

ABSTRACT
Many viral envelope proteins are modified by asparagine (N)-linked glycosylation, which can influence their structure, physicochemical properties, intracellular transport, and function. Here, we systematically analyzed the functional relevance of N-linked glycans in the alphaherpesvirus pseudorabies virus (PrV) glycoprotein H (gH), which is an essential component of the conserved core herpesvirus fusion machinery. Upon gD-mediated receptor binding, the heterodimeric complex of gH and gL activates gB to mediate fusion of the viral envelope with the host cell membrane for viral entry. gH contains five potential N-linked glycosylation sites at positions 77, 162, 542, 604, and 627, which were inactivated by conservative mutations (asparagine to glutamine) singly or in combination. The mutated proteins were tested for correct expression and fusion activity. Additionally, the mutated gH genes were inserted into the PrV genome for analysis of function during virus infection. Our results demonstrate that all five sites are glycosylated. Inactivation of the PrV-specific N77 or the conserved N627 resulted in significantly reduced in vitro fusion activity, delayed penetration kinetics, and smaller virus plaques. Moreover, substitution of N627 greatly affected transport of gH in transfected cells, resulting in endoplasmic reticulum (ER) retention and reduced surface expression. In contrast, mutation of N604, which is conserved in the Varicellovirus genus, resulted in enhanced in vitro fusion activity and viral cell-to-cell spread. These results demonstrate a role of the N-glycans in proper localization and function of PrV gH. However, even simultaneous inactivation of all five N-glycosylation sites of gH did not severely inhibit formation of infectious virus particles.
IMPORTANCE Herpesvirus infection requires fusion of the viral envelope with cellular membranes, which involves the conserved fusion machinery consisting of gB and the heterodimeric gH/gL complex. The bona fide fusion protein gB depends on the presence of the gH/gL complex for activation. Viral envelope glycoproteins, such as gH, usually contain N-glycans, which can have a strong impact on their folding, transport, and functions. Here, we systematically analyzed the functional relevance of all five predicted N-linked glycosylation sites in the alphaherpesvirus pseudorabies virus (PrV) gH. Despite the fact that mutation of specific sites affected gH transport, in vitro fusion activity, and cell-to-cell spread and resulted in delayed penetration kinetics, even simultaneous inactivation of all five N-glycosylation sites of gH did not severely inhibit formation of infectious virus particles. Thus, our results demonstrate a modulatory but nonessential role of N-glycans for gH function.
KEYWORDS: herpesvirus, pseudorabies virus, glycoprotein gH, membrane fusion, virus entry, N-linked glycosylation
INTRODUCTION
Infectious entry of herpesviruses into cells depends on fusion of the viral envelope with host cell membranes. To accomplish this task, herpesviruses use specialized surface glycoproteins allowing them to bind to an appropriate cellular receptor and to execute membrane fusion. Whereas many enveloped viruses rely on a single protein for receptor binding and entry, multiple viral glycoproteins are involved in herpesvirus-mediated membrane fusion.
Glycoprotein B (gB) and the heterodimeric complex of membrane-bound glycoprotein H and anchorless glycoprotein L (gH/gL) constitute the core fusion machinery and are conserved across the Herpesviridae, a large family of enveloped double-stranded DNA viruses infecting mammals, birds, and reptiles (reviewed in references 1 and 2). Binding of nonconserved herpesvirus subfamily-specific glycoproteins to cellular receptors is thought to trigger activation of the conserved fusion machinery. However, the exact molecular mechanism of this process remains to be elucidated. According to the current model, the entry glycoproteins are activated in a cascade-like fashion, beginning with binding of, e.g., alphaherpesvirus gD to a cellular receptor. Receptor binding was shown to lead to a conformational change in the C-terminal region of gD, presumably enabling gD to interact with gH/gL (3, 4). gH/gL in turn is thought to activate gB by direct interaction of their respective ectodomains (5–9). gB then inserts its two fusion loops (FL) into the target membrane to subsequently merge the viral and cellular membranes by a large conformational change (5, 8). gB is a type I transmembrane protein and is considered the bona fide fusion protein, since crystal structures of gB homologs from pseudorabies virus (PrV) (10, 11), herpes simplex virus 1 (HSV-1) (12), human cytomegalovirus (HCMV) (13, 14), and Epstein-Barr virus (EBV) (15) resemble those of typical class III postfusion trimers. However, despite its homology to, e.g., the vesicular stomatitis virus fusion protein G (16) or baculovirus gp64 (17), gB is not able to mediate membrane fusion on its own but requires the conserved gH/gL complex.
Although the specific contribution of the gH/gL complex to herpesvirus membrane fusion remains largely unknown, functional analysis points to a regulatory role (5). In contrast to that of gB, the crystal structures of soluble gH/gL of HSV-2 (7), EBV (18), and varicella-zoster virus (VZV) (19) and of a core fragment of PrV gH (20) (Fig. 1) revealed no structural homology to any known viral fusion protein. However, despite poor sequence conservation, the crystallographic studies revealed a strikingly similar domain organization of the four gH homologs.
FIG 1.

Positions of potential N-linked glycosylation sites in PrV gH. (A) The primary translation product of PrV gH (686 aa) is schematically shown. The first amino acids of the predicted signal peptide (SP), the ectodomain parts (I to IV), the transmembrane domain (TMD), and the cytoplasmic domain (CD), as well the modified asparagine (N) residues, are indicated. (B) Crystal structure of the gH core fragment (aa 107 to 639) (20). The N and C termini are labeled. In domain II, the fence (dark blue) and the syntaxin-like bundle (SLB) (light blue) are indicated. Domain III is colored yellow and domain IV red, with the flap highlighted in blue. Predicted glycosylation sites at N162, N542, and N627 are indicated by green spheres. N604 lies within a flexible region (dashed green line), which was not solved in the crystal structure. N77 was not included in the construct used for crystallization. The image was generated using the UCSF Chimera package (version 1.11.2) (65).
The least-conserved domain I, located at the N terminus of gH, is associated with gL, and the presence of both proteins seems to be critical for the tertiary structure of this domain in HSV and EBV (7, 18, 21). However, in PrV, Bovine herpesvirus 4, and Murid herpesvirus 4, gL is not required for correct folding, transport, or virion incorporation of gH (22–27). Moreover, infection by PrV can occur in the absence of gL and the gL-binding domain of gH when compensatory mutations in other glycoproteins are present (28–30). In addition, the absence of gL obviously facilitates maturation of certain N-glycans of PrV gH, which are possibly masked during wild-type (WT) replication (25). Interestingly, domain I of PrV gH, which was not included in the crystallized core fragment, contains one of the predicted N-glycosylation sites at an asparagine (N) at amino acid (aa) position 77 (Fig. 1).
Domain II contains two conserved elements (Fig. 1), the “fence,” a sheet of antiparallel beta-chains, and a bundle of three alpha-helices which is tightly packed against the fence and was designated syntaxin-like bundle (SLB) due to its structural similarities to a specific domain of cellular syntaxins (20). The side of the fence which packs against the SLB is very hydrophobic, whereas the opposite side, including an N-glycosylation site at position 162, displays only polar residues (20). The integrity and flexibility of the SLB were recently shown to be relevant for the function of PrV gH in membrane fusion (31).
Domain III, which contains no N-glycosylation sites, is composed of eight alpha-helices (Fig. 1) and contains a highly conserved amino acid stretch (serine-proline-cysteine) which is important for regulation of membrane fusion (32).
