

ABSTRACT
Viruses often encompass overlapping reading frames and unconventional translation mechanisms in order to maximize the output from a minimum genome and to orchestrate their timely gene expression. Hepatitis C virus (HCV) possesses such an unconventional open reading frame (ORF) within the core-coding region, encoding an additional protein, initially designated ARFP, F, or core+1. Two predominant isoforms of core+1/ARFP have been reported, core+1/L, initiating from codon 26, and core+1/S, initiating from codons 85/87 of the polyprotein coding region. The biological significance of core+1/ARFP expression remains elusive. The aim of the present study was to gain insight into the functional and pathological properties of core+1/ARFP through its interaction with the host cell, combining in vitro and in vivo approaches. Our data provide strong evidence that the core+1/ARFP of HCV-1a stimulates cell proliferation in Huh7-based cell lines expressing either core+1/S or core+1/L isoforms and in transgenic liver disease mouse models expressing core+1/S protein in a liver-specific manner. Both isoforms of core+1/ARFP increase the levels of cyclin D1 and phosphorylated Rb, thus promoting the cell cycle. In addition, core+1/S was found to enhance liver regeneration and oncogenesis in transgenic mice. The induction of the cell cycle together with increased mRNA levels of cell proliferation-related oncogenes in cells expressing the core+1/ARFP proteins argue for an oncogenic potential of these proteins and an important role in HCV-associated pathogenesis.
IMPORTANCE This study sheds light on the biological importance of a unique HCV protein. We show here that core+1/ARFP of HCV-1a interacts with the host machinery, leading to acceleration of the cell cycle and enhancement of liver carcinogenesis. This pathological mechanism(s) may complement the action of other viral proteins with oncogenic properties, leading to the development of hepatocellular carcinoma. In addition, given that immunological responses to core+1/ARFP have been correlated with liver disease severity in chronic HCV patients, we expect that the present work will assist in clarifying the pathophysiological relevance of this protein as a biomarker of disease progression.
KEYWORDS: cyclin D1, HCV, Rb, cell cycle, core+1/ARFP, hepatocellular carcinoma, oncogenes
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common human cancers, accounting for more than 700,000 deaths per year, while infection with hepatitis B virus (HBV) or hepatitis C virus (HCV) remains the major cause of HCC worldwide (1, 2). HCV is associated mostly with chronic hepatitis, which often progresses to severe liver diseases, including fibrosis, cirrhosis, and HCC (3). Although no prophylactic vaccine exists to prevent HCV infection, interferon-free antiviral therapies based on direct-acting antiviral agents (DAAs) have shown significant effectiveness in all HCV genotypes (4, 5). However, access to such treatment is limited due to its high cost. Moreover, the risk of HCC development remains increased after virus elimination for patients who had liver cirrhosis at the time of treatment (6). Thus, antiviral treatment may not be sufficient to combat HCV-associated HCC, especially when diagnosed in advanced stages, suggesting the need for alternative anticancer approaches. In this regard, understanding the molecular mechanisms underlying HCV-induced HCC remains critical.
HCV is a member of the Flaviviridae family belonging to the Hepacivirus genus that replicates exclusively in the cytoplasm (7). It is a small enveloped virus with a 9.6-kb single-stranded, positive-sense RNA genome which encodes a polyprotein precursor of approximately 3,000 amino acids. Host and viral proteases process the immature polyprotein into at least 10 mature structural and nonstructural proteins (C, E1, E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (8–10). Notably, HCV is the only member of the Flaviviridae family that is linked to oncogenesis (7, 11).
Several studies from independent laboratories, including our own, have shown that an alternative open reading frame (ORF) overlapping the core coding region in the +1 frame of genotype 1a synthesizes another viral protein, named ARFP (12), F (13), or core+1 (14) protein. In HCV isolates that contain 10 consecutive adenosine residues at codons 8 to 11 of the core coding region, the core+1/ARFP protein was shown to be synthesized by a ribosomal frameshift mechanism within the A-rich area (13, 14). However, in the absence of this repetitive sequence (as it applies for the majority of HCV isolates of genotype 1), no frameshift is detected and the prevailing isoforms of core+1/ARFP in transfected cells are generated by internal translation initiation at codons 85/87 (core+1/S) (15) or codon 26 (core+l/L) (16). Importantly, recent work from our laboratory verified the expression of the two core+1/ARFP isoforms in the context of replicons derived from the genotype 2a HCV isolate JFH1 and revealed differences in the expression kinetics of the core+1/ARFP isoforms during infection (17). Importantly, the detection of core+1/ARFP-specific antibodies (18–25) and T-cell responses in HCV-infected patients (26–29) reported by several laboratories worldwide suggest that this protein is also expressed in vivo. However, core+1/ARFP is not required for JFH1 HCV (genotype 2a) replication in cultured cells (30) and for HCV genotype 1a replication in vivo in chimpanzees (31). To date, the biological significance of this protein remains elusive.
In the present report, we provide evidence that the core+1/ARFP protein promotes cell proliferation both in the context of Huh7-based cell lines expressing either core+1/S or core+1/L isoforms of HCV-1a and in transgenic mice expressing core+1/S protein in the liver. Our data indicate that both isoforms of core+1/ARFP enhance cell proliferation, increase the levels of cyclin D1 and Rb phosphorylation, and induce the expression of several oncogenes. These results lend support to the participation of core+1/ARFP in the oncogenic potential of HCV.
RESULTS
core+1/ARFP induces cell proliferation.
Previous studies using the HCV genotype 2a JFH1/Huh7.5 infectious system or JFH1-infected mice with humanized livers have shown that nonsense mutations in core+1/ARFP do not affect JFH1 replication (30, 31). In order to gain insight into the biological function of HCV core+1/ARFP, we established Huh7.5 cell lines that constitutively express core+1/L or core+1/S isoforms of HCV genotype 1a. A myc tag was added for the efficient detection of the proteins. Stable expression of core+1/L or core+1/S was achieved using the pWPI-GUN lentiviral vector (Fig. 1A and B). The expression of core+1/L and core+1/S proteins in the newly generated cell lines was confirmed by immunofluorescence using anti-myc antibody (Fig. 1C and D). Detection by Western blotting using myc tag and a homemade core+1/ARFP antibody verified the expression of core+1/L, while core+1/S was detected only at the mRNA level, possibly due to increased instability of the protein (32).
FIG 1.

Construction of Huh-7-based cell lines stably expressing core+1/S or core+1/L protein. (A) A schematic representation of the parental HCV-1a genome is shown at the top. The 5′ and 3′ nontranslated regions (NTRs) are indicated with thick black lines. The region encoding core protein and core+1/L and core+1/S constructs are drawn below. (B) A schematic representation of the myc-tagged core+1/L and core+1/S constructs and transcription features that are present in Huh7-based cell lines. Control construct was the empty pWPI-GUN vector. (C) Detection of core+1/L protein by Western blotting using myc tag and a homemade core+1 antibody (HCV-1a strain). (D) Detection of core+1/L and core+1/S protein by immunofluorescence using myc tag antibody. TO-PRO3 is used to stain nucleic acid.
Passaging the newly established cell lines as either individual colonies or the initial pool, we observed that Huh7.5c+1/L and Huh7.5c+1/S cells proliferated more rapidly than control cells. To confirm our observation, we initially performed MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] and colony-forming assays (Fig. 2A and B). Cell lines that expressed core+1/L or core+1/S protein demonstrated enhanced proliferation compared to the control. In addition, both Huh7.5c+1/L and Huh7.5c+1/S cells generated more colonies than control cells that differed in size and shape.
