

Abstract
Goblet cells (GCs) are the predominant secretory epithelial cells lining the luminal surface of the mammalian gastrointestinal (GI) tract. Best known for their apical release of mucin 2 (Muc2), which is critical for the formation of the intestinal mucus barrier, GCs have often been overlooked for their active contributions to intestinal protection and host defense. In part, this oversight reflects the limited tools available to study their function but also because GCs have long been viewed as relatively passive players in promoting intestinal homeostasis and host defense. In light of recent studies, this perspective has shifted, as current evidence suggests that Muc2 as well as other GC mediators are actively released into the lumen to defend the host when the GI tract is challenged by noxious stimuli. The ability of GCs to sense and respond to danger signals, such as bacterial pathogens, has recently been linked to inflammasome signaling, potentially intrinsic to the GCs themselves. Moreover, further work suggests that GCs release Muc2, as well as other mediators, to modulate the composition of the gut microbiome, leading to both the expansion as well as the depletion of specific gut microbes. This review will focus on the mechanisms by which GCs actively defend the host from noxious stimuli, as well as describe advanced technologies and new approaches by which their responses can be addressed. Taken together, we will highlight current insights into this understudied, yet critical, aspect of intestinal mucosal protection and its role in promoting gut defense and homeostasis.
Keywords: goblet cell, gut infections, inflammatory bowel disease, microbes, mucus
INTRODUCTION TO INTESTINAL GOBLET CELLS
The mammalian gastrointestinal (GI) tract is a unique and complex environment inhabited by vast numbers of commensal bacteria (81). The containment of this enormous microbial burden within the gut lumen, without triggering overt inflammation, is a remarkable task and critical to the maintenance of intestinal health. Many studies have demonstrated that the intestinal epithelium plays a primary role in the segregation of luminal commensals, food antigens, and other luminal content from the underlying immune system. Intestinal epithelial cells (IECs), by virtue of their location, are ideally situated to block these luminal factors from escaping the GI tract. This protection is mediated by the intestinal epithelium creating a combined cellular and paracellular barrier through its interconnected intracellular junctions, as well as through specialized epithelial secretory cells that release an array of proteins, lectins, and other factors that physically and biochemically restrict luminal antigens. These secretory cells include tuft cells, enteroendocrine cells, Paneth cells, and goblet cells (GCs). Each of these secretory cell types plays a different, yet key, role in GI host defense and are together reviewed in detail elsewhere (40, 82, 125). Based on our expertise, as well as the array of novel findings arising over the last several years, this review will focus on GCs and their contribution to intestinal homeostasis, as well as protection against pathogenic bacteria.
GCs are the most abundant secretory epithelial cells in the GI. Their primary function involves the secretion of mucins that self assemble into a protective mucus layer, coating the apical surface of epithelial cells. This mucus layer not only limits commensal microbe contact with the epithelium but also reduces mechanical stress on the epithelium through lubrication of the luminal bolus comprised of food contents or feces (24). Notably, mucus and other epithelial-derived factors play a critical role in host defense against invading bacterial pathogens, although this defense has often been considered relatively passive and readily subverted by pathogenic microbes (90). Recent studies have begun to challenge this concept, noting the capacity of GCs to sense and respond to infections actively, as well as secrete additional products, besides mucins, that are vital in promoting intestinal defense (19, 81). GCs have thus recently emerged as key regulators of intestinal homeostasis at the host-microbe interface within the GI tract.
Based on their primary role as secretory cells, GCs possess a unique structure, which facilitates their production and unidirectional secretion of mucins (18). Unlike absorptive IECs, the GC nuclei, cytoplasm, and organelles are located in the basolateral region of the cell. Directly above are densely packed granules containing the large molecular weight mucin glycoproteins that are separated from the lateral plasma membrane by the theca, a layer of filament-rich cytoplasm (18). Once released into the lumen, these mucins expand to form a dense, carbohydrate-rich matrix, assembling into homo-oligomers that give mucus its viscous properties (18, 121). The mucus barrier is dynamically regulated, not only by continuous low-level mucin release by the GCs but also by its continuous degradation via the mucinolytic actions of commensal, as well as pathogenic, bacteria (59). Moreover, the aboral movements of the gut help flush excess mucus and luminal contents out with the feces (14, 19, 39, 117, 121, 122). The oligosaccharide content in the mucus layer can influence the microbiota of the GI tract by the enhancement or inhibition of adherence of specific bacteria (79). Mucins are also direct sources of carbohydrates that provide nutrients for bacteria (111). Whereas best known for synthesis and secretion of mucin 2 (MUC2; Muc2 in mice; to avoid confusion, the murine nomenclature will be used, except when the topic clearly relates to human MUC2), GCs are also capable of producing other mediators, including the cytoprotective trefoil factor 3 (Tff3) and the proinflammatory resistin-like molecule (Relm)-β, which play their own, unique roles in modulating gut health and disease (6, 75, 81). This review seeks to highlight new studies, demonstrating that GCs play an active and pivotal role in controlling host-microbe interactions under baseline, as well as during disease conditions. By releasing specific mediators, GCs can promote host defense during enteric infections, but they can also drive inflammation and intestinal pathology as part of the aberrant host response to microbes seen in patients with inflammatory bowel disease (IBD).
GC DEVELOPMENT AND THE INFLUENCE OF MICROBIAL COLONIZATION
Intestinal GCs develop relatively early during mammalian embryogenesis, around embryonic days 15–16 in the mouse intestine and at roughly 9 wk in human embryos (106). At this time, the first epithelial precursor cells arise from intestinal leucine-rich repeat-containing G protein-coupled receptor 5-positive stem cells in the pseudo-crypt region and migrate up the villi (106). The intestinal stem cells are responsible for the constant replenishment of the intestinal epithelium. Immature epithelial cells, produced by the stem cells, are capable of differentiating along both the absorptive or secretory lineages. Differentiation of these precursor cells into the secretory lineage initially depends on the inhibition of Notch signaling (100) and subsequent repression of the hairy and enhancer of split (30) transcription factor that would otherwise drive them toward the absorptive lineage (120). As the secretory precursor moves out of the crypt, it experiences a reduction in Wnt signaling, which begins to dedicate the precursor toward a GC fate rather than becoming a Paneth cell (28). Moreover, inhibition of Notch signaling leads to activation of the transcription factor Math1, a key factor in GC differentiation (120). Recently, it has been suggested that mammalian target of rapamycin signaling negatively regulates GC differentiation by promoting the activation of the Notch signaling pathway and therefore, driving cells down the absorptive lineage (133). The transcription factor, SAM pointed domain containing ETS transcription factor (Spdef), is also known to control GC terminal differentiation by inducing the expression of the GC mediators Muc2 and Relm-β (84). Several other factors, including bacteria, diet, and immune cells, have also been shown to influence GC differentiation (8, 21, 44). GCs are also highly susceptible to changes in homeostasis of the intestinal epithelial layer, as significant changes in the development/differentiation of immature epithelial cells toward enterocytes/colonocytes will concurrently affect the numbers and maturation of GCs (26, 38).