The membrane-proximal domain IV is the most conserved domain of gH. It consists of a beta-sandwich comprising two opposed four-stranded beta-sheets, which in PrV contain one and two predicted N-glycosylation sites, respectively, at aa 554, 604, and 627 (Fig. 1). The two sheets are connected by an extended polypeptide chain, which is designated “flap” (20). Interestingly, the flap, supported by the N-glycan at position 627, covers a patch of hydrophobic amino acid residues which is conserved in PrV, HSV, and EBV. Movement of the flap during a receptor-triggered conformational change of gH is thought to enable interaction of this underlying hydrophobic surface with the viral envelope (20, 33). This hypothesis was supported by studies revealing that disruption of conserved disulfide bonds important for positioning of the flap, prevention of flap movement by introduction of artificial disulfide bonds, or multiple alanine substitutions within the flap or the hydrophobic patch led to significant defects in gH function (33). Moreover, recent studies demonstrated functional conservation of domain IV by construction of a chimeric gH consisting of PrV domains I to III and HSV-1 domain IV. This chimera supported replication of gH-deleted PrV and was able to promote membrane fusion when coexpressed with HSV-1 or PrV gB and PrV gD and gL (34).
N-linked glycosylation is one of the most common types of membrane protein modification and involves the transfer of oligosaccharides to the asparagine residue of the sequon N-X-threonine (T) or serine (S) in the endoplasmic reticulum (ER), frequently followed by trimming and substitution of mannose residues by various other sugars in the Golgi apparatus (35, 36). Unlike sugar chains of the complex type, the initially added high-mannose chains are sensitive to digestion with endoglycosidase H (endo H), which permits differentiation (37). Many viral envelope proteins contain N-linked glycans, which can play major roles in correct folding, physicochemical properties, intracellular transport, function during entry, and also immune evasion (38–40). Disruption of N-glycosylation sites from envelope proteins of a variety of viruses, such as HSV-2 (41), Ebola virus (40), influenza virus (42), respiratory syncytial virus (43) Zika virus (44), or Hendra and Nipah viruses (45), has been shown to have a strong impact on viral entry, cell-cell fusion, or shielding from neutralizing antibodies. N-glycans on other envelope proteins, such as HSV-1 gD, were shown to be structurally important. In contrast, N-glycosylation of gD was not required for its function during virus entry or spread (46–48).
While PrV gH contains five potential N-linked glycosylation sites (49) (Fig. 1), its complex partner gL is modified only by O-linked carbohydrates (23). Earlier studies indicated that N-linked glycans contribute approximately 19 kDa to the molecular mass of mature PrV gH and provided no evidence for the presence of O-linked sugars (49). Occupation of two (N162 and N627) of the five potential N-linked glycosylation sites was confirmed by crystal structure analysis of the core fragment of PrV gH (20). The site at aa 627 is highly conserved among the Herpesviridae and was suggested to play a role in the fusion process by partially masking the highly conserved hydrophobic patch in domain IV (20, 33). Mutation of this site affected the function of gH, although it was not abolished (33). However, more-detailed studies on the relevance of this site and of the four other potential PrV gH N-glycosylation sites were required.
To this end, we have now inactivated all potential N-linked glycosylation sites singly or in various combinations by conservative amino acid replacements of N by glutamine (Q) using site-directed mutagenesis of the plasmid-cloned PrV gH gene. The resulting gH expression plasmids were tested in a virus-free transfection-based cell fusion assay. Furthermore, the mutated gH genes were transferred into the PrV genome. Protein expression and glycosylation as well as in vitro replication properties, including penetration, growth kinetics, and plaque formation, of the obtained virus mutants were investigated.
RESULTS
Effect of N-glycosylation site mutations on gH expression and transport.
The deduced amino acid sequence of PrV gH contains five potential N-linked glycosylation sites matching the consensus motif N-X-T/S, where X can represent any amino acid except proline (36). They are all located within the gH ectodomain at aa 77, 162, 542, 604, and 627 (Fig. 1). To investigate their occupation and functional relevance, the codons encoding asparagine were replaced by glutamine codons via site-directed mutagenesis of the expression plasmid-cloned gH gene of PrV strain Kaplan (PrV-Ka). The resulting plasmids, containing one to five mutations, were used for in vitro expression studies and fusion assays, as well as for generation of virus recombinants by mutagenesis of the PrV genome cloned as a bacterial artificial chromosome (BAC) in Escherichia coli (31).
Western blot (not shown) and indirect immunofluorescence (IIF) analyses (Fig. 2A) of rabbit kidney (RK13) cells cotransfected with gH and gL expression plasmids demonstrated that all generated gH mutants were stably expressed at similar levels. However, comparative IIF tests of permeabilized and nonpermeabilized RK13 cells revealed that whereas wild-type (WT) gH and most of the gH mutants were readily found at the cell surface, gH mutants containing the amino acid substitution N627Q were barely detectable on nonpermeabilized cells (Fig. 2A). These findings were confirmed by fluorescence-activated cell sorter (FACS) analysis of cotransfected cells showing that significantly decreased proportions of gHN627Q and gHN77/162/542/604/627Q were present on the cell surface compared to wild-type gH (Fig. 2B).
FIG 2.

Expression and surface localization of gH. RK13 cells were cotransfected with expression plasmids for either wild-type (WT) or mutant gH in combination with gL. After 24 h, cells were either permeabilized or not and were stained with a gH-specific rabbit antiserum and Alexa Fluor 488-conjugated secondary antibodies. Total and cell surface expression of gH was analyzed either by fluorescence microscopy (Nikon Eclipse Ti-S) (A) or by flow cytometry (B). Bars represent the ratio between cells expressing gH at the surface and total gH-positive cells. Mean values and standard deviations from three independent experiments are shown, and the significance of differences from results obtained with wild-type gH is marked. (**, P ≤ 0.01). Size bar in panel A, 100 μm.
Analysis of the distribution of gH in the cytoplasm of plasmid transfected cells by laser scanning confocal microscopy revealed wild-type-like speckled patterns for most of the gH single-site mutants (Fig. 3). However, all variants, including that with the mutation N627Q showed a more perinuclear localization indicative of the endoplasmic reticulum (ER). For confirmation, colocalization of gH with an ER marker (pmTurquoise2ER; Addgene) was analyzed. As shown in Fig. 4, both gHN627Q and the quintuple mutant colocalized with the ER marker, whereas wild-type gH was found mainly in speckles distinct from the ER, indicating that N627Q impaired ER export of PrV gH to the plasma membrane.
FIG 3.

Subcellular localization of gH. RK13 cells were cotransfected with plasmids encoding wild-type (WT) or mutated gH in combination with gL. One day after transfection, cells were fixed with 3% paraformaldehyde and permeabilized with 0.1% Triton X-100. gH was detected using a monospecific rabbit antiserum and Alexa Fluor 488-conjugated secondary antibodies. Green fluorescence was excited at 488 nm and recorded with a laser scanning confocal microscope (Leica SP5). Size bar, 10 μm.
FIG 4.

Colocalization of gH mutants with the endoplasmic reticulum. RK13 cells were cotransfected with expression plasmids for wild-type (WT) or mutated gH in combination with gL and the ER marker pmTurquoise2ER (Addgene). At 24 h posttransfection, cells were fixed with 3% paraformaldehyde and permeabilized with 0.1% Triton X-100. gH was detected using a monospecific rabbit antiserum and Alexa Fluor 633-conjugated secondary antibodies. Green (ER) and red (gH) fluorescence was excited at 488 nm and 633 nm, respectively, and recorded with a laser scanning confocal microscope (Leica SP5). Channels were merged to visualize ER localization of gH. Size bars, 10 μm.
Effects of gH N-glycosylation site mutations on in vitro fusion activity.
To analyze the impact of the gH N-glycosylation sites on in vitro fusion activity, the different gH mutants were tested in transient-transfection-based fusion assays (Fig. 5). RK13 cells were cotransfected with expression plasmids encoding wild-type or mutated gH in combination with gB, gD, and gL of PrV-Ka as described previously (50). In addition, an enhanced green fluorescent protein (EGFP) expression plasmid was cotransfected to facilitate evaluation of the syncytium formation by fluorescence microscopy (51). The results of assays with wild-type gH were set as 100%. While fusion activities of mutants gHN77Q and gHN162Q were reduced by around 20% and 10%, respectively, no significant reduction was observed for gHN542Q (Fig. 5A). Interestingly, mutation of the N-glycosylation site at position 604, which is conserved in members of the Varicellovirus genus of alphaherpesviruses, led to a significant increase in in vitro fusion activity of approximately 30% compared to that with WT gH. In contrast, disruption of the highly conserved N627 completely abolished fusion activity. In line, the double, triple, quadruple, and quintuple mutants, which all contain mutation N627Q, exhibited no fusogenic potential (Fig. 5A).