FIG 2.
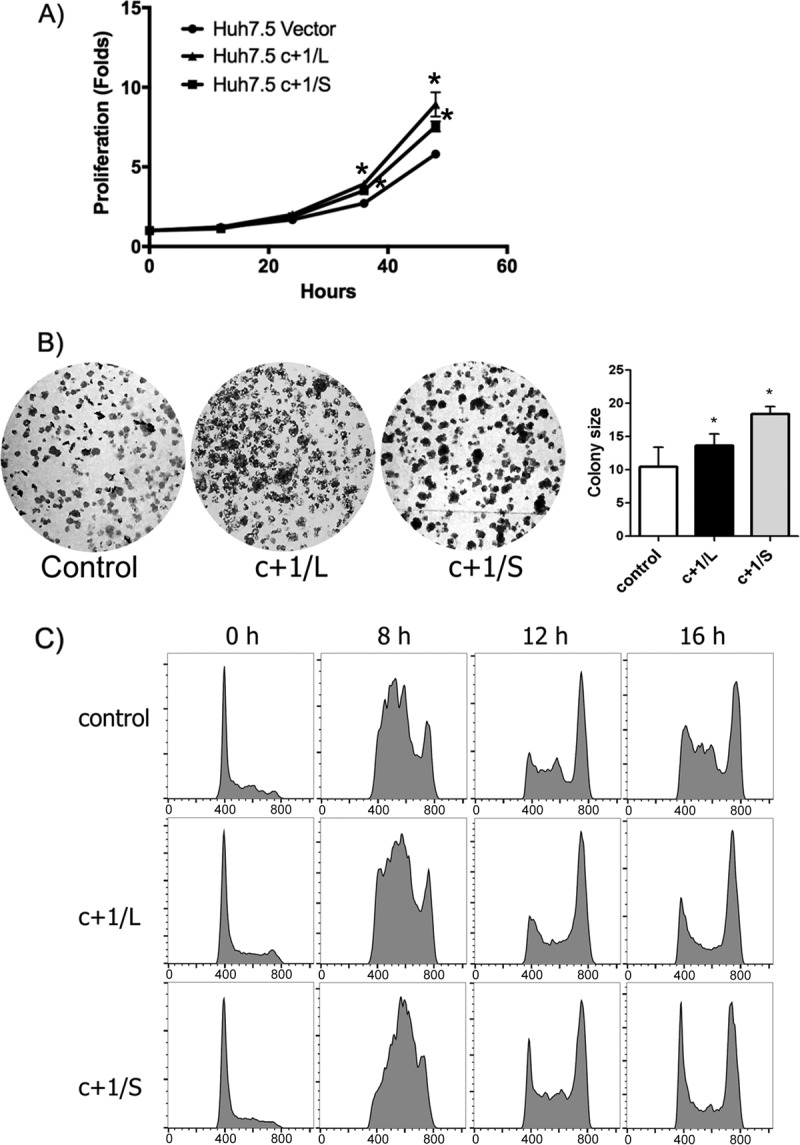
core+1/ARFP induces cell proliferation. (A) MTT assay. Huh7.5c+1/L, Huh7.5c+1/S, and control cells were seeded in triplicates into 96-well plates the previous day, and 0, 12, 24, 36, 48 h later cell proliferation was evaluated by MTT assay. Cell pools were used. The results are presented as the means ± standard deviations (SD) from three separate experiments and are expressed as fold difference relative to the 0-h value for each cell line. (B) Colony formation assay of Huh7.5c+1/L, Huh7.5c+1/S, and control cells. Both Huh7.5c+1/L and Huh7.5/S cells generated more colonies than control cells that differed in size and shape (left and right, respectively). (C) Cell cycle analysis of Huh7.5c+1/L, Huh7.5c+1/S, and control cells. Propidium iodide staining was performed and samples were analyzed by flow cytometry at 0, 8, 12, and 16 h postsynchronization with hydroxyurea.
To investigate further the hypothesis that core+1/ARFP expression induces the proliferation of hepatocytes, we studied the effect of core+1/ARFP isoforms on the progression of the cell cycle by flow cytometry. Huh7.5c+1/L, Huh7.5c+1/S, and control cells were synchronized to the same extent with hydroxyurea (HU) at G1/S phase, and the cell cycle was analyzed using propidium iodide (PI) at 0, 8, 16, and 24 h after release.
At 8 h postsynchronization, Huh7.5c+1/S cells showed increased S-phase activity compared to the control, while Huh7.5c+1/L cells showed a less marked increase of the S phase (Fig. 2C). At 16 h after release, almost all Huh7.5c+1/S and Huh7.5c+1/L cells had entered the G2/M phase, while control cells were still accumulating in the S phase (Fig. 2C). These results suggest that the expression of core+1/L or core+1/S affects cell cycle progression. The results were also recapitulated in Huh-7-based cell lines stably expressing separately the core+1 isoforms (data not shown).
core+1/ARFP enhances cyclin D1 expression and induces phosphorylation of Rb protein.
In order to elucidate the role of core+1/ARFP expression on cell cycle progression, we examined the expression levels of G1/S cell cycle checkpoint key proteins, such as cyclin D1, cyclin E, CDK2, CDK4, p21, p18, and pRb. Huh7.5c+1/L and Huh7.5c+1/S cells were synchronized at G1/S phase with 2 mM HU for 24 h and then were lysed at 0 h and 16 h after the treatment. Immediately after release (0 h) the levels of cyclin D1 were substantially increased in Huh7.5c+1/S cells compared to those in control cells, indicating an advantage in G1/S transition (Fig. 3A). On the contrary, cyclin D1 levels were only slightly increased in the Huh7.5c+1/L cells (Fig. 3A). Cyclin E, which is also associated with G1/S transition, did not present significant alterations in the presence of core+1/ARFP proteins. In terms of the respective kinases, CDK4 levels remained constant, whereas CDK2 expression was slightly affected in both Huh7.5c+1/L and Huh7.5c+1/S cells. Notably, p21, the regulatory protein of cyclin D1, demonstrated a concomitant cyclin D1 increase in the Huh7.5c+1/S cell line, whereas p18 expression was similar in all cell lines (Fig. 3A). At 16 h after release, cyclin D1 expression was remarkably induced in core+1/L cells, while its levels returned to basal levels in core+1/S cells. Cyclin E remained relatively constant, as previously found, while CDK2 expression was augmented in Huh7.5c+1/S cells. At this time point p18, p21, and CDK4 remain stable (Fig. 3A). These results indicate a role for core+1/ARFP in modulating the cyclin D1 pathway of the G1/S transition. To further investigate the effect of core+1/ARFP on cyclin D1, we performed quantitative PCR (qPCR) analysis of ccnd1 mRNA that demonstrated increased abundance in Huh7.5c+1/L and Huh7.5c+1/S cell lines (Fig. 3B). It should be mentioned that the results were consistent in both the initial pool and the selected colonies.
FIG 3.

core+1/ARFP induces the expression of cyclin D1 and pRB. (A) Expression of cell cycle regulatory proteins was determined by Western blotting. Cells were synchronized with hydroxyurea, and cell lysates were generated at 0 h and 16 h after the synchronization. (B) Expression of pRB was determined by Western blotting. Vp, V1, and V2 lanes are independent single colonies of Huh7.5 vector cells. Lp, L1, and L2 lanes are independent colonies of Huh7.5c+1/L cells, and Sp, S1, and S2 lanes are independent colonies of Huh7.5c+1/S cells. (C) mRNAs from Huh7.5 vector, Huh7.5 c+1/L, and Huh7.5 c+1/S cells were analyzed by qPCR for ccnd1, hras, vav1, c-jun, c-fos, msh3, pten, and xiap. The results are presented as fold difference relative to control cells and as the means ± SD from three independent experiments.