Although GCs are first detected during embryogenesis, they undergo dramatic postnatal development and maturation during the neonatal period. This includes significant increases in the total number of GCs present in the gut, as well as an increase in their mucin expression and glycosylation, to the levels seen in the adult intestine (10, 103). In part, this GC maturation may reflect changes in the function and use of the intestinal tract, since in utero, the fetus does not use the gut for nutrient acquisition, instead receiving nutrients through its mother’s placenta. Following birth, there is a dramatic switch to the GI tract as the primary source of nutrition. Whereas the flow of milk and other factors through the gut may, on their own, induce intestinal and GC maturation, much of this maturation is also thought to depend on exposure to the microbiota, since in utero, the gut lumen is considered largely sterile and devoid of bacteria (91). Bacterial colonization of the GI tract begins at birth, and subsequently, this exposure to bacterial products promotes neonatal maturation and differentiation of the intestinal epithelium (86). This differentiation includes a dramatic increase in the expression of Muc2, as well as Relm-β (Fig. 1A) (49). Germ-free mice are often considered useful models for examining the influence of bacterial colonization on GI maturation and development. When compared with conventionally housed mice, the intestines of germ-free mice contain significantly fewer GCs that express only modest levels of Muc2 and virtually no Relm-β, and their mucus layer is very thin (10). However, when germ-free mice are colonized with commensal bacteria, their intestinal GCs quickly increase in both size and number, and they produce significantly higher levels of Relm-β and Muc2, resulting in a much thicker mucus layer (49). Furthermore, studies have demonstrated that the expression of key enzymes (glycosyltransferases) involved in Muc2 glycosylation are modest in germ-free mice, directly leading to shorter Muc2 glycans and likely contributing to the thinner mucus layer found in these mice (5). Taken together, these results strongly support microbial colonization as a key step toward GC development and maturation. Exactly how this occurs is unclear, but it may reflect direct sensing of gut commensals and/or their metabolites by the intestinal epithelium. It is tempting to speculate that this process enables the epithelium to adapt rapidly to luminal stimuli, thereby promoting both mucosal homeostasis and defense.
Fig. 1.

The role of intestinal goblet cells (GCs) and their mediators in health and disease. Each panel represents several crypts within a region of colonic mucosa. Light green shows the outer mucus layer, whereas dark green shows the inner mucus layer, and light blue represents the lamina propria, as labeled in A. Relative expression/release of mediators by GCs is represented by arrows of different sizes (large arrows, heavy or increased secretion; smallest arrows, minimal or decreased secretion; medium arrows, baseline secretion state). A: bacterial colonization after birth triggers an increase in GC numbers and differentiation (rose-colored cells). This leads to dramatically increased production/secretion of Muc2 (green arrows), as well as Relm-β (purple arrows), into the gut lumen, whereas Tff3 (black arrows) also increases but to a lesser degree. B: under healthy (homeostatic) conditions, there is limited Relm-β secretion (purple arrows), whereas Tff3 is constantly secreted (black arrows), and Muc2 secretion (green arrows) is heavy and equivalent to the loss of mucus through defecation and its degradation by commensal bacteria (bacteria within white circle). C: during many enteric infections, GCs release significant amounts of Muc2 (green arrows), whereas Relm-β (purple arrows) as well Tff3 (black arrows) production and release also increase. An infection-induced increase in Muc2 secretion promotes the flushing of pathogens, as well as commensals from the gut (brown arrow). Relm-β secretion into the submucosa promotes immune cell (CD4+ T cell) recruitment that drives a variety of host defense mechanisms, including increased epithelial cell proliferation. D: in IBD and experimental colitis, host-driven changes in GC function lead to GC depletion, a reduction in Muc2 secretion (green arrows), and a reduction in overall mucus-layer thickness (light and dark green layers). Whereas Tff3 expression may undergo only modest changes in expression (black arrows), Relm-β secretion (purple arrows) is increased, which leads to the induction, production, and release of antimicrobial peptides and lectins (RegIIIβ and RegIIIγ; blue dots) by epithelial cells into the gut lumen. This increased antimicrobial activity leads to intestinal microbial dysbiosis, including loss of beneficial microbes, and the overgrowth of bacterial pathobionts associated with IBD.
MUC2 STRUCTURE, FUNCTION, AND INTESTINAL HOMEOSTASIS
Mucins constitute a broad class of glycoproteins, with Muc2 being the major gel-forming mucin secreted within the GI tract and thus the major mucin component of the intestinal mucus layer. Whereas Muc5AC and Muc5B are also secreted, their expression is modest and localized predominantly to the stomach (53, 96). In contrast, the other mucins expressed by GCs and enterocytes within the gut (Muc1, Muc3, Muc4, Muc13, Muc17) are membrane bound, making up part of the glycocalyx of mucosal tissues and also playing varying roles in cell signaling (56). Muc2 is the protein core of the mucin and is largely made up of serine- and threonine-rich variable number tandem repeat domains and proline/threonine/serine-rich domains, each of which is heavily O-glycosylated, whereas both the COOH- and NH2-terminal ends contain N-glycosylated, cysteine-rich domains, which can form disulfide links with individual mucin proteins, leading to the formation of large, interconnected networks (56). Before Muc2 is secreted, its immature or core form is found within the Golgi apparatus of GCs, where the mucin N-glycans are modified by Golgi resident glycosyltransferases (4), following which the mucin domains become O-glycosylated and some of the sugars are sulfated to achieve their mature form (56).
These oligosaccharides constitute ~80% of the mucin mass and increase its size by more than five times (7). Although representing potential ligands for microbial adhesins and/or potential food sources for some commensal microbes, these oligosaccharides also protect MUC2/Muc2 against the actions of bacterial proteases (4). There are several “core” O-glycan structures, with human MUC2 primarily bearing core 3-derived structures, catalyzed by the core 3 β-1,3-N-acetylglucosaminyltransferase 6 (B3gnt6) enzyme, whereas murine Muc2 predominantly contains core 1- and 2-derived structures catalyzed by the core 1 synthase, glycoprotein-N-acetylgalactosamine 3-β-galactosyltransferase 1 (C1galt1) and core 2 β-1,6-N-acetylglucosaminyltransferase 2 (53, 115). These oligosaccharides can be further modified by the addition of fucose, sulfate groups, or sialic acid, which is mediated by specific fucosyl transferases (Fut1 and Fut2), sulfotransferases, and sialotransferases, generating different acidomucins, mainly sialomucins and sulfomucins (76, 115). Notably, MUC2 glycosylation patterns vary depending on their location in the GI tract or mammalian species. For instance, sialomucins are found predominantly in the human distal colon but not in the small intestine (76). In contrast, sialomucins are expressed at higher levels in the murine proximal colon but not in the distal colon. Moreover, sialic acid in terminal glycans is found at much higher levels in humans than in mice. Because the longitudinal gradient for sialomucin expression in the human colon is reversed in mice, sialomucins are expressed at higher levels in the murine proximal colon rather than distally (53, 76, 117). Differences in acidomucin patterns have been observed in patients suffering from colorectal cancer and IBD (25, 31, 102). Moreover, Croix and colleagues (27) showed a close relationship between the growth of some enteric sulfate-reducing bacteria and differential patterns of acidomucins. Specifically, sulfate-reducing bacteria, known to be increased in ulcerative colitis (UC) patients, were found in the colonic mucus that displays a higher level of sulfomucins compared with sialomucins (27). This finding highlights the importance of the consideration of acidomucin patterns in the context of understanding host-microbe interactions in intestinal disease.
