection for virologic and immunologic analysis.
AGS-009 is a humanized IgG4 monoclonal antibody (MAb) developed by Argos Therapeutics to block IFN signaling for the treatment of systemic lupus erythematosus. In preclinical studies, Argos demonstrated through surface plasmon resonance that AGS-009 has an affinity comparable to those of human and macaque IFN-αs (data not shown). Furthermore, AGS-009 neutralized 11 of 13 cynomolgus macaque IFN-αs (cyIFNA2, cyIFNA6, cyIFNA14, cyIFNA16, cyIFNA17, cyIFNA21, cyIFNA41, cyIFNA42, cyIFNA43, cyIFNA44, and cyIFNA45, but not cyIFNA1 and cyIFNA8) subtypes in a reporter gene assay (data not shown).
Plasma RNA and cell-associated DNA viral quantification.
Plasma viral quantification was performed as described previously (28). For the quantification of cell-associated viral DNA in CD4+ peripheral blood cells, frozen PBMCs were thawed in a 37°C water bath and immediately washed in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine, and penicillin-streptomycin. CD4+ peripheral blood cells were then separated by positive selection using CD4 microbeads for nonhuman primates (Miltenyi Biotec) on a Miltenyi Biotec LD column according to the manufacturer's specifications. The CD4+ peripheral blood cells were counted and extracted using a blood DNA minikit (Qiagen). Quantitative real-time PCR (qPCR) was then performed on the extracted cell-associated DNA, as previously described (29). Viral genome copy numbers were normalized to the number of copies of albumin.
Immunophenotyping by flow cytometry.
Blood was collected in EDTA tubes. Plasma was obtained by centrifugation. PBMCs were separated by density gradient centrifugation using 90% lymphocyte separation medium from Lonza. LN biopsy specimens were cut in half, and one half was put in 4% paraformaldehyde and then embedded in paraffin. LN-derived cells were obtained by grinding the other half over a 70-μm cell strainer. RB were digested in 0.75 mg/ml collagenase (Sigma-Aldrich) and 0.15 μl DNase in 10% fetal calf serum (FCS), 1% penicillin-streptomycin, and 1% l-glutamine at 37°C with gentle shaking. After 2 h, tissues were mechanically separated using a plastic cannula and run over a 70-μm filter. Multicolor flow cytometric analysis was performed using predetermined optimal concentrations of the following fluorescently conjugated MAbs: anti-Ki67-fluorescein isothiocyanate (FITC) (B56), anti-CD62L-phycoerythrin (PE) (SK11), anti-CD123-PE (7G3), anti-CD95-PE-Cy5 (DX2), anti-HLA-DR-PE-Cy5 (L243), anti-CCR7-PE-Cy7 (3D12), anti-CD14-PE-Cy7 (M5E2), anti-CD11c-allophycocyanin (APC) (S-HCL-3), anti-CD3-Alexa 700 (SP34-2), anti-CD69-APC-Cy7 (FN50), and anti-CD20-APC-H7 (L27) from BD Biosciences; anti-CD28-ECD (CD28.2) and anti-CD16-ECD (3G8) from Beckman Coulter; anti-CD4-Pacific Blue (OKT4) from Biolegend; anti-CD8-QDot705 (3B5) and the Aqua Blue LIVE/DEAD Discriminator from Invitrogen; and anti-PD-1–APC (J105) from eBioscience. Flow cytometric acquisition was performed on an LSRII flow cytometer driven by the FACSDiva software package (BD Biosciences). Analysis of the acquired data was performed using FlowJo version 7.6.5 software (TreeStar). Any subpopulation whose parent population contained less than 100 events was excluded from analysis.
Interferon-stimulated-gene expression analyses by oligonucleotide microarray.
Whole blood for RNA gene expression analysis was collected into PAXgene tubes (Preanalytix) and frozen at −80°C until further processing, as previously described (30, 31). Total RNA was extracted using PAXgene blood RNA kits (Qiagen) according to the manufacturer's instructions. Globin was removed using a Globinclear kit (Ambion), and the quantity and quality of the RNA were confirmed using a NanoDrop 2000c (Thermo Fisher Scientific) and a Bioanalyzer 2100 (Agilent). Samples (50 ng) were amplified using Illumina TotalPrep RNA amplification kits (Ambion). The microarray analysis was conducted using 750 ng of biotinylated cRNA hybridized to HumanHT-12_V4 BeadChips (Illumina) at 58°C for 20 h. The arrays were scanned using Illumina's iScan system. Transcript expression values were estimated using quantile normalization. Log2-transformed data were used for subsequent analyses. Normalization of sample batch effects was achieved using ComBat with the default settings (32). Nineteen ISGs were selected for expression analyses based on significantly downregulated genes from other IFN blockade experiments (20).
Plasma cytokine measurement by cytometric bead array.
Plasma cytokine levels were measured on a Bioplex 200 system (Bio-Rad) using the 23-plex Milliplex nonhuman primate cytokine magnetic bead kit (Millipore) following the manufacturer's instructions.