Finally, we studied the phosphorylation status of retinoblastoma protein (Rb), a key molecule of cell cycle progression that is initially phosphorylated by cyclin D1/CDK4/CDK6 complex. Western blot data from pooled Huh7.5c+1/L and Huh7.5c+1/S and respective isolated clones show that pRb levels are remarkably increased in the presence of core+1/ARFP isoforms, in accordance with the cell cycle progression that we described above (Fig. 3C).
core+1/ARFP induces the expression of oncogenes hras, vav1, c-jun, and c-fos.
As cell cycle acceleration is tightly associated with oncogenesis and cancer progression, we aimed to analyze the expression of selected oncogenes associated with cell proliferation and HCC, such as hras (33, 34), vav1 (35, 36), c-jun, c-fos, msh3 (37), pten (38), and xiap (39). In order to investigate possible alterations in the expression of the above-described genes by core+1, we performed qPCR analysis on total mRNA from Huh7.5c+1/L and Huh7.5c+1/S cells in either the initial pool or the individual clones (Fig. 3B). Both core+1/ARFP isoforms induced the mRNA expression of hras, vav1, c-jun, and c-fos (Fig. 3C). The mRNA levels of msh3, pten, and xiap remained unaffected.
core+1/S affects liver homeostasis.
In order to assess the function of core+1/ARFP in the context of liver physiology, we generated transgenic mice that express core+1/S under the control of the transthyretin (TTR) promoter. The TTR promoter drives expression in a liver-specific manner. A myc tag was used for the detection of protein synthesis. Transgenic mice were examined by Western blotting and qPCR for the liver-specific expression of the transgene (Fig. 4A). As core+1/ARFP was reported to be an intrinsically disordered protein (40), which may result in cell stress, we assessed the potential contribution of core+1/S to hepatocyte death. We applied an established tumor necrosis factor (TNF)-dependent acute liver failure model using lipopolysaccharide (LPS)–d-galactosamine (DGAL) in 15 TTRc+1/S and 9 wild-type littermate mice (41). Kaplan-Meier survival curves after administration of LPS-DGAL showed that core+1/S did not affect death from acute liver failure (Fig. 4B). In order to assess the effect of core+1/ARFP on liver regeneration, we applied a 2/3 partial hepatectomy (PH) model in 4 TTRc+1/S and 4 wild-type mice. During partial hepatectomy, hepatocytes are forced to enter G1 phase from steady-state G0. Incorporation of bromodeoxyuridine (BrdU) during recovery in cycling nuclei showed that the liver of TTRcore+1/S mice had 31% (P < 0.05) more BrDU+ nuclei than their wild-type littermates (Fig. 4C). In order to investigate the possible alterations in mRNA expression of hras, vav1, c-fos, and ccdn1 after liver hepatectomy, we performed qPCR analysis. Hepatectomized mice expressing core+1/S have accelerated expression of ccnd1 and c-fos genes (Fig. 5). These data support our in vitro findings that strongly suggest that core+1/ARFP enhances cell proliferation.
FIG 4.

core+1/Short affects liver homeostasis and enhances chemically induced oncogenesis. (A) Liver-specific expression of core+1/Short in TTRc+1 mice is validated through qPCR analysis of mRNAs from various animal tissues and Western blot analysis of liver tissue lysate (left and right, respectively). (B) TNF-dependent acute liver failure model using lipopolysaccharide (LPS)–d-galactosamine (DGAL). n = 15 TTRc+1/S mice and n = 9 control mice. (C) Bromodeoxyuridine (BrdU) staining after partial hepatectomy. Hepatocyte proliferation was determined by BrdU staining 40 h posthepatectomy. n = 4 TTRc+1/S mice and n = 4 control mice. (D) Model of DEN-TCPOBOP-induced carcinogenesis. Eight months after DEN administration, cancer foci had developed in both TTRcore+1/S and littermate control mice. n = 10 TTRc+1/S mice and n = 8 control mice. *, P < 0.05 versus control.
FIG 5.
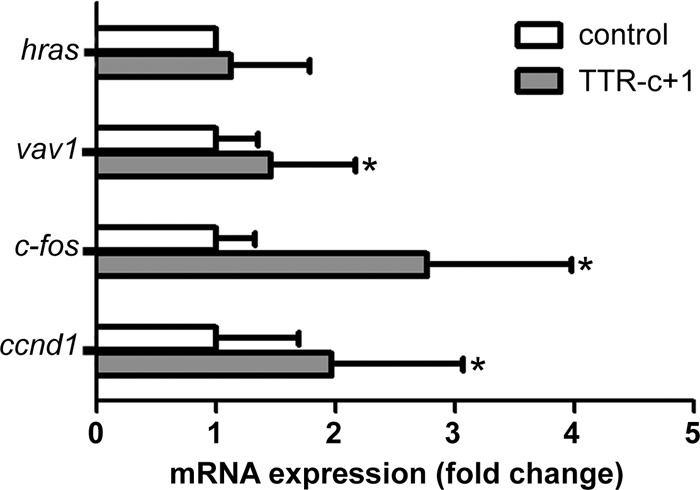
ccnd1 and c-fos expression is induced in core+1/S hepatectomized mice. mRNAs from core+1/S hepatectomized mice were analyzed by qPCR for hras, vav1, and c-fos. The results are presented as fold difference relative to control cells and represent the means ± SD from three independent experiments. n = 11 TTRc+1/S mice and n = 10 control mice. *, P < 0.05 versus control.
core+1/S enhances chemically induced oncogenesis.
While cell culture-based assays provide a mechanistic insight into the function of core+1/ARFP, assessment of its role in the physiology of the liver required an in vivo model. To assess core+1/S protein's effect on oncogenesis, TTRcore+1/S mice up to the age of 1.5 years were examined for spontaneous carcinogenesis (10 TTRc+1/S and 8 wild-type mice were used). No cancerous foci were observed either macroscopically or histologically. In order to assess the potential additive effect of core+1/S in the development of hepatocellular carcinoma, we applied the inducible model of diethyl nitrosamine (DEN)–3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene (TCPOBOP) -induced carcinogenesis. The model involves a single dose of the DEN mutagen and bimonthly continuous administration of TCPOBOP. Eight months after DEN administration, cancer foci had developed in both TTRcore+1/S and littermate control mice (Fig. 4D). TTRcore+1/S mice developed nearly twice as many tumor foci, while mean focus diameter or tumor grade did not vary between the groups. Thus, core+1/S expression enhanced oncogenesis in the presence of a de facto carcinogen.
DISCUSSION
Many mammalian positive-sense RNA viruses share distinct ORF features in their genomes. The presence of such ORFs raises a number of intriguing questions regarding their functional significance during infection, as often they are not directly involved in virus replication but may contribute to virus-cell–organism interaction (42–44).
The HCV core+1/ARF protein is encoded by an unconventional small ORF overlapping the core coding region, and it is conserved among the different HCV genotypes (18). Although core+1/ARFP expression and anti-core+1/ARFP humoral or cellular immune responses in HCV-infected individuals have been well characterized, the biological role of the protein has not been established yet. core+1/ARFP expression is dispensable for virus infection in the established JFH-1/Huh7.5 infectious system and for HCV genotype 1a replication in vivo in chimpanzees (31), a finding that has hindered studies related to core+1/ARFP (30, 31). However, the JFH1 strain is a unique HCV-2a clinical isolate with robust replication capacity in Huh7.5 cells and low pathogenicity in chimpanzees, and it demonstrates high sequence divergence from other 2a genotypes (31, 45).
