

ABSTRACT
Serotype 3 (T3) reoviruses induce substantially more type 1 interferon (IFN-I) secretion than serotype 1 (T1) strains. However, the mechanisms underlying differences in IFN-I production between T1 and T3 reoviruses remain undefined. Here, we found that differences in IFN-I production between T1 and T3 reoviruses correlate with activation of interferon regulatory factor 3 (IRF3), a key transcription factor for the production of IFN-I. T3 strain rsT3D activated IRF3 more rapidly and to a greater extent than the T1 strain rsT1L, in simian virus 40 (SV40) immortalized endothelial cells (SVECs). Differences in IRF3 activation between rsT1L and rsT3D were observed in the first hours of infection and were independent of de novo viral RNA and protein synthesis. NF-κB activation mirrored IRF3 activation, with rsT3D inducing more NF-κB activity than rsT1L. We also found that IRF3 and NF-κB are activated in a mitochondrial antiviral-signaling protein (MAVS)-dependent manner. rsT1L does not suppress IRF3 activation, as IRF3 phosphorylation could be induced in rsT1L-infected cells. Transfected rsT1L and rsT3D RNA induced IRF3 phosphorylation, indicating that genomic RNA from both strains has the capacity to activate IRF3. Finally, bypassing the normal route of reovirus entry by transfecting in vitro-generated viral cores revealed that rsT1L and rsT3D core particles induced equivalent IRF3 activation. Taken together, our findings indicate that entry-related events that occur after outer capsid disassembly, but prior to deposition of viral cores into the cytoplasm, influence the efficiency of IFN-I responses to reovirus. This work provides further insight into mechanisms by which nonenveloped viruses activate innate immune responses.
IMPORTANCE Detection of viral nucleic acids by the host cell triggers type 1 interferon (IFN-I) responses, which are critical for containing and clearing viral infections. Viral RNA is sensed in the cytoplasm by cellular receptors that initiate signaling pathways, leading to the activation of interferon regulatory factor 3 (IRF3) and NF-κB, key transcription factors required for IFN-I induction. Serotype 3 (T3) reoviruses induce significantly more IFN-I than serotype 1 (T1) strains. In this work, we found that differences in IFN-I production by T1 and T3 reoviruses correlate with differential IRF3 activation. Differences in IRF3 activation are not caused by a blockade of the IRF3 activation by a T1 strain. Rather, differences in events during the late stages of viral entry determine the capacity of reovirus to activate host IFN-I responses. Together, our work provides insight into mechanisms of IFN-I induction by nonenveloped viruses.
KEYWORDS: IRF3, type 1 interferon, reovirus, double-stranded RNA virus, recombinant virus
INTRODUCTION
The type 1 interferon (IFN-I) response is a critical component of innate immunity that functions to limit viral replication and initiate adaptive responses that clear viral infections (1). IFN-I responses are initiated by the detection of viral nucleic acids by cellular pattern recognition receptors (PRRs) (2). PRRs for RNA viruses include the cytosolic retinoic acid inducible gene I (RIG-I)-like receptors (RLRs) and membrane-bound Toll-like receptors (TLRs) (2). RLRs detect viral RNA in the cytoplasm, whereas TLRs detect viral RNA in endosomal compartments (3). Detection of viral RNA by RLRs or TLRs activates signaling pathways that culminate in the phosphorylation and activation of interferon regulatory factor 3 (IRF3), a transcription factor that controls IFN-I production (3, 4). The RLRs RIG-I and melanoma differentiation-associated protein 5 (MDA5) recruit the adaptor molecule mitochondrial antiviral-signaling protein (MAVS) (4), which activates TANK-binding kinase 1 (TBK1), which phosphorylates and activates IRF3 (1). TLR3 detects double-stranded RNA (dsRNA) in the endosome and signals through Toll-like receptor adaptor molecule 1 (TICAM-1/TRIF) and TBK-1, leading to IRF3 activation (1). RLR and TLR signaling also activates NF-κB, which is required for IFN-I production (3). NF-κB is held inactive in the cytoplasm by inhibitor of kappa B (IκB) proteins (3). RLR signaling through MAVS leads to the activation of IκB kinases α and β (IKKα/β), which phosphorylate IκBα, causing it to be degraded by the proteasome, thereby freeing NF-κB (p65-p50 heterodimers) to translocate to the nucleus (3). Activated IRF3 also translocates to the nucleus, where it coordinates with NF-κB and AP-1 to induce expression and secretion of IFN-I (1). Secreted IFN-I binds to the interferon alpha (IFN-α) receptor (IFNAR) in an autocrine and paracrine manner to trigger Janus-activated kinases (JAKs), JAK-1 and tyrosine kinase 2 (TYK-2), which phosphorylate signal transducer and activator of transcription 1 (STAT1) and 2 (STAT2) (1). Phosphorylated STAT1 and STAT2 dimerize and associate with interferon regulatory factor 9 (IRF9) to form the interferon-stimulated gene factor 3 (ISGF3) transcription factor complex, which translocates to the nucleus to bind interferon-stimulated response elements (ISREs) and induce expression of numerous interferon-stimulated genes (ISGs) (1). ISGs perform a variety of effector functions, such as inhibition of cellular translation and activation of RNases, to produce an antiviral state that restricts viral replication and programs adaptive responses that mediate viral clearance (1).
Mammalian orthoreoviruses (reoviruses) are nonenveloped viruses with segmented dsRNA genomes (5). Reovirus virions are composed of two protein shells, the outer capsid and the inner core, which encapsulate 10 segments of viral genomic dsRNA. Reoviruses spread by oral or respiratory routes, and most people are infected by at least one of three reovirus serotypes during childhood (6). Although reovirus infection of humans is largely asymptomatic, recent evidence suggests that reovirus has the capacity to break oral immune tolerance, which can lead to celiac disease (7). In neonatal mice, reoviruses exhibit serotype-specific patterns of dissemination and disease (5). Following oral or nasal inoculation, reoviruses disseminate from the intestine or lung to every organ system in the body, including the central nervous system (CNS) (5). Serotype 1 (T1) reoviruses spread to the CNS by hematogenous routes and infect ependymal cells, causing nonlethal hydrocephalus (5). In contrast, serotype 3 (T3) reoviruses disseminate to the CNS by hematogenous and neural routes, infect neurons, and cause lethal encephalitis (5).
Like other RNA viruses, reoviruses potently induce IFN-I in cell culture and in vivo (8, 9). IFN-I is critical for the control of reovirus infection in mouse models of infection. Although adult mice are normally resistant to reovirus disease, mice lacking IFN-α receptor 1 (IFNAR1) succumb to reovirus infection (10–12). In addition to serotype-specific differences in routes of viral dissemination and CNS cell tropism and disease, T1 and T3 reoviruses differ in the induction of, and sensitivity to, IFN-I (8, 14). T3 reoviruses induce more IFN-I than T1 reoviruses (8). Although T1 viruses elicit less IFN-I than T3 strains, T1 reoviruses are more resistant to the effects of IFN-I, at least in cultured cells (14). In vivo, however, virulence of the T3 strain T3SA+ is minimally affected in mice lacking IRF3 (15). In contrast, T1L kills IRF3-deficient mice but not wild-type animals (15). These findings indicate that in vivo, IFN-I responses may be more important for control of T1 reoviruses than for that of T3 strains. Thus, T1 strains may minimize IFN-I induction as a means of protection from the antiviral effects of IFN-I. Despite the dramatic differences in IFN-I induction between T1 and T3 reoviruses, the mechanisms that dictate differences in IFN-I induction between reovirus serotypes remain undefined.
In this study, we characterized the IFN-I response to T1 and T3 reovirus in simian virus 40 (SV40) immortalized endothelial cells (SVECs), a mouse endothelial cell line. Endothelial cells are critical targets for reovirus infection in mice and are a key cell type for reovirus dissemination via the blood (16, 17). We found that similar to other cell types, the T3 reovirus rsT3D induced substantially more IFN-β secretion from SVECs that the T1 strain rsT1L. IRF3 and NF-κB activation by the two strains correlated with the level of IFN-β production, as rsT3D activated IRF3 and NF-κB to a far greater extent than rsT1L. Minimal IRF3 activation by rsT1L does not result from impairment of IRF3 induction by rsT1L. Rather, our data indicate that detection of incoming viral genomic RNA differs between rsT1L and rsT3D. Our work further reveals that late steps in reovirus entry mediate serotype-specific differences in reovirus IFN-I induction. Together, these results suggest that slight differences in entry events can lead to significant differences in innate immune detection and response, which may alter pathogenesis.
RESULTS
rsT3D induces more IFN-β than rsT1L in SVECs.
To assess reovirus-mediated IFN-I responses in endothelial cells, we measured IFN-β production from SVECs following infection with rsT1L or rsT3D. Infection with rsT3D induced IFN-β with more rapid kinetics and to higher levels than rsT1L (Fig. 1A). These findings are consistent with studies in other cells, including cardiac myocytes, HeLa cells, and L929 cells (8, 12). Also consistent with other cell types, we found that rsT1L and rsT3D produced comparable viral progeny yields from SVECs despite differences in IFN-I production (Fig. 1B). These findings indicate that reoviruses have mechanisms that enable replication in the presence of IFN-I responses.
FIG 1.

