

Abstract
Airway remodeling is an important process in response to repetitive inflammatory‐mediated airway wall injuries. This is characterized by profound changes and reorganizations at the cellular and molecular levels of the lung tissue. It is of particular importance to understand the mechanisms involved in airway remodeling, as this is strongly associated with severe asthma leading to devastating airway dysfunction. In this study, we have investigated the transforming growth factor‐β (TGF β, a proinflammatory mediator)‐activated fibroblast to myofibroblast transdifferentiation pathway, which plays a key role in asthma‐related airway remodeling. We show that TGF β induces fibroblast to myofibroblast transdifferentiation by the expression of α SMA, a specific myofibroblast marker. Furthermore, Smad2/Smad3 gene and protein expression patterns are different between fibroblasts and myofibroblasts. Such a change in expression patterns reveals an important role of these proteins in the cellular phenotype as well as their regulation by TGF β during cellular transdifferentiation. Interestingly, our data show a myofibroblastic TGF β‐mediated increase in glucocorticoid receptor (GR) expression and a preferential localization of GR in the nucleus, compared to in fibroblasts. Furthermore, the GR β (nonfunctional GR isoform) is increased relative to GR α (functional isoform) in myofibroblasts. These results are interesting as they support the idea of a GR β‐mediated glucocorticoid resistance observed in the severe asthmatic population. All together, we provide evidence that key players are involved in the TGF β‐mediated fibroblast to myofibroblast transdifferentiation pathway in a human lung fibroblast cell line. These players could be the targets of new treatments to limit airway remodeling and reverse glucocorticoid resistance in severe asthma.
Keywords: Airway remodeling, glucocorticoid resistance, myofibroblast, TGFβ, transdifferentiation
Introduction
Airway remodeling refers to the structural modifications of the normal architecture of the airway wall. Such tissue reorganization involves changes in the composition and the organization of its cellular and molecular constituents contributing to the thickening of airway walls (Kuwano et al. 1993; James 1997). Airway remodeling has been attributed to repetitive injury to the airway wall arising from cycles of inflammation such as during repair, and asthma (Ganesan and Sajjan 2013). One of the main structural changes observed during remodeling includes the transdifferentiation of fibroblasts into myofibroblasts. Since myofibroblasts are practically absent in normal airways, fibroblast to myofibroblast transdifferentiation is therefore one of the key events contributing to the chronic sequel of asthma (Hackett et al. 2009), ultimately leading to permanently impaired pulmonary function (Pascual and Peters 2005).
Transforming growth factor‐β (TGFβ) exerts multiple essential biological functions. Importantly, it plays a major role in lung function and in the pathogenesis of pulmonary diseases. Dysregulations in TGFβ cellular pathway have been implicated in numerous chronic lung conditions, including asthma. Indeed, its expression has been shown to be increased in chronic lung diseases (Vignola et al. 1997). Interestingly, studies suggest that TGFβ expression is upregulated in asthmatic lungs (Redington et al. 1997; Vignola et al. 1997; Tillie‐Leblond et al. 1999; Chu et al. 2000). In the lung, TGFβ is involved in airway remodeling (Desmouliere 1995; Doherty and Broide 2007). It has been shown to promote fibroblast to myofibroblast transdifferentiation and to trigger their proliferation (Michalik et al. 2009). Therefore, taken together, these studies suggest that TGFβ is tightly associated with asthma‐related airway remodeling.
The major signaling pathway of TGFβ is through its transmembrane serine/threonine kinase receptor (Nakao et al. 1997; Attisano and Wrana 2000; ten Dijke et al. 2000; Massague and Chen 2000; Massague and Wotton 2000; Shi and Massague 2003). Activated TGFβ receptor stimulates the phosphorylation of cytoplasmic receptor‐regulated Smad proteins. The activated Smad2 and Smad3 proteins form complexes, which subsequently translocate and accumulate in the nucleus, where they regulate the transcription of target genes. Recent studies show that TGFβ‐induced transdifferentiation of fibroblasts into myofibroblasts is characterized by the induction of alpha‐smooth muscle actin (αSMA) by the Smad complex (Desmouliere et al. 1993; Ronnov‐Jessen and Petersen 1993; Roy et al. 2001). Therefore, αSMA is commonly used to define the myofibroblastic phenotype, whereas vimentin, an intermediate cytoplasmic filament protein, is routinely used as nonspecific marker for both fibroblasts and myofibroblasts (McAnulty 2007).
Glucocorticoids are the most potent anti‐inflammatory drugs used in the treatment of inflammatory diseases such as asthma (Donohue and Ohar 2004; Raissy et al. 2013). Their cellular effects are mediated by the glucocorticoid receptor (GR). In the absence of ligand, GR is primarily cytoplasmic; while following ligand binding, it translocates to the nucleus, where it induces or represses the transcription of specific target genes (Galon et al. 2002; Lu et al. 2007; Ren et al. 2012). Glucocorticoids are the most effective anti‐inflammatory medication for the treatment of asthma. Unfortunately, asthma resistance to glucocorticoids has been observed and such glucocorticoid resistance is a major barrier to the treatment of severe asthma (Holgate and Polosa 2006; Jarjour et al. 2012).
Numerous studies have proposed a role for GR isoforms in steroid resistance (Hamid et al. 1999; Sousa et al. 2000). The two major GR isoforms, GRα and GRβ, are generated through alternative splicing. While GRα mediates the actions of glucocorticoids leading to GR‐mediated cellular response, GRβ does not bind glucocorticoid agonists, limiting glucocorticoids‐mediated target gene transcription regulation (Bamberger et al. 1995; Pujols et al. 2007). Therefore, when both GR splice variants are coexpressed, GRβ acts as a dominant negative inhibitor and antagonizes the activity of GRα (Bamberger et al. 1995; Yudt et al. 2003). This ability of GRβ to inhibit GRα activity suggests that increased levels of GRβ could lead to glucocorticoid resistance (Sousa et al. 2000; Boardman et al. 2014).
Given the importance of fibroblast to myofibroblast transdifferentiation in airway diseases, we set out to investigate the TGFβ molecular pathways in fibroblasts and myofibroblasts using a human lung fibroblast cell line (WI‐38). Here we use WI‐38 treated with TGFβ to induce fibroblast to myofibroblast transdifferentiation or left untreated to demonstrate that TGFβ signaling pathway is affected in myofibroblasts. TGFβ‐treated myofibroblasts exhibit decreased Smad3 gene and protein expression. We also provide molecular evidence for a role of GRβ in glucocorticoid resistance in asthma‐like cells.
Materials and Methods
Antibodies
The primary antibodies for the following proteins were used for western blot analysis and immunofluorescence (IF): αSMA (ab7817, Abcam), α/β‐Tubulin (2148, Cell Signaling Technology), GR (sc‐8992, Santa Cruz Biotechnology), Smad2/3 (sc‐133098, Santa Cruz Biotechnology), Vimentin (sc‐5565, Santa Cruz Biotechnology).
Cell culture
Human embryonic lung fibroblasts, WI‐38 cell line, were obtained from the European Collection of Cell Cultures (ECACC, Ref No 90020107). Cells were grown in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Bovogen, Australia) and antibiotics (penicillin, streptomycin, neomycin; Life Technologies) at 37°C in a humidified atmosphere of 5% CO2.