In the present study, we assessed the effect of core+1/ARFP from HCV genotype 1a in hepatocyte homeostasis using stable cell lines and a transgenic mouse model. Huh-7.5 cell lines expressing either core+1/L or core+1/S showed increased proliferation activity. Huh7.5c+1/S and Huh7.5c+1/L showed enhanced cell cycle progression but with different kinetics, as the progression in Huh7.5c+1/S was much faster. The enhancement of cell cycle progression was associated with induction of cyclin D1 expression and Rb phosphorylation. Cyclin D1 promotes inactivation of tumor suppressor protein Rb through phosphorylation, which allows dissociation of the transcription factor E2F from the Rb-E2F complex, and the transcription of several target genes responsible for cycle progression through the G1/S phase (46). On the other hand, cyclin E, which also functions during the G1/S transition by activating CDK2, is not significantly affected by core+1/ARFP. However, it is known that while activation of cyclin D1 is required for G1/S transition, cyclin E participation may be dispensable (47). Intriguingly, p21 CDK inhibitor is enhanced in the presence of both core+1/ARPF isoforms. It is well known that p21 inhibits progression of the cell cycle by blocking the formation of cyclin-CDK complexes. However, under certain conditions, p21 appears to enhance assembly and activity of cyclin D1/CDK4/CDK6 complexes, suggesting a dual role for p21 (48). It is possible that core+1/ARFP promotes the oncogenic form of p21.
Cell cultures may not reflect directly the effect of core+1/ARFP in hepatocytes within the tissue, where most of the cells are in G0 phase and under liver-specific signals. In order to evaluate core+1/ARFP function in vivo, we developed transgenic mice that express HCV-1a core+1/S under a liver-specific promoter. Two-thirds partial hepatectomy, which is an established model of liver regeneration, was applied as a trigger of hepatocyte transition from G0 to cycling. core+1/ARFP-expressing mice showed increased proliferation of hepatocytes that correlated with increased cyclin D1 expression. A previous study using transgenic mice expressing F protein of HCV-1b that may correspond to core+1/L showed an increase in liver size following administration of phenobarbital (49). This increase in liver size was postulated to be dependent on Wnt signaling enhancement by F protein, as shown in cell culture. In our study, β-catenin, the main effector of Wnt signaling, showed no alteration in either Ser552 or Ser675 phosphorylation.
Cell cycle perturbation is one of the hallmarks of carcinogenesis, and oncogenic viruses often target proteins that are central regulators of these processes, such as Rb, p53, cyclins, and their respective kinases (50). Remarkably, HCV modulates the cell cycle in multiple ways, enhancing cycling in the early phases and inhibiting transition to G2/M later (51) using several of its proteins, including core, NS3, NS5A, and NS5B. HCV core protein has a pro-proliferative role through increase of c-myc stability, activation of the Wnt/β-catenin pathway, targeting of the Rb/E2F pathway, modulating p21 expression, and decreasing the p16 CDK inhibitor (52–54). NS3 and NS5A proteins appear to promote cell proliferation through repression of p21 expression, downregulation of abnormal spindle-like microcephaly-associated protein (ASPM), and upregulation of proliferating cell nuclear antigen (PCNA) (55–58). Interestingly, NS5B was shown to induce pRB ubiquitination and subsequent proteasome-mediated degradation in HCV-infected Huh7.5 cells (57).
Furthermore, we showed that both core+1/ARFP isoforms induced the transcription of several cancer-related genes, including hras, vav1, c-jun, ccnd1, and c-fos in Huh7.5 cells expressing core+1/ARFP. ccnd1 and c-fos were also shown to be overexpressed in core+1-expressing hepatectomized mice. Induction of c-fos in the model of partial hepatectomy has been well documented (59). An increase in c-fos ccnd1 transcription in the context of the liver further supports an increased liver regeneration activity and signifies an increased oncogenic potential (59).
The oncogenic potential of core+1/ARFP that can be hypothesized by the transcriptional output of cancer related genes was further evaluated in vivo. Spontaneous carcinogenesis, as reported before for HCV core protein (60), was not observed, suggesting that core+1/ARFP does not act as a carcinogenesis trigger. However, when the role of core+1/ARFP in carcinogenesis was assessed within an established chemically induced mouse model of HCC, enhancement of the oncogenic potential was observed. After administration of DEN-TCPOBOP, core+1/S-expressing mice developed 87% more tumor foci than littermate mice, although the size of the tumors and the grade were similar. The fact that core+1/ARFP did not induce the formation of spontaneous cancer foci, similar to core transgenic mice (61, 62), signifies it is not a cause of genomic instability but rather an enhancing agent that possibly acts in conjunction with a bona fide oncogenic protein, such as core (61). Future crossing of core+1/ARFP- and core-expressing mice would elaborate on such a possibility.
It should be mentioned that several clinical studies have suggested a role of core+1/ARFP in advanced stages of disease. Results of sequence analysis of clinical viral isolates derived from a microdissection of tumor and nontumor hepatocytes collected from the same subjects showed that mutations in the core-core+1/ARFP region exhibit a stronger selective pressure on the putative core+1/ARFP protein than on core protein (63). Phylogenetic analysis has shown a clustering of core+1/ARFP variants in HCC for most of the samples studied, suggesting distinct functional properties of core+1/ARFP variants in HCV-induced HCC. In addition, recent studies from our laboratory have shown high prevalence of anti-core+1/ARFP antibodies in patients with HCV-induced HCC or advanced cirrhosis compared to control groups (20, 64), suggesting an association of core+1/ARFP with virus pathogenesis of the late stages of infection. Finally, a number of previous studies that involved transient expression of the F protein in Huh7 and HepG2 cell lines have suggested an effect of the F protein on the expression of a number of cancer-related genes as well as proinflammatory cytokines and chemokines (65–69).
Overall, our data provide, for the first time, strong evidence that core+1/ARFP plays a role in HCV pathogenesis by inducing liver cell proliferation and enhancing oncogenic signals that contribute to HCC development. Clearly, however, these data need verification in appropriate infectious model systems for HCV pathogenesis when such experimental models become available.
MATERIALS AND METHODS
Cell culture.
Huh-7.5 (kindly provided by C. Rice) (70), Huh-7 (kindly provided by R. Bartenschlager) (71), and HEK293T (ATCC) cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific) supplemented with nonessential amino acids, 2 mM l-glutamine (Thermo Fisher Scientific), 100 μg/ml penicillin-streptomycin (Thermo Fisher Scientific), and 10% fetal bovine serum (Thermo Fisher Scientific) at 37°C and 5% CO2.
Plasmid construction.