rsT3D more potently induces IFN-β than rsT1L in SVECs. (A) SVECs were mock infected or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. At 4, 8, 12, and 24 h, IFN-β levels in the cell culture supernatant were measured by IFN-β-specific ELISA. Results are presented as the mean of duplicate samples ± standard deviation (SD). *, P < 0.05; ****, P < 0.0001 (as determined by two-way analysis of variance [ANOVA]). (B) SVECs were infected with rsT1L or rsT3D at an MOI of 1 PFU/cell, and viral titers were quantified at 0, 24, 48, and 72 h on L929 cells. Data are presented as mean viral yields for triplicate samples from three independent experiments ± SD. (C) SVECs were mock infected (M), treated with purified IFN-β (IFN) (200 U/ml), or infected with rsT1L (T1) or rsT3D (T3) at an MOI of 100 PFU/cell. At 2, 4, 6, and 8 h postinfection (hpi), whole-cell lysates were prepared and proteins were separated by SDS-PAGE. Immunoblot analysis was performed for phosphorylated IRF3 (p-IRF3), total IRF3, phosphorylated STAT1 (p-STAT1), total STAT1, phosphorylated STAT2 (p-STAT2), total STAT2, or β-actin. (D and E) SVECs were mock infected or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. At 8 (D) and 24 (E) h, Oas1b and IFIT1 mRNA levels were quantified by RT-qPCR. Results are presented as the mean of triplicate samples from two independent experiments ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (as determined by Student's t test).
Differences in IFN-β secretion correlated with differences in IFNAR signaling and ISG expression. In comparison to rsT1L-infected cells, rsT3D induced higher levels of phosphorylated STAT1 and STAT2 (Fig. 1C). At 8 h, rsT3D induced markedly higher levels of Oas1b and IFIT1 than those induced by rsT1L (Fig. 1D). By 24 h, ISG transcript levels had normalized between rsT1L- and rsT3D-infected cells, although OAS1b levels remained higher for rsT3D than rsT1L (Fig. 1E). These results indicate that IFN-β produced from SVECs in response to reovirus infection is biologically active. Further, although rsT3D induces high levels of ISGs at 8 h, ISG levels are reduced by 24 h. It is unclear whether rsT3D actively represses ISG induction or if reduced ISG levels are due to intrinsic down-modulation of the IFN-I response associated with prolonged IFN-I exposure (18). The reduction in ISGs at late times could account for the observation that rsT1L and rsT3D replicate comparably in SVECs (Fig. 1B). We also noted that rsT3D induced substantially more phosphorylation of IRF3 on Ser396 than rsT1L. Phosphorylation of IRF3 on Ser396 is a marker for transcriptionally active IRF3 (19, 20). Phosphorylated IRF3 was detected in rsT3D-infected cells as early as 2 h postinfection, and phospho-IRF3 levels increased over the time course. In contrast, rsT1L induced little, if any, phospho-IRF3. Together, these data indicate that rsT3D more potently elicits IFN-I responses than rsT1L in SVECs. These findings further suggest that differential IFN-I activation between rsT1L and rsT3D is elicited at the early stages of reovirus infection and prior to induction of IFN-I gene expression.
rsT3D and rsT1L differentially activate IRF3 in SVECs.
To characterize IRF3 activation by rsT1L and rsT3D in SVECs, we assessed IRF3 phosphorylation following infection with rsT1L and rsT3D at a range of multiplicities as determined by titer on L929 cells (Fig. 2A). Importantly, the particle-to-PFU ratios for the rsT1L and rsT3D stocks were comparable. Therefore, the total number of virus particles used for experiments did not vary due to the presence of excessive defective particles for either strain. At 6 h, high levels of phosphorylated IRF3 were detected in cells infected with rsT3D at a multiplicity of infection (MOI) of 100 PFU/cell. However, little phosphorylated IRF3 was induced by rsT1L at any MOI tested. At 24 h, IRF3 phosphorylation was detected at MOIs of 10 and 100 PFU/cell for rsT3D but only at an MOI of 100 PFU/cell for rsT1L. The reovirus replication cycle in cell culture is 18 to 24 h, and IRF3 phosphorylation at 24 h likely results from secondary infection by new progeny virions. To confirm that phosphorylated IRF3 translocates to the nucleus in SVECs, we assessed IRF3 in cytoplasmic and nuclear fraction lysates at 6 h (Fig. 2B). Consistent with results from whole-cell lysates, substantially less phosphorylated IRF3 was detected in nuclear fractions from rsT1L-infected cells than from rsT3D-infected cells. To determine whether IRF3 is transcriptionally active, we transfected SVECs with a firefly luciferase reporter under the control of tandem IRF-binding sites (Fig. 2C). At 24 h posttransfection, cells were mock infected, transfected with the dsRNA analog poly I·C, or infected with rsT1L or rsT3D. Consistent with greater nuclear phospho-IRF3, rsT3D induced significantly more luciferase activity than did rsT1L. Similar results were observed in 293T cells (data not shown). Together, these data indicate that although rsT3D and rsT1L have the capacity to activate IRF3, rsT3D activates IRF3 more effectively than rsT1L.
FIG 2.

rsT3D induces higher levels of IRF3 activation than rsT1L in SVECs. (A) SVECs were mock infected (M) or infected with rsT1L or rsT3D at an MOI of 1, 10, or 100 PFU/cell. Whole-cell lysates were prepared at the indicated times, and immunoblot analysis was performed to detect phosphorylated IRF3 (p-IRF3), total IRF3, β-actin, or reovirus proteins. (B) SVECs were mock infected or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell, and cytoplasmic and nuclear fraction lysates were prepared at 6 h. Immunoblot analysis was performed to detect phosphorylated IRF3 (p-IRF3), total IRF3, β-actin, α-tubulin, lamin A/C, or reovirus proteins. (C) SVECs were transfected with an IRF3 firefly luciferase reporter (p55C1BLuc) and a Renilla luciferase control. At 24 h, cells were mock infected, transfected with poly I·C (2 μg), or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. Luciferase expression was measured at 24 h posttransfection. Results are presented as the mean ratio of firefly luciferase activity to Renilla luciferase activity for triplicate samples from three independent experiments ± SD. **, P < 0.01; ***, P < 0.001 (as determined by one-way ANOVA test). (D) SVECs were infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. At 24 h, cells were fixed and stained for reovirus antigen (green). Nuclei were stained with DAPI (blue). (E) Quantification of the percentage of infected cells in panel D. Results are presented as the mean number of infected cells from triplicate samples from two independent experiments. (F) SVECs were infected with rsT1L or rsT3D at an MOI of 100 or 1,000 PFU/cell. At 6 h, whole-cell lysates were prepared and immunoblot analysis was performed to detect phosphorylated IRF3 (p-IRF3), β-actin, or reovirus proteins.
T3 reovirus strains that bind sialic acid (SA), including T3D, can infect cells more efficiently than T1 strains (21). To assess viral infectivity in SVECs, cells were adsorbed with rsT1L or rsT3D at an MOI of 100 PFU/cell and viral antigen-positive cells were quantified at 24 h (Fig. 2D and E). We found a slightly higher percentage of cells infected with rsT3D than rsT1L. To determine whether differences in infectivity between rsT1L and rsT3D lead to differences in IFN-I induction, we increased the MOI to 1,000 PFU/cell (Fig. 2F). At an MOI of 1,000 PFU/cell, rsT1L induced slight IRF3 phosphorylation. However, the levels of phospho-IRF3 induced by rsT1L at this dose did not reach the levels induced by 10-fold less rsT3D. These results indicate that differences in infectivity do not cause differential IFN-I induction by rsT1L and rsT3D.
RIG-I, MDA5, MAVS, and TBK1 mediate reovirus-induced IRF3 phosphorylation in SVECs.
To define the cellular sensors and kinases that detect reovirus and activate IRF3 in SVECs, we used RNA interference (RNAi) to knock down endogenous RIG-I, MDA5, MAVS, TBK1, IKKε, and TLR3 (Fig. 3). Knockdown of RIG-I, MDA5, MAVS, TBK1, and IKK protein levels were confirmed by immunoblotting (Fig. 3A to C and E). Knockdown of TLR3 transcript levels were confirmed by reverse transcription-quantitative PCR (RT-qPCR) (Fig. 3D). MAVS-specific siRNA transfection abolished rsT3D-induced IRF3 phosphorylation (Fig. 3A and B). Transfection of siRNA specific for RIG-I or MDA5 alone had no effect on reovirus-induced IRF3 phosphorylation. However, cotransfection of RIG-I- and MDA5-specific siRNAs reduced IRF3 phosphorylation to background levels (Fig. 3C). Transfection of TBK1 siRNA substantially reduced IRF3 phosphorylation, whereas transfection of siRNA specific for IKKε had a moderate effect on IRF3 phosphorylation (Fig. 3E). These results indicate that both TBK1 and IKKε have the capacity to phosphorylate IRF3 on Ser396. However, TBK1 appears to be the predominant kinase responsible. In contrast, TLR3-specific siRNA did not affect IRF3 phosphorylation in response to rsT3D infection (Fig. 3B). Together, these results indicate that reovirus activates IRF3 via the RIG-I/MDA5–MAVS–TBK1/IKKε axis in SVECs but that TLR3 does not contribute to reovirus-induced IFN-I induction in this cell line.
FIG 3.

RIG-I, MDA5, MAVS, and TBK1 mediate reovirus-induced IRF3 phosphorylation. SVECs were transfected with negative-control siRNA or siRNA specific for MAVS (A), MAVS or TLR3 alone or in combination (B), RIG-I or MDA5 alone or in combination (C), or TBK1 or IKKε (E). At 48 h, cells were mock infected (M), infected with rsT3D (T3) or rsT1L (T1) at an MOI of 100 PFU/cell, or transfected with poly(I·C) (2 μg). At 6 h postinfection, whole-cell lysates were prepared and immunoblot analysis was performed for phosphorylated IRF3 (p-IRF3), total IRF3, RIG-I, MDA5, MAVS, β-actin, TBK1, IKKε, or reovirus proteins, as indicated. (D) At 48 h posttransfection with negative-control siRNA or TLR3-specific siRNA, TLR3 mRNA levels were quantified by RT-qPCR. Results are presented as the mean of triplicate samples ± SD. *, P < 0.01 (as determined by Student's t test).
rsT3D activates NF-κB more potently than rsT1L in SVECs.