TGFβ1 treatment
At day 0 cells were either seeded in 35 mm Petri dishes (for day 1 collection, D1) or maintained in 25 cm2 flasks for later collection (when myofibroblast transdifferentiation was obtained). Cells were incubated in DMEM with 10% FBS, antibiotics and 2–5 ng/mL TGFβ1 (R&D systems, Minneapolis, MN). TGFβ1 supplemented media was changed every second day until collection. Myofibroblast phenotype was confirmed by western blot and immunofluorescence for αSMA. Once transdifferentiation was achieved cells were collected for further analysis with respective untreated controls (see below for determination of fibroblast to myofibroblast transdifferentiation). Samples were labelled as follow D1 or D20 WI‐38 +TGFβ (with) or ‐TGFβ (without).
Western blot analysis
Overnight confluent cultures of WI‐38 ± TGFβ1 were collected for protein extraction at the specified time points. Whole‐cell lysates were collected in RIPA buffer (150 mmol/L NaCl, 1% Triton X‐100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/L Tris, with protease and phosphatase inhibitors). Then, protein extracts were heated at 90°C for 5 min in Laemmli buffer prior to SDS‐PAGE. Cell lysates were subjected to SDS‐polyacrylamide electrophoresis followed by transfer to nitrocellulose membranes in Tris‐glycine‐ethanol buffer (25 mmol/L Tris‐HCl, 192 mmol/L glycine, 20% ethanol). Blots were stained with Ponceau S (Sigma, St Louis, MO) to confirm proteins transfer and then blocked for 1 h in PBS Odyssey Blocking Buffer containing 0.1% Tween 20 or 2.5% skim milk in PBS‐T (10 mmol/L Na2HPO4, 1.7 mmol/L KH2PO4, 2.7 mmol/L KCl, 137 mmol/L NaCl, 0.1% Tween 20), prior to incubation with different primary antibodies diluted in PBS Odyssey Blocking Buffer containing 0.1% Tween 20 or 2.5% skim milk in PBS‐T overnight at 4°C. Primary antibodies were detected using LI‐COR IRDye Infrared Dye (1:15000) or horseradish peroxidase (HRP; 1:5000) secondary antibodies. Detection of HRP bound antibodies was performed with enhanced chemiluminescence (Perkin Elmer, Waltham, MA). Blots were visualized using the Odyssey Fc Infrared Imager (LI‐COR Biotechnology, NE) and quantified by densitometry using Image Studio software (LI‐COR, NE). Results are expressed as relative to corresponding α/β‐tubulin and normalized to D1 WI‐38 ‐TGFβ, which constitutes our control group. Where required, blots were stripped (2% SDS, 62.5 mmol/L Tris‐HCl, 114.4 mmol/L β‐mercaptoethanol) at 50°C for 10 min, washed and reprobed.
mRNA analysis by real‐time PCR
Total RNA was extracted from overnight confluent cultures of WI‐38 ± TGFβ1 using TRI Reagent (Sigma), first‐strand cDNA synthesized from 1 μg RNA (High‐Capacity cDNA Reverse Transcription Kit, Life Technologies) and cDNA for αSMA, GAPDH, GR, Smad2, Smad3 (see Table 1 for description of primer sequences) estimated by semiquantitative real‐time PCR using Bio‐Rad CFX96 (Bio‐Rad, CA). Data were analyzed using CFX Manager Software version 3.1 (Bio‐Rad), and is presented relative to GAPDH and normalized to D1 WI‐38 ‐TGFβ.
Table 1.
Primer sequences used for semiquantitative real‐time PCR analyses
| Gene | Forward primer | Reverse primer |
|---|---|---|
| αSMA | TTATGTTTGAGACTTTCAATGTC | GTCCAGAGGCATAGAGAG |
| GAPDH | TGGTATGACAACGAATTTGG | TCTACATGGCAACTGTGAGG |
| GR | GATGTCATTATGGAGTCTTAACTT | TTGTGCTGTCTACCTTCC |
| Smad2 | GCTTTACAGACCCATCAAAT | CCTCTTCCTATATGCCTTCT |
| Smad3 | CTACCAGTTGACCCGAAT | CAGTCTGTCTCCTGTACTC |
Immunofluorescence (IF)
Overnight confluent cultures of WI‐38 ± TGFβ1 were grown on glass coverslips. Cells were fixed in 4% formaldehyde for 10 min and blocked/permeabilized in 2% BSA containing 0.5% Triton X‐100 for 10 min. Cells were then incubated with primary antibodies overnight at 4°C and bound antibodies were detected by Alexa Fluor conjugated secondary antibodies (1:1000; Life Technologies). Coverslips were mounted on slides in ProLong Gold reagent with DAPI (Life Technologies). Samples were examined under a Nikon Ti Eclipse confocal laser‐scanning microscope (CLSM) with Nikon 60x/1.40 oil immersion lens (Plan Apo VC OFN25 DIC N2; optical section of 0.5 μm) and the NIS Elements AR software (Nikon Corporation, Japan). ImageJ 1.48v shareware (courtesy of Wayne Rasband, NIH) was used to analyze the digital images to determine the relative intensity of fluorescence in the nucleus (Fn) and in the cytoplasm (Fc) in order to estimate the nucleus‐cytoplasm fluorescence ratio (Fn/c ratio) after the subtraction of background fluorescence. Fn and Fc intensities were measured from 40 cells in each experimental condition.
Myofibroblast determination
It is well accepted that cell expression of αSMA is a specific marker of fibroblast to myofibroblast transdifferentiation, whereas vimentin is considered as a nonspecific fibroblast and myofibroblast marker (McAnulty 2007). Therefore, we used αSMA and vimentin antibodies to determine fibroblast to myofibroblast transdifferentiation by IF and western blot analysis. In this study, we defined fibroblast to myofibroblast transdifferentiation using the following criteria: (1) coexpression of αSMA and vimentin should be observed in cells using IF staining and (2) TGFβ‐treated samples with positive αSMA IF staining should be associated with at least a fivefold increase in αSMA expression.
Statistical analyses
Data are presented as mean ± SEM for all experiments. From these experiments, statistical analysis was performed setting the level of statistical significance at P < 0.05. Statistical analysis was performed using two‐way ANOVA followed by correction for multiple comparisons using Sidak's test in Prism 6 software (GraphPad software, CA). Data were grouped by treatment and time.
Results
Effect of TGFβ treatment on αSMA and vimentin expression in human lung fibroblasts and myofibroblasts
TGFβ plays a key role in airway remodeling, partly due to its capacity to induce fibroblast to myofibroblast transdifferentiation. Therefore, we first examined that TGFβ treatment could induce such a transdifferentiation in WI‐38 cell line. Vimentin protein expression was measured at all timepoints (D1, D10, D15, and D20) and no significant change in its expression was observed through the treatment period (Two‐way ANOVA: P = 0.85; n = 3 in each group; Fig. 1A). Upon TGFβ stimulation, WI‐38 showed a significant change in αSMA protein expression compared to untreated cells (Two‐way ANOVA: P = 0.0006; n = 3 in each group; Fig. 1B). Furthermore, higher αSMA protein expression was observed at all timepoints with an 8.1‐fold increase measured at D20 (Sidak's test, P < 0.001; n = 3 in each group; Fig. 1B), which is higher than the fivefold threshold we set to determine cell transdifferentiation.
Figure 1.