HCV-1a core+1/L and HCV-1a core+1/S sequences were PCR amplified from pFK_JFH1/H77/C-842_dg plasmid (72) (nucleotides 417 to 828 and 598 to 828, respectively) and were fused with the myc epitope at the 3′ end by inserting the fragment into the pcDNA3.1(−)/myc-His B vector (XbaI restriction site). Myc-tagged fragments were amplified by PCR and cloned into lentiviral vector pWPI-GUN (73), a derivative of the bicistronic lentiviral vector pWPI. In this vector the expression of the transgene is controlled by an internal human elongation factor 1 alpha (EF1-α) promoter. The pWPI-GUN vector contains an encephalomyocarditis virus internal ribosome entry site (EMCV IRES) element that allows internal initiation of translation of green fluorescent protein (GFP)-ubiquitin-neomycin phosphotransferase fusion protein as selectable markers. Primer sequences used for PCR were the following: core+1_1a_shortF, 5′-GCGCTCTAGATATCGCCATGGTAATGAGGG-3′; core+1_1a_longF, 5′-GGATCTAGACCATGGTGGCGGTCAGAT-3′; core+1_1a_R, 5′-CGGGTCTAGAGCCGCCGTCTTCCAGAACCC-3′; core+1shortmycF, 5′-GCGGGATCCACCATGGCAATGAGGGTTGCGGGTGGGC-3′; core+1longmycF, 5′-GCGGGATCCACCATGGTGGCGGTCAGAT-3′; and core+1mycR, 5′-CGGGGATATCACAGATCCTCTTCTGAGATG-3′. The generated plasmids pWPI-C+1/L-GUN and pWPI-C+1/S-GUN were sequenced to verify their integrity.
Production of lentiviral particles.
Retroviral particles were produced by Lipofectamine cotransfection of HEK293T cells with pczVSV-G, pCMVR8.74 (pCMVR8.74 was a gift from Didier Trono, École Polytechnique Fédérale de Lausanne), and transducing lentivirus vector pWPI-GUN at a ratio of 3:3:1. Initially, 1.5 × 106 HEK293T cells were seeded in 6-cm plates 24 h prior to transfection with the three plasmids. The medium was replaced 6 h after transfection. Supernatants containing the lentiviral particles were harvested 48 h later.
Generation of stable cell lines.
Retroviral particles that encode core+1/L, core+1/S, or empty vector were used to infect 4 × 104 Ηuh7.5 and Huh7 cells. Supernatants were replaced with fresh medium 6 h postinoculation. Transduced cells were selected using 1 mg/ml G418 48 h postinoculation. Single clones were generated from polyclonal stable cell lines by colony formation and expansion. Of these subclones, clones 1 and 2 and pooled cells were used throughout this study.
Cell cycle analysis.
To synchronize cells at the G1/S transition, growing cells were treated with 2 mM HU (Sigma-Aldrich) for 24 h. Subsequently cells were washed with phosphate-buffered saline (PBS) and fresh medium was added. Following the release (0 h), cells were trypsinized, washed with PBS, and centrifuged (500 × g, 5 min). Cells were fixed overnight (4°C) with 100% ice-cold ethanol. The fixed cells were washed and then treated with 25 g/ml propidium iodide (PI; Sigma-Aldrich) and 50 g/ml RNase A (Sigma-Aldrich) for 1 h at 37°C. DNA content was determined by flow cytometry with a FACSCalibur instrument (BD Biosciences), and data were analyzed with FlowJo software (TreeStar).
MTT assay.
Huh7.5c+1/L and Huh7.5c+1/S cells (1. 2 × 104 in a 96-well plate) were seeded the previous day, and cell proliferation was evaluated by MTT assay (Sigma-Aldrich). Cells were treated with MTT at 0, 12, 24, 36, and 48 h postseeding, the product was resuspended in dimethyl sulfoxide (DMSO), and absorbance was determined at 570 nm using a microplate reader. The experiment was performed in triplicate.
Western blotting.
Cells were harvested at the indicated times and lysed in radioimmunoprecipitation assay (RIPA) buffer containing phosphatase inhibitors and protease inhibitors. Total protein concentration was determined by the bicinchoninic acid (BCA) protocol (as described by the manufacturer [Thermo Fisher Scientific]), and 50 μg of total protein was separated on SDS-PAGE gels. Each gel was transferred to nitrocellulose transfer membrane (Protran; Sigma-Aldrich). The membrane was blocked with 5% nonfat dry milk or 2.5% bovine serum albumin in PBS and 0.1% Tween for 1 h before the addition of specific antibodies. The following antibodies were used: cyclin D1, cyclin E, p21, CDK4, CDK2, cyclin D1, pRb, Rb, phospho-β-catenin (S552), phospho-β-catenin (S675), β-catenin, myc tag, and actin (Cell Signaling Technology). For the detection of core+1a, homemade rabbit polyclonal antibody against core+1/ARFP protein of genotype 1a was used. Following washing and incubation with secondary mouse monoclonal or rabbit polyclonal horseradish peroxidase-linked antibodies (Cell Signaling Technology), proteins were detected using Lumisensor horseradish peroxidase substrate (GenScript) by following the manufacturer's recommendations.
Immunofluorescence analysis.
Stably transfected Huh7.5 cells were cultured on 10-mm coverslips. One day after seeding, the cells were fixed with 3.7% paraformaldehyde for 30 min at room temperature and neutralized for 10 min with 0.1 M glycine (Sigma-Aldrich) in PBS. Cells were then washed twice with PBS and permeabilized with 0.1% Triton-X (Sigma) in PBS (permeabilization buffer) in the presence of 2 mg of bovine serum albumin per ml for 30 min at room temperature. Cells next were incubated overnight at 4°C with the myc tag antibody in the permeabilization buffer. After three washes with the permeabilization buffer, cells were incubated with anti-rabbit Alexa Fluor 488-conjugated secondary antibody (Thermo Fisher Scientific), diluted 1:2,000 in the permeabilization buffer for 1 h at room temperature. After two washes with the permeabilization buffer and one last wash with PBS containing TO-PRO-3 fluorescent stain, coverslips were mounted with Mowiol (Sigma-Aldrich) and examined by laser scanning confocal microscopy (Leica TCS-SP microscope equipped with Leica confocal software).
qPCR.
For quantitative PCR (qPCR), total RNA was extracted from cells by TRIzol reagent (Thermo Fischer Scientific) according to the manufacturer's protocol. Two μg of the RNA extract was used as the template for reverse transcription by murine leukemia virus reverse transcriptase (MLV RT) (Promega) with oligo(dT)s at 42°C for 60 min. Real-time PCR was performed in a Corbett Research Rotor-Gene 6000 under the following conditions: 95°C for 3 min to heat activate the polymerase (KAPA SYBR FAST), followed by 40 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 15 s.
Transgenic mice.
Transgenic C57BL/6 mice that express liver-specific core+1/S protein were generated by BSRC Fleming. The core+1/S gene amplified by PCR from pHPI-1495 plasmid (74) of HCV-1a was placed downstream of a TTR liver-specific promoter. core+1/S was inserted into the StuI site of the pTTR1-ExV3 plasmid, and the transgene was prepared by purifying the HindIII fragment containing the TTR promoter and core+1/S coding sequence (75). Transgenic mice were produced by pronuclear injection at the Transgenesis Facility of the Biomedical Research Foundation Academy of Athens. TTR promoter was kindly provided by Ioannis Talianidis, Institute of Molecular Biology and Biotechnology of FORTH in Crete, Heraklion, Greece. Mice were maintained under specific-pathogen-free conditions. The expression of core+1/S protein was confirmed by Western blotting with the homemade rabbit polyclonal antibody against core+1/ARFP, and the tissue-specific expression of core+1/S was determined by real-time PCR analysis of cDNAs from various transgenic mouse tissues.
PH.
Partial hepatectomy (PH) was performed on 8-week-old female and male mice according to previously described methods (76). Forty hours after PH, 100 to 200 μl (1 to 2 mg) of bromodeoxyuridine (BrdU) solution was administered intraperitoneally in mice. Partial hepatectomy was performed under ketamine (100 mg/kg of body weight) and xylazine (16 mg/kg) anesthesia.
Histological analysis.