At present, the regulation of Muc2 release is poorly understood; however, there appears to be at least two major forms of Muc2 secretion by GCs. Under homeostatic conditions, Muc2 is constitutively secreted at a low and relatively uniform level through simple exocytosis (Fig. 1B) (121). This baseline mucin secretion is controlled by a number of stimuli, including intracellular Ca2+ ions and PGE2 (46). However, when exposed to a noxious stimulus, all mucin granules within a GC are rapidly expelled together in a process, termed “compound exocytosis” (18). This accelerated secretion of mucins into the gut lumen is observed only when the epithelium is exposed to severe stress, such as enteric infections and/or disease conditions. In brief, compound exocytosis of mucin granules begins when the granules fuse with the GC plasma membrane, followed by the diffusion of water and cations into the granule space, triggering a chain reaction that drives the dramatic release of the secretory granule products and the rapid hydration of the resulting mucin network (121). This aspect of mucin secretion has been studied extensively in pulmonary GC and has been reviewed in detail elsewhere (121). Whereas the exact triggering mechanisms behind this rapid mucin release are currently under investigation, recent studies have revealed that intestinal tissues exposed to bacterial products (LPS, flagellin, etc.) and/or cytokines (IFN-γ, IL-22) induce compound exocytosis, suggesting a possible role for this process in innate host defense (104, 116).
Upon its secretion, the negatively charged Muc2 becomes rapidly hydrated, with the glycosylation and fucosylation of Muc2 determining the composition/structure of the resulting mucus. Notably, the latter has been demonstrated to aid in mucus expansion, since O-glycosylated mucin domains attract water, resulting in the formation of large Muc2 sheets, which cover the apical surface of IECs. Expansion of Muc2 is also dependent on pH, as well as an oxidative environment, where bicarbonate and iron play important roles (2). As Muc2 is released, these sheets form stratified layers, which are hypothesized to be linked by disulfide bonds (76). In the small bowel and cecum, these Muc2 networks form a single loose and soluble layer that can be easily penetrated by bacteria. In contrast, mucus in the colon comprises two layers: an outer, loose layer—much like that found in the upper GI tract—as well as an inner mucus layer that is firmly attached to the epithelium, acting as a stratified, polyanionic, physicochemical barrier that can directly inhibit the adherence of Gram-negative bacteria to the underlying epithelium (56, 60). This inner layer is dense, stratified, and insoluble and reflects a higher Muc2 concentration than that of the outer layer. In mice, the inner mucus layer is ~50 µm thick and is compact and sterile, whereas the outer layer is considerably thicker (100–150 µm), loose, and permeable and houses mucosa-associated microbes (60). Whereas it remains unclear how two distinct mucus layers are continuously generated and maintained in the colon, the permeable outer layer may be generated by proteolysis of the inner mucus layer through the actions of endogenous host and bacterial enzymes (105). These enzymes are thought to destabilize the mucus, as well as liberate terminal sugars found on glycans, thereby creating the overlying permeable mucus layer (105). Production of these two mucus layers likely provides several benefits, with the firm, inner layer preventing the massive numbers of nearby luminal bacteria from contacting and colonizing underlying IECs, thereby triggering inflammation (47). In contrast, the outer, loose mucus layer helps lubricate the fecal stream, as well as provide a flushing action to remove cellular debris and transient luminal populations of microbes. Moreover, it also provides a niche, as well as an energy source for specific mucus-associated bacteria, such as Akkermansia muciniphila and Mucispirillum spp (70).
GUT MICROBE INTERACTIONS WITH INTESTINAL MUCUS
Interestingly, even though GCs and mucus are present in the embryonic gut, the specific glycosylation characteristics of intestinal mucus are acquired only after birth. Thus in the fetal intestine, Muc2 glycans are not terminally decorated with either sialic acid or fucose—the last components added during the glycosylation process of Muc2 (99). In a similar fashion, the inner mucus layer in the colons of germ-free mice appears far more permeable than that found in conventionally housed mice (58). These findings thus support the hypothesis that bacterial colonization in the gut plays a critical role in the determination of the glycosylation patterns of Muc2, in keeping with the finding that expression of various glycosyltransferases is significantly reduced in germ-free mice (5). By changing the glycosylation patterns of Muc2, it is likely that bacteria can directly influence the functional capability of the mucus layer. Correspondingly, it has been hypothesized that the structure/glycosylation of intestinal mucus can influence the makeup of the gut microbiota, by impacting the expression of binding sites for specific microbes, as well as the availability of key nutrients. Although an attractive idea, at present, there is only limited evidence supporting a direct role for mucus in controlling the makeup of the gut microbiome. Thus further studies are required to help define the relationship between mucus and the establishment of resident gut microbes. During its maturation, Muc2 is highly glycosylated and provides a wide range of different sugars on its terminal O-glycans, thereby acting as a major nutrient source for the commensal microbes found in the gut (35, 87, 111). Many commensal bacterial species possess enzymes, such as fucosidases, mucinases (mucin-degrading enzymes), glycosylsulfatases, sialidases, sialate O-acetylesterases, and N-acetylneuraminate lyases, which are able to cleave the terminal glycans found on Muc2 (74). This release of sugars from the Muc2 protein is an essential step in their use as a nutrient source by gut microbes.
Notably, several studies have revealed that microbes expressing these glycosidases are not the only microbes that benefit from the release of mucin-associated glycans (23, 87, 107). Other commensal and pathogenic bacteria that express relevant sugar metabolism pathways can use these liberated glycans as food sources, despite lacking the mucinases themselves (87). For example, one of the most prevalent microbial species found within the human gut, Bacteroides thetaiotaomicron, has been found to increase glycan fucosylation in host cells (36). In turn, B. thetaiotaomicron produces a plethora of fucosidases and other glycosidases, which cleave these glycans (87). Whereas these glycans can be subsequently used by B. thetaiotaomicron through its wide-ranging and diverse metabolic pathways, other microbes, including many enteric pathogens, can use the newly released glycans, assuming they possess the relevant metabolic pathways, bypassing the need for their own glycosidases (23). Specific examples include a recent study showing that fucose availability and sensing directly affect the virulence of the bacterial pathogen enterohemorrhagic Escherichia coli (EHEC) (87). Pacheco et al. (87) demonstrated that fucose sensing by EHEC impacts its virulence gene expression, as well as its ability to form attaching and effacing lesions on host cells. Moreover, they showed that EHEC benefited from the ability of B. thetaiotaomicron to cleave fucose from host glycans, resulting in high-fucose availability for EHEC and decreasing its virulence (87). A similar study by Stahl et al. (107) on Campylobacter jejuni found that certain C. jejuni strains possessed a fucose metabolic pathway that aids its colonization and pathogenesis. However, these C. jejuni strains lacked any fucosidases, leaving them reliant on other bacteria to cleave fucose from host mucins before they could scavenge the nutrient for themselves (107).
Although the degradation of mucins has the potential to fuel the growth of pathogenic bacteria, their beneficial aspects should not be overlooked. These mucin-derived glycans also promote the proliferation of beneficial commensals, as well as providing the raw materials for the production of beneficial metabolites, in particular, the short-chain fatty acids: acetate, butyrate, and propionate, which are the products of sugar fermentation by certain members of the microbiota (110). These metabolites are capable of diffusing through the mucus, where they are absorbed by IECs and used as a major energy source, as well as released basolaterally, where they can act as immunomodulatory factors within the mucosa (110).
Until now, only a handful of studies has investigated whether these bacterial metabolites can directly affect GC function (22, 126). In a germ-free rat model, the authors inoculated rats with B. thetaiotaomicron and Faecalibacterium prausnitzii and then assessed their subsequent intestinal development (126). They demonstrated that B. thetaiotaomicron, which produces acetate as a byproduct of fermentation, leads to the increased differentiation of GCs, as well as elevated mucin expression. Conversely, F. prausnitzii, which consumes acetate to produce butyrate, another product of bacterial fermentation, was shown to attenuate the effect of B. thetaiotaomicron on GCs (126). Another recent paper demonstrated that butyrate stimulated the HT29 cell line to produce and secrete cathelicidin, an antimicrobial peptide found in the small intestinal mucus layer (22). Notably, the effect of butyrate on cathelicidin depends on the production and secretion of Muc2, since small interfering RNA knockdown of Muc2 abrogated the effect of butyrate. Correspondingly, the authors showed that Muc2 deficient (−/−) mice expressed lower levels of this antimicrobial peptide compared with wild-type mice (22). Thus these studies demonstrated that GC differentiation and function could be impacted, not only by the actions of a commensal bacterium but also by the relative abundance of different microbes within the commensal community.