FIG 5.

In vitro fusion activity of gH mutants. RK13 cells were cotransfected with expression plasmids for EGFP, gD, gL, wild-type (WT) or mutated gH, and wild-type gB (A) or C-terminally truncated gB-008 (B). Assays without gH served as negative controls. One day after transfection, the relative fusion activity was determined by multiplication of the mean syncytium area by the number of syncytia in 10 fields of view. Fusion activities in assay mixtures containing wild-type gH were set as 100%. Shown are the mean relative values and corresponding standard deviations from four independent experiments. Results significantly differing from those obtained with WT gH are marked (*, P ≤ 0.05; **, P ≤ 0.01, ***, P ≤ 0.001; ****, P ≤ 0.0001). NS, not significant.
To investigate whether the gH mutants with an inactivated glycosylation site at position 627 are able to trigger fusion in combination with a more fusogenic gB, we replaced native gB with a C-terminally truncated protein (gB-008). In comparison to fusion assays with the WT glycoproteins, replacement of WT gB by gB-008 leads to a 3-fold increase in fusion activity (52) (Fig. 5B). Assays with plasmids encoding WT gH, gB-008, gL, and gD served as positive controls, and results were set as 100%. Assays conducted with the empty expression vector (pcDNA3) instead of a gH plasmid served as negative controls.
In relative terms, fusion activities obtained for gHN77Q, gHN162Q, gHN542Q, and gHN604Q in combination with the hyperfusogenic gB-008 (Fig. 5B) were similar to those obtained with wild-type gB (Fig. 5A). However, in contrast to fusion assays with wild-type gB, in which no activity could be observed for gH N627Q mutants, assays with gB-008 revealed that all these mutants mediated cell-cell fusion, although at ≥70% lower levels than WT gH (Fig. 5B). These results indicate that the carbohydrate moiety on N627 plays an important role in fusion or proper localization of gH.
Effects of N-glycosylation site mutations on processing and virion incorporation of gH.
To investigate the relevance of the N-glycosylation sites for processing and virion incorporation of gH and virus replication, the mutated gH genes were introduced into the PrV genome by BAC mutagenesis in E. coli as described previously (31). PCR amplification and subsequent sequencing of the entire gH open reading frames (ORF) of all virus recombinants confirmed that neither reversions of the desired mutations nor additional mutations elsewhere in the gene had occurred.
None of the analyzed N-glycosylation site mutations of gH abrogated formation of infectious PrV (see below), and Western blot analyses of purified virions revealed that all mutated proteins were incorporated into virus particles (Fig. 6A). Moreover, no obvious reduction of gH amounts compared to those of other envelope glycoproteins such as gB (not shown) or gD (Fig. 6B) could be observed. As expected, all glycosylation site mutants of gH exhibited higher electrophoretic mobilities than WT gH, which possessed an apparent molecular mass of approximately 90 kDa (Fig. 6A, first lane). Considerable differences could be observed between the apparent masses of the single mutants, indicating that the sizes of carbohydrate chains of gH differ site specifically. Interestingly, removal of the highly conserved glycosylation site at gH position 627 had the least effect on electrophoretic mobility (Fig. 6A and 7F). The apparent molecular masses of the double (gHN162/627Q), triple (gHN77/162/627Q), quadruple (gHN77/162/604/627Q), and quintuple (gHN77/162/542/604/627Q) mutants gradually decreased as expected (Fig. 6A). These results demonstrate that all five predicted N-glycosylation sites are occupied by carbohydrates in PrV gH. Since N77 is located in PrV gH domain I, which was recently confirmed to be required for gL binding and virion incorporation (30), virus particles were also analyzed for the presence of gL (Fig. 6C). Despite moderate variations in signal strength, gL was detectable in virions of all investigated gH mutants, including the N77Q substitution mutants. Thus, N-glycosylation of gH is not required for interaction with gL.
FIG 6.
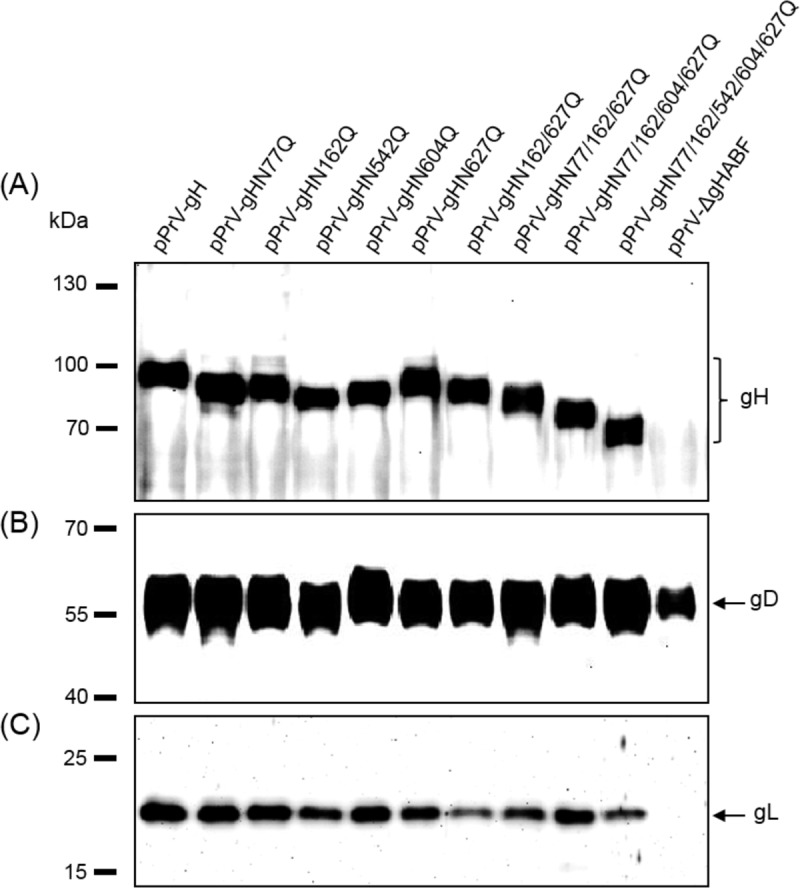
Western blot analyses of purified PrV particles. Virion proteins of the indicated gH mutants were separated by SDS-PAGE. Blots were probed with monospecific rabbit antisera against gH (A), gD (B), or gL (C). Molecular masses of marker proteins are indicated.
FIG 7.

Analysis of N-linked carbohydrates in gH. Purified virion proteins of the indicated PrV recombinants were either left untreated (lanes 1), digested with PNGase F (lanes 2) or endo H (lanes 3), or incubated with reaction buffer without enzymes (lanes 4). Samples were separated by SDS-PAGE, and blots were tested with a gH-specific rabbit antiserum. Molecular masses of marker proteins are indicated. Arrows indicate gH mutants exhibiting reduced or no endo H sensitivity.
To further analyze the glycosylation state of gH, purified virions of the different mutants were treated with either endo H to remove only high-mannose-type N-glycans (Fig. 7, lanes 3) or peptide-N-glycosidase F (PNGase F) to cleave off all N-linked carbohydrates (Fig. 7, lanes 2). As expected, WT gH and all gH mutants, except the quintuple mutant, were sensitive to digestion with PNGase F, which resulted in a shift from the different sizes of the untreated proteins (Fig. 7, lanes 1 and 4) to a similar apparent molecular mass of approximately 70 kDa (Fig. 7, lanes 2). This size corresponded to that of the glycosidase-resistant quintuple mutant of gH (Fig. 7H) and was close to the 69-kDa calculated molecular mass of the unmodified polypeptide after removal of the predicted signal peptide. These results confirmed that all predicted N-glycosylation sites on wild-type gH are occupied and that they are inactivated in the quintuple mutant.