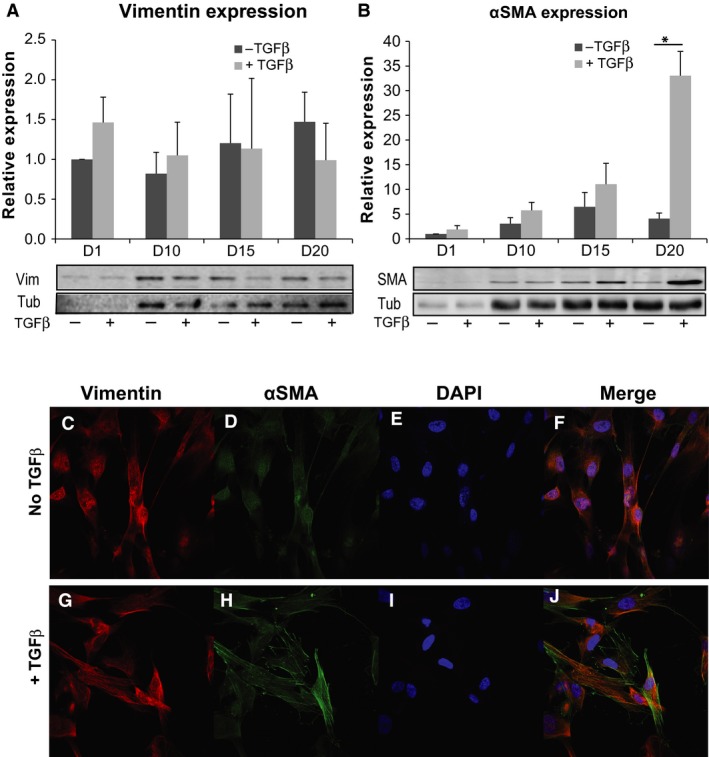
TGF β induces fibroblast to myofibroblast transdifferentiation. (A and B) Graphs show the relative protein expression for vimentin and α SMA in TGF β‐untreated and ‐treated cells at indicated timepoints. Representative western blots for vimentin and α SMA in the different experimental conditions are shown below the graphs; Tub = tubulin used as loading control. Note the significant increase in α SMA protein expression at D20 in B. (C–J) Immunofluorescence staining for vimentin, α SMA and DAPI in TGF β‐untreated (top) and ‐treated (bottom) cells. +TGF β: TGF β‐treated cells; ‐ TGF β: TGF β‐untreated cells; *P < 0.05; data are mean ± SEM from three independent experiments.
In parallel, indirect IF was performed, where monolayers of WI‐38 ± TGFβ grown on coverslips were fixed at D20 and probed for vimentin (Fig. 1C and G) in combination with αSMA (Fig. 1D and H) and counterstained with DAPI (Fig. 1E and I). Consistent with the western blot analysis, indirect IF showed that WI‐38 cells treated with TGFβ induced fibroblast to myofibroblast transdifferentiation, indicated by the presence of positive αSMA staining in the treated cells (Fig. 1H and J) and the presence of vimentin (Fig. 1G) similar to the untreated cells (Fig. 1C). Taken together, these data suggest that treatment of WI‐38 with TGFβ induced fibroblast to myofibroblast transdifferentiation at D20. Thus, following experiments will describe specific differences observed between fibroblasts (D1 WI‐38 ± TGFβ, D20 WI‐38 ‐TGFβ) and transdifferentiated myofibroblasts (D20 WI‐38 + TGFβ).
Effects of TGFβ treatment on Smad2/3 expression in human lung fibroblasts and transdifferentiated myofibroblasts
The mRNA levels of Smad2 and Smad3 were lower in TGFβ‐treated fibroblast (D1 WI‐38 + TGFβ) compared to untreated cells (D1 WI‐38 ‐ TGFβ), with approximately two and sixfold decrease, respectively (Fig. 2A and B). Expression of Smad2 and Smad3 genes was lower in D20 WI‐38 ‐ TGFβ compared to D1 WI‐38 ‐ TGFβ, whereas it remained similar in D1 WI‐38 + TGFβ compared to D20 WI‐38 + TGFβ. Overall Smad2 mRNA levels were higher and Smad3 mRNA levels lower in myofibroblast (D20 WI‐38 + TGFβ) compared to their respective control (D20 WI‐38 ‐ TGFβ).
Figure 2.
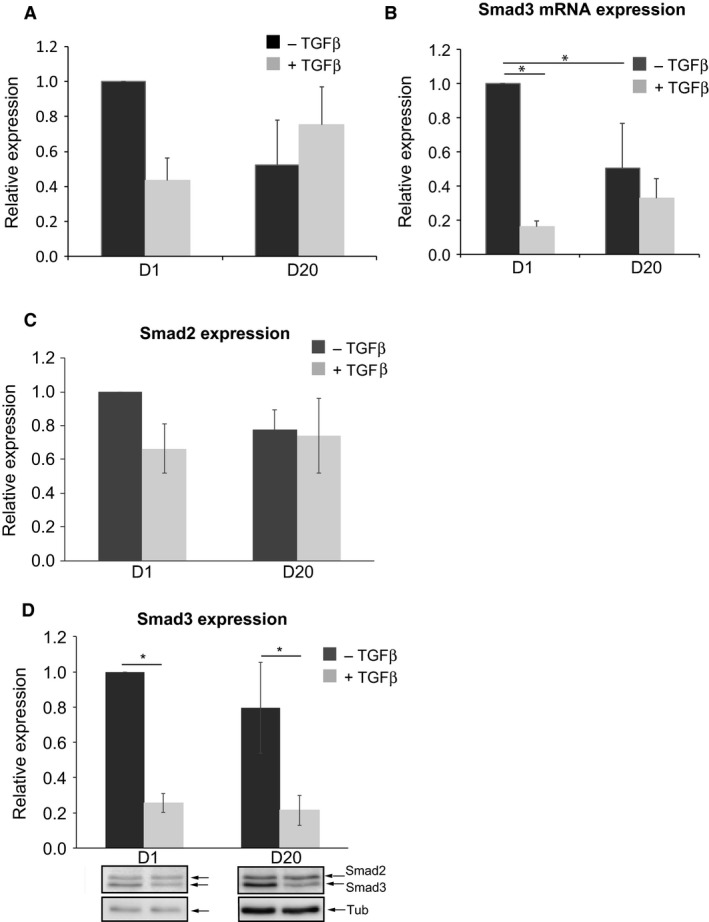
Characterization of Smad2 and Smad3 expression in fibroblast and myofibroblast during TGF β treatment. (A and B) Graphs show the relative expression of Smad2 and Smad3 mRNAs in fibroblast and myofibroblast in TGF β‐untreated and ‐treated cells. (C and D) Graphs show the relative expression of Smad2 and Smad3 proteins in fibroblast and myofibroblast in TGFβ‐untreated and ‐treated cells. Representative western blot for Smad2/3 in the different experimental conditions is illustrated below (D) Tub = tubulin used as loading control. +TGFβ: TGFβ‐treated cells; ‐TGFβ: TGFβ‐untreated cells. Data shown are mean ± SEM from four independent experiments; *P < 0.05.