BEYOND MUC2: PROTECTIVE FACTORS WITHIN INTESTINAL MUCUS
Whereas gut microbes and the intestinal mucus layer clearly modulate each other, it remains unclear to what extent the protective effects of intestinal mucus reflect the actions of the Muc2 molecule alone or if other proteins found in the mucus layer could contribute to its protective functions. In the small intestine, the presence of Paneth cell-derived, antimicrobial peptides and the family of regenerating islet-derived 3 (RegIII) lectins impart direct antimicrobial activity to the mucus (29). Interestingly, Paneth cells are absent in the colon; however, the inner colonic mucus layer remains largely free of commensal microbes. We and others (14) have not detected overt antimicrobial activity in the colonic mucus from naïve mice, suggesting that other mechanisms are likely at play. The mucin layer also contains secretory antibodies, which are transported across IECs via the polymeric Ig receptor (pIgR; Table 1) (98). Interestingly, despite its presence at the apical cell surface, the effect of IgA on intestinal homeostatic control of commensal microbes appears to lie within the outer mucus layer. In a Pigr−/− mouse, fluorescence in situ hybridization against bacterial 16S rRNA revealed that the outer mucus layer was penetrated by bacteria as normal, whereas the inner layer remained sterile (98).
Table 1.
Novel goblet cell secretory factors
| Mediator | Goblet Cell Secreted | Secreted by Other Cell Types | Functions | Reference |
|---|---|---|---|---|
| Muc2 | Yes | No | Creates intestinal mucus barrier | (4, 7, 61, 92, 97) |
| Muc3 | Yes | Yes (enterocytes) | Membrane-bound mucin, creates glycocalyx | (61) |
| Muc5Ac | Yes | Yes (enterocytes) | Creates gastric mucus barrier | (53, 93, 96) |
| Relm-β | Yes | No | Varied immune/antimicrobial functions | (15, 16, 29, 92) |
| RegIIIβ | Yes | Yes (enterocytes, immune cells) | Antimicrobial functions | (29, 81) |
| RegIIIγ | Yes | Yes (enterocytes, immune cells) | Antimicrobial functions | (29, 81) |
| Tff3 | Yes | No | Promotes mucosal repair | (37, 73, 95, 127) |
| IgA | No | Yes (B cells) | Antimicrobial and immunomodulatory functions | (98) |
| pIgR | ? | Yes | Transports antibodies across the intestinal epithelium into mucus | (98) |
| Fcgbp | Yes | No | Crosslinks Muc2 | (62, 96) |
| Agr2 | Yes | Yes | ER protein, proposed extracellular function in the mucus | (11, 89, 131) |
| ZG16 | Yes | ? | Binds to Gram+ve bacteria | (12, 96) |
| Lypd8 | No | Yes | Binds to Gram−ve bacteria | (85) |
?, unknown secretion from this cell type.
Proteomics studies by Hansson and colleagues (60, 62, 97) have identified several host proteins within the Muc2 matrix that appear to strengthen the mucus layer’s ability to constrain gut microbes. A list of the factors found within the intestinal mucus layer is presented in Table 1. One of the components identified in both human and mouse mucus was the Fcγ-binding protein (Fcgbp) (62). It is produced by GCs and is linked to Muc2 by a reduction-insensitive and chaotropic salt-resistant linkage, most likely through a covalent bond (62). The authors proposed this bond formed between the C-terminus of Muc2 and the N-terminus of Fcgbp, where Muc2 undergoes autocatalytic cleavage under the acidic conditions of the secretory granule, forming an anhydride that readily attaches to hydroxyl groups or the primary amine of cleaved Fcgbp (62). Here, Fcgbp can act as a crosslinker of the Muc2 network through covalent, as well as disulfide, bonds.
The disulfide isomerase-like protein anterior gradient protein 2 homolog (Agr2) is also present in secreted mucus throughout the murine GI tract, although it does not directly bind Muc2 (11). In humans, AGR2 is encoded on the chromosomal region 7p21.3, which is linked to an increased risk of developing IBD (132). It is also downregulated in UC patients compared with healthy controls, whereas Agr2−/− mice have proven highly susceptible to dextran sulfate sodium (DSS)-induced colitis, a model of IBD (89, 132). The function of this endoplasmic reticulum (ER) protein has not yet been defined, but current studies suggest it plays a role in GC differentiation through its regulatory actions on the GC-specific transcription factors, forkhead box proteins A1 and A2 (132). This concept is supported by the observation that Agr2−/− mice possess fewer mature colonic GCs, as well as a poorly developed inner colonic mucus layer (89). Interestingly, the molar levels of Agr2 within the murine mucus layer appear to be on par or even higher than Muc2, suggesting that in addition to its role in the ER, it plays an extracellular function within the mucus layer (11). Further study is required to elucidate fully the functions of Agr2 and its role in the ER, GC differentiation, and mucus layer function.
Another abundant component within the mucus layer is the lectin-like zymogen granulae protein 16 (ZG16; Table 1) (12). ZG16 was found to bind and aggregate Gram-positive bacteria by binding to the peptidoglycan, abundant in their bacterial cell walls. Zg16−/− mice presented with a colonic mucus layer of normal thickness; however, commensal bacteria were found to penetrate the mucus barrier and colonize the colonic epithelium (12). Moreover, the authors noted a significant increase in translocated Firmicutes microbes to systemic tissues and an increase in serum levels of the cytokines IL-4 and IFN-γ (12). Another mucus-bound factor, namely the Ly6/plasminogen activator urokinase receptor domain containing 8 (Lypd8), provides the colonic mucus layer with a similar protective function but instead, against Gram-negative bacteria Lypd8 protein, is expressed at the apex of murine colonic crypts and is secreted by IECs into the colonic lumen and mucus layer, where it was found to bind to Gram-negative, flagellated bacteria, including Proteus mirabilis, aggregating them and thereby suppressing their motility (85). Strikingly, healthy Lypd8−/− mice carried commensals within their inner colonic mucus layer, whereas many flagellated bacteria were found to have invaded the colonic epithelium (85). Lypd8−/− mice were also found to be highly sensitive to DSS-induced colitis (85). Interestingly, intestinal inflammation, triggered by DSS treatment, was ameliorated upon administration of an antibiotic active against Gram-negative bacteria (85). Thus ZG16 and Lypd8 appear to act within the intestinal mucus layer to promote the segregation between commensals and epithelial cells, likely as a means to preserve intestinal homeostasis. Curiously, the potential impact of these factors in promoting host defense against enteric bacterial pathogens has yet to be tested.