In addition to IRF3, production of IFN-β also requires NF-κB activation. To assess reovirus-induced NF-κB activation in SVECs, we monitored phosphorylation of IKKα/β, IκBα degradation, and nuclear translocation of p65 (RelA) following rsT1L or rsT3D infection (Fig. 4). In rsT3D-infected cells, phosphorylated IKKα/β was detected as early as 2 h and peaked at 6 h (Fig. 4A). However, in rsT1L-infected cells, only low levels of phosho-IKKα/β were detected. IκBα degradation was observed following infection with rsT3D but not rsT1L (Fig. 4A). Finally, p65 was detected in the nucleus of rsT1L- and rsT3D-infected cells, although substantially higher levels of nuclear p65 were detected for rsT3D (Fig. 4B). These results indicate that rsT3D induces higher levels of NF-κB signaling and activation than rsT1L.
FIG 4.
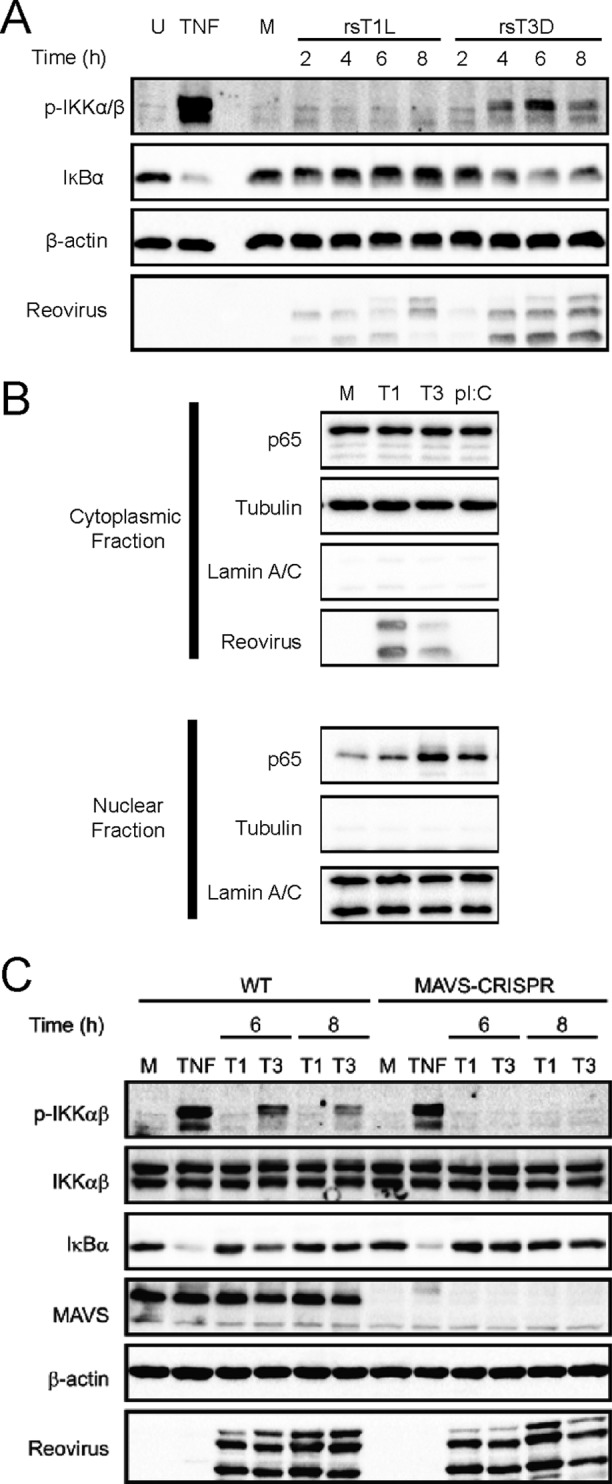
rsT3D elicits more NF-κB activation than rsT1L in SVECs. SVECs were left untreated (U) or were treated with TNF-α (TNF) for 15 min, mock infected (M), or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. Whole-cell lysates were prepared at the indicated times, and immunoblot analysis was performed to detect phosphorylated IKKα/β (p-IKKα/β), IκBα, β-actin, or reovirus proteins. (B) SVECs were mock infected, infected with rsT1L or rsT3D at an MOI of 100 PFU/cell, or transfected with poly(I·C). At the indicated times, cytoplasmic and nuclear fraction lysates were prepared and immunoblot analysis was performed to detect p65, β-actin, α-tubulin, lamin A/C, or reovirus proteins. (C) Wild-type SVECs or MAVS-CRISPR SVECs were mock infected (M), treated with TNF-α (TNF) for 15 min, or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell. Whole-cell lysates were prepared at the indicated times, and immunoblot analysis was performed to detect phosphorylated IKKα/β (p-IKKα/β), total IKKα/β, IκBα, MAVS, β-actin, or reovirus proteins.
Recruitment and oligomerization of MAVS can lead to NF-κB activation (3). To determine whether MAVS mediates reovirus-induced NF-κB activation, we compared NF-κB activation in wild-type SVECs and SVECs in which MAVS were deleted using CRISPR/Cas technology (MAVS-CRISPR). As before, we found higher levels of phospho-IKKα/β and degradation of IκB in wild-type SVECs infected with rsT3D (Fig. 4C). However, rsT3D did not induce IKKα/β phosphorylation or IκB degradation in MAVS-CRISPR SVECs. Importantly, deletion of MAVS had no effect on tumor necrosis factor alpha (TNF-α)-induced NF-κB activation. Together, these findings indicate that NF-κB activation by rsT1L and rsT3D mirror strain-specific differences in IRF3 induction. These results also indicate that early reovirus-induced NF-κB activation is mediated in a MAVS-dependent manner in SVECs.
Viral protein synthesis is dispensable for differential IRF3 activation by rsT1L and rsT3D.
To determine whether viral protein synthesis is required for differential IRF3 activation by rsT1L and rsT3D in SVECs, we tested the capacity of reovirus to activate IRF3 in the presence of ribavirin (Fig. 5). Ribavirin is a guanosine analog that is incorporated into RNA and introduces mutations that make viral mRNAs untranslatable (22, 23). SVECs were incubated with a range of ribavirin concentrations, and reovirus protein synthesis was analyzed at 6 h (Fig. 5). Viral protein production by rsT3D was completely blocked at all ribavirin concentrations tested. We found that rsT1L was more resistant to ribavirin, with complete inhibition of rsT1L protein synthesis observed only at 40 μM ribavirin. IRF3 was phosphorylated in response to rsT3D at each ribavirin concentration tested. However, no dose of ribavirin tested enabled rsT1L to activate IRF3. This finding indicates that rsT1L does not synthesize a gene product that blocks IRF3 activation.
FIG 5.

Viral protein synthesis and RNA synthesis are not required for reovirus-induced IRF3 phosphorylation. (A) SVECs were mock infected (M) or infected with rsT1L or rsT3D at an MOI of 100 PFU/cell in the absence or presence of ribavirin at the indicated concentrations. At 6 h, whole-cell lysates were prepared and immunoblot analysis was performed for phosphorylated-IRF3 (p-IRF3), total IRF3, β-actin, or reovirus proteins. (B) SVECs were infected with rsT1L or rsT3D at an MOI of 100 PFU/cell in the absence or presence of 40 μM ribavirin. At 6 h, total RNA was extracted and reovirus S4 mRNA levels were determined by RT-qPCR. Results are presented as the mean of triplicate samples ± SD and are representative of three independent experiments. *, P < 0.05; **, P < 0.01 (as determined by Student's t test).
Ribavirin also inhibits reovirus RNA synthesis at concentrations that do not interfere with cellular RNA synthesis (14, 15). Using RT-qPCR, we found that 40 μM ribavirin reduced the amount of S4 mRNA synthesized by rsT1L and rsT3D to similar levels detected at time zero hour postinfection (Fig. 5B). As 40 μM ribavirin had no effect on reovirus-induced IRF3 phosphorylation (Fig. 5A), this result suggests that reovirus RNA synthesis is not required for IRF3 phosphorylation by rsT3D. These data are consistent with the hypothesis that incoming viral genomic dsRNA is sufficient to activate IRF3 in SVECs, at least for T3 reoviruses.
Entry-related IRF3 activation is not suppressed by rsT1L.
To determine whether rsT1L blocks induction of the IFN-I response that is triggered during early stages of infection, SVECs were infected with rsT1L for 4 h and then transfected with poly I·C to trigger IRF3 activation (Fig. 6). We found that cells infected with rsT1L retained the capacity to activate IRF3 in response to poly I·C during the early stages of infection. We also noted that levels of RIG-I and MDA5 were comparable in rsT1L- and rsT3D-infected cells. This finding indicates that rsT1L does not impair IRF3 activation by causing the degradation of IRF3 or RNA sensors. Together, our results indicate that differences in IRF3 activation between rsT1L and rsT3D do not result from rsT1L suppressing the signaling pathway that leads to IRF3 activation. Rather, this finding suggests that events early in the replication cycle related to the detection of incoming viral genomic RNA differ between rsT1L and rsT3D, leading to differences in IRF3 activation.
FIG 6.