A similar pattern was observed in Smad2 and Smad3 protein expression with approximately 1.5 and a significant, fourfold decreased expression in TGFβ‐treated fibroblast (D1 WI‐38 + TGFβ) compared to untreated fibroblasts (D1 WI‐38 ‐ TGFβ), respectively (Fig. 2C,D). Smad2 protein expression was unchanged in D20 WI‐38 ‐TGFβ compared to D20 WI‐38 + TGFβ, whereas Smad3 expression was significantly decreased 4.1‐fold in D20 WI‐38 + TGFβ compared to D20 WI‐38 ‐ TGFβ (Two‐way ANOVA, P = 0.009; Sidak's test, P = 0.036). These results indicate that TGFβ treatment induces changes in Smad2 and Smad3 gene and protein expression patterns in both fibroblasts and myofibroblasts. These changes in protein and gene patterns might underlie a differential TGFβ‐induced signaling pathway between fibroblast and myofibroblast.
TGFβ‐induced transdifferentiation upregulates GRβ expression in human lung myofibroblasts
There was a significant change in GR mRNA levels in TGFβ‐treated fibroblasts (D1 WI‐38 + TGFβ) compared to untreated fibroblasts but this was not reflected in the protein data (D1 WI‐38 ‐ TGFβ; Fig. 3A and B). We observed a 2.0‐fold (nonsignificant) increase in GR mRNA and total protein expression in TGFβ‐treated myofibroblasts (D20 WI‐38 + TGFβ) compared to cells left untreated (D20 WI‐38 ‐ TGFβ; Fig. 3A and B). Interestingly, a time‐dependent increase in GR protein levels was observed on TGFβ treatment (D1 WI‐38 + TGFβ compared to D20 WI‐38 + TGFβ; Fig. 3B). GR is known to be expressed in two different isoforms (GRα and GRβ) due to differential splicing. Thus, we then sought to establish which GR isoform was present in fibroblasts and myofibroblasts (±TGFβ). We found that GRα was present in both fibroblasts and myofibroblasts, regardless of TGFβ treatment (Fig. 3C). However, its expression was increased 1.3 and 1.5‐fold in TGFβ‐treated cells (D1 WI‐38 + TGFβ and D20 WI‐38 + TGFβ, respectively) compared to their respective untreated controls (D1 WI‐38‐TGFβ and D20 WI‐38 ‐ TGFβ). Surprisingly, a 1.5‐fold increase in GRβ expression was also observed in D1 WI‐38 + TGFβ compared to the untreated group (D1 WI‐38 ‐ TGFβ, Fig. 3D). Moreover, GRβ was significantly expressed in D20 WI‐38 + TGFβ compared to D20 WI‐38 ‐ TGFβ (Fig. 3D), with a 3.0‐fold increase in the TGFβ‐treated group compared to the untreated group at D20 (Two‐way ANOVA, P = 0.038, n = 3 in each treatment group; Sidak's test, P = 0.01). In summary, increased GRα and GRβ protein levels were mainly observed in TGFβ‐treated cells, regardless of the cell phenotype (Fig. 3C and D). GRα and GRβ proteins are the active and the inactive isoforms of GR, respectively. Furthermore, their respective level in cells is thought to be involved in the glucocorticoids cell response. So, we then focused on the ratio of GRα and GRβ isoforms under our experimental conditions. Interestingly, our results show that despite an increase in both GRα and GRβ isoforms, GRβ is the predominant isoform in TGFβ‐treated cells at both D1 and D20 compared to their respective controls (D1: GRβ:GRα = 1.2; D20: GRβ:GRα = 1.6; Fig. 3E).
Figure 3.
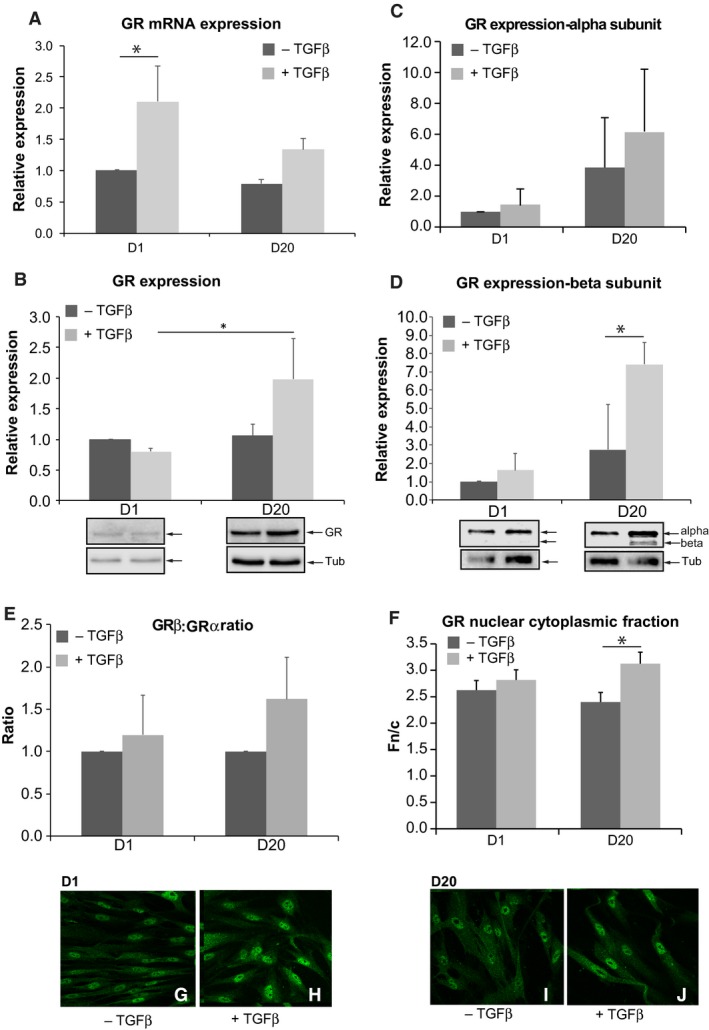
Glucocorticoid receptor expression in fibroblast and myofibroblast following TGF β treatment. (A) Graph shows the relative expression of glucocorticoid receptor (GR) mRNA in fibroblast and myofibroblast in TGF β‐untreated and ‐treated cells. +TGF β: TGF β‐treated cells; ‐TGF β: TGF β‐untreated cells. Data are mean ± SEM from three independent experiments; *P < 0.05. (B) Graph shows the relative expression of GR proteins in fibroblast and myofibroblast in TGF β‐untreated and ‐treated cells. Below the graph, representative western blots are illustrated for the different experimental conditions; Tub = tubulin used as loading control. +TGF β: TGF β‐treated cells; ‐ TGF β: TGF β‐untreated cells. Data are mean ± SEM from three independent experiments; *P < 0.05. (C and D) Graphs show GR alpha subunit (C) and beta subunit (D) in fibroblast and myofibroblast in TGF β‐untreated and ‐treated cells. The representative western blot for both isoforms is illustrated below (D). +TGF β: TGF β‐treated cells; ‐ TGF β: TGF β‐untreated cells. Data shown are mean ± SEM from three independent experiments; *P < 0.05. (E) Graph shows the ratio of GR β:GR α isoforms for each experimental condition. Note that TGF β increases GR β isoform. Data were normalized to D1 –TGF β. +TGF β: TGF β‐treated cells; ‐ TGF β: TGF β‐untreated cells. (F) Graph shows the relative fluorescence in the nucleus compared to that in the cytoplasm (Fn/c) of GR in both fibroblast and myofibroblast cells in different experimental conditions. Note the increased Fn/c at D20 in presence of TGF β. Each datapoint represents mean ± SEM of data from 40 cells; *P < 0.05. +TGF β: TGF β‐treated cells; ‐ TGF β: TGF β‐untreated cells. (G–J) Immunofluorescence images for GR expression in different experimental conditions: at D1–TGF β (G), at D1+TGF β (H), at D20–TGF β (I), at D20+TGF β (J).