Statistical analysis.
Most statistical analyses were performed in GraphPad Prism 6. Mixed-effects regression analysis of immunophenotype time series data were performed in R. The slopes of each IFN-α blockade group were compared with that of the sham blockade group to determine the statistical significance of the impact of treatment on the immunophenotype.
Accession number(s).
Expression data are available through the National Center for Biotechnology Information and the Gene Expression Omnibus (GEO) database under accession number GSE110617.
ACKNOWLEDGMENTS
We thank Barbara Cervasi, Kiran Gill, and Melon Nega for technical support; Stephanie Ehnert and the Yerkes Research Resources technicians for study organization and scheduling; and the YNPRC veterinary staff, especially Sherrie Jean, for caring for the animals.
This work was funded by P01 AI76174 to Michael Lederman and Guido Silvestri, P51 OD011132 to the YNPRC, and P30 AI050409 to the Emory CFAR.
REFERENCES
- 1.Evans DT, Silvestri G. 2013. Nonhuman primate models in AIDS research. Curr Opin HIV AIDS 8:255–261. doi: 10.1097/COH.0b013e328361cee8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon SN, Dunham RM, Engram JC, Estes J, Wang Z, Klatt NR, Paiardini M, Pandrea IV, Apetrei C, Sodora DL, Lee HY, Haase AT, Miller MD, Kaur A, Staprans SI, Perelson AS, Feinberg MB, Silvestri G. 2008. Short-lived infected cells support virus replication in sooty mangabeys naturally infected with simian immunodeficiency virus: implications for AIDS pathogenesis. J Virol 82:3725–3735. doi: 10.1128/JVI.02408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boulougoura A, Sereti I. 2016. HIV infection and immune activation: the role of coinfections. Curr Opin HIV AIDS 11:191–200. doi: 10.1097/COH.0000000000000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deeks SG, Tracy R, Douek DC. 2013. Systemic effects of inflammation on health during chronic HIV infection. Immunity 39:633–645. doi: 10.1016/j.immuni.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fauci AS, Folkers GK, Dieffenbach CW. 2013. HIV-AIDS: much accomplished, much to do. Nat Immunol 14:1104–1107. doi: 10.1038/ni.2735. [DOI] [PubMed] [Google Scholar]
- 6.Hsu DC, Sereti I. 2016. Serious non-AIDS events: therapeutic targets of immune activation and chronic inflammation in HIV infection. Drugs 76:533–549. doi: 10.1007/s40265-016-0546-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosinger SE, Li Q, Gordon SN, Klatt NR, Duan L, Xu L, Francella N, Sidahmed A, Smith AJ, Cramer EM, Zeng M, Masopust D, Carlis JV, Ran L, Vanderford TH, Paiardini M, Isett RB, Baldwin DA, Else JG, Staprans SI, Silvestri G, Haase AT, Kelvin DJ. 2009. Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J Clin Invest 119:3556–3572. doi: 10.1172/JCI40115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacquelin B, Mayau V, Targat B, Liovat AS, Kunkel D, Petitjean G, Dillies MA, Roques P, Butor C, Silvestri G, Giavedoni LD, Lebon P, Barre-Sinoussi F, Benecke A, Muller-Trutwin MC. 2009. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest 119:3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doyle T, Goujon C, Malim MH. 2015. HIV-1 and interferons: who's interfering with whom? Nat Rev Microbiol 13:403–413. doi: 10.1038/nrmicro3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kane M, Zang TM, Rihn SJ, Zhang F, Kueck T, Alim M, Schoggins J, Rice CM, Wilson SJ, Bieniasz PD. 2016. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe 20:392–405. doi: 10.1016/j.chom.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Havenar-Daughton C, Kolumam GA, Murali-Krishna K. 2006. Cutting edge: the direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol 176:3315–3319. doi: 10.4049/jimmunol.176.6.3315. [DOI] [PubMed] [Google Scholar]
- 13.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. 2005. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 174:4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]