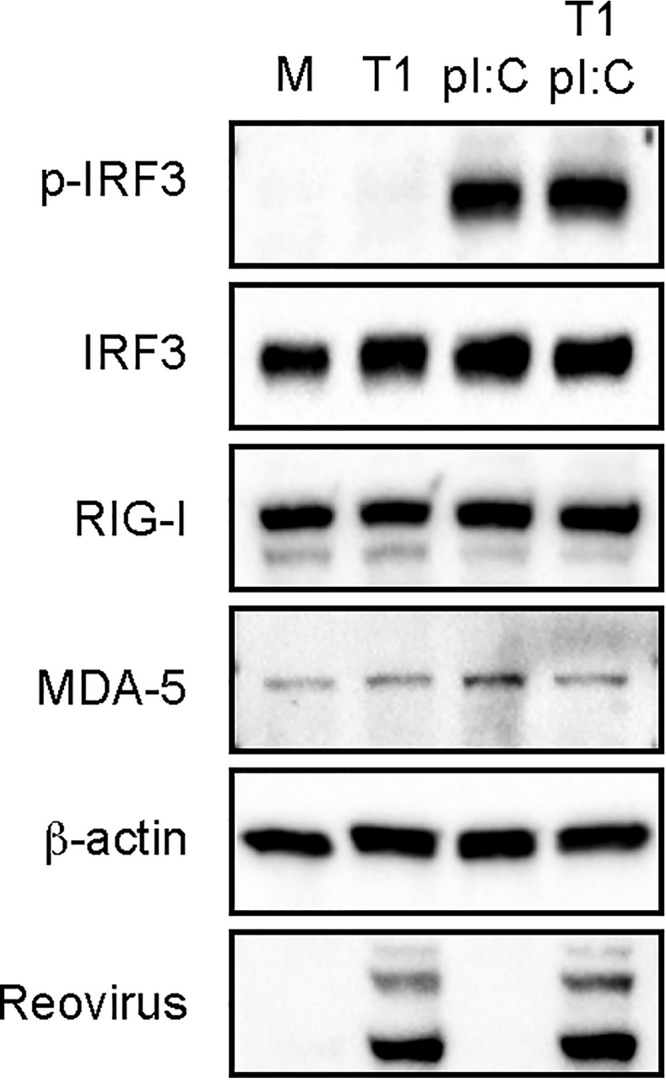
rsT1L does not block entry-related IRF3 phosphorylation. SVECs were mock-infected (M), infected with rsT1L (T1) at an MOI of 100 PFU/cell, or transfected with poly I·C (2 μg). At 4 h postinfection, rsT1L-infected cells were transfected with poly I·C (2 μg). At 6 h postinfection, whole-cell lysates were prepared and proteins were separated by SDS-PAGE. Western blot analysis was performed for phosphorylated IRF3, total IRF3, RIG-I, MDA-5, β-actin, or reovirus proteins.
rsT3D and rsT1L genomic RNAs induce IRF3 phosphorylation in SVECs.
Our data suggest that rsT1L does not actively antagonize IRF3 activation. Thus, the minimal IRF3 activation by rsT1L could be due to (i) differential detection of rsT1L and rsT3D genomic RNA or (ii) differing events leading to detection of viral RNA between rsT1L and rsT3D. To assess the detection of T1L and T3D reovirus genomic RNA by cellular sensors, SVECs were transfected with RNA isolated from purified rsT1L or rsT3D virions and IRF3 phosphorylation was assessed by immunoblotting. At the lower dose (0.25 μg), transfected genomic RNA from rsT1L and rsT3D induced comparable levels of IRF3 phosphorylation (Fig. 7A). We noted that at the higher dose (0.5 μg), rsT1L genomic RNA induced IRF3 activation more potently than rsT3D RNA. These data indicate that T1 and T3 reovirus genomes are detected by cytoplasmic RNA sensors and activate IRF3 in SVECs.
FIG 7.

Reovirus genomic RNA induces IRF3 phosphorylation in SVECs. (A) SVECs were transfected with 0.25 μg or 0.5 μg of rsT1L or rsT3D genomic RNA or with 2.0 μg of poly(I·C). (B) SVECs were transfected with siRNA specific for MAVS or TLR3 alone or in combination for 48 h prior to transfection with 0.5 μg of genomic RNA or poly(I·C) (2 μg). At 6 h, immunoblot analysis was performed for phosphorylated IRF3 (p-IRF3), total IRF3, β-actin, or MAVS. **, P < 0.001 (as determined by Student's t test).
Transfected RNA can be taken up into endosomes and lead to TLR3 activation (24). Our results indicate that TLR3 does not mediate IRF3 activation during reovirus infection (Fig. 3). To ensure that transfected rsT1L genomic RNA has the capacity to activate the same cytoplasmic RNA sensors that detect rsT3D during infection, we performed siRNA knockdown of MAVS or TLR3 for 48 h prior to transfection with 0.5 μg of rsT1L or rsT3D genomic RNA or 2 μg of poly I·C. MAVS- or TLR3-specific siRNAs alone modestly reduced IRF3 phosphorylation by genomic RNA from both strains. However, cotransfection of MAVS- and TLR3-specific siRNAs reduced IRF3 phosphorylation to near background levels (Fig. 7B). These findings indicate that transfected reovirus genomic RNA activates cytoplasmic sensors and TLR3. Although TLR3 is artificially triggered by RNA transfection, RIG-I and MDA5, both of which are triggered by reovirus infection, have the capacity to detect T1L and T3D genomic RNA in the cytoplasm. Thus, differential IRF3 phosphorylation by T1L and T3D does not result from differences in RNA structure or sequence. Rather, our findings suggest that events leading to the delivery of viral RNA into the cytosol likely differ between rsT1L and rsT3D, resulting in differential IRF3 induction.
Virion disassembly is required for IRF3 phosphorylation.
To determine whether reovirus disassembly is required for IRF3 activation in SVECs, we tested the effect of ammonium chloride and E64 on reovirus-induced IRF3 phosphorylation. Ammonium chloride inhibits endosomal acidification, and E64 inhibits cysteine proteases cathepsin B and L, both of which are required for reovirus outer capsid protein disassembly (25, 26). Infection with rsT1L and rsT3D was completely inhibited by 10 mM ammonium chloride or 100 μM E64 (data not shown), and neither compound was toxic to SVECs at these concentrations (data not shown). The effectiveness of ammonium chloride or E64 treatment was confirmed by the lack of μ1 cleavage into the δ fragment (Fig. 8A). Ammonium chloride completely blocked IRF3 phosphorylation by rsT3D (Fig. 8A). E64 markedly reduced IRF3 activation, although not as effectively as ammonium chloride (Fig. 8A). This is likely because E64 is not membrane permeable and must be taken up by cells through endocytosis. These data indicate that reovirus outer capsid disassembly is required for IRF3 activation in SVECs.
FIG 8.

Reovirus disassembly is required for IRF-3 phosphorylation. (A) SVECs were infected with rsT1L (T1) or rsT3D (T3) at an MOI of 100 PFU/cell in the absence or presence of ammonium chloride or E64 at the indicated concentrations. At 6 h, immunoblot analysis was performed for phosphorylated IRF3 (p-IRF3), total IRF3, β-actin, or reovirus proteins. (B) To generate ISVPs, 2 × 1010 rsT1L or rsT3D particles were incubated with chymotrypsin (CHT). Proteins were resolved by SDS-PAGE and stained with Coomassie blue. Viral proteins are indicated to the right of the image. (C) SVECs were infected with rsT1L or rsT3D virions at an MOI of 100 PFU/cell or rsT1L or rsT3D ISVPs at an MOI of 10 PFU/cell. At 24 h, cells were fixed with methanol, stained with DAPI (blue) and reovirus polyclonal antiserum (green), and visualized by indirect immunofluorescence. (D) SVECs were infected with rsT1L or rsT3D virions or ISVPs at an MOI of 100 PFU/cell in the absence or presence of 20 mM ammonium chloride. At 6 h, immunoblot analysis was performed for phosphorylated IRF3, total IRF3, β-actin, or reovirus proteins.
To determine whether differences in IRF3 activation between rsT1L and rsT3D result from differences in disassembly, we infected cells with infectious subvirion particles (ISVPs). ISVPs are formed in the gut and lung by tissue-resident proteases or in endosomes by cathepsin proteases (27, 28). We generated ISVPs in vitro by chymotrypsin digestion of rsT1L and rsT3D virions (Fig. 8B). Consistent with other cell types (29–32), ISVPs were more infectious than virions in SVECs (Fig. 8C). In contrast to rsT1L virions, rsT1L ISVPs induced IRF3 phosphorylation (Fig. 8D). The difference is likely due to the increased infectivity of ISVPs relative to that of virions. However, IRF3 phosphorylation levels induced by rsT3D ISVPs remained greater than those elicited by rsT1L ISVPs. These findings suggest that differences in outer capsid processing do not lead to differential IRF3 activation by rsT1L and rsT3D.
rsT1L and rsT3D cores induce IRF3 phosphorylation.
Our data suggest that differential IRF3 activation between rsT1L and rsT3D results from differences at steps following viral disassembly but prior to viral gene expression. Following disassembly, ISVPs mediate penetration of the endosomal membrane and release of the transcriptionally active core into the cytosol (16–18). We generated reovirus core particles in vitro by chymotrypsin digestion with the addition of cesium chloride (Fig. 9A). Reovirus cores are not infectious because they lack the viral attachment protein σ1 and membrane penetration protein μ1 (21). To functionally confirm the generation of core particles, cells were exposed to rsT1L or rsT3D cores with or without transfection reagent (Fig. 9B). Few cells stained positive for viral antigen when cores were added to cells without transfection reagent. However, transfection of cores led to an increase in infected cell numbers. This result demonstrates that core particles were not infectious unless assisted across the cell membrane. To determine whether rsT1L and rsT3D cores activate IRF3, cells were exposed to rsT1L or rsT3D cores at MOIs of 10,000 or 100,000 particles/cell with or without transfection reagent. In the absence of transfection reagent, IRF3 phosphorylation was not detected (Fig. 9C). However, transfected rsT1L and rsT3D cores induced comparable levels of IRF3 phosphorylation. Together, our results indicate that rsT1L and rsT3D cores have the capacity to activate IFN-I signaling when deposited in the cytoplasm. Thus, differences in IRF3 activation do not result from native differences in the capacity of capsids to release innate immune agonists. Rather, our results suggest that entry-related events that occur prior to delivery of viral cores into the cytoplasm dictate differences in reovirus serotype-specific IRF3 activation.