We then investigated if GR localization was altered in TGFβ‐treated cells. GR activation occurs in the cytoplasm, whereas its cellular action occurs in the nucleus, via a translocation mechanism. As shown in Figure 3G–J, GR nuclear localization was increased in TGFβ‐treated myofibroblast phenotype cells (D20 WI‐38 + TGFβ) compared to untreated cells (D20 WI‐38 ‐ TGFβ). Image analysis confirmed this observation, with a significant difference in the relative fluorescence in the nucleus compared to that in the cytoplasm (Fn/c) between treated and untreated cells at D20 (Two‐way ANOVA, P = 0.017, n = 40; Fig. 3F, I and J). No significant change was detected at D1 between TGFβ‐treated and ‐untreated fibroblasts (Two‐way ANOVA: P = 0. 71; n = 40; Fig. 3F–H).
Taken together, these data show that TGFβ treatment in WI‐38 cells increases both GRα and GRβ expression irrespective of the time of treatment. Furthermore, we demonstrate that GR is predominantly localized in the nucleus of transdifferentiated myofibroblast upon TGFβ treatment.
Discussion
Fibroblast to myofibroblast transdifferentiation is one of the pivotal events contributing to chronic asthma sequels, which can result in severe impaired lung function. In this study, we focus our attention on the key molecular components involved in this transdifferentiation process. Our data describe for the first time that TGFβ treatment of human lung fibroblast cell line (WI‐38) induces fibroblast to myofibroblast transdifferentiation. This mechanism is accompanied by an altered TGFβ signaling pathway, involving at least Smad2/3. Importantly, we have also demonstrated that GRβ expression is increased in myofibroblasts, which is potentially responsible for the glucocorticoid resistance observed in severe asthma.
Airway remodeling in asthma
Airway remodeling is a key process leading to the progression of the symptoms associated with impaired pulmonary function in asthma (Lazaar and Panettieri 2003; Nihlberg et al. 2006; Yamauchi 2006). Airway remodeling is defined as a response of the airway wall to repetitive tissue injury leading to chronic inflammation and partial repair process (Nihlberg et al. 2006; Postma and Timens 2006; Yamauchi 2006). This remodeling is characterized by complex structural changes from cellular to molecular level, altering airway wall function and its constituents, among them: epithelium destruction, goblet cells hyperplasia, angiogenesis, and increase in basement membrane thickness due to extracellular matrix deposition ‐the so‐called subepithelial fibrosis (McDonald 2001; Postma and Timens 2006; Yamauchi 2006).
During asthma, inflammatory cells invade the airway wall and in combination with airway wall cells, they secrete inflammatory mediators like cytokines, including TGFβ, which is described as playing an important role in regulating the airway remodeling process (Minshall et al. 1997; Panettieri 2003; Xu et al. 2003; Kay et al. 2004). Indeed, TGFβ has numerous effects depending on cellular environment and cell condition (Makinde et al. 2007). Among the different responses to TGFβ stimulation, the subepithelial layer fibrosis is one of the important processes occurring during airway remodeling. This fibrosis corresponds to the deposition of extracellular matrix (ECM) leading to the thickening of the basement membrane and the subepithelial layer.
TGFβ induces fibroblast to myofibroblast transdifferentiation
Myofibroblasts are almost absent in normal lung tissue. It has been shown that fibroblast to myofibroblast transdifferentiation is a critical event in the development of progressive lung fibrosis (Kuhn and McDonald 1991; Zhang et al. 1994; Phan 2002). Indeed, fibroblast to myofibroblast transdifferentiation is an important process leading to the subepithelial fibrosis (Makinde et al. 2007). TGFβ is classically described as an important fibrogenic factor and it has been shown to trigger fibroblast to myofibroblast transdifferentiation in vitro as well as in vivo (Ronnov‐Jessen and Petersen 1993; Sime et al. 1997; Hashimoto et al. 2001). Furthermore, studies show that TGFβ is involved in the pathogenesis of asthma. Indeed, TGFβ upregulation in asthmatic airways triggers fibroblast to myofibroblast transdifferentiation (Redington et al. 1997; Vignola et al. 1997; Tillie‐Leblond et al. 1999; Chu et al. 2000).
Our data show that treatment with TGFβ for 20 days induces the expression of αSMA in WI‐38 cell line (see Fig. 1). This intermediate filament protein is classically described as the principal feature in characterizing myofibroblastic phenotype (McAnulty 2007). Thus, we show that TGFβ induces WI‐38 cell line transdifferentiation into myofibroblasts. Our data demonstrate that WI‐38 human lung fibroblast cell line can be used as a model of TGFβ‐mediated myofibroblast transdifferentiation. This result is important as this model enables us to study molecular aspects of fibroblast to myofibroblast transdifferentiation‐related diseases in vitro, and more particularly TGFβ‐associated lung conditions, like asthma.
TGFβ‐induced Smad3 downregulation in myofibroblast
The main intracellular signaling pathway of TGFβ is well characterized. This pathway involves the activation of the transmembrane serine/threonine kinase TGFβ receptor, leading to Smad2 and Smad3 phosphorylation (receptor‐regulated Smads or R‐Smads). They then form a heteromeric complex with Smad4 (common‐mediator Smad or Co‐Smad), which translocates to the nucleus where it regulates the transcription of specific target genes. This pathway is regulated by inhibitory Smads (including Smad7), which inhibit Smad2/3 phosphorylation during TGFβ stimulation (Attisano and Wrana 2002; Shi and Massague 2003).
Our results demonstrate that TGFβ differentially regulates Smad2 and Smad3 expression in WI‐38 cells. Indeed, Smad2 expression is not affected by TGFβ at D20 (see Fig. 2A and C). Whereas, Smad3 gene and protein expressions are strongly downregulated following TGFβ treatment at both D1 and D20 (see Fig. 2B and D).
Hu et al. (2003) have shown that TGFβ treatment increases Smad3 expression in primary rat lung fibroblasts. Furthermore, Gu et al. (2007) found that TGFβ‐induced αSMA gene upregulation is under the control of Smad3 in vitro. In our study, we show that αSMA gene expression is increased in TGFβ‐treated cells (data not shown), similar to its protein expression (Fig. 1). This would suggest a role for Smad3 in fibroblast to myofibroblast transdifferentiation in WI‐38 cells. Interestingly, our data indicate a decrease in Smad3 expression following TGFβ treatment (see Fig. 2D). This discrepancy with previous studies can be explained by the complex nature of the Smads signaling pathway. Indeed, αSMA gene promoter is also under the control of Smad2 in rodent vascular cells treated with TGFβ (Corjay et al. 1989; Mack and Owens 1999). Our data do not indicate changes in Smad2 protein in TGFβ‐transdifferentiated myofibroblast at D20. So, we can hypothesize that αSMA expression during TGFβ treatment is mediated through Smad2. Furthermore, it is also possible that other TGFβ‐activated signaling pathways may contribute to the transcriptional regulation of αSMA in myofibroblast. Indeed, it has been described that non‐Smad pathways, such as the phosphatidylinositol 3‐kinase (PI3K)/Akt pathway, participates in TGFβ‐induced myofibroblast formation during epithelial to mesenchymal transition (Kim et al. 2006; Lamouille and Derynck 2007). Further experiments should be performed in order to clarify the cellular pathway(s) involved in the fibroblast to myofibroblast transdifferentiation and more precisely in the regulation of αSMA gene expression.