ENTERIC BACTERIAL PATHOGEN INTERACTIONS WITH THE INTESTINAL MUCUS LAYER
Together, the epithelial and mucus barriers are largely successful at restricting commensal microbes within the gut lumen and in maintaining mucosal homeostasis. However, in their coevolution with their human hosts, enteric bacterial pathogens have developed various strategies for navigating through and subverting the intestinal mucus layer. Each enteric pathogen has adapted convergent or unique virulence strategies to colonize the underlying IECs to establish a successful infection that subsequently results in disease. Among these, enteropathogenic E. coli (EPEC) and EHEC possess virulence factors that allow them not only to breach the sterility of the inner mucus layer and colonize the epithelium, but as adherent pathogens, they also express virulence factors that enable them to thrive and multiply within the intestinal mucus layer itself. EHEC can use the mono- and disaccharides contained within mucus as carbon sources (35). EHEC possesses several metabolic pathways shared with other strains of E. coli, as well as unique prophage-encoded pathways for the highly efficient degradation of N-acetyl-d-glucosamine, N-acetyl neuraminic acid, galactose, and fucose (35, 42). It is important to note that EPEC and EHEC most likely rely on nearby commensal microbes for the release of these glycans, readily importing them and using them as an energy source to fuel their quick cell division. This would aid these adherent microbes in outcompeting their commensal counterparts and establishing their infectious niche.
Although EHEC infection is known to stimulate MUC2 production by GCs, instead of increasing the distance between luminal EHEC and the IECs, in vitro infection has been shown to decrease mucus thickness overlying murine and human cells (51, 55, 128). This suggests that EHEC infection reduces colonic mucus thickness, facilitating the ability of EHEC microbes to reach the apical intestinal surface and infect additional IECs. The mechanism by which EHEC reduces mucus thickness is still under investigation, with two papers publishing conflicting roles for the metalloprotease StcE (51, 55). The method by which EHEC propels itself through the mucin layer is also controversial, as both EPEC and EHEC have flagella (H6 and H7, respectfully) that can bind mucin, but it is unclear if this property would trap these pathogens within the mucus or if they use their flagella to cross the mucus layer (34, 51, 55). The former postulate is more likely, since EHEC downregulates its flg genes in the presence of porcine mucin, and its motility is reduced in a mucin-swarming assay (66). Another potential means by which enteric pathogens cross through the mucus layer includes mucolytic enzymes. Citrobacter rodentium, the murine relative of EPEC and EHEC, expresses a serine protease autotransporter, termed Pic, which possesses mucinase activity in vitro (17, 48). Pic homologues are expressed by several other enteric pathogens, including Shigella flexneri, enteroaggregative E. coli, and uropathogenic E. coli (48, 88). Despite its in vitro activity, a Pic-C. rodentium mutant (ΔpicC) did not affect overall intestinal mucin secretion compared with wild-type C. rodentium and surprisingly, led to an increase in host mortality (17). More work is required to elucidate the factors used by adherent pathogens to cross the mucus barrier, as well as the potential for nonmotile pathogens, such as C. rodentium, to use mucolytic enzymes with potentially novel proteolytic and glycohydrolytic function(s).
Aside from nonmotile pathogens, several motile microbes, such as C. jejuni and Salmonella enterica serovar Typhimurium, use their flagella to swim through the intestinal mucus layer. S. Typhimurium is known to use virulence factors encoded within its S. Typhimurium pathogenicity island 1 to infect and/or translocate across epithelial cells. Studies have shown that S. Typhimurium pathogenicity island 1 is not essential for Salmonella to navigate through mucus, since avirulent strains of these bacteria are still able to cross the mucus layer and colonize the cecal epithelium (83). A rfaJ mutant (lacking a glucosyltransferase responsible for connecting the O-antigen of Salmonella to its LPS outer core) bound tightly to murine cecal mucus and was impaired in its motility within the mucus, and although it was still able to colonize the intestinal epithelium, it was easily outcompeted when its wild-type counterpart was added simultaneously (77, 83). Despite these findings, few studies have examined how S. Typhimurium interacts with and ultimately crosses the intestinal mucus layer to reach the underlying epithelium. Mice lacking a mucus layer (Muc2−/−) show heightened susceptibility to wild-type S. Typhimurium infection, carrying higher pathogen burdens and suffering exaggerated barrier disruption (129).
C. jejuni is also a major cause of infectious gastroenteritis. This microbe uses L-fucose, found in the mucus, as a nutrient but also as a chemoattractant and a promoter of biofilm formation (32, 107). Interestingly, the cellular spiral shape of C. jejuni is essential for it to swim effectively through the mucus layer in the gut. Mutant rod (uncurved)-shaped strains were found to be unable to cross the mucus layer and colonize the underlying intestinal crypts (108). These data indicate that the helical morphology of C. jejuni enables it to traverse the mucus layer and colonize the intestinal epithelium to trigger inflammation. Although the mechanisms by which bacterial pathogens cross the intestinal mucus layer are still under investigation, it is evident that this barrier has the capacity to influence their pathogenicity directly. Taken together, these results support the concept that the mucus barrier provides essential protection against both motile and nonmotile bacterial pathogens.
GCs AND THEIR ACTIVE RESPONSE TO ENTERIC INFECTIONS
When an intestinal pathogen, such as C. rodentium, breaks through the inner mucus layer, the host responds with an increase in mucin secretion by GCs, potentially as a defense mechanism to push the pathogen away simultaneously from the mucosal surface, as well as flush it out of the colon (Figs. 1C and 2) (14, 19, 43, 124, 128, 129). An intact mucin barrier is essential in restricting enteric infection, with Bergstrom and colleagues (14) demonstrating that Muc2−/− mice were highly susceptible to C. rodentium infection, undergoing rapid colonization and carrying 10- to 100-fold greater pathogen burdens in their intestines than wild-type mice. This susceptibility led to mucosal ulcers, severe epithelial barrier disruption, and high mortality rates. Interestingly, whereas the C. rodentium, found on the mucosal surface of wild-type mice, were intimately attached to the epithelium, there was a marked increase in C. rodentium that were only loosely adherent to the mucosa in the highly colonized Muc2−/− mice. It appears that mucus dramatically limits pathogen burdens at the intestinal surface by removing many of the loosely attached bacteria from the mucosal surface. Moreover, this protective role was not limited to C. rodentium only, since Muc2−/− mice were also found to be highly susceptible to S. Typhimurium infection, carrying up to 100-fold higher pathogen burdens than wild-type mice (129). Thus intestinal mucus promotes host defense against enteric pathogens with diverse virulence strategies.
Fig. 2.
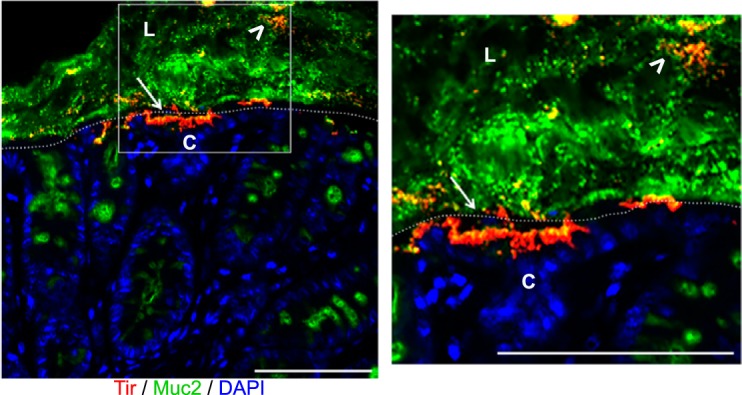
Citrobacter rodentium localizes within the mucus layer and on the apical surface of the intestinal epithelium. Immunofluorescent staining of colonic tissue from a C57BL/6 mouse, 6 days post-C. rodentium infection. Antibodies were used against the translocated intimin receptor (Tir; red) to identify C. rodentium, mucus (Muc2; green), and DNA [4′,6-diamidino-2-phenylindole (DAPI); blue]. During infection, C. rodentium is found within the intestinal mucus layer (white arrowheads), as well as infecting the apical surface of the intestinal epithelium (white arrows). L, lumen; C, crypts, tissue surface, dotted, white lines. Original scale bars, 50 μm.