As shown previously (49), PrV wild-type virions still contain endo H-sensitive carbohydrates (Fig. 7A). To investigate which of the five glycosylation sites of mature wild-type gH might be modified by high mannose N-glycans, virions of the gH mutants were also treated with endo H. All mutants containing deletions of only one N-glycosylation site were found to be sensitive, and faster-migrating gH species were detected (Fig. 7, lanes 3). However, the shifts compared to untreated gH (Fig. 7, lanes 1 and 4) were different, and least in pPrV-gHN162Q and pPrV-gHN627Q virions (Fig. 7C and F, arrows). Western blot analysis of virions of the double mutant pPrV-gHN162/627Q revealed that this gH mutant was no longer sensitive to endo H digestion (Fig. 7G, arrow). These results strongly suggest that high-mannose-type N-glycans are retained only at aa 162 and 627 of mature gH.
Effect of gH N-glycosylation site mutations on virus entry and spread.
To determine the effect of N-glycosylation in PrV gH on virus replication, the penetration and growth kinetics, as well as direct viral cell-to-cell spread of all obtained virus mutants, were analyzed in RK13 cells. Penetration kinetics studies revealed that entry of WT gH-expressing PrV (pPrV-gHK) and pPrV-gHN162Q occurred at a similar high velocity, with almost 100% of infectious input virus protected from inactivation by low-pH treatment after 10 min at 37°C (Fig. 8). Penetration of pPrV-gHN77Q, pPrV-gHN542Q, and pPrV-gHN604Q was slightly delayed, with approximately 80% of virions being internalized within 10 min. However, after 30 min, almost complete penetration was achieved. In contrast, only 70% of infectious pPrV-gHN627Q and pPrV-gHN162/627Q particles were able to penetrate within 10 min, and 2 h was required for complete penetration (Fig. 8). The defect in entry was even more pronounced for the triple, quadruple, and quintuple virus mutants, of which only 30% of the infectious particles were internalized within 10 min. However, after 2 h, almost complete penetration was observed for all mutants (Fig. 8).
FIG 8.

Penetration kinetics of PrV gH mutants. RK13 cells grown in 6-well plates were infected with approximately 250 PFU/well of pPrV-gHK or the different gH mutants. After adsorption at 4°C, the cells were incubated for 0, 5, 10, 15, 30, 60, and 120 min at 37°C, followed by acid inactivation of nonpenetrated virus. After 48 h at 37°C under semisolid medium, plaques were counted and compared to plaque numbers found without inactivation. Mean percentages from three independent experiments and corresponding standard deviations are shown.
In contrast to the effects of N-glycosylation site mutations on the kinetics of virus entry, the impact on total replication was less striking (Fig. 9). Multistep growth analyses performed after infection at a multiplicity of infection (MOI) of 0.01 revealed no significant effects on maximum titers of any of the virus recombinants possessing single gH mutations compared to WT gH-rescued pPrV-gHK, demonstrating that none of the N-glycosylation sites is essential for efficient production of infectious virions. The multiply mutated viruses exhibited an approximately 5- to maximum 10-fold reduction of final titers compared to pPrV-gHK (Fig. 9). Moreover, the quintuple mutant showed a delayed increase of virus titers (Fig. 9). These results indicate that although none of the N-glycosylation sites is essential, complete absence of N-glycosylation impairs gH function during productive viral replication.
FIG 9.

In vitro growth kinetics of PrV gH mutants. RK13 cells were infected with pPrV-gHK or the different gH mutants at an MOI of 0.01. Cells and supernatants were harvested after 0, 6, 12, 24, 48, and 72 h at 37°C and lysed by freeze-thawing. Progeny virus titers were determined on RK13 cells. Shown are mean values from four independent experiments and corresponding standard deviations.
The most striking differences between the investigated virus mutants were observed in direct cell-to-cell spread, and the results from plaque assays generally paralleled the findings from transient-transfection fusion assays (Fig. 5). Whereas plaque sizes of pPrV-gHN162Q and pPrV-gHN542Q were similar to those of WT gH-expressing pPrV-gHK, plaque areas of pPrV-gHN604Q were significantly enlarged to 135% (Fig. 10). In contrast, pPrV-gHN77Q (65%) and pPrV-gHN627Q (45%) formed significantly smaller plaques than wild-type virus. Deletion of multiple glycosylation sites further reduced plaque sizes, and the quintuple mutant pPrV-gHN77/162/542/604/627Q formed only very tiny plaques reaching 20% of the pPrV-gHK size (Fig. 10).
FIG 10.

Plaque sizes of PrV gH mutants. RK13 cells were infected with the indicated PrV recombinants and subsequently incubated under plaque assay conditions. After 48 h, areas of 30 plaques per virus were measured. Plaque areas of wild-type gH-expressing pPrV-gHK were set as 100%. Mean values and standard deviations from four independent experiments are shown. Results significantly differing from those obtained with pPrV-gHK are marked (*, P ≤ 0.05; **, P ≤ 0.01, ***, P ≤ 0.001). NS, not significant.
In summary, the results indicate that the N-glycosylation of the partially conserved site at position 77, positioned in the gL binding domain, and of the highly conserved site at position 627, located within the hydrophobic patch in domain IV, is important but not absolutely required for gH function during virus entry and cell-to-cell spread. In contrast, deletion of the PrV-specific glycosylation site at gH position 604 seems to be beneficial for virus spread in cell culture, like for in vitro membrane fusion.
DISCUSSION
N-linked glycosylation of envelope glycoproteins of a variety of viruses, such as, e.g., gB from HSV-2 (41), has been shown to play an important role in viral infection. However, little is known about the role of N-linked glycosylation in the function of herpesvirus gH. In the present study, we investigated the role of N-linked glycosylation of the alphaherpesvirus PrV gH for its function during entry and spread. To this end, the five predicted N-glycosylation sites in PrV gH (N77, N162, N542, N604, and N627) (Fig. 1) were inactivated singly or in combination by introduction of conservative amino acid substitutions (N→Q). The effect of the mutations on protein expression, maturation, virion incorporation, and, in particular, function in membrane fusion was investigated.
Western blot analysis of purified virions showed that the apparent molecular masses of all mutated gH variants were lower than that of wild type, demonstrating that the five potential glycosylation sites are indeed modified by N-glycans in PrV gH. Furthermore, gH of a quintuple mutant was no longer sensitive to N-glycosidases (Fig. 7H) and showed a molecular mass as predicted for the nonglycosylated precursor protein. This demonstrates that no other (e.g., O-linked) glycans are present in gH, which is in line with previous results (49). Only two of the sites (N162 and N627) were found to be glycosylated in the crystallized gH core fragment, because they were either absent from the analyzed protein (N77) or located in insufficiently resolved parts of the determined structure (N542 and N604) (20). Interestingly, the five single mutants exhibited apparent molecular masses that were different from each other (Fig. 7A to F), which could be due to differences between the oligosaccharide chains attached to the individual N-glycosylation sites. It has been reported that other glycoproteins containing more than one N-X-T/S sequon per molecule are also often heterogeneous with respect to their content of complex N-glycans on different sequons (53, 54). It is conceivable that the glycans attached to the different glycosylation sites in gH are site specific and that the observed N-glycan diversity is caused by the surrounding amino acid sequence or conformation affecting substrate availability for Golgi glycosidases or glycosyltransferases (53).
Previous studies revealed that mature PrV gH, as found in virions, is still sensitive to endo H digestion, indicating that N-linked carbohydrates on gH are not completely processed to complex structures in the Golgi apparatus (49). Here, we show that only the two glycosylation sites at N162 and N627 carry high-mannose sugars. N162 is located in gH domain II (Fig. 1) and is part of the “fence,” a sheet of antiparallel beta-chains. The side of the fence which includes glycan N162 displays only polar residues (20). In HSV-2 gH, which contains no equivalent glycosylation site, the corresponding side of the fence is partly covered by gL, which was not crystallized with the PrV gH core fragment (7, 20). Thus, N162 in PrV gH may also be covered by gL, which limits accessibility for glycosidases or glycosyltransferases in the Golgi apparatus, leading to incomplete modification of this glycosylation site and endo H sensitivity. This is in line with previous data showing that the carbohydrates incorporated into gH of PrV-ΔgL virions are completely endo H resistant (25).