Glucocorticoid receptor upregulation in TGFβ induced myofibroblast
Glucocorticoids play an important role in many physiological functions essential for life, such as growth, inflammation, tissue repair, reproduction, metabolism, immune, cardiovascular, and nervous system functions (Kadmiel and Cidlowski 2013). In the cell, glucocorticoid responses are mediated by the activation and nuclear translocation of the cytoplasmic glucocorticoid receptor (GR). This activated receptor then regulates the transcription of target genes. In humans, alternate splicing of GR mRNA results in the synthesis of two functionally different isoforms. GRα is known to be the predominant isoform responsible for GR cellular response (Reichardt and Schutz 1998). While GRβ is a dominant negative isoform of GR, whose presence induces inhibition of glucocorticoid activity (Bamberger et al. 1995). Thus, the cellular ratio of the two isoforms of GR could influence the cellular responses to glucocorticoids.
Studies show that GR is expressed in lung fibroblasts as well as myofibroblasts (Eickelberg et al. 1999; Baouz et al. 2005). Consistent with these studies, our data indicate that GR is expressed in WI‐38 cell line regardless of time and TGFβ treatment. However, TGFβ treatment induces a 2.0 and 1.5‐fold increase in GR mRNA and protein expression at D20, respectively (Fig. 3A and B). Our results demonstrate for the first time a differential expression of GR in WI‐38 fibroblast and myofibroblast. The expression of GR in cells, especially in fibroblasts and myofibroblasts, is important for cell homeostasis. Unfortunately, there is a lack of evidence correlating the observed increase in GR expression and fibroblast to myofibroblast transdifferentiation. Further investigation is required to elucidate the cellular mechanisms involved in GR regulation during TGFβ‐induced fibroblast to myofibroblast transdifferentiation. Thus, our study leads to important questions on the role of GR and its regulation in myofibroblast‐related disease (such as asthma).
Our data show a significant increase in GR nuclear localization in TGFβ‐transdifferentiated myofibroblast (D20, Fig. 3E–I). We also show that GRβ isoform expression is strongly increased in transdifferentiated myofibroblast (D20, Fig. 3D). While we do not directly demonstrate the nature of the nuclear GR isoform, it is tempting to postulate that the observed increased GR in the nucleus is due to GRβ isoform. Indeed, studies show that GRβ isoform is constitutively localized in the nucleus of cells (Lewis‐Tuffin and Cidlowski 2006; Kino et al. 2009). In our study, the accumulation of GRβ in the nucleus could be due to (1) a decrease in GRβ degradation, (2) an increase in GRβ expression and/or 3) an increase in nucleo‐cytoplasmic transport of this protein. Current research in the group is focused on assessing how TGFβ regulates GR isoform expression and activity in the WI‐38 cell line.
All together, our data indicate that TGFβ‐treated WI‐38 cell line is a relevant model to investigate the cellular and molecular mechanisms involved in fibroblast to myofibroblast transdifferentiation occurring in asthma.
Furthermore, our data indicate that GRβ expression is predominant compared to GRα in TGFβ‐transdifferentiated myofibroblast, as indicated by the increase in GRβ:GRα ratio (D20, Fig. 3E). Interestingly, numerous studies demonstrate that increased GRβ is associated with lung diseases, such as asthma (Hamid et al. 1999; Christodoulopoulos et al. 2000; Sousa et al. 2000; Pujols et al. 2004; Goleva et al. 2006). These studies also indicate that increased expression of the nonfunctional GRβ isoform may account for glucocorticoid insensitivity in inflammatory diseases, among them asthma (Leung et al. 1997; Hamid et al. 1999; Sousa et al. 2000; Goleva et al. 2006). We can therefore hypothesize that TGFβ‐transdifferentiated myofibroblasts from WI‐38 cell line present a resistance to glucocorticoid treatment. This cellular model represents a critical tool in the understanding of the underlying mechanisms of glucocorticoid insensitivity observed in steroid‐resistant severe asthma.
Conclusion
In conclusion, the results of this study show that TGFβ induces (1) fibroblast to myofibroblast transdifferentiation in a human lung fibroblast cell line, (2) decrease in Smad3 expression in myofibroblasts, 3) increase in GRβ expression in myofibroblasts.
TGFβ plays an important role in airway remodeling. This remodeling involves transdifferentiation of fibroblasts into myofibroblasts, a key event initiating tissue fibrosis observed in many lung diseases, such as asthma. This transdifferentiation mechanism implicates Smad3, a component of the TGFβ signaling pathway. While Smad3 seems to be affected during WI‐38 myofibroblast transdifferentiation, the exact role of Smad3 in fibroblast to myofibroblast transdifferentiation is still not fully understood. Attention has been given to a potential role for GR to inhibit TGFβ signaling pathway through a regulation of Smad3 (Song et al. 1999). So, further studies should focus on the interaction between TGFβ, Smad3, GR and possible glucocorticoid insensitivity in WI‐38 human lung fibroblast cell line to provide greater insights in asthma‐related fibroblast to myofibroblast transdifferentiation and glucocorticoid resistance.
The human lung fibroblast WI‐38 cell line used in this study represents a promising model to study lung diseases and more particularly asthma and glucocorticoid‐resistant asthma in providing better understanding in the cellular and molecular aspects of such disease after myofibroblast transdifferentiation.
Conflict of Interest
The authors declare no conflict of interest.
Breton J.‐D., Heydet D., Starrs L. M., Veldre T., Ghildyal R.. Molecular changes during TGFβ‐mediated lung fibroblast‐myofibroblast differentiation: implication for glucocorticoid resistance. Physiol Rep, 6 (7), 2018, e13669, https://doi.org/10.14814/phy2.13669
Funding Information
This work is supported by the University of Canberra Strategic Research Funds (grant to R. Ghildyal, postdoctoral fellowship to D. Heydet) and an Early Career Grant from Centre for Research in Therapeutic Solutions (to D. Heydet).