The discovery that bacterial infection leads to a rapid increase in intestinal mucin production, as well as a dramatic increase in mucus thickness in the distal colon, indicates that the host actively responds to infection with increased mucin release. This suggests that mucin production/release could be driven through an innate immune response. Bhinder et al. (16) examined the impact of myeloid differentiation primary response 88 (MyD88) signaling, demonstrating that mice lacking MyD88 in their IECs showed decreased Muc2 staining during enteric C. rodentium and S. Typhimurium infections. These results suggested that innate immune signaling could control the response of GCs to a pathogen infection. In agreement with this concept, Flavell and colleagues (123) discovered that mice impaired in inflammasome signaling, specifically lacking the canonical inflammasome component, nucleotide oligomerization domain-like receptor family pyrin domain containing 6 (Nlrp6), displayed impaired mucus production under baseline conditions. Moreover, numerous mucin-filled GCs were found within the colonic lumens of Nlrp6−/− mice, suggesting that normal mucin release by GCs was impaired. Nlrp6−/− mice also displayed impaired clearance of C. rodentium infection, albeit only at late stages of infection. Expanding on this finding, a study by the Hansson group (19) identified a novel subpopulation of GCs located at the top of colonic crypts, which secreted Muc2 in an Nlrp6-dependent manner. With the use of a unique colonic explant approach, these authors demonstrated that exposure of these explants to bacteria and/or their products led to endocytosis of Toll-like receptor (TLR)1/2, TLR4, and TLR5 ligands (19). This activated the Nlrp6 inflammasome and induced reactive oxygen species production, which culminated in a massive expulsion/exocytosis of mucin from these GCs, along with the death of these cells in a Ca2+-dependent manner (19). Moreover, the calcium signals were found to pass through gap junctions between cells, thereby promoting mucin secretion from adjacent GCs, as well as bacterial expulsion from the crypts. The authors termed these cells “sentinel GCs,” due to their novel ability to sense nearby bacteria directly, leading to mucin expulsion and the spread of signals to surrounding GCs, resulting in a coordinated mucin secretion response that flushed the bacterial stimulus away. Interestingly, the authors reported that two components of bacterial peptidoglycan—diaminopimelic acid and muramyl dipeptide—failed to elicit any secretory response. A similar finding was observed by Wang et al. (122), in that treatment with either of these compounds did not have an effect on GCs; however, when both diaminopimelic acid and muramyl dipeptide were administered to mice in parallel, it led to an increase in GC numbers by 1.5-fold, whereas similar exposure increased Muc2 expression in LS174T cells.
The ability of sentinel GCs to respond to bacterial products required inflammasome-mediated signaling. Until recently, the majority of inflammasome research has focused on its function in immune cell lineages. Two publications, by Knodler et al. (67) and Sellin et al. (101), shifted this paradigm by establishing that IECs use inflammatory caspases (caspase-1 and -4/11) to restrict intracellular pathogens. Inflammasome activation also triggered an epithelial form of pyroptosis that physically extruded the S. Typhimurium-infected cells from the epithelial barrier, both in vitro and in vivo. Activation of caspase-4/11 was also linked with IEC processing and release of the cytokine IL-18 (67). Together, these findings suggest that GCs themselves likely contain a functional inflammasome and use it to respond to bacterial products, signal to adjacent mucosal cells, and potentially influence the number of GCs within intestinal crypts. The discovery that a subgroup of GCs uses innate immune signaling to respond to bacterial products has raised an exciting, new area of focus—exploring how the mucosal barrier responds to enteric infection—and will undoubtedly uncover novel pathway(s) by which GCs use nucleotide oligomerization domain, TLR, and inflammasome-mediated mechanisms to coordinate mucosal barrier defense.
ALTERED GC FUNCTION IN RESPONSE TO SUCCESSFUL INFECTIONS
Whereas Muc2 clearly plays a protective role against pathogenic microbes invading the GI tract, in many cases, the mucus layer and/or the compound exocytosis of Muc2 by GCs are overcome by pathogens successfully infecting the intestinal mucosa (14, 78, 108). This raises the question of how GCs respond to pathogens that have successfully crossed the mucus layer. As previously shown with studies of absorptive enterocytes following infection or other proinflammatory events, the epithelium undergoes changes in its function that helps to limit the threat and/or subsequent damage. For example, when absorptive enterocytes are stimulated by bacterial products or by inflammatory mediators, this can alter their physiological function, causing diarrhea, as well as leading to the elevated production of nitric oxide and increased expression of other antimicrobial and proinflammatory mediators, including chemokines (65, 69). This switch in epithelial cell function to a more active proinflammatory role has been described in the context of many infections, both in vitro and in vivo (65). In contrast, the potential for intestinal GCs to undergo immune-driven changes in function and thereby play a more active and prolonged role in host defense has received comparatively less attention.
Aside from Muc2, intestinal GCs also produce Tff3 and Relm-β (Table 1). Previous studies, as well as results from our laboratory, showed that Tff3 and Relm-β display an immunostaining pattern similar to that seen with the Muc2 protein, which is found within GC granules (6, 66, 109). These results support the concept that Tff3 and Relm-β are also localized within the secretory granules of GC; however, further studies will be necessary to define the exact intracellular location(s) of these secreted factors. Moreover, it is unclear whether all intestinal GCs are equally capable of expressing all three mediators (Muc2, Tff3, and Relm-β) or if there are GC subsets that are specialized in expressing only a subset of these mediators, i.e., Muc2 alone, Muc2 and only Relm-β, or Muc2 and only Tff3. Additionally, it is unknown how these mediators are arranged within the theca of intestinal GCs; i.e., are the granules mixed together, or are they localized within distinct/segregated compartments? Further studies will be necessary to define how GCs control the production and secretion of these and other mediators (Fig. 3). The mediator Tff3 has been described as playing several tissue-protective roles, including inhibiting apoptosis, as well as aiding cell migration and angiogenesis, thereby actively promoting epithelial restitution and mucosal repair during an intestinal injury (75, 95, 112). Curiously, despite the fact that Tff3 is strongly expressed by GCs under baseline conditions, mice lacking Tff3 were not found to have any overt defects in baseline GI function (30). Studies of Tff3 have noted that it is modestly reduced in expression during the course of infection by C. rodentium, as well as other pathogens (16, 73, 124). Its direct impact on host defense against C. rodentium or S. Typhimurium has not yet been assessed directly; however, TFF3 was shown to play a protective role in human HT29 epithelial cells infected by Shigella dysenteriae, by upregulating the expression of MUC5Ac (93). In contrast, Tff3−/− mice showed greater protection from infection with the parasite Toxoplasma gondii in concert with a reduction in IFN-γ and IL-12 responses; however, the potential for Tff3 to recruit immune cells was not addressed (37).
Fig. 3.