The N-glycosylation site at aa 627 in PrV gH is located within a highly conserved hydrophobic patch which is covered by a negatively charged surface loop, designated the “flap” (20). Therefore, it is conceivable that the flap might interfere with glycosylation at position N627, leading to endo H sensitivity of the respective glycans in mature gH.
N-glycans on other entry glycoproteins, such as, e.g., gB from HSV-2, were shown to be important for correct intracellular trafficking and maturation (41). N-glycans on HSV-1 gD were found to be critical for the formation and maintenance of the gD structure. However, in contrast to what was observed for gB, no effect on protein processing or trafficking could be observed for HSV-1 gD N-glycosylation site mutants. Moreover, none of the investigated N-glycans on HSV-1 gD were required for gD function during entry and spread (46–48).
In PrV gH, the introduced N-glycosylation site mutations had no effect on expression level, and the subcellular localization of most single mutants was similar to that of wild-type gH. However, inactivation of the highly conserved N627 greatly affected efficient transport, resulting in ER retention and reduced cell surface expression as demonstrated by colocalization of gHN627Q with an ER marker as well as by IIF and FACS analysis of nonpermeabilized cells cotransfected with gH and gL expression plasmids (Fig. 2 to 4). Moreover, in transient-transfection assays no fusion activity could be observed for gH N627 mutants in combination with wild-type gB, gD, and gL (Fig. 5A), and surface expression of mutant gH was still barely detectable (data not shown). This indicates that none of the entry glycoproteins is able to assist gHN627Q in efficient trafficking to the cell surface. However, in contrast to the results of fusion assays conducted with wild-type gB, gHN627Q exhibited moderate fusion activity with hyperfusogenic gB-008 (Fig. 5B), as reported earlier (33). Thus, due to the enhanced surface expression of the C-terminally truncated gB-008 (52), smaller amounts of gH might be sufficient to trigger membrane fusion.
Although a great majority of gHN627Q seemed to be retained in the ER of transfected cells, Western blot analysis of corresponding PrV recombinants demonstrated that gHN627Q, as well as multiplex mutants including this amino acid substitution, is efficiently incorporated into virus particles (Fig. 6A). Thus, intracellular transport of gH during virus infection may differ from the situation after plasmid transfection, e.g., due to concomitant transport of other membrane proteins to the secondary envelopment site in the trans-Golgi network (55). For HSV and EBV it was shown that gL is critical for correct folding, transport, and virion incorporation of gH (7, 18). However, in PrV, gL is not required for these processes (21–27), and other protein interactions involved in gH transport remain to be analyzed.
As demonstrated by in vitro replication studies, none of the introduced single or multiple glycosylation site mutations abolished gH function in the viral context (Fig. 8 to 10). Most PrV recombinants reached maximum titers similar to those of wild-type PrV, and only the multiple mutants exhibited 5- to 10-fold reductions in final titers and a slight delay in replication kinetics (Fig. 9). Whereas formation of infectious progeny was barely affected, cell-to-cell spread of several of the PrV mutants was significantly impaired (Fig. 10). Plaque sizes of PrV-gHN77Q and PrV-gHN627Q were reduced by approximately 30% and 50%, respectively, while plaque sizes of the triple, quadruple, and quintuple mutants including both amino acid substitutions were further reduced by 70% to 85% compared to that of wild-type PrV. Consistently, mutations N77Q and N627Q led to reduced fusion activity in transfected cells, and simultaneous removal of both sites decreased fusion activity even further (Fig. 5B). N77 is located in the structurally uncharacterized gH domain I, which is important for binding of gL (30). Removal of the carbohydrates from this site might affect optimal interaction of gH with gL and thus interfere with efficient membrane fusion. However, Western blot analysis of purified pPrV-gHN77Q virions showed that gL was efficiently incorporated into particles (Fig. 6A), demonstrating that gH/gL binding per se is not disturbed.
N627 is located within a highly conserved hydrophobic patch of domain IV which was suggested to play a role in membrane fusion by temporally regulated interaction with the viral envelope (20, 33). In line with our previous results (33), N-glycosylation of gH at position 627 was found to be important, although not essential, for PrV-mediated membrane fusion. The homologous glycosylation site in HSV-1 gH was also shown to be nonessential for its function (56). Thus, in the absence of the carbohydrates, the underlying hydrophobic patch might still be sufficiently masked by the charged “flap” to prevent its premature movement into the membrane. However, as outlined above, inactivation of the glycosylation site at position N627 severely affected gH transport and surface expression, which might also contribute to the observed spreading defects of the virus mutants. However, we cannot completely exclude that the mutation of the amino acid rather than the absence of the glycan is relevant for the observed phenotype.
Whereas mutations N77Q and N627Q led to a decrease in plaque sizes, an increase in plaque size by 30% was observed after substitution of N604, and, consistent with this, the in vitro fusion activity of gHN604Q was enhanced (Fig. 5). The N-glycosylation site N604 is located in gH domain IV (Fig. 1) and is conserved between members of the Varicellovirus genus, whereas members of the Simplexvirus genus lack a corresponding site (20). Previous studies using chimeric proteins consisting of domains of PrV and the simplexvirus HSV-1 gH demonstrated functional conservation of domain IV and indicated species-specific interactions of this domain with gB (34). Possibly, the absence of the carbohydrate at aa 604 positively influences this interaction between gH and gB, resulting in the observed phenotype.
In line with the observed defects in cell-to-cell spread and in vitro fusion activity, penetration of the triple, quadruple, and quintuple PrV mutants was significantly delayed (Fig. 8). pPrV-gHN627Q showed a less pronounced but also significant lag in penetration, whereas penetration of the single mutants with substitutions of N77, N162, N542, and N604 was not or only marginally affected. Although gHN604Q resulted in a significantly increased fusogenicity and plaque size, it exhibited a slight delay in penetration, again indicating that the mechanisms of membrane fusion during entry of free virus particles and direct viral cell-to-cell spread are similar but not identical. This is also highlighted by the different protein requirements for the two membrane fusion events. Whereas PrV gD is dispensable for plaque formation, it is required for entry, functionally separating the two events (50, 57, 58).
In summary, our results demonstrate that all five predicted N-glycosylation sites in PrV gH are modified by N-glycans, which play a modulating role in proper localization and function of PrV gH during membrane fusion. However, even simultaneous inactivation of all five sites did not severely inhibit formation of infectious virus particles, demonstrating that modification of gH by N-glycans is not necessary for efficient PrV replication. Although in vitro replication of the mutants was not severely affected, the relevance of the N-glycosylation sites in vivo remains to be determined. N-linked glycosylation of viral proteins expressed on the surface of virions may interfere with the host antibody response (59). For influenza and human immunodeficiency viruses, it is well documented that glycan modifications can serve as a “shield” which covers essential viral epitopes, thereby protecting the viruses from antibody-mediated neutralization (60, 61). The same might apply to PrV gH, but we cannot exclude that the N-glycans in PrV gH represent parts of epitopes, which can be targeted by neutralizing antibodies.
MATERIALS AND METHODS
Cells and viruses.
Rabbit kidney (RK13) cells were grown in Eagle's minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) at 37°C and 5% CO2. The viruses described in this study were derived from pPrV-ΔgHABF (31), which is a gH deletion mutant of PrV strain Kaplan (PrV-Ka) cloned as a bacterial artificial chromosome (BAC) (33). The gH-rescued viruses were propagated on RK13 cells, while pPrV-ΔgHABF was cultivated on RK13-gH/gL (62).
Mutagenesis of the cloned gH gene and generation of PrV N-glycosylation mutants.