References
- Attisano, L. , and Wrana J. L.. 2000. Smads as transcriptional co‐modulators. Curr. Opin. Cell Biol. 12:235–243. [DOI] [PubMed] [Google Scholar]
- Attisano, L. , and Wrana J. L.. 2002. Signal transduction by the TGF‐beta superfamily. Science 296:1646–1647. [DOI] [PubMed] [Google Scholar]
- Bamberger, C. M. , Bamberger A. M., de Castro M., and Chrousos G. P.. 1995. Glucocorticoid receptor beta, a potential endogenous inhibitor of glucocorticoid action in humans. J. Clin. Invest. 95:2435–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baouz, S. , Giron‐Michel J., Azzarone B., Giuliani M., Cagnoni F., Olsson S., et al. 2005. Lung myofibroblasts as targets of salmeterol and fluticasone propionate: inhibition of alpha‐SMA and NF‐kappaB. Int. Immunol. 17:1473–1481. [DOI] [PubMed] [Google Scholar]
- Boardman, C. , Chachi L., Gavrila A., Keenan C. R., Perry M. M., Xia Y. C., et al. 2014. Mechanisms of glucocorticoid action and insensitivity in airways disease. Pulm. Pharmacol. Ther. 29:129–143. [DOI] [PubMed] [Google Scholar]
- Christodoulopoulos, P. , Leung D. Y., Elliott M. W., Hogg J. C., Muro S., Toda M., et al. 2000. Increased number of glucocorticoid receptor‐beta‐expressing cells in the airways in fatal asthma. J. Allergy Clin. Immunol. 106:479–484. [DOI] [PubMed] [Google Scholar]
- Chu, H. W. , Trudeau J. B., Balzar S., and Wenzel S. E.. 2000. Peripheral blood and airway tissue expression of transforming growth factor beta by neutrophils in asthmatic subjects and normal control subjects. J. Allergy Clin. Immunol. 106:1115–1123. [DOI] [PubMed] [Google Scholar]
- Corjay, M. H. , Thompson M. M., Lynch K. R., and Owens G. K.. 1989. Differential effect of platelet‐derived growth factor‐versus serum‐induced growth on smooth muscle alpha‐actin and nonmuscle beta‐actin mRNA expression in cultured rat aortic smooth muscle cells. J. Biol. Chem. 264:10501–10506. [PubMed] [Google Scholar]
- Desmouliere, A. 1995. Factors Influencing Myofibroblast Differentiation during Wound‐Healing and Fibrosis. Cell Biol. Int. 19:471–476. [DOI] [PubMed] [Google Scholar]
- Desmouliere, A. , Geinoz A., Gabbiani F., and Gabbiani G.. 1993. Transforming growth factor‐beta 1 induces alpha‐smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke, P. , Miyazono K., and Heldin C. H.. 2000. Signaling inputs converge on nuclear effectors in TGF‐beta signaling. Trends Biochem. Sci. 25:64–70. [DOI] [PubMed] [Google Scholar]
- Doherty, T. , and Broide D.. 2007. Cytokines and growth factors in airway remodeling in asthma. Curr. Opin. Immunol. 19:676–680. [DOI] [PubMed] [Google Scholar]
- Donohue, J. F. , and Ohar J. A.. 2004. Effects of corticosteroids on lung function in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 1:152–160. [DOI] [PubMed] [Google Scholar]
- Eickelberg, O. , Roth M., Lorx R., Bruce V., Rudiger J., Johnson M., et al. 1999. Ligand‐independent activation of the glucocorticoid receptor by beta2‐adrenergic receptor agonists in primary human lung fibroblasts and vascular smooth muscle cells. J. Biol. Chem. 274:1005–1010. [DOI] [PubMed] [Google Scholar]
- Galon, J. , Franchimont D., Hiroi N., Frey G., Boettner A., Ehrhart‐Bornstein M., et al. 2002. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 16:61–71. [DOI] [PubMed] [Google Scholar]
- Ganesan, S. , and Sajjan U. S.. 2013. Repair and remodeling of airway epithelium after injury in chronic obstructive pulmonary disease. Curr. Respir. Care Rep. 2:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goleva, E. , Li L. B., Eves P. T., Strand M. J., Martin R. J., and Leung D. Y.. 2006. Increased glucocorticoid receptor beta alters steroid response in glucocorticoid‐insensitive asthma. Am. J. Respir. Crit. Care Med. 173:607–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, L. , Zhu Y. J., Yang X., Guo Z. J., Xu W. B., and Tian X. L.. 2007. Effect of TGF‐beta/Smad signaling pathway on lung myofibroblast differentiation. Acta Pharmacol. Sin. 28:382–391. [DOI] [PubMed] [Google Scholar]
- Hackett, T. L. , Warner S. M., Stefanowicz D., Shaheen F., Pechkovsky D. V., Murray L. A., et al. 2009. Induction of epithelial‐mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor‐beta1. Am. J. Respir. Crit. Care Med. 180:122–133. [DOI] [PubMed] [Google Scholar]
- Hamid, Q. A. , Wenzel S. E., Hauk P. J., Tsicopoulos A., Wallaert B., Lafitte J. J., et al. 1999. Increased glucocorticoid receptor beta in airway cells of glucocorticoid‐insensitive asthma. Am. J. Respir. Crit. Care Med. 159:1600–1604. [DOI] [PubMed] [Google Scholar]
- Hashimoto, S. , Gon Y., Takeshita I., Matsumoto K., Maruoka S., and Horie T.. 2001. Transforming growth Factor‐beta1 induces phenotypic modulation of human lung fibroblasts to myofibroblast through a c‐Jun‐NH2‐terminal kinase‐dependent pathway. Am. J. Respir. Crit. Care Med. 163:152–157. [DOI] [PubMed] [Google Scholar]
- Holgate, S. T. , and Polosa R.. 2006. The mechanisms, diagnosis, and management of severe asthma in adults. Lancet 368:780–793. [DOI] [PubMed] [Google Scholar]
- Hu, B. , Wu Z., and Phan S. H.. 2003. Smad3 mediates transforming growth factor‐beta‐induced alpha‐smooth muscle actin expression. Am. J. Respir. Cell Mol. Biol. 29:397–404. [DOI] [PubMed] [Google Scholar]
- James, A. L. 1997. Relationship between airway wall thickness and airway hyperresponsiveness. Airway Wall Remodelling in Asthma. Pp. 1–27. [Google Scholar]
- Jarjour, N. N. , Erzurum S. C., Bleecker E. R., Calhoun W. J., Castro M., Comhair S. A., et al. 2012. Severe asthma: lessons learned from the National Heart, Lung, and Blood Institute Severe Asthma Research Program. Am. J. Respir. Crit. Care Med. 185:356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadmiel, M. , and Cidlowski J. A.. 2013. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 34:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay, A. B. , Phipps S., and Robinson D. S.. 2004. A role for eosinophils in airway remodelling in asthma. Trends Immunol. 25:477–482. [DOI] [PubMed] [Google Scholar]
- Kim, K. K. , Kugler M. C., Wolters P. J., Robillard L., Galvez M. G., Brumwell A. N., et al. 2006. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl Acad. Sci. USA 103:13180–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino, T. , Su Y. A., and Chrousos G. P.. 2009. Human glucocorticoid receptor isoform beta: recent understanding of its potential implications in physiology and pathophysiology. Cell. Mol. Life Sci. 66:3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, C. , and McDonald J. A.. 1991. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- Kuwano, K. , Bosken C. H., Paré P. D., Bai T. R., Wiggs B. R., and Hogg J. C.. 1993. Small airways dimensions in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 148:1220–1225. [DOI] [PubMed] [Google Scholar]
- Lamouille, S. , and Derynck R.. 2007. Cell size and invasion in TGF‐beta‐induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178:437–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaar, A. L. , and Panettieri R. A. Jr. 2003. Is airway remodeling clinically relevant in asthma? Am. J. Med. 115:652–659. [DOI] [PubMed] [Google Scholar]