FIG 9.
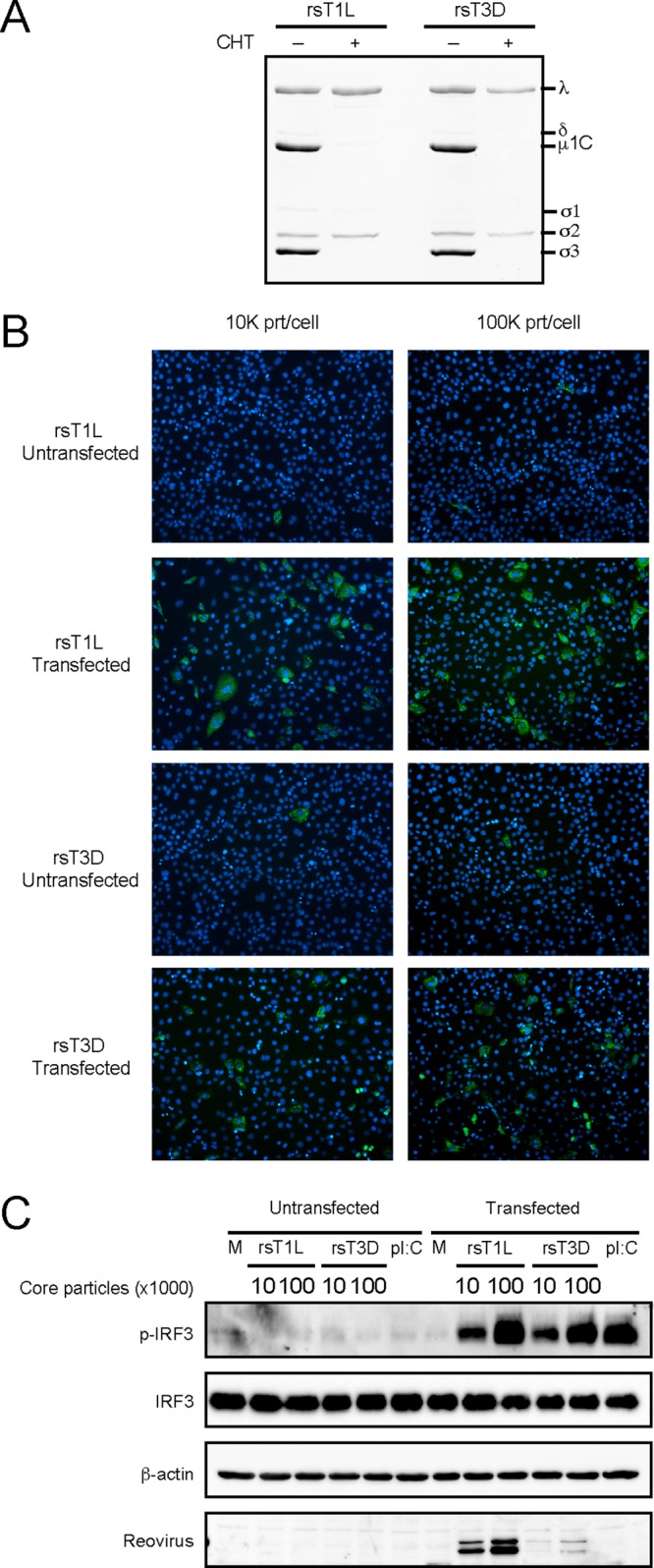
rsT1L and rsT3D core particles induce IRF3 phosphorylation. (A) To generate core particles, rsT1L and rsT3D virions were incubated with chymotrypsin (200 μg/ml) and cesium chloride (400 mM). A total of 2 × 1010 particles were resolved by SDS-PAGE, and proteins were stained with Coomassie blue. Viral proteins are indicated to the right of the image. (B and C) SVECs were exposed to rsT1L or rsT3D cores at an MOI of 10,000 or 100,000 particles (prt)/cell with or without transfection reagent. (B) At 24 h, cells were fixed with methanol, stained with DAPI (blue) and reovirus polyclonal antiserum (green), and visualized by indirect immunofluorescence. (C) At 6 h, immunoblot analysis was performed for phosphorylated IRF3 (p-IRF3), total IRF3, β-actin, or reovirus proteins.
DISCUSSION
Here, we characterized how T1 and T3 reoviruses activate IFN-I responses in a mouse endothelial cell line (SVECs). Endothelial cells are a critical target for reovirus infection in vivo and facilitate viral spread via the bloodstream (16, 17). Consequently, determining how reovirus activates IFN-I responses in endothelial cells is important for understanding reovirus pathogenesis. Consistent with studies in other cell types (8), we found that a T3 reovirus (rsT3D) induced substantially more IFN-β than a T1 strain (rsT1L) in SVECs. We further found that the difference in IFN-β induction between the two strains correlated with IRF3 activation; rsT3D induced higher levels of IRF3 phosphorylation than rsT1L in SVECs. Differences in IRF3 phosphorylation between rsT1L and rsT3D were apparent as early as 2 h postinfection, indicating that differential IFN-I responses to T1 and T3 strains originate early during reovirus infection. This time frame corresponds to the window of reovirus entry (25, 33). Similar to IRF3 activation, we found that rsT3D induced NF-κB activation more potently than rsT1L. Moreover, NF-κB activation is mediated by a MAVS-dependent pathway (Fig. 4). Consistent with other studies, our findings indicate that incoming genomic reovirus RNA is sufficient to activate the IFN-I pathway in SVECs (34–37), as transfected genomic rsT1L or rsT3D RNA had the capacity to activate IRF3 (Fig. 7). This result indicates that physical or chemical differences between genomic RNA from the two strains do not cause differential activation of the IFN-I pathway. Instead, our data indicate that entry-related events lead to differential detection of T1 and T3 reovirus genomic RNA and cause differences in IFN-I induction between reovirus serotypes.
Reovirus infection is initiated by attachment to cell surface receptors, including carbohydrates, Nogo receptor 1 (NgR1), and junctional adhesion molecule A (JAM-A) (26, 38, 39). Virions are internalized into the endocytic pathway and converted to ISVPs through the activity of acid-dependent cathepsin proteases that degrade σ3 and cleave μ1 to produce the μ1N, μ1δ, and μ1ϕ fragments (25, 26). Blocking ISVP conversion with ammonium chloride or E64 prevents IRF3 activation (Fig. 8). This finding indicates that the initial outer capsid disassembly steps leading to ISVP formation are required to trigger IFN-I responses. However, rsT3D ISVPs induced higher levels of IRF3 phosphorylation than rsT1L ISVPs, indicating that differences in outer capsid disassembly do not mediate differential IRF3 activation (Fig. 8). Thus, events downstream of ISVP formation determine the efficiency of IFN-I induction by reovirus. ISVPs are subsequently modified to produce a disassembly species termed ISVP*, which is characterized by separation of the μ1N and μ1ϕ domains from the particle (40, 41). The σ1 protein also dissociates from its docking site with the λ2 protein, opening the channel that allows mRNA exit during transcription (42). ISVP* formation also requires interaction with cellular lipids (43, 44). Following ISVP* formation, the viral core is deposited in the cytoplasm (42). The liberated μ1N and μ1ϕ fragments are hypothesized to mediate pore formation in the endosomal membrane. However, the mechanism by which reovirus cores cross cellular membranes is poorly understood. We found that bypassing the normal route of entry by transfecting in vitro-generated rsT1L and rsT3D viral core particles leads to comparable levels of IRF3 phosphorylation (Fig. 9). It is important to note that a limitation of using in vitro-generated reovirus cores is that the μ1-δ fragment can remain associated and is hypothesized to be removed by cellular Hsc70 during entry (45–47). Future work will focus on understanding how this step in viral disassembly contributes to reovirus innate immune activation. The finding that cores do not differ in IRF3 activation indicates that once in the cytoplasm, rsT1L and rsT3D cores have similar capacities to activate the IFN-I pathway. These data also indicate that events after ISVP formation, but prior to deposition of the viral core into the cytoplasm, determine the capacity of reovirus to activate IFN-I responses. As both rsT1L and rsT3D genomic RNAs are capable of activating IRF3, our findings suggest that rsT1L and rsT3D genomes are delivered to host cells in a different manner.
It is possible that differential IFN-I induction between rsT1L and rsT3D could relate to differences in ISVP* formation or membrane penetration. In vitro-generated T3D ISVPs convert to ISVP*s more rapidly than T1L ISVPs (40, 48). Faster conversion to ISVP*s could allow T3D to undergo the subsequent entry steps more rapidly than T1L. Initiating downstream entry events at a higher rate may allow T3D to become accessible to RLRs with more rapid kinetics than T1L. It also is possible that more rapid virion disassembly could lead to earlier exit from the endosomal pathway for T3D than for T1L. T3D cores may be deposited in the cytoplasm in closer proximity to RLRs than T1L cores, thus allowing more efficient detection of viral RNA. Once ISVP* formation is complete, the μ1 fragments mediate exit of the core from the endosome into the host cell cytosol by promoting formation of pores in the endosomal membrane (5). T3D has a greater capacity to cause membrane disruption than T1L. In addition, mutations in the μ1 protein that confer resistance to 70% ethanol also reduce IRF3 activation by T3D (49).