The expression plasmid pcDNA-gHKDE (31) containing the gH ORF (UL22) of PrV-Ka was used for site-directed mutagenesis (QuikChange II XL kit; Agilent). The oligonucleotide primers used for mutagenesis leading to inactivation of the potential N-glycosylation sites are indicated in Table 1. The resulting expression plasmids were digested with DrdI, and a 3,461-bp fragment containing the modified gH gene together with a downstream kanamycin resistance (Kanr) gene was purified from an agarose gel and used for Red-mediated mutagenesis of pPrV-ΔgH in E. coli as described previously (30, 31, 33). Desired BAC recombinants were selected on LB agar plates supplemented with kanamycin (50 μg/ml) and chloramphenicol (30 μg/ml). DNA was prepared from overnight liquid cultures of single bacterial colonies by alkaline lysis, phenol-chloroform extraction, isopropanol precipitation, and RNase treatment. BAC DNA was used for transfection (FuGene HD transfection reagent; Promega) of RK13 cells, and progeny virus isolated from single plaques was propagated and characterized. Correct mutagenesis and recombination were verified by sequencing using the primers listed in Table 1, the BigDye Terminator v1.1 cycle sequencing kit, and a 3130 genetic analyzer (Applied Biosystems).
TABLE 1.
Oligonucleotide primers for mutagenesis, PCR, and sequence analysesa
| Primer name | Sequence (5′ to 3′)b | Nucleotide positionsc |
|---|---|---|
| PGHN77Q-F | CTGGGGGCGCTCCAGGACACGCGCATC | 60986–61012 |
| PGHN162Q-F | GCGGCCGTCTTCCAGGTGACGCTGGGC | 61241–61267 |
| PGHN542Q-F | GCCATCGTCAGCCAGGACAGCGCCGCG | 62381–62407 |
| PGHN604Q-F | CATGGCCGGCGCCCAGTCCACCATCCC | 62566–62592 |
| PGHN627Q-F | TGATGCTCTTCCCCCAAGGCACCGTGGTC | 62634–62662 |
| PgH-PSF | TTCACGTCGGAGATGGGG | 60611–60628 |
| PgH-PSF2 | GGAAGCCCTTCGACCAG | 61875–61891 |
| PgHK-PSR | CGCGAGCCCATTTATACCC | (Kanr) |
| PgH-PSR2 | GTCGAGCAGGCTGAAGG | 62055–62071 (r) |
| PgH-WH3 | TGCACGAGAGCGACGACTACC | 61479–61499 |
Only forward-strand mutagenesis primers (F) are listed, since the reverse-strand primers were exactly complementary.
Nonmatching nucleotides are shown in boldface.
IIF analysis.
For IIF analyses, RK13 cell monolayers grown in 24-well plates were cotransfected with 200 ng of wild-type or mutant gH and gL expression plasmids in 50 μl Opti-MEM using 1 μl Lipofectamine 2000 (Thermo Fisher Scientific). For colocalization studies, cells were additionally transfected with 200 μl of the ER marker pmTurquoise2ER (Addgene). The transfection mixture was incubated for 20 min at room temperature and added to the cells. After 3 h at 37°C, the cells were washed with phosphate-buffered saline (PBS) and incubated in MEM supplemented with 2% FBS for another 24 h at 37°C. The cells were fixed with 3% paraformaldehyde (PFA) in PBS for 20 min and, optionally, permeabilized in PBS containing 0.1% Triton X-100 for 10 min at room temperature. Afterwards, cells were washed with PBS, blocked with 0.25% fat-free powdered milk in PBS, and incubated with the rabbit antiserum specific for PrV gH (28) at a dilution of 1:200 in PBS. After 1 h at room temperature, bound antibody was detected with Alexa 488- or 633-conjugated goat anti-rabbit antibodies (Invitrogen) at a dilution of 1:1,000 in PBS. After each step, cells were washed repeatedly with PBS. Green fluorescence was excited at 488 nm and red fluorescence at 633 nm, and the cells were analyzed with an Eclipse Ti-S fluorescence microscope (Nikon) or with a laser scanning confocal microscope (SP5; Leica, Mannheim, Germany).
Flow cytometry.
RK13 cells cotransfected with gH and gL expression plasmids were detached after 24 h with 2 mM EDTA in PBS for 1 h at 4°C. Thereafter, cells were fixed with 4% PFA for 20 min at 4°C and subsequently washed with PBS supplemented with 2% bovine serum albumin (BSA) and 2 mM EDTA. Each sample was split into two FACS tubes. One aliquot was permeabilized with 0.5% saponin in PBS with 0.2% BSA for 30 min at 4°C, while the second was incubated without saponin. Cells were probed with the monoclonal antibody 13c2 directed against gH (28) at a dilution of 1:5 in PBS with 2% BSA for 1 h at 4°C. After a washing step with 2 mM EDTA in PBS, cells were incubated for 1 h at 4°C with a secondary antibody (Alexa Fluor 488, goat anti-mouse IgG; Invitrogen) at a dilution of 1:1,000 in PBS with 2% BSA. Samples were analyzed using a MACSQuant analyzer (Miltenyi Biotec). Surface expression of gH was quantified as follows: percent fluorescein isothiocyanate (FITC)-positive, nonpermeabilized cells/percent FITC-positive permeabilized cells × 100. Mean values and standard deviations from three independent assays were determined.
Western blot analyses.
RK13 cells were harvested 2 days after PrV infection at an MOI of 2, and virions were purified from culture supernatants as described previously (25). Protein samples (5 μg protein per lane) were separated by discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to nitrocellulose membranes, and incubated with antibodies (63). Monospecific rabbit antisera against PrV gD (62), gH (28), and gL (25) were used at a dilution of 1:1,000 (gL) or 1:10,000 (gD and gH).
Enzymatic deglycosylation.
Purified PrV particles were incubated in 1% SDS and 10% β-mercaptoethanol for 10 min at 100°C. The pretreated purified virion preparations (15 μg each) were digested either with 500 U of endo H to remove high-mannose or hybrid forms of N-linked glycans, with 500 U of PNGase F to remove all N-linked glycans, or without enzymes for 2 h at 37°C under buffer conditions according to the manufacturer's (New England BioLabs) instructions. After digestion, samples (5 μg/lane) were separated by SDS-PAGE and analyzed by Western blotting.
In vitro fusion assays.
The fusogenic properties of the gH mutants were analyzed using a transient-transfection-based cell fusion assay (30, 51). Briefly, approximately 3 × 105 RK13 cells per well were seeded onto 12-well cell culture plates and after 20 h at 37°C transfected with 400 ng each of expression plasmids for EGFP (pEGFP-N1; Clontech) and PrV-Ka glycoproteins gB (or C-terminally truncated gB-008), gD, gL, and wild-type or mutagenized gH (29, 31, 50, 52) in 100 μl Opti-MEM using 2 μl Lipofectamine 2000 as described above. The empty expression vector (pcDNA3; Thermo Fisher Scientific) served as a negative control. After 24 h at 37°C, the cells were washed with PBS and fixed with 3% PFA for 20 min. An Eclipse Ti-S fluorescence microscope and the NIS-Elements imaging software (Nikon) were used to analyze syncytium formation. Total fusion activity was determined by multiplication of the area of syncytia with three or more nuclei by the number of syncytia counted within 10 fields of view (5.5 mm2 each). Mean values and standard deviations from four independent assays were determined.
In vitro replication studies.
For analysis of growth kinetics, confluent monolayers of RK13 cells in 96-well plates were infected with the wild-type gH revertant pPrV-gHK (31) or the different PrV gH mutants at an MOI of 0.01 and were subsequently incubated for 1 h on ice to permit virus adsorption. After 2 h at 37°C, the inoculum was removed and nonpenetrated virus was inactivated by low-pH treatment (64). The cells were then washed with PBS, and fresh medium was added. Immediately thereafter and after 6, 12, 24, 48, and 72 h at 37°C, cells were harvested together with the supernatants and lysed by freeze-thawing (−70°C and 37°C). Progeny virus titers were determined by plaque assays on RK13 cells. After 48 h at 37°C under semisolid medium containing 6 g/liter methylcellulose, areas of 30 plaques were measured microscopically, and percentages of wild-type (pPrV-gHK) sizes were calculated. Mean results and standard deviations from four independent growth kinetic studies and comparative plaque assays were determined.