- Leung, D. Y. , Hamid Q., Vottero A., Szefler S. J., Surs W., Minshall E., et al. 1997. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor beta. J. Exp. Med. 186:1567–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis‐Tuffin, L. J. , and Cidlowski J. A.. 2006. The physiology of human glucocorticoid receptor beta (hGRbeta) and glucocorticoid resistance. Ann. N. Y. Acad. Sci. 1069:1–9. [DOI] [PubMed] [Google Scholar]
- Lu, N. Z. , Collins J. B., Grissom S. F., and Cidlowski J. A.. 2007. Selective regulation of bone cell apoptosis by translational isoforms of the glucocorticoid receptor. Mol. Cell. Biol. 27:7143–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, C. P. , and Owens G. K.. 1999. Regulation of smooth muscle alpha‐actin expression in vivo is dependent on CArG elements within the 5' and first intron promoter regions. Circ. Res. 84:852–861. [DOI] [PubMed] [Google Scholar]
- Makinde, T. , Murphy R. F., and Agrawal D. K.. 2007. The regulatory role of TGF‐beta in airway remodeling in asthma. Immunol. Cell Biol. 85:348–356. [DOI] [PubMed] [Google Scholar]
- Massague, J. , and Chen Y. G.. 2000. Controlling TGF‐beta signaling. Genes Dev. 14:627–644. [PubMed] [Google Scholar]
- Massague, J. , and Wotton D.. 2000. Transcriptional control by the TGF‐beta/Smad signaling system. EMBO J. 19:1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAnulty, R. J. 2007. Fibroblasts and myofibroblasts: their source, function and role in disease. The international journal of biochemistry & cell biology 39:666–671. [DOI] [PubMed] [Google Scholar]
- McDonald, D. M. 2001. Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am. J. Respir. Crit. Care Med. 164:S39–S45. [DOI] [PubMed] [Google Scholar]
- Michalik, M. , Pierzchalska M., Legutko A., Ura M., Ostaszewska A., Soja J., et al. 2009. Asthmatic bronchial fibroblasts demonstrate enhanced potential to differentiate into myofibroblasts in culture. Med. Sci. Monit. 15: BR194–201. [PubMed] [Google Scholar]
- Minshall, E. M. , Leung D. Y., Martin R. J., Song Y. L., Cameron L., Ernst P., et al. 1997. Eosinophil‐associated TGF‐beta1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 17:326–333. [DOI] [PubMed] [Google Scholar]
- Nakao, A. , Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., et al. 1997. TGF‐beta receptor‐mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 16:5353–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nihlberg, K. , Larsen K., Hultgardh‐Nilsson A., Malmstrom A., Bjermer L., and Westergren‐Thorsson G.. 2006. Tissue fibrocytes in patients with mild asthma: a possible link to thickness of reticular basement membrane? Respir. Res. 7:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panettieri, R. A. Jr . 2003. Airway smooth muscle: immunomodulatory cells that modulate airway remodeling? Respir. Physiol. Neurobiol. 137:277–293. [DOI] [PubMed] [Google Scholar]
- Pascual, R. M. , and Peters S. P.. 2005. Airway remodeling contributes to the progressive loss of lung function in asthma: an overview. J. Allergy Clin. Immunol. 116:477–486; quiz 487. [DOI] [PubMed] [Google Scholar]
- Phan, S. H. 2002. The myofibroblast in pulmonary fibrosis. CHEST J. 122:286S–289S. [DOI] [PubMed] [Google Scholar]
- Postma, D. S. , and Timens W.. 2006. Remodeling in asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 3:434–439. [DOI] [PubMed] [Google Scholar]
- Pujols, L. , Xaubet A., Ramirez J., Mullol J., Roca‐Ferrer J., Torrego A., et al. 2004. Expression of glucocorticoid receptors alpha and beta in steroid sensitive and steroid insensitive interstitial lung diseases. Thorax 59:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujols, L. , Mullol J., and Picado C.. 2007. Alpha and beta glucocorticoid receptors: relevance in airway diseases. Curr. Allergy Asthma Rep. 7:93–99. [DOI] [PubMed] [Google Scholar]
- Raissy, H. H. , Kelly H. W., Harkins M., and Szefler S. J.. 2013. Inhaled corticosteroids in lung diseases. Am. J. Respir. Crit. Care Med. 187:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redington, A. E. , Madden J., Frew A. J., Djukanovic R., Roche W. R., Holgate S. T., et al. 1997. Transforming growth factor‐beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am. J. Respir. Crit. Care Med. 156:642–647. [DOI] [PubMed] [Google Scholar]
- Reichardt, H. M. , and Schutz G.. 1998. Glucocorticoid signalling–multiple variations of a common theme. Mol. Cell. Endocrinol. 146:1–6. [DOI] [PubMed] [Google Scholar]
- Ren, R. , Oakley R. H., Cruz‐Topete D., and Cidlowski J. A.. 2012. Dual role for glucocorticoids in cardiomyocyte hypertrophy and apoptosis. Endocrinology 153:5346–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnov‐Jessen, L. , and Petersen O. W.. 1993. Induction of alpha‐smooth muscle actin by transforming growth factor‐beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Invest. 68:696–707. [PubMed] [Google Scholar]
- Roy, S. G. , Nozaki Y., and Phan S. H.. 2001. Regulation of alpha‐smooth muscle actin gene expression in myofibroblast differentiation from rat lung fibroblasts. The international journal of biochemistry & cell biology 33:723–734. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , and Massague J.. 2003. Mechanisms of TGF‐beta signaling from cell membrane to the nucleus. Cell 113:685–700. [DOI] [PubMed] [Google Scholar]
- Sime, P. J. , Xing Z., Graham F. L., Csaky K. G., and Gauldie J.. 1997. Adenovector‐mediated gene transfer of active transforming growth factor‐beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Invest. 100:768–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, C. Z. , Tian X., and Gelehrter T. D.. 1999. Glucocorticoid receptor inhibits transforming growth factor‐beta signaling by directly targeting the transcriptional activation function of Smad3. Proc. Natl Acad. Sci. USA 96:11776–11781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, A. R. , Lane S. J., Cidlowski J. A., Staynov D. Z., and Lee T. H.. 2000. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor beta‐isoform. J. Allergy Clin. Immunol. 105:943–950. [DOI] [PubMed] [Google Scholar]
- Tillie‐Leblond, I. , Pugin J., Marquette C. H., Lamblin C., Saulnier F., Brichet A., et al. 1999. Balance between proinflammatory cytokines and their inhibitors in bronchial lavage from patients with status asthmaticus. Am. J. Respir. Crit. Care Med. 159:487–494. [DOI] [PubMed] [Google Scholar]
- Vignola, A. M. , Chanez P., Chiappara G., Merendino A., Pace E., Rizzo A., et al. 1997. Transforming growth factor‐beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 156:591–599. [DOI] [PubMed] [Google Scholar]
- Xu, Y. D. , Hua J., Mui A., O'Connor R., Grotendorst G., and Khalil N.. 2003. Release of biologically active TGF‐beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 285:L527–L539. [DOI] [PubMed] [Google Scholar]
- Yamauchi, K. 2006. Airway remodeling in asthma and its influence on clinical pathophysiology. Tohoku J. Exp. Med. 209:75–87. [DOI] [PubMed] [Google Scholar]
- Yudt, M. R. , Jewell C. M., Bienstock R. J., and Cidlowski J. A.. 2003. Molecular origins for the dominant negative function of human glucocorticoid receptor beta. Mol. Cell. Biol. 23:4319–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, K. , Rekhter M. D., Gordon D., and Phan S. H.. 1994. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am. J. Pathol. 145:114–125. [PMC free article] [PubMed] [Google Scholar]