Two hours following PH, animals were sacrificed and the liver tissue harvested. Liver tissues were fixed overnight in 10% neutral buffered formalin (NBF) solution at 4°C, embedded in paraffin, and cut into 4-mm-thick sections. The sections were deparaffinized and rehydrated before the immunochemical staining with anti-BrdU antibody (BU1; Millipore). Secondary detection was performed with the Vectastain-ABC kit (Vector Laboratories). Visualization of slides and images were taken by an Axiovert 135 (Carl Zeiss Microscopy).
Murine model of chemically induced carcinogenesis.
Male and female C57BL/6 mice were given a single dose of diethyl nitrosamine (DEN) intraperitoneally (5 μg/g; Sigma-Aldrich) at the age of 3 weeks, and every 2 weeks an intraperitoneal injection of TCPOBOP (3 μg/g; Sigma-Aldrich) was given. Mice were then sacrificed at 8 months. Livers were fixed in 10% NBF.
Murine model of acute liver failure.
Eight-week-old male and female mice were administered LPS–d-galactosamine as described previously (77).
Ethics statement.
All animal protocols used in this study were reviewed and approved in accordance with the Regulatory Guide of the Department of Animal Experiments, in line with Directive 2010/63/EU and National Decree 56/2013 requirements. Animal handling was conducted under the 6619/16-10-2014 license provided by the Prefecture of Attica Ethics Committee.
ACKNOWLEDGMENTS
We are grateful to R. Bartenschlager for kindly providing us the pFK-JFH1/H77/C842-dg plasmid and valuable discussions. We also thank N. Vassilaki for helpful suggestions.
This work was funded by the National ERC (ARISTEIA)-1560 grant (ESPA2007-2013) and State Scholarship Foundation (IKY) ARISTEIA research grant.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Farazi PA, DePinho RA. 2006. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer 6:674–687. doi: 10.1038/nrc1934. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. 2015. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN. 2012. Int J Cancer 136:E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Bandiera S, Billie Bian C, Hoshida Y, Baumert TF, Zeisel MB. 2016. Chronic hepatitis C virus infection and pathogenesis of hepatocellular carcinoma. Curr Opin Virol 20:99–105. doi: 10.1016/j.coviro.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. 2015. From non-A, non-B hepatitis to hepatitis C virus cure. J Hepatol 62:S87–S99. doi: 10.1016/j.jhep.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 5.Gotte M, Feld JJ. 2016. Direct-acting antiviral agents for hepatitis C: structural and mechanistic insights. Nat Rev Gastroenterol Hepatol 13:338–351. doi: 10.1038/nrgastro.2016.60. [DOI] [PubMed] [Google Scholar]
- 6.Wirth TC, Manns MP. 2016. The impact of the revolution in hepatitis C treatment on hepatocellular carcinoma. Ann Oncol 27:1467–1474. doi: 10.1093/annonc/mdw219. [DOI] [PubMed] [Google Scholar]
- 7.International Committee on Taxonomy of Viruses. 2000. Virus taxonomy. Classification and nomenclature of viruses. Seventh report of the International Committee on Taxonomy of Viruses. Academic Press, San Diego, CA. [Google Scholar]
- 8.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 9.Poenisch M, Bartenschlager R. 2010. New insights into structure and replication of the hepatitis C virus and clinical implications. Semin Liver Dis 30:333–347. doi: 10.1055/s-0030-1267535. [DOI] [PubMed] [Google Scholar]
- 10.Moradpour D, Penin F. 2013. Hepatitis C virus proteins: from structure to function. Curr Topics Microbiol Immunol 369:113–142. [DOI] [PubMed] [Google Scholar]
- 11.Lin MV, King LY, Chung RT. 2015. Hepatitis C virus-associated cancer. Annu Rev Pathol 10:345–370. doi: 10.1146/annurev-pathol-012414-040323. [DOI] [PubMed] [Google Scholar]
- 12.Walewski JL, Keller TR, Stump DD, Branch AD. 2001. Evidence for a new hepatitis C virus antigen encoded in an overlapping reading frame. RNA 7:710–721. doi: 10.1017/S1355838201010111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Z, Choi J, Yen TS, Lu W, Strohecker A, Govindarajan S, Chien D, Selby MJ, Ou J. 2001. Synthesis of a novel hepatitis C virus protein by ribosomal frameshift. EMBO J 20:3840–3848. doi: 10.1093/emboj/20.14.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varaklioti A, Vassilaki N, Georgopoulou U, Mavromara P. 2002. Alternate translation occurs within the core coding region of the hepatitis C viral genome. J Biol Chem 277:17713–17721. doi: 10.1074/jbc.M201722200. [DOI] [PubMed] [Google Scholar]
- 15.Vassilaki N, Mavromara P. 2003. Two alternative translation mechanisms are responsible for the expression of the HCV ARFP/F/core+1 coding open reading frame. J Biol Chem 278:40503–40513. doi: 10.1074/jbc.M305504200. [DOI] [PubMed] [Google Scholar]
- 16.Baril M, Brakier-Gingras L. 2005. Translation of the F protein of hepatitis C virus is initiated at a non-AUG codon in a +1 reading frame relative to the polyprotein. Nucleic Acids Res 33:1474–1486. doi: 10.1093/nar/gki292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotta-Loizou I, Karakasiliotis I, Vassilaki N, Sakellariou P, Bartenschlager R, Mavromara P. 2015. Expression of the novel hepatitis C virus core+1/ARF protein in the context of JFH1-based replicons. J Virol 89:5164–5170. doi: 10.1128/JVI.02351-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vassilaki N, Mavromara P. 2009. The HCV ARFP/F/core+1 protein: production and functional analysis of an unconventional viral product. IUBMB Life 61:739–752. doi: 10.1002/iub.201. [DOI] [PubMed] [Google Scholar]
- 19.Dalagiorgou G, Vassilaki N, Foka P, Boumlic A, Kakkanas A, Kochlios E, Khalili S, Aslanoglou E, Veletza S, Orfanoudakis G, Vassilopoulos D, Hadziyannis SJ, Koskinas J, Mavromara P. 2011. High levels of HCV core+1 antibodies in HCV patients with hepatocellular carcinoma. J Gen Virol 92:1343–1351. [DOI] [PubMed] [Google Scholar]
- 20.Karamitros T, Kakkanas A, Katsoulidou A, Sypsa V, Dalagiorgou G, Mavromara P, Hatzakis A. 2012. Detection of specific antibodies to HCV-ARF/CORE+1 protein in patients treated with pegylated interferon plus ribavirin. J Viral Hepatitis 19:182–188. doi: 10.1111/j.1365-2893.2011.01502.x. [DOI] [PubMed] [Google Scholar]
- 21.Gao DY, Zhang XX, Hou G, Jin GD, Deng Q, Kong XF, Zhang DH, Ling Y, Yu DM, Gong QM, Zhan Q, Yao BL, Lu ZM. 2008. Assessment of specific antibodies to F protein in serum samples from Chinese hepatitis C patients treated with interferon plus ribavirin. J Clin Microbiol 46:3746–3751. doi: 10.1128/JCM.00612-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ajorloo M, Bamdad T, Hashempour T, Alborzi AM, Mozhgani SH, Asadi R, Haj-sheykholeslami A, Merat S. 2015. Detection of specific antibodies to HCV-ARF/CORE+1 protein in cirrhotic and non-cirrhotic patients with hepatitis C: a possible association with progressive fibrosis. Arch Iran Med 18:304–307. [PubMed] [Google Scholar]