Another possibility to explain differences in IFN-I induction between T1 and T3 reoviruses is that T1 strains antagonize the activation of the response. Many viruses cleave or degrade components of the IFN-I induction pathway (50, 51). However, levels of RIG-I, MDA5, MAVS, and IRF3 did not differ between rsT1L and rsT3D infections (Fig. 3 and 6). Thus, the key RNA sensing and signal transducing molecules that activate IRF3 are not degraded by rsT1L. Moreover, IRF3 could be phosphorylated in response to poly I·C in cells infected with rsT1L (Fig. 6), indicating that rsT1L does not functionally inactivate the IFN-I pathway. Finally, IRF3 phosphorylation was not induced by rsT1L in the presence of ribavirin, indicating that an rsT1L gene product does not block IRF3 activation. Together, these findings indicate that minimal IRF3 activation by rsT1L does not result from viral antagonism of the IRF3 signaling pathway.
Previous work indicates that reovirus activates NF-κB via a MAVS-independent mechanism (34). However, NF-κB activity was measured by a luciferase reporter assay at late times during infection (24 h). Reovirus induces at least two phases of NF-κB activation during infection (52), and assays quantifying NF-κB at late times could detect activity from multiple rounds of NF-κB induction. Indeed, using a reporter assay, we found that rsT3D induced more NF-κB activity than rsT1L did at 12 h, but activity at 24 h did not differ between rsT1L and rsT3D in HeLa cells (data not shown). Our results indicate that NF-κB can be activated at early stages of infection through MAVS. However, it is possible that cell-specific differences also contribute to reovirus-induced NF-κB activation.
Although rsT1L induces IFN-I production and ISG induction less potently than rsT3D, rsT1L does induce IFN-I responses (Fig. 1 and 2). It could be predicted that greater induction of the IFN-I pathway would limit viral replication of rsT3D relative to that of rsT1L. We found that rsT3D induced higher ISG levels than rsT1L did at early times during infection (8 h) (Fig. 1). However, ISG induction by rsT3D is reduced to the levels induced by rsT1L at late times. It is unclear whether reduced ISG results from repression by rsT3D or normal cellular down-modulation of the response (18). Reduced ISG levels in rsT3D-infected cells may explain why rsT1L and rsT3D replicate comparably in many cell types, including SVECs (Fig. 1). To replicate efficiently in spite of strong IFN-I responses suggests that T3 strains have the capacity to more effectively resist the effects IFN-I. However, T1 strains are more resistant to IFN-Is than T3 viruses, a phenotype that maps to the M1 gene-encoded μ2 protein (53). The T1 μ2 protein blocks IRF9 recycling, which impairs IFN-I signaling (53). Recent work revealed that reovirus blocks IFN-I production by sequestering IRF3 in viral factories during the late stages of infection (54). Thus, reoviruses have the capacity to overcome the IFN-I response and replicate efficiently, regardless of the level of IFN-I production.
In this study, we characterized the activation of IFN-I responses to reovirus infection in endothelial cells. A T3 reovirus strain induced markedly more IFN-I from endothelial cells than the T1 strain tested. Concomitant with IFN-I production, the T3 strain activated IRF3 more rapidly and to higher levels than the T1 strain. Differences in IRF3 activation occur during the early stages of infection but prior to deposition of reovirus cores in the cytoplasm. Together, our results indicate that differential delivery of the viral genome leads to serotype differences in detection of reovirus genomic RNA by cellular RNA sensors. Understanding how reoviruses elicit IFN-I responses provides insight into the mechanisms by which nonenveloped viruses are sensed by the host innate immune system.
MATERIALS AND METHODS
Cell lines.
Murine SV40 immortalized endothelial cells (SVEC4-10; SVECs) 293T cells, HeLa cells, and HEK293T cells (ATCC, Rockville, MD) were maintained in Dulbecco's modified Eagle's media (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA) and 2 mM l-glutamine (Invitrogen, Carlsbad, CA). Spinner-adapted murine L929 cells were maintained in Joklik's minimum essential medium (JMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 5% FBS, 2 mM l-glutamine, 100 U/ml penicillin–100 μg/ml streptomycin mixture (Invitrogen, Carlsbad, CA), and 250 ng/ml amphotericin B (Sigma-Aldrich, St. Louis, MO). Baby hamster kidney cells constitutively expressing bacteriophage T7 RNA polymerase (BHK-T7) were maintained in DMEM supplemented with 10% FBS, 2 mM l-glutamine, 100 U/ml penicillin–100 μg/ml streptomycin mixture, 250 ng/ml amphotericin B, and 1 mg/ml Geneticin (Invitrogen, Carlsbad, CA) (55).
Generation of CRISPR/Cas9 knockout SVECs.
The target guide sequence specific for MAVS was designed and cloned into the lentiCRISPRv2 plasmid. LentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid 52961). Cloning of the MAVS guide sequence was performed using the protocol provided by the Zhang lab (56). Briefly, the lentiCRISPRv2 plasmid was digested using BsmB1 and FastAP. Oligonucleotides specific for MAVS (forward, 5′-CACCGAGGAAGCCCGCAGTCGATCC-3′; reverse, 5′-AAACGGATCGACTGCGGGCTTCCTC-3′) were annealed and ligated into digested plentiCRISPR v2. Lentivirus was generated by transfection of HEK293T cells with lentiCRISPR-MAVS (4,000 ng), pSPAX2, and pCMV-G at a 6:4:2 ratio using Lipofectamine 2000. After 24 h, filtered supernatant from HEK29T cells was added to SVECs in 6-well plates in combination with Polybrene. At 48 h postransduction, lentiCRISPR v2-MAVS-containing cells were selected with puromycin (2 μg/ml). Loss of MAVS expression in SVECs were confirmed by immunoblotting.
Viruses.
Recombinant strains (rs) of T1L (rsT1L) and T3D (rsT3D) were generated by plasmid-based reverse genetics (57, 58). Purified reovirus virions were generated using second- or third-passage L929 cell lysate stocks of twice-plaque-purified reovirus as described previously (59). Viral particles were extracted with Vertrel (TMC Industries, Waconia, MN) from infected cell lysates, layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 29,000 rpm for 18 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed against virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]). The concentration of reovirus virions in purified preparations was determined from the following equivalence: 1 optical density at 260 nm (OD260) unit = 2.1 × 1012 virions (60). Viral titers were determined by plaque assay using L929 cells (61).
ISVPs were generated by incubating 2 × 1011 virions with 200 μg/ml of TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone)-treated α-chymotrypsin (CHT) in a total volume of 0.1 ml virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) at 37°C for 1 h (rsT1L) or 20 min (rsT3D) (62, 63). CHT activity was terminated by adding 2 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich, St. Louis, MO) on ice. Core particles were generated by incubation of 5 × 1011 virions with 200 μg/ml of CHT and 400 mM CsCl in a total volume of 0.1 ml virion storage buffer at 37°C (rsT3D) or 42°C (rsT1L) for 90 min. CHT protease activity was terminated by the addition of 2 mM PMSF on ice. Core particles were dialyzed against virion storage buffer prior to use. Generation of ISVPs and core particles was confirmed by separating 2 × 1010 particles by SDS-PAGE, followed by Coomassie brillant blue staining. Core particle formation was further confirmed by overlaying cells with cores combined with or without Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Virus replication.
Monolayers of cells (1 × 105 cells/well) in 24-well plates (Corning Inc., Corning, NY) were adsorbed in triplicate with reovirus at the MOIs indicated in the figure legends for 1 h at room temperature in 1× phosphate-buffered saline (PBS) containing calcium chloride and magnesium chloride (PBS+/+). Cells were washed twice with PBS+/+ and incubated at 37°C in supplemented medium for various time intervals. Cells were frozen and thawed twice prior to the determination of viral titer by plaque assay on L929 cells (61). Viral yields were calculated according to the equation log10 yieldtx = log10(PFU/ml)tx – log10(PFU/ml)t0, where tx is the time postinfection.
Immunoblot analysis.
Monolayers of cells (1 × 106 cells/well) in 6-well plates (Corning Inc., Corning, NY) were infected or transfected as indicated in the figure legends. At the times indicated in the figure legends, the cells were washed twice with ice-cold PBS, and total cellular protein was extracted using 1× RIPA buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Igepal, 0.1% SDS, 0.1% deoxycholate) containing cocktails of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Cell lysates were transferred to tubes and incubated on ice for 30 min, followed by centrifugation at 14,800 rpm at 4°C for 15 to 30 min to remove cellular debris. For extraction of cytoplasmic and nuclear proteins, cells in 6-well plates (1 × 106 cells/well) were washed twice with ice-cold PBS, collected using 18-cm cell lifters, and transferred to tubes. Following centrifugation at 4,000 rpm at 4°C for 5 min, the supernatant was removed and cytoplasmic lysates were prepared using cytoplasmic extract (CE) buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1 mM EDTA, 0.3% Igepal) containing a cocktail of protease and phosphatase inhibitors. After 5 min of incubation on ice, lysates were centrifuged at 3,000 rpm at 4°C for 5 min, and supernatant was harvested as the cytoplasmic extract. The pellet was resuspended and washed in CE buffer without NP-40 and centrifuged at 3,000 rpm at 4°C for 5 min, and the supernatant was discarded. Nuclear lysates were prepared by adding nuclear extract buffer (20 mM HEPES [pH 7.9], 0.4 M NaCl, 1 mM EDTA, 25% glycerol) containing a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO) to the pellet, followed by incubation on ice for 10 min and centrifugation at 14,000 rpm at 4°C for 10 min. Supernatant was harvested as nuclear extract. Protein concentrations were determined by the DC protein assay (Bio-Rad, Hercules, CA), and 30 μg of protein per lysate was heated at 99°C for 5 min in Laemmli sample buffer and subjected to SDS-PAGE and immunoblot analysis. Immunoblots were visualized with the enhanced chemiluminescent (ECL) assay (ThermoFisher Scientific, Waltham, MA) using a ChemiDoc MP imaging system (Bio-Rad, Hercules, CA).