- 14.Brewitz A, Eickhoff S, Dahling S, Quast T, Bedoui S, Kroczek RA, Kurts C, Garbi N, Barchet W, Iannacone M, Klauschen F, Kolanus W, Kaisho T, Colonna M, Germain RN, Kastenmuller W. 2017. CD8+ T cells orchestrate pDC-XCR1+ dendritic cell spatial and functional cooperativity to optimize priming. Immunity 46:205–219. doi: 10.1016/j.immuni.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ng CT, Sullivan BM, Teijaro JR, Lee AM, Welch M, Rice S, Sheehan KC, Schreiber RD, Oldstone MB. 2015. Blockade of interferon beta, but not interferon alpha, signaling controls persistent viral infection. Cell Host Microbe 17:653–661. doi: 10.1016/j.chom.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng L, Ma J, Li J, Li D, Li G, Li F, Zhang Q, Yu H, Yasui F, Ye C, Tsao LC, Hu Z, Su L, Zhang L. 2017. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest 127:269–279. doi: 10.1172/JCI90745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J, Carrillo M, Martin H, Kasparian S, Syed P, Rice N, Brooks DG, Kitchen SG. 2017. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest 127:260–268. doi: 10.1172/JCI89488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sandler NG, Bosinger SE, Estes JD, Zhu RT, Tharp GK, Boritz E, Levin D, Wijeyesinghe S, Makamdop KN, del Prete GQ, Hill BJ, Timmer JK, Reiss E, Yarden G, Darko S, Contijoch E, Todd JP, Silvestri G, Nason M, Norgren RB Jr, Keele BF, Rao S, Langer JA, Lifson JD, Schreiber G, Douek DC. 2014. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511:601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petricoin EF III, Ito S, Williams BL, Audet S, Stancato LF, Gamero A, Clouse K, Grimley P, Weiss A, Beeler J, Finbloom DS, Shores EW, Abraham R, Larner AC. 1997. Antiproliferative action of interferon-alpha requires components of T-cell-receptor signalling. Nature 390:629–632. doi: 10.1038/37648. [DOI] [PubMed] [Google Scholar]
- 22.Le Saout C, Hasley RB, Imamichi H, Tcheung L, Hu Z, Luckey MA, Park JH, Durum SK, Smith M, Rupert AW, Sneller MC, Lane HC, Catalfamo M. 2014. Chronic exposure to type-I IFN under lymphopenic conditions alters CD4 T cell homeostasis. PLoS Pathog 10:e1003976. doi: 10.1371/journal.ppat.1003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, Klatt NR, Brenchley JM, Vaccari M, Gostick E, Price DA, Waldmann TA, Restifo NP, Franchini G, Roederer M. 2013. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Invest 123:594–599. doi: 10.1172/JCI66327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Utay NS, Douek DC. 2016. Interferons and HIV infection: the good, the bad, and the ugly. Pathog Immun 1:107–116. doi: 10.20411/pai.v1i1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Veazey RS, Pilch-Cooper HA, Hope TJ, Alter G, Carias AM, Sips M, Wang X, Rodriguez B, Sieg SF, Reich A, Wilkinson P, Cameron MJ, Lederman MM. 2016. Prevention of SHIV transmission by topical IFN-beta treatment. Mucosal Immunol 9:1528–1536. doi: 10.1038/mi.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harper MS, Guo K, Gibbert K, Lee EJ, Dillon SM, Barrett BS, McCarter MD, Hasenkrug KJ, Dittmer U, Wilson CC, Santiago ML. 2015. Interferon-alpha subtypes in an ex vivo model of acute HIV-1 infection: expression, potency and effector mechanisms. PLoS Pathog 11:e1005254. doi: 10.1371/journal.ppat.1005254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, Barbian HJ, Russell RM, Gondim MV, Bahari CY, Shaw CM, Li Y, Decker T, Haynes BF, Shaw GM, Sharp PM, Borrow P, Hahn BH. 2017. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc Natl Acad Sci U S A 114:E590–E599. doi: 10.1073/pnas.1620144114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmann-Lehmann R, Swenerton RK, Liska V, Leutenegger CM, Lutz H, McClure HM, Ruprecht RM. 2000. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res Hum Retroviruses 16:1247–1257. doi: 10.1089/08892220050117014. [DOI] [PubMed] [Google Scholar]
- 29.Cartwright EK, McGary CS, Cervasi B, Micci L, Lawson B, Elliott ST, Collman RG, Bosinger SE, Paiardini M, Vanderford TH, Chahroudi A, Silvestri G. 2014. Divergent CD4+ T memory stem cell dynamics in pathogenic and nonpathogenic simian immunodeficiency virus infections. J Immunol 192:4666–4673. doi: 10.4049/jimmunol.1303193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barouch DH, Ghneim K, Bosche WJ, Li Y, Berkemeier B, Hull M, Bhattacharyya S, Cameron M, Liu J, Smith K, Borducchi E, Cabral C, Peter L, Brinkman A, Shetty M, Li H, Gittens C, Baker C, Wagner W, Lewis MG, Colantonio A, Kang HJ, Li W, Lifson JD, Piatak M Jr, Sekaly RP. 2016. Rapid inflammasome activation following mucosal SIV infection of rhesus monkeys. Cell 165:656–667. doi: 10.1016/j.cell.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fukazawa Y, Park H, Cameron MJ, Lefebvre F, Lum R, Coombes N, Mahyari E, Hagen SI, Bae JY, Reyes MD III, Swanson T, Legasse AW, Sylwester A, Hansen SG, Smith AT, Stafova P, Shoemaker R, Li Y, Oswald K, Axthelm MK, McDermott A, Ferrari G, Montefiori DC, Edlefsen PT, Piatak M Jr, Lifson JD, Sekaly RP, Picker LJ. 2012. Lymph node T cell responses predict the efficacy of live attenuated SIV vaccines. Nat Med 18:1673–1681. doi: 10.1038/nm.2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]