- 23.Morice Y, Ratinier M, Miladi A, Chevaliez S, Germanidis G, Wedemeyer H, Laperche S, Lavergne JP, Pawlotsky JM. 2009. Seroconversion to hepatitis C virus alternate reading frame protein during acute infection. Hepatology 49:1449–1459. doi: 10.1002/hep.22821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shehat MG, Bahey-El-Din M, Kassem MA, Farghaly FA, Abdul-Rahman MH, Fanaki NH. 2015. Recombinant expression of the alternate reading frame protein (ARFP) of hepatitis C virus genotype 4a (HCV-4a) and detection of ARFP and anti-ARFP antibodies in HCV-infected patients. Arch Virol 160:1939–1952. doi: 10.1007/s00705-015-2465-4. [DOI] [PubMed] [Google Scholar]
- 25.Tobler LH, Stramer SL, Chien DY, Lin S, Arcangel P, Phelps BH, Cooper SL, Busch MP. 2007. Antibodies to a novel antigen in acute hepatitis C virus infections. Vox Sang 92:1–7. doi: 10.1111/j.1423-0410.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 26.Gao DY, Jin GD, Yao BL, Zhang DH, Gu LL, Lu ZM, Gong Q, Lone YC, Deng Q, Zhang XX. 2010. Characterization of the specific CD4+ T cell response against the F protein during chronic hepatitis C virus infection. PLoS One 5:e14237. doi: 10.1371/journal.pone.0014237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bain C, Parroche P, Lavergne JP, Duverger B, Vieux C, Dubois V, Komurian-Pradel F, Trepo C, Gebuhrer L, Paranhos-Baccala G, Penin F, Inchauspe G. 2004. Memory T-cell-mediated immune responses specific to an alternative core protein in hepatitis C virus infection. J Virol 78:10460–10469. doi: 10.1128/JVI.78.19.10460-10469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Troesch M, Jalbert E, Canobio S, Boulassel MR, Routy JP, Bernard NF, Bruneau J, Lapointe N, Boucher M, Soudeyns H. 2005. Characterization of humoral and cell-mediated immune responses directed against hepatitis C virus F protein in subjects co-infected with hepatitis C virus and HIV-1. AIDS 19:775–784. doi: 10.1097/01.aids.0000168971.57681.6e. [DOI] [PubMed] [Google Scholar]
- 29.Park SB, Seronello S, Mayer W, Ojcius DM. 2016. Hepatitis C virus frameshift/alternate reading frame protein suppresses interferon responses mediated by pattern recognition receptor retinoic-acid-inducible gene-I. PLoS One 11:e0158419. doi: 10.1371/journal.pone.0158419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vassilaki N, Friebe P, Meuleman P, Kallis S, Kaul A, Paranhos-Baccala G, Leroux-Roels G, Mavromara P, Bartenschlager R. 2008. Role of the hepatitis C virus core+1 open reading frame and core cis-acting RNA elements in viral RNA translation and replication. J Virol 82:11503–11515. doi: 10.1128/JVI.01640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMullan LK, Grakoui A, Evans MJ, Mihalik K, Puig M, Branch AD, Feinstone SM, Rice CM. 2007. Evidence for a functional RNA element in the hepatitis C virus core gene. Proc Natl Acad Sci U S A 104:2879–2884. doi: 10.1073/pnas.0611267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z, Choi J, Lu W, Ou J-H. 2003. Hepatitis C virus F protein is a short-lived protein associated with the endoplasmic reticulum. J Virol 77:1578–1583. doi: 10.1128/JVI.77.2.1578-1583.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newell P, Toffanin S, Villanueva A, Chiang DY, Minguez B, Cabellos L, Savic R, Hoshida Y, Lim KH, Melgar-Lesmes P, Yea S, Peix J, Deniz K, Fiel MI, Thung S, Alsinet C, Tovar V, Mazzaferro V, Bruix J, Roayaie S, Schwartz M, Friedman SL, Llovet JM. 2009. Ras pathway activation in hepatocellular carcinoma and anti-tumoral effect of combined sorafenib and rapamycin in vivo. J Hepatol 51:725–733. doi: 10.1016/j.jhep.2009.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vigil D, Cherfils J, Rossman KL, Der CJ. 2010. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10:842–857. doi: 10.1038/nrc2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ura S, Honda M, Yamashita T, Ueda T, Takatori H, Nishino R, Sunakozaka H, Sakai Y, Horimoto K, Kaneko S. 2009. Differential microRNA expression between hepatitis B and hepatitis C leading disease progression to hepatocellular carcinoma. Hepatology (Baltimore, MD) 49:1098–1112. doi: 10.1002/hep.22749. [DOI] [PubMed] [Google Scholar]
- 36.Denicola G, Tuveson DA. 2005. VAV1: a new target in pancreatic cancer? Cancer Biol Ther 4:509–511. doi: 10.4161/cbt.4.5.1781. [DOI] [PubMed] [Google Scholar]
- 37.Machida K, Cheng KT, Sung VM, Shimodaira S, Lindsay KL, Levine AM, Lai MY, Lai MM. 2004. Hepatitis C virus induces a mutator phenotype: enhanced mutations of immunoglobulin and protooncogenes. Proc Natl Acad Sci U S A 101:4262–4267. doi: 10.1073/pnas.0303971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Golob-Schwarzl N, Krassnig S, Toeglhofer AM, Park YN, Gogg-Kamerer M, Vierlinger K, Schroder F, Rhee H, Schicho R, Fickert P, Haybaeck J. 2017. New liver cancer biomarkers: PI3K/AKT/mTOR pathway members and eukaryotic translation initiation factors. Eur J Cancer 83:56–70. doi: 10.1016/j.ejca.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Shi YH, Ding WX, Zhou J, He JY, Xu Y, Gambotto AA, Rabinowich H, Fan J, Yin XM. 2008. Expression of X-linked inhibitor-of-apoptosis protein in hepatocellular carcinoma promotes metastasis and tumor recurrence. Hepatology 48:497–507. doi: 10.1002/hep.22393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boumlic A, Nomine Y, Charbonnier S, Dalagiorgou G, Vassilaki N, Kieffer B, Trave G, Mavromara P, Orfanoudakis G. 2010. Prevalence of intrinsic disorder in the hepatitis C virus ARFP/core+1/S protein. FEBS J 277:774–789. [DOI] [PubMed] [Google Scholar]
- 41.Hirono S, Nakama T, Tsubouchi H. 2001. Molecular mechanisms of d-galactosamine/lipopolysaccharide-induced fulminant hepatic failure in mice and therapeutic the effects of agents. Trends Gastroenterol Hepatol 2001:59–62. doi: 10.1007/978-4-431-67895-3_8. [DOI] [Google Scholar]
- 42.Youn S, Cho H, Fremont DH, Diamond MS. 2010. A short N-terminal peptide motif on flavivirus nonstructural protein NS1 modulates cellular targeting and immune recognition. J Virol 84:9516–9532. doi: 10.1128/JVI.00775-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Muller DA, Young PR. 2013. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res 98:192–208. doi: 10.1016/j.antiviral.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Fan ZC, Bird RC. 2012. An alternative −1/+2 open reading frame exists within viral N(pro)(1-19) region of bovine viral diarrhea virus SD-1. Virus Res 163:341–351. doi: 10.1016/j.virusres.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. 1999. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gladden AB, Diehl JA. 2003. Cell cycle progression without cyclin E/CDK2: breaking down the walls of dogma. Cancer Cell 4:160–162. doi: 10.1016/S1535-6108(03)00217-4. [DOI] [PubMed] [Google Scholar]
- 48.Abbas T, Dutta A. 2009. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu W-T, Li H-C, Lee S-K, Ma H-C, Yang C-H, Chen H-L, Lo S-Y. 2013. Both core and F proteins of hepatitis C virus could enhance cell proliferation in transgenic mice. Biochem Biophys Res Commun 435:147–152. doi: 10.1016/j.bbrc.2013.04.059. [DOI] [PubMed] [Google Scholar]