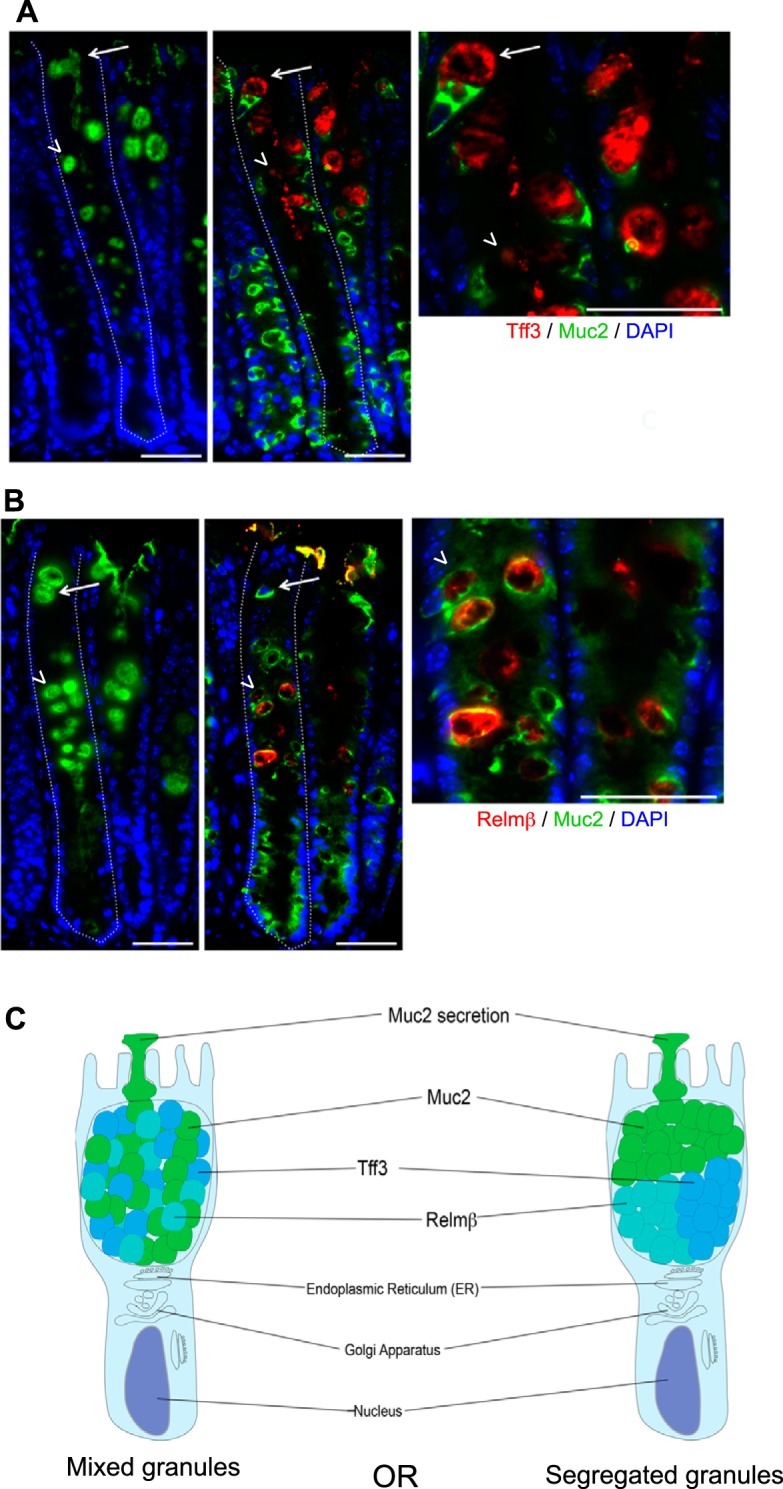
Expression of Tff3 and Relm-β in colonic goblet cells (GCs) during Citrobacter rodentium infection. Immunofluorescent staining of colonic tissue from a C57BL/6 mouse, 6 days post-C. rodentium infection. A, left: presence of GCs stained by using Santa Cruz anti-Muc2 (H300) antibody (green), targeting the mature form of Muc2. Middle: the same GCs on a serial cut section expressing both the Tff3 (red) and precursor (immature) form of Muc2, stained by using a Santa Cruz anti-Muc2 (P18) antibody (green; white arrows). Host cell nuclei are stained with DAPI (blue). Right: the higher magnification inset confirms that GCs express both Tff3 and Muc2 (white arrow). Colonic crypts (dashed white lines), GCs expressing Tff3 (white arrows), and GCs expressing Relm-β from B (arrowheads). Original scale bars, 50 μm. B, left: presence of GCs stained by using Santa Cruz anti-Muc2 (H300) antibody (green), targeting the mature form of Muc2. Middle: the same GCs on a serial cut section expressing both Relm-β (red) and the precursor form of Muc2, stained by using a Santa Cruz anti-Muc2 (P18) antibody (green; white arrowheads). Host cell nuclei are stained with DAPI (blue). Right: the higher magnification inset confirms that GCs express both Relm-β and Muc2 (white arrowhead). Colonic crypts (dashed white lines), GCs expressing Tff3 from A (white arrows), and GCs expressing Relm-β (arrowheads). Original scale bars, 50 μm. C: a cartoon outlining the possibility that intestinal GCs produce secretory granules containing Muc2, Relm-β, and Tff3 that are intermixed throughout the theca or an alternative possibility: that the different granules are segregated within the body of the theca.
Relm-β is a member of the RELM/found in inflammatory zone gene family of cysteine-rich cytokines (109). Whereas other members are expressed by a variety of cells and possess a broad range of activities, including insulin regulation, Relm-β is a GC mediator secreted apically into the intestinal lumen and basolaterally into the underlying mucosa (109). In contrast to Tff3, Relm-β expression by intestinal GCs is modest under baseline; however, Bergstrom et al. (15) found that it increased 100- to 200-fold at the transcript and protein level in response to infection by C. rodentium. Likewise, Bhinder et al. (16) found that Relm-β expression in the cecum and colon was upregulated in both C. rodentium and S. Typhimurium infections in a manner dependent on MyD88 signaling within the intestinal epithelium. In the study by Bergstrom et al. (15), Relm-β−/− mice [resistin-like β−/− (Retnlb−/−)] were found to suffer increased mortality and overt mucosal ulceration due to deep C. rodentium penetration of colonic crypts. Notably, the susceptibility of the infected Retnlb−/− mice was found to reflect impairment in their infection-induced IEC proliferation, rather than differences in pathogen burdens. Whereas Relm-β was not found to trigger epithelial cell proliferation directly, it did act as a CD4+ T cell chemoattractant. Correspondingly, Retnlb−/− mice suffered impaired recruitment of CD4+ T cells to their infected colons, along with reduced production of IL-22, a multifunctional cytokine that directly increases epithelial cell proliferation (15, 81). Relm-β enemas were able to restore CD4+ T cell recruitment in the colons of infected Retnlb−/− mice, reducing colonic pathology, along with increasing IL-22 levels and epithelial cell proliferation (81). These findings demonstrate that GCs (through Relm-β) can play an unexpected role in the promotion of immune cell recruitment to the colon to protect against an enteric pathogen (Fig. 1C). Notably, a recent study found that Relm-β can also exhibit bactericidal activity against specific gut bacteria under conditions of low salt and acidic pH (92). Mouse Relm-β and the related human resistin protein were shown to kill selectively Gram-negative bacteria, including Pseudomonas aeruginosa and C. rodentium, by damaging bacterial membranes (92). The authors of this study also demonstrated that mice lacking Relm-β carry more γ- and ε-proteobacteria within their inner colonic mucus layer (92). Thus Relm-β may exhibit antimicrobial, as well as immunomodulatory, functions that protect the host under both homeostatic and infectious conditions.