Antibodies.
Antibodies included rabbit monoclonal anti-phospho-IRF3(Ser396) antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-IRF3 antibody (1:1,000) (Abcam, Cambridge, United Kingdom), rabbit polyclonal anti-STAT1 antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-phospho-STAT1(Tyr701) antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit polyclonal anti-STAT2 antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit polyclonal anti-STAT2(phospho Y690) antibody (1:1,000) (Abcam, Cambridge, United Kingdom), mouse monoclonal anti-α-tubulin antibody (1:1,000) (Cell Signaling, Danvers, MA), mouse monoclonal anti-lamin A/C antibody (1:1000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-Rig-I antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-MDA5 antibody (1:1,000) (Cell Signaling, Danvers, MA), anti-MAVS antibody (1:1,000) (Cell Signaling, Danvers, MA), mouse monoclonal anti-IκBα antibody (1:2000) (Santa Cruz Inc., Santa Cruz, CA), rabbit monoclonal anti-phospho-IKKα/β antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-IKKαβ antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-IKKβ antibody (1:1,000), rabbit monoclonal anti-NF-κB p65 antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-TBK-1 antibody (1:1,000) (Cell Signaling, Danvers, MA), rabbit monoclonal anti-IKKε antibody (1:1,000) (Cell Signaling, Danvers, MA), mouse monoclonal anti-β actin antibody (1:5,000) (Sigma-Aldrich, St. Louis, MO), goat polyclonal horseradish peroxidase (HRP)-conjugated anti-mouse antibody (1:2,000), and goat polyclonal HRP-conjugated anti-rabbit antibody (1:2,000) (Jackson ImmunoResearch, West Grove, PA). Reovirus-specific rabbit polyclonal antiserum (1:2500) was provided by Terence Dermody (University of Pittsburgh School of Medicine).
Luciferase reporter assay.
Monolayers of SVECs (1 × 105 cells/well) in 12-well culture plates (Corning, Corning, NY) were transfected with 0.45 μg of IRF3 reporter plasmid (p55C1B-Luc) and 0.05 μg of Renilla luciferase internal control plasmid (pRL-TK) using Lipofectamine LTX with Plus reagent (Invitrogen, Carlsbad, CA). At 24 h posttransfection, cells were transfected with the dsRNA analog polyinosinic:poly(C) (poly I·C) (Sigma-Aldrich, St. Louis, MO), mock infected, or absorbed with rsT1L or rsT3D at an MOI of 100 PFU/cell at room temperature for 1 h. Cells were harvested 24 h postinfection in luciferase passive lysis buffer, and luciferase expression was measured using the dual-luciferase reporter assay system (Promega, Madison, WI) and a FLUOstar Omega luminometer (BMG Labtech, Cary, NC). Firefly luciferase activity was normalized to the Renilla luciferase control. The average of triplicate samples was presented as the fold induction of firefly luciferase activity over that of Renilla luciferase activity.
Quantification of IFN-β by ELISA.
Monolayers of SVECs (1 × 105 cells/well) in 24-well plates were mock infected or adsorbed with rsT1L or rsT3D at an MOI of 100 PFU/cell at room temperature for 1 h. At 4, 8, 12, and 24 h, IFN-β in cell culture medium was measured using the VeriKine mouse IFN-β ELISA kit (PBL Assay Science, Piscataway Township, NJ) according to the manufacturer's instructions.
Treatment with ammonium chloride (NH4Cl), E64, ribavirin, or IFN-β.
Monolayers of SVECs (1 × 106 cells/well) in 6-well plates were incubated in the presence or absence of 10 mM NH4Cl (ThermoFisher Scientific, Waltham, MA), 100 μM E64 (Sigma-Aldrich, St. Louis, MO), or 5 to 40 μM ribavirin (Sigma-Aldrich, St. Louis, MO) at 37°C for 1 h prior to viral adsorption. Cells were mock infected or adsorbed with rsT1L or rsT3D at an MOI of 100 PFU/cell at room temperature for 1 h. Following adsorption, treatments were readministered and cells were incubated at 37°C for various time intervals. For experiments using IFN-β or TNF-α, cells were treated with 200 U of purified mouse IFN-β (PBL Assay Science, Piscataway Township, NJ) or 20 ng/ml of TNF-α for 15 min (ThermoFisher Scientific, Waltham, MA) immediately following a viral adsorption period. Cells were harvested for immunoblot analysis at the times indicated in the figure legend.
Transfection of core particles.
The concentration of core particles was determined by OD260. Cells were transfected with 10,000 or 100,000 particles/cell. In separate tubes, core particles were diluted in a final volume of 250 μl of Opti-MEM I reduced serum medium (Invitrogen, Carlsbad, CA). A volume of 10 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was also diluted in Opti-MEM I. Tubes were incubated at room temperature for 5 min, and mixtures were combined and incubated at room temperature for 20 min. Monolayers of SVECs grown on 6-well plates to 50 to 70% confluence were washed twice with PBS, and the Lipofectamine-core particle mixture was added. After 4 h of incubation at 37°C, 2 ml of supplemented DMEM was added. At 8 h posttransfection, whole-cell lysates were prepared for immunoblot analysis, and at 24 h, posttransfection cells were fixed for indirect immunofluorescence (64).
RT-qPCR.
For reverse transcription-quantitative PCR (RT-qPCR), monolayers of SVECs (1 × 106 cells/well) in 6-well plates were mock infected or adsorbed in triplicate with rsT1L or rsT3D at an MOI of 100 PFU/cell. At the indicated time points, total RNA was extracted using the RNeasy Plus kit (Qiagen, Hilden, Germany). RNA was eluted into a final volume of 30 μl, and 100 ng/μl was used for cDNA synthesis. RNA was converted to cDNA by RT using an Eppendorf thermal cycler (Eppendorf, Hamburg, Germany) and amplified by PCR using the Step One Plus real-time PCR system (Applied Biosystems, Foster City, CA). RT-qPCR was performed with the TaqMan fast virus 1-step master mix (Applied Biosystems, Foster City, CA), and reaction conditions were generated according to the manufacturer's instructions. Forward (S4 83F, 5′-CGCTTTTGAAGGTCGTGTATCA-3′) and reverse (S4 153R, 5′-CTGGCTGTGCTGAGATTGTTTT-3′) primers corresponding to the viral S4 gene were used for reverse transcription and quantitative PCR amplification. The S4-specific fluorogenic probe used was 5′-56-FAM-AGCGCGCAAGAGGGATGG GA-3BHQ-1-3′ (IDT Technologies, Coralville, IA). Reactions were performed in triplicate. The levels of S4 mRNA were normalized to the expression of the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) housekeeping gene, and the relative quantification in gene expression was determined using the 2−ΔΔCT method (65). For analysis of murine TLR3 mRNA, forward (5′-TTCCTGCTGGAAAACTGGATGG-3′) and reverse (5′-TCAGCCTGAAAGTGAAACTCGC-3′) primers corresponding to murine TLR3 were used for reverse transcription and quantitative PCR amplification. Oas1b and IFIT1 mRNA expression was determined using the TaqMan gene expression assay (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions.
Indirect immunofluorescence.
Monolayers of SVECs (5 × 104 cells/well) grown on 8-well Lab-Tek chamber slides (ThermoFisher Scientific, Waltham, MA) were adsorbed with rsT1L or rsT3D reovirus at an MOI of 100 PFU/cell at room temperature for 1 h. Following the removal of the inoculum, cells were washed with PBS and incubated with supplemented medium at 37°C. At 24 h, monolayers were fixed with ice-cold methanol at −20°C for at least 30 min, washed twice with PBS, and blocked with 2% bovine serum albumin (BSA) in PBS for 15 min. For detection of reovirus proteins, cells were stained with rabbit polyclonal reovirus-specific antiserum at a 1:500 dilution in PBS–0.5% Triton X-100 at room temperature for 30 min. Monolayers were washed twice with PBS and incubated with a 1:1,000 dilution of anti-rabbit Alexa 488-labeled antibody and 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, MO). Monolayers were washed twice with PBS, and infected cells were visualized by indirect immunofluorescence using an Eclipse-Ti confocal fluorescence microscope (Nikon Instruments, Tokyo, Japan).
Small interfering RNA and transient transfections.
SVECs were grown on 6-well plates in supplemented medium to 50 to 70% confluence. Cells were transfected with murine MAVS, TLR3, RIG-I, MDA5 Stealth RNAi siRNA, TBK-1, IKKε Silencer duplex oligoribonucleotides, or negative-control medium GC duplex (ThermoFisher Scientific, Waltham, MA) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. The transfection mixture consisted of 200 pmol of siRNA and 10 μl of Lipofectamine 2000 reagent. After 48 h, cells were transfected with poly I·C (2 μg) (Sigma-Aldrich, St. Louis, MO), mock infected, or absorbed with rsT3D at an MOI of 100 PFU/cell at room temperature for 1 h. Cells were harvested at 6 h for immunoblot analysis. The sequences for Stealth RNAi siRNAs are as follows: MAVS sense, 5′-GGCUGAUCAAGUGACUCGAGUUUAU-3′; MAVS antisense, 5′-AUAAACUCGAGUCACUUGAUCAGCC-3′; TLR3 sense, 5′-CCUGAUGAUCUUCCCUCUAACAUAA-3′; TLR3 antisense, 5′-UUAUGUUAGAGGGAAGAUCAUCAGG-3′; RIGI sense, 5′-GGAAGUCAUGCAACAUAUCUGUAAA-3′; RIGI antisense, 5′-UUUACAGAUAUGUUGCAUGGCUUCC-3′; MDA5 sense, 5′-CCGAACGGGAGAUUGUUAAUGAUUU-3′; MDA5 antisense, 5′-AAAUCAUUAACAAUCUCCCGUUCGG-3′.