In vitro penetration kinetics.
For determination of penetration kinetics, confluent monolayers of RK13 cells in in 6-well plates were infected on ice with approximately 250 PFU of PrV-gHK or the different PrV gH mutants. After 1 h, the inoculum was replaced by prewarmed MEM supplemented with 5% FBS, and cells were incubated at 37°C. Before and 5, 10, 15, 30, 60, and 120 min after the temperature shift, remaining extracellular virus particles were inactivated by low-pH treatment. Cells were washed two times with PBS and overlaid with semisolid medium containing 6 g/liter methylcellulose. For 100% penetration controls, infected cells were washed with PBS only after 2 h at 37°C and overlaid with semisolid medium. After 48 h at 37°C, plaques were counted, and the penetration rate was calculated by comparison with corresponding controls. The experiment was repeated four times, and mean values and standard deviations were determined.
Statistical analyses.
The statistical significance of differences observed in FACS analyses, transient fusion assays, and in vitro replication studies was evaluated using an unpaired t test with Welch correction provided by GraphPad Prism 7 software (GraphPad Software, Inc., San Diego, CA).
ACKNOWLEDGMENTS
These studies were supported by the Deutsche Forschungsgemeinschaft (DFG grant Me 854/11-2). Molecular graphics were obtained with the UCSF Chimera package; Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, supported by the National Institutes of Health (NIGMS P41-GM103311).
We thank B. Wanner for providing plasmids for BAC mutagenesis and G. Strebelow for performing sequence analyses. The technical assistance of A. Landmesser and K. Biebl is greatly appreciated.
REFERENCES
- 1.Harrison SC. 2015. Viral membrane fusion. Virology 479-480:498–507. doi: 10.1016/j.virol.2015.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. 2012. Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Di Giovine P, Settembre EC, Bhargava AK, Luftig MA, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C, Carfi A. 2011. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog 7:e1002277. doi: 10.1371/journal.ppat.1002277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazear E, Whitbeck JC, Zuo Y, Carfi A, Cohen GH, Eisenberg RJ, Krummenacher C. 2014. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 448:185–195. doi: 10.1016/j.virol.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J Virol 84:12292–12299. doi: 10.1128/JVI.01700-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gianni T, Amasio M, Campadelli-Fiume G. 2009. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J Biol Chem 284:17370–17382. doi: 10.1074/jbc.M109.005728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17:882–888. doi: 10.1038/nsmb.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper RS, Heldwein EE. 2015. Herpesvirus gB: a finely tuned fusion machine. Viruses 7:6552–6569. doi: 10.3390/v7122957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84:3825–3834. doi: 10.1128/JVI.02687-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vallbracht M, Brun D, Tassinari M, Vaney MC, Pehau-Arnaudet G, Guardado-Calvo P, Haouz A, Klupp BG, Mettenleiter TC, Rey FA, Backovic M. 18 October 2017. Structure-function dissection of the pseudorabies virus glycoprotein B fusion loops. J Virol doi: 10.1128/JVI.01203-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Yang F, Hu X, Tan F, Qi J, Peng R, Wang M, Chai Y, Hao L, Deng J, Bai C, Wang J, Song H, Tan S, Lu G, Gao GF, Shi Y, Tian K. 2017. Two classes of protective antibodies against pseudorabies virus variant glycoprotein B: implications for vaccine design. PLoS Pathog 13:e1006777. doi: 10.1371/journal.ppat.1006777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. doi: 10.1126/science.1126548. [DOI] [PubMed] [Google Scholar]
- 13.Chandramouli S, Ciferri C, Nikitin PA, Calo S, Gerrein R, Balabanis K, Monroe J, Hebner C, Lilja AE, Settembre EC, Carfi A. 2015. Structure of HCMV glycoprotein B in the postfusion conformation bound to a neutralizing human antibody. Nat Commun 6:8176. doi: 10.1038/ncomms9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burke HG, Heldwein EE. 2015. Crystal structure of the human cytomegalovirus glycoprotein B. PLoS Pathog 11:e1005227. doi: 10.1371/journal.ppat.1005227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc Natl Acad Sci U S A 106:2880–2885. doi: 10.1073/pnas.0810530106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. doi: 10.1126/science.1127683. [DOI] [PubMed] [Google Scholar]
- 17.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat Struct Mol Biol 15:1024–1030. doi: 10.1038/nsmb.1484. [DOI] [PubMed] [Google Scholar]
- 18.Matsuura H, Kirschner AN, Longnecker R, Jardetzky TS. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc Natl Acad Sci U S A 107:22641–22646. doi: 10.1073/pnas.1011806108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xing Y, Oliver SL, Nguyen T, Ciferri C, Nandi A, Hickman J, Giovani C, Yang E, Palladino G, Grose C, Uematsu Y, Lilja AE, Arvin AM, Carfi A. 2015. A site of varicella-zoster virus vulnerability identified by structural studies of neutralizing antibodies bound to the glycoprotein complex gHgL. Proc Natl Acad Sci U S A 112:6056–6061. doi: 10.1073/pnas.1501176112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Backovic M, DuBois RM, Cockburn JJ, Sharff AJ, Vaney MC, Granzow H, Klupp BG, Bricogne G, Mettenleiter TC, Rey FA. 2010. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc Natl Acad Sci U S A 107:22635–22640. doi: 10.1073/pnas.1011507107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutchinson L, Browne H, Wargent V, Davis-Poynter N, Primorac S, Goldsmith K, Minson AC, Johnson DC. 1992. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J Virol 66:2240–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaye JF, Gompels UA, Minson AC. 1992. Glycoprotein H of human cytomegalovirus (HCMV) forms a stable complex with the HCMV UL115 gene product. J Gen Virol 73:2693–2698. doi: 10.1099/0022-1317-73-10-2693. [DOI] [PubMed] [Google Scholar]
- 23.Klupp BG, Baumeister J, Karger A, Visser N, Mettenleiter TC. 1994. Identification and characterization of a novel structural glycoprotein in pseudorabies virus, gL. J Virol 68:3868–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu DX, Gompels UA, Nicholas J, Lelliott C. 1993. Identification and expression of the human herpesvirus 6 glycoprotein H and interaction with an accessory 40K glycoprotein. J Gen Virol 74:1847–1857. doi: 10.1099/0022-1317-74-9-1847. [DOI] [PubMed] [Google Scholar]
- 25.Klupp BG, Fuchs W, Weiland E, Mettenleiter TC. 1997. Pseudorabies virus glycoprotein L is necessary for virus infectivity but dispensable for virion localization of glycoprotein H. J Virol 71:7687–7695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lete C, Machiels B, Stevenson PG, Vanderplasschen A, Gillet L. 2012. Bovine herpesvirus type 4 glycoprotein L is nonessential for infectivity but triggers virion endocytosis during entry. J Virol 86:2653–2664. doi: 10.1128/JVI.06238-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gillet L, May JS, Colaco S, Stevenson PG. 2007. Glycoprotein L disruption reveals two functional forms of the murine gammaherpesvirus 68 glycoprotein H. J Virol 81:280–291. doi: 10.1128/JVI.