As infection by an enteric pathogen progresses, GC depletion is a commonly observed pathology. This is characterized by a reduction in intestinal mucus thickness, as well as in the number of mature, mucin-filled GCs found in the intestine (9, 16, 21, 33, 63, 78). Whereas this may, in part, reflect the appearance of GCs after they have undergone compound exocytosis, the thinner mucus layer suggests that GC function is overtly impaired. We (16) and others (104) have studied this pathology in the context of enteric bacterial infection models and noted that it occurs between 6 and 10 days post-C. rodentium infection and between 3 and 5 days post-S. Typhimurium-induced colitis. Interestingly, mucus thinning occurs concurrent with increased epithelial cell proliferation, suggesting that it may reflect the increased turnover of the epithelium, resulting in less time for GCs to mature properly. It is also possible that increased epithelial turnover leads to more GC shedding into the gut lumen, thereby depleting their numbers. In keeping with this concept, Relm-β protein expression by GCs was also found to decrease by day 10 post-C. rodentium infection (15). Several studies have shown that infection-induced GC depletion depends on the presence of immune cells, including CD4+ T lymphocytes, as well as the cytokines IFN-γ and IL-22 (15, 21). As outlined above, IL-22 can directly promote epithelial cell proliferation; however, this is not the case with IFN-γ. In contrast, IFN-γ can promote ER stress, which is seen most overtly in secretory cell types (50). ER stress is caused by the accumulation of misfolded proteins within the ER lumen, which in GCs, would impair their ability to produce normal levels of Muc2, lead to a decrease in secretory vesicles, and impair their ability to produce and release their characteristic mediators (71). As demonstrated with the aforementioned Agr2−/− mice, which lack an important ER player for protein folding, these mice produce a thinner-than-normal inner mucus layer associated with fewer mature colonic GCs than in wild-type mice (89, 131). Therefore, IFN-γ, by promoting ER stress, could lead to GC depletion by impairing their ability to fold proteins properly, thereby reducing their secretory functions (50). The impact of ER stress on GC function is highlighted by the Winnie mouse, which carries a nonsense mutation in the Muc2 gene, resulting in an accumulation of misfolded Muc2 proteins within the ER lumen of GCs, leading to GC depletion and spontaneous colitis (50, 94).
Whereas GC depletion appears to be a maladaptive host response, it is likely beneficial under certain circumstances. The host could benefit by the reduction of the release of mucins that may provide nutrients for invading microbes and through the impact of increasing the sloughing of infected epithelial cells, along with the invading pathogens into the gut lumen. Although increased epithelial cell turnover may be considered a generalized host defense mechanism against invading pathogens, it is important to note that epithelial cell proliferation, on its own, may not overcome other genetic factors that increase susceptibility to infection. For example, Tnf-α−/− and Muc2−/− mice, despite exhibiting increased epithelial turnover, still demonstrate increased susceptibility to C. rodentium infection (14, 41), suggesting that although beneficial, such a response cannot, on its own, overcome a severe genetic susceptibility to clear an infection successfully.
GC RESPONSES IN IBD/EXPERIMENTAL COLITIS
In recent years, significant attention has been paid to the potential role played by GCs and their mediators in controlling susceptibility to IBD, namely, Crohn’s disease and UC. Intestinal biopsies taken from the inflamed regions of IBD patients often display relatively thin mucus layers, as well as GC depletion. Likewise, a thin mucus layer, highly penetrable by bacteria, is a common feature of mouse models of chemical, as well as spontaneous, colitis (61). For example, DSS-induced colitis, a mouse model of human IBD, has been shown to be triggered by the DSS, causing a breakdown in the colonic mucus barrier, thus leading to bacterial contact with the intestinal epithelium that subsequently triggers overt inflammation (57). Taken together, these findings suggest that the intestinal mucus layer is structurally and functionally deficient in human and murine forms of IBD/experimental colitis (Fig. 1D). Until recently, it was unclear whether these defects were simply a consequence of IBD or a prerequisite for the development and/or chronicity of the disease. Whereas this is difficult to address in humans, a recent study demonstrated that pediatric UC patients displayed a significant reduction in GCs, as well as mucin production, in their noninflamed tissues compared with healthy tissues (1). This observation was associated with impaired expression of the innate receptor NLRP6 in the colonic epithelium, as well as increased bacterial penetration into the mucus layers. Since these results were found in noninflamed tissues, it suggests that defects in GC function may be present in individuals before disease development and thus predispose these individuals to IBD.
Correspondingly, mutations in the MUC2 gene, as well as in other genes impacting GC function, have been linked to IBD (20, 114). In a seminal study by Velcich and colleagues (118), the critical role of Muc2 in maintaining intestinal homeostasis and mucosal protection was demonstrated through the generation of Muc2−/− mice. At birth, on a 129SV background, Muc2−/− pups present with healthy GI tracts but develop spontaneous colitis within 8–12 wk. This spontaneous colitis was delayed and milder in Muc2−/− mice generated on a C57BL/6 background (118). These Muc2−/− C57BL/6 mice also developed intestinal epithelial adenocarcinomas, with both their colitis and cancer development being commensal microbe dependent. Notably, humans that develop IBD still produce an intestinal mucus layer but often display reduced Muc2 glycosylation (114). This is also consistent with the finding that B3gnt6−/− mice (previously named C3GnT−/−) are more susceptible to DSS-induced colitis, whereas mice lacking the C1galt1 enzyme within their IECs (IEC C1galt1−/− mice) developed spontaneous colitis, much like Muc2−/− mice (3). Thus defective glycosylation of MUC2 could predispose to IBD, likely by increasing microbe interactions with the intestinal epithelium and the mucosal immune system.
Reduced TFF3 expression is another common finding in IBD patient tissues, whereas the loss of Tff3 in mice has been shown to increase susceptibility to diverse forms of colitis by impairing tissue repair (Fig. 1D) (75). In particular, the role of Tff3 in intestinal restitution was first demonstrated in mice lacking Tff3, as they showed impaired mucosal healing with poor epithelial regeneration in the DSS mouse model of colitis (75). In addition, treatment with recombinant Tff3 protected these mice by increasing epithelial cell restitution and mucosal healing (75). A similar protective role was observed in Tff3−/− mice in a radiation-induced intestinal inflammation model (127). The wound-healing properties attributed to Tff3 likely reflect its ability to modulate the epithelial cell tight-junction proteins claudin-1 and -2 and zona occludens 1 (127). With the increase of junctional protein expression, it assures a more impermeable epithelium that constitutes one of the key steps in intestinal wound healing. Moreover, in a mouse model of trinitrobenzene sulfonic acid colitis, treatment with human recombinant TFF3 ameliorated the intestinal inflammation by reducing the activation of the innate signaling molecules TLR4, NF-κB, and TNF-α (113). With the reduction of innate immune signaling activation, TFF3 limited the degree and progression of the intestinal inflammation and subsequent damage to the epithelium.
In contrast, the role played by RELM-β in colitis appears more complex. Whereas few studies have examined RELM-β expression in IBD patients, reports by Hogan et al. (52) and McVay et al. (80) both determined that intestinal Relm-β expression in mice was strongly elevated during experimental gut inflammation, including in a spontaneous mouse model of Crohn’s disease (SAMP1/YitFc mice). Whereas both studies demonstrated that colitic Retnlb−/− mice suffered significantly less mucosal damage and inflammation than wild-type mice, they disagreed over the underlying mechanisms (52, 80). The paper by Hogan et al. (52) found that during DSS- and trinitrobenzene sulfonic acid-induced colitis, Retnlb−/− mice suffered reduced epithelial barrier dysfunction and expressed less of the antimicrobial lectin RegIIIγ. McVay et al. (80) also used DSS to induce colitis in Retnlb−/− mice, similarly showing that Retnlb−/− mice are less susceptible to DSS-induced colitis. In contrast, they found no role for Relm-β in modulating IEC barrier function or proliferation, although they tested relatively few parameters. Instead, they hypothesized that luminal Relm-β was able to leak across the epithelium, activating macrophages within the intestinal mucosa, although they