- 50.Luo GG, Ou J-HJ. 2015. Oncogenic viruses and cancer. Virol Sin 30:83–84. doi: 10.1007/s12250-015-3599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kannan RP, Hensley LL, Evers LE, Lemon SM, McGivern DR. 2011. Hepatitis C virus infection causes cell cycle arrest at the level of initiation of mitosis. J Virol 85:7989–8001. doi: 10.1128/JVI.00280-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SH, Lim JS, Lim SY, Tiwari I, Jang KL. 2011. Hepatitis C virus Core protein stimulates cell growth by down-regulating p16 expression via DNA methylation. Cancer Lett 310:61–68. doi: 10.1016/j.canlet.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 53.Ruggieri A, Murdolo M, Harada T, Miyamura T, Rapicetta M. 2004. Cell cycle perturbation in a human hepatoblastoma cell line constitutively expressing hepatitis C virus core protein. Arch Virol 149:61–74. doi: 10.1007/s00705-003-0202-x. [DOI] [PubMed] [Google Scholar]
- 54.Liu J, Ding X, Tang J, Cao Y, Hu P, Zhou F, Shan X, Cai X, Chen Q, Ling N, Zhang B, Bi Y, Chen K, Ren H, Huang A, He TC, Tang N. 2011. Enhancement of canonical Wnt/beta-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS One 6:e27496. doi: 10.1371/journal.pone.0027496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghosh AK, Steele R, Meyer K, Ray R, Ray RB. 1999. Hepatitis C virus NS5A protein modulates cell cycle regulatory genes and promotes cell growth. J Gen Virol 80(Part 5):1179–1183. [DOI] [PubMed] [Google Scholar]
- 56.Banerjee A, Ray RB, Ray R. 2010. Oncogenic potential of hepatitis C virus proteins. Viruses 2:2108–2133. doi: 10.3390/v2092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munakata T, Liang Y, Kim S, McGivern DR, Huibregtse J, Nomoto A, Lemon SM. 2007. Hepatitis C virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog 3:1335–1347. doi: 10.1371/journal.ppat.0030139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu S-C, Chang SC, Wu H-Y, Liao P-J, Chang M-F. 2008. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J Biol Chem 283:29396–29404. doi: 10.1074/jbc.M802821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morello D, Fitzgerald MJ, Babinet C, Fausto N. 1990. c-myc, c-fos, and c-jun regulation in the regenerating livers of normal and H-2K/c-myc transgenic mice. Mol Cell Biol 10:3185–3193. doi: 10.1128/MCB.10.6.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanaka N, Moriya K, Kiyosawa K, Koike K, Aoyama T. 2008. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: implications for HCV-associated hepatocarcinogenesis. Int J Cancer 122:124–131. doi: 10.1002/ijc.23056. [DOI] [PubMed] [Google Scholar]
- 61.Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. 1998. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med 4:1065–1067. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- 62.Maass T, Marquardt J, Lee JS, Krupp M, Scholz-Kreisel P, Mogler C, Schirmacher P, Muller M, Westphal H, Galle PR, Teufel A. 2016. Increased liver carcinogenesis and enrichment of stem cell properties in livers of Dickkopf 2 (Dkk2) deleted mice. Oncotarget 7:28903–28913. doi: 10.18632/oncotarget.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sobesky R, Feray C, Rimlinger F, Derian N, Dos Santos A, Roque-Afonso AM, Samuel D, Brechot C, Thiers V. 2007. Distinct hepatitis C virus core and F protein quasispecies in tumoral and nontumoral hepatocytes isolated via microdissection. Hepatology 46:1704–1712. doi: 10.1002/hep.21898. [DOI] [PubMed] [Google Scholar]
- 64.Kassela K, Karakasiliotis I, Charpantidis S, Koskinas J, Mylopoulou T, Mimidis K, Sarrazin C, Grammatikos G, Mavromara P. 2017. High prevalence of antibodies to core+1/ARF protein in HCV-infected patients with advanced cirrhosis. J Gen Virol 98:1713–1719. doi: 10.1099/jgv.0.000851. [DOI] [PubMed] [Google Scholar]
- 65.Fiorucci M, Boulant S, Fournillier A, Abraham JD, Lavergne JP, Paranhos-Baccala G, Inchauspe G, Bain C. 2007. Expression of the alternative reading frame protein of hepatitis C virus induces cytokines involved in hepatic injuries. J Gen Virol 88:1149–1162. doi: 10.1099/vir.0.82575-0. [DOI] [PubMed] [Google Scholar]
- 66.Samrat SK, Li W, Singh S, Kumar R, Agrawal B. 2014. Alternate reading frame protein (F protein) of hepatitis C virus: paradoxical effects of activation and apoptosis on human dendritic cells lead to stimulation of T cells. PLoS One 9:e86567. doi: 10.1371/journal.pone.0086567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Basu A, Steele R, Ray R, Ray RB. 2004. Functional properties of a 16 kDa protein translated from an alternative open reading frame of the core-encoding genomic region of hepatitis C virus. J Gen Virol 85:2299–2306. [DOI] [PubMed] [Google Scholar]
- 68.Wu WB, Shao SW, Zhao LJ, Luan J, Cao J, Gao J, Zhu SY, Qi ZT. 2007. Hepatitis C virus F protein up-regulates c-myc and down-regulates p53 in human hepatoma HepG2 cells. Intervirology 50:341–346. doi: 10.1159/000107271. [DOI] [PubMed] [Google Scholar]
- 69.Ma HC, Lin TW, Li H, Iguchi-Ariga SM, Ariga H, Chuang YL, Ou JH, Lo SY. 2008. Hepatitis C virus ARFP/F protein interacts with cellular MM-1 protein and enhances the gene trans-activation activity of c-Myc. J Biomed Sci 15:417–425. doi: 10.1007/s11373-008-9248-9. [DOI] [PubMed] [Google Scholar]
- 70.Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol 76:13001–13014. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Friebe P, Boudet J, Simorre JP, Bartenschlager R. 2005. Kissing-loop interaction in the 3′ end of the hepatitis C virus genome essential for RNA replication. J Virol 79:380–392. doi: 10.1128/JVI.79.1.380-392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, Negro F, Dreux M, Cosset FL, Bartenschlager R. 2006. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 103:7408–7413. doi: 10.1073/pnas.0504877103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Binder M, Kochs G, Bartenschlager R, Lohmann V. 2007. Hepatitis C virus escape from the interferon regulatory factor 3 pathway by a passive and active evasion strategy. Hepatology 46:1365–1374. doi: 10.1002/hep.21829. [DOI] [PubMed] [Google Scholar]
- 74.Vassilaki N, Boleti H, Mavromara P. 2007. Expression studies of the core+1 protein of the hepatitis C virus 1a in mammalian cells. The influence of the core protein and proteasomes on the intracellular levels of core+1. FEBS J 274:4057–4074. [DOI] [PubMed] [Google Scholar]
- 75.Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. 2010. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol 30:565–577. doi: 10.1128/MCB.00927-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitchell C, Willenbring H. 2008. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc 3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 77.Mignon A, Rouquet N, Fabre M, Martin S, Pages JC, Dhainaut JF, Kahn A, Briand P, Joulin V. 1999. LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am J Resp Crit Care Med 159:1308–1315. doi: 10.1164/ajrccm.159.4.9712012. [DOI] [PubMed] [Google Scholar]