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klupp BG, Mettenleiter TC. 1999. Glycoprotein gL-independent infectivity of pseudorabies virus is mediated by a gD-gH fusion protein. J Virol 73:3014–3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schröter C, Vallbracht M, Altenschmidt J, Kargoll S, Fuchs W, Klupp BG, Mettenleiter TC. 2015. Mutations in pseudorabies virus glycoproteins gB, gD, and gH functionally compensate for the absence of gL. J Virol 90:2264–2272. doi: 10.1128/JVI.02739-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vallbracht M, Rehwaldt S, Klupp BG, Mettenleiter TC, Fuchs W. 2017. Functional relevance of the N-terminal domain of pseudorabies virus envelope glycoprotein H and its interaction with glycoprotein L. J Virol 91:e00061-. doi: 10.1128/JVI.00061-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Böhm SW, Eckroth E, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2015. Structure-based functional analyses of domains II and III of pseudorabies virus glycoprotein H. J Virol 89:1364–1376. doi: 10.1128/JVI.02765-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schröter C, Klupp BG, Fuchs W, Gerhard M, Backovic M, Rey FA, Mettenleiter TC. 2014. The highly conserved proline at position 438 in pseudorabies virus gH is important for regulation of membrane fusion. J Virol 88:13064–13072. doi: 10.1128/JVI.01204-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs W, Backovic M, Klupp BG, Rey FA, Mettenleiter TC. 2012. Structure-based mutational analysis of the highly conserved domain IV of glycoprotein H of pseudorabies virus. J Virol 86:8002–8013. doi: 10.1128/JVI.00690-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Böhm SW, Backovic M, Klupp BG, Rey FA, Mettenleiter TC, Fuchs W. 2016. Functional characterization of glycoprotein H chimeras composed of conserved domains of the pseudorabies virus and herpes simplex virus 1 homologs. J Virol 90:421–432. doi: 10.1128/JVI.01985-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cherepanova N, Shrimal S, Gilmore R. 2016. N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr Opin Cell Biol 41:57–65. doi: 10.1016/j.ceb.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kornfeld R, Kornfeld S. 1985. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem 54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 37.Maley F, Trimble RB, Tarentino AL, Plummer TH Jr. 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 180:195–204. doi: 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 38.Helenius A, Aebi M. 2004. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 39.Helle F, Vieyres G, Elkrief L, Popescu CI, Wychowski C, Descamps V, Castelain S, Roingeard P, Duverlie G, Dubuisson J. 2010. Role of N-linked glycans in the functions of hepatitis C virus envelope proteins incorporated into infectious virions. J Virol 84:11905–11915. doi: 10.1128/JVI.01548-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lennemann NJ, Walkner M, Berkebile AR, Patel N, Maury W. 2015. The role of conserved N-linked glycans on Ebola virus glycoprotein 2. J Infect Dis 212(Suppl 2):S204–S209. doi: 10.1093/infdis/jiv201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo S, Hu K, He S, Wang P, Zhang M, Huang X, Du T, Zheng C, Liu Y, Hu Q. 2015. Contribution of N-linked glycans on HSV-2 gB to cell-cell fusion and viral entry. Virology 483:72–82. doi: 10.1016/j.virol.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi Y, Suzuki Y. 2012. Evidence for N-glycan shielding of antigenic sites during evolution of human influenza A virus hemagglutinin. J Virol 86:3446–3451. doi: 10.1128/JVI.06147-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald TP, Jeffree CE, Li P, Rixon HW, Brown G, Aitken JD, MacLellan K, Sugrue RJ. 2006. Evidence that maturation of the N-linked glycans of the respiratory syncytial virus (RSV) glycoproteins is required for virus-mediated cell fusion: the effect of alpha-mannosidase inhibitors on RSV infectivity. Virology 350:289–301. doi: 10.1016/j.virol.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 44.Annamalai AS, Pattnaik A, Sahoo BR, Muthukrishnan E, Natarajan SK, Steffen D, Vu HLX, Delhon G, Osorio FA, Petro TM, Xiang SH, Pattnaik AK. 20 September 2017. Zika virus encoding nonglycosylated envelope protein is attenuated and defective in neuroinvasion. J Virol doi: 10.1128/JVI.01348-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bradel-Tretheway BG, Liu Q, Stone JA, McInally S, Aguilar HC. 2015. Novel functions of Hendra virus G N-glycans and comparisons to Nipah virus. J Virol 89:7235–7247. doi: 10.1128/JVI.00773-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sodora DL, Cohen GH, Eisenberg RJ. 1989. Influence of asparagine-linked oligosaccharides on antigenicity, processing, and cell surface expression of herpes simplex virus type 1 glycoprotein D. J Virol 63:5184–5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sodora DL, Cohen GH, Muggeridge MI, Eisenberg RJ. 1991. Absence of asparagine-linked oligosaccharides from glycoprotein D of herpes simplex virus type 1 results in a structurally altered but biologically active protein. J Virol 65:4424–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sodora DL, Eisenberg RJ, Cohen GH. 1991. Characterization of a recombinant herpes simplex virus which expresses a glycoprotein D lacking asparagine-linked oligosaccharides. J Virol 65:4432–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klupp BG, Visser N, Mettenleiter TC. 1992. Identification and characterization of pseudorabies virus glycoprotein H. J Virol 66:3048–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klupp BG, Nixdorf R, Mettenleiter TC. 2000. Pseudorabies virus glycoprotein M inhibits membrane fusion. J Virol 74:6760–6768. doi: 10.1128/JVI.74.15.6760-6768.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vallbracht M, Schröter C, Klupp BG, Mettenleiter TC. 2017. Transient transfection-based fusion assay for viral proteins. Bio-protocol 7:e2162. doi: 10.21769/BioProtoc.2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nixdorf R, Klupp BG, Karger A, Mettenleiter TC. 2000. Effects of truncation of the carboxy terminus of pseudorabies virus glycoprotein B on infectivity. J Virol 74:7137–7145. doi: 10.1128/JVI.74.15.7137-7145.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanley PSH, Taniguchi N. 2009. N-glycans, essentials of glycobiology. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 54.Bieberich E. 2014. Synthesis, processing, and function of N-glycans in N-glycoproteins. Adv Neurobiol 9:47–70. doi: 10.1007/978-1-4939-1154-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mettenleiter TC. 2006. Intriguing interplay between viral proteins during herpesvirus assembly or: the herpesvirus assembly puzzle. Vet Microbiol 113:163–169. doi: 10.1016/j.vetmic.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 56.Galdiero M, Whiteley A, Bruun B, Bell S, Minson T, Browne H. 1997. Site-directed and linker insertion mutagenesis of herpes simplex virus type 1 glycoprotein H. J Virol 71:2163–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rauh I, Mettenleiter TC. 1991. Pseudorabies virus glycoproteins gII and gp50 are essential for virus penetration. J Virol 65:5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peeters B, de Wind N, Hooisma M, Wagenaar F, Gielkens A, Moormann R. 1992. Pseudorabies virus envelope glycoproteins gp50 and gII are essential for virus penetration, but only gII is involved in membrane fusion. J Virol 66:894–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vigerust DJ, Shepherd VL. 2007. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol 15:211–218. doi: 10.1016/j.tim.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wanzeck K, Boyd KL, McCullers JA. 2011. Glycan shielding of the influenza virus hemagglutinin contributes to immunopathology in mice. Am J Respir Crit Care Med 183:767–773. doi: 10.1164/rccm.201007-1184OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, Komarova NL, Nowak MA, Hahn BH, Kwong PD, Shaw GM. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 62.Klupp B, Altenschmidt J, Granzow H, Fuchs W, Mettenleiter TC. 2008. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J Virol 82:6299–6309. doi: 10.1128/JVI.00386-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pavlova SP, Veits J, Keil GM, Mettenleiter TC, Fuchs W. 2009. Protection of chickens against H5N1 highly pathogenic avian influenza virus infection by live vaccination with infectious laryngotracheitis virus recombinants expressing H5 hemagglutinin and N1 neuraminidase. Vaccine 27:773–785. doi: 10.1016/j.vaccine.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 64.Mettenleiter TC. 1989. Glycoprotein gIII deletion mutants of pseudorabies virus are impaired in virus entry. Virology 171:623–625. doi: 10.1016/0042-6822(89)90635-1. [DOI] [PubMed] [Google Scholar]
- 65.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 66.Grimm KS, Klupp BG, Granzow H, Muller FM, Fuchs W, Mettenleiter TC. 2012. Analysis of viral and cellular factors influencing herpesvirus-induced nuclear envelope breakdown. J Virol 86:6512–6521. doi: 10.1128/JVI.00068-12. [DOI] [PMC free article] [PubMed] [Google Scholar]