ACKNOWLEDGMENTS
We thank Emily Simon and Morgan Howells for experimental assistance. We thank Jennifer Anstadt for reading the manuscript.
This research was supported by Public Health awards K22 AI90497 (to K.W.B.), R01 AI118801 (to K.W.B.), and R15 AI094440 (to G.H.H.). J.D.S. was supported by the UAMS Initiative for Maximizing Student Diversity (IMSD) (R25 GM83247). Additional support was provided by the Center for Microbial Pathogenesis and Host Inflammatory Response (P20 GM103625).
REFERENCES
- 1.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schlee M. 2013. Master sensors of pathogenic RNA—RIG-I like receptors. Immunobiology 218:1322–1335. doi: 10.1016/j.imbio.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O, Akira S. 2010. Pattern recognition receptors and inflammation. Cell 140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 4.Goubau D, Deddouche S, Reis e Sousa C. 2013. Cytosolic sensing of viruses. Immunity 38:855–869. doi: 10.1016/j.immuni.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dermody TS, Parker JSL, Sherry B. 2013. Orthoreovirus, p 1304–1346. In Knipe DM, Howley PM (ed), Fields virology, 6th ed, vol 2 Lippincott, Williams, & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE Jr, Dermody TS. 2005. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis 191:1221–1224. doi: 10.1086/428911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bouziat R, Hinterleitner R, Brown JJ, Stencel-Baerenwald JE, Ikizler M, Mayassi T, Meisel M, Kim SM, Discepolo V, Pruijssers AJ, Ernest JD, Iskarpatyoti JA, Costes LM, Lawrence I, Palanski BA, Varma M, Zurenski MA, Khomandiak S, McAllister N, Aravamudhan P, Boehme KW, Hu F, Samsom JN, Reinecker HC, Kupfer SS, Guandalini S, Semrad CE, Abadie V, Khosla C, Barreiro LB, Xavier RJ, Ng A, Dermody TS, Jabri B. 2017. Reovirus infection triggers inflammatory responses to dietary antigens and development of celiac disease. Science 356:44–50. doi: 10.1126/science.aah5298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sherry B, Torres J, Blum MA. 1998. Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Noah DL, Blum MA, Sherry B. 1999. Interferon regulatory factor 3 is required for viral induction of beta interferon in primary cardiac myocyte cultures. J Virol 73:10208–10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansson C, Wetzel JD, He J, Mikacenic C, Dermody TS, Kelsall BL. 2007. Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J Exp Med 204:1349–1358. doi: 10.1084/jem.20061587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luethy LN, Pfeiffer JK. 2012. Viral infection brings mitochondrial traffic to a standstill. Cell Host Microbe 11:420–421. doi: 10.1016/j.chom.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dionne KR, Galvin JM, Schittone SA, Clarke P, Tyler KL. 2011. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J Neurovirol 17:314–326. doi: 10.1007/s13365-011-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reference deleted.
- 14.Jacobs BL, Ferguson RE. 1991. The Lang strain of reovirus serotype 1 and the Dearing strain of reovirus serotype 3 differ in their sensitivities to beta interferon. J Virol 65:5102–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm GH, Pruijssers AJ, Li L, Danthi P, Sherry B, Dermody TS. 2010. Interferon regulatory factor 3 attenuates reovirus myocarditis and contributes to viral clearance. J Virol 84:6900–6908. doi: 10.1128/JVI.01742-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antar AA, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. 2009. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe 5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai CM, Boehme KW, Pruijssers AJ, Parekh VV, Van Kaer L, Parkos CA, Dermody TS. 2015. Endothelial JAM-A promotes reovirus viremia and bloodstream dissemination. J Infect Dis 211:383–393. doi: 10.1093/infdis/jiu476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shuai K, Liu B. 2003. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol 3:900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 19.Suhara W, Yoneyama M, Iwamura T, Yoshimura S, Tamura K, Namiki H, Aimoto S, Fujita T. 2000. Analyses of virus-induced homomeric and heteromeric protein associations between IRF-3 and coactivator CBP/p300. J Biochem 128:301–307. doi: 10.1093/oxfordjournals.jbchem.a022753. [DOI] [PubMed] [Google Scholar]
- 20.Lin R, Mamane Y, Hiscott J. 1999. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19:2465–2474. doi: 10.1128/MCB.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- 22.Witkowski JT, Robins RK, Sidwell RW, Simon LN. 1972. Design, synthesis, and broad spectrum antiviral activity of 1-beta–d-ribofuranosyl-1,2,4-triazole-3-carboxamide and related nucleosides. J Med Chem 15:1150–1154. doi: 10.1021/jm00281a014. [DOI] [PubMed] [Google Scholar]
- 23.Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. 1972. Broad-spectrum antiviral activity of Virazole: 1-beta-d-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 177:705–706. doi: 10.1126/science.177.4050.705. [DOI] [PubMed] [Google Scholar]
- 24.Zhou Y, Guo M, Wang X, Li J, Wang Y, Ye L, Dai M, Zhou L, Persidsky Y, Ho W. 2013. TLR3 activation efficiency by high or low molecular mass poly I:C. Innate Immun 19:184–192. doi: 10.1177/1753425912459975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebert DH, Deussing J, Peters C, Dermody TS. 2002. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- 26.Guglielmi KM, Johnson EM, Stehle T, Dermody TS. 2006. Attachment and cell entry of mammalian orthoreovirus. Curr Top Microbiol Immunol 309:1–38. [DOI] [PubMed] [Google Scholar]
- 27.Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. 1990. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J Virol 64:1830–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodkin DK, Nibert ML, Fields BN. 1989. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J Virol 63:4676–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. 1979. Two modes of entry of reovirus particles into L cells. J Gen Virol 45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- 30.Borsa J, Sargent MD, Copps TP, Long DG, Chapman JD. 1973. Specific monovalent cation effects on modification of reovirus infectivity by chymotrypsin digestion in vitro. J Virol 11:1017–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spendlove RS, McClain ME, Lennette EH. 1970. Enhancement of reovirus infectivity by extracellular removal or alteration of the virus capsid by proteolytic enzymes. J Gen Virol 8:83–94. doi: 10.1099/0022-1317-8-2-83. [DOI] [PubMed] [Google Scholar]
- 32.Spendlove RS, Schaffer FL. 1965. Enzymatic enhancement of infectivity of reovirus. J Bacteriol 89:597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mainou BA, Zamora PF, Ashbrook AW, Dorset DC, Kim KS, Dermody TS. 2013. Reovirus cell entry requires functional microtubules. mBio 4:e00405-13. doi: 10.1128/mBio.00405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm GH, Zurney J, Tumilasci V, Leveille S, Danthi P, Hiscott J, Sherry B, Dermody TS. 2007. Retinoic acid-inducible gene-I and interferon-beta promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem 282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- 35.Berger AK, Hiller BE, Thete D, Snyder AJ, Perez E Jr, Upton JW, Danthi P. 2017. Viral RNA at two stages of reovirus infection is required for the induction of necroptosis. J Virol 91:e02404-. doi: 10.1128/JVI.02404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis ESC. 2014. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514:372–375. doi: 10.1038/nature13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 39.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Odegard AL, Chandran K, Zhang X, Parker JS, Baker TS, Nibert ML. 2004. Putative autocleavage of outer capsid protein micro1, allowing release of myristoylated peptide micro1N during particle uncoating, is critical for cell entry by reovirus. J Virol 78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schiff LA, Nibert ML, Tyler KL. 2007. Orthoreoviruses and their replication, p 1853–1915. In Knipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2 Lippincott, Williams, & Wilkins, Philadelphia. [Google Scholar]
- 43.Snyder AJ, Danthi P. 2015. Lipid membranes facilitate conformational changes required for reovirus cell entry. J Virol 90:2628–2638. doi: 10.1128/JVI.02997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder AJ, Danthi P. 2016. Lipids cooperate with the reovirus membrane penetration peptide to facilitate particle uncoating. J Biol Chem 291:26773–26785. doi: 10.1074/jbc.M116.747477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein micro 1 undergoes conformational change and release from reovirus particles during cell entry. J Virol 77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanovic T, Agosto MA, Chandran K, Nibert ML. 2007. A role for molecular chaperone Hsc70 in reovirus outer capsid disassembly. J Biol Chem 282:12210–12219. doi: 10.1074/jbc.M610258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madren JA, Sarkar P, Danthi P. 2012. Cell entry-associated conformational changes in reovirus particles are controlled by host protease activity. J Virol 86:3466–3473. doi: 10.1128/JVI.06659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan YK, Gack MU. 2016. Viral evasion of intracellular DNA and RNA sensing. Nat Rev Microbiol 14:360–373. doi: 10.1038/nrmicro.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ploegh HL. 1998. Viral strategies of immune evasion. Science 280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- 52.Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. 2007. IkappaB kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. J Virol 81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. 2009. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol 83:2178–2187. doi: 10.1128/JVI.01787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stanifer ML, Kischnick C, Rippert A, Albrecht D, Boulant S. 2017. Reovirus inhibits interferon production by sequestering IRF3 into viral factories. Sci Rep 7:10873. doi: 10.1038/s41598-017-11469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi T, Antar AA, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. 2007. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe 1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 61.Virgin HW IV, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nibert ML, Chappell JD, Dermody TS. 1995. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved sigma 1 protein. J Virol 69:5057–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thete D, Danthi P. 2015. Conformational changes required for reovirus cell entry are sensitive to pH. Virology 483:291–301. doi: 10.1016/j.virol.2015.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang J, Coombs KM. 2005. Infectious entry of reovirus cores into mammalian cells enhanced by transfection. J Virol Methods 128:88–92. doi: 10.1016/j.jviromet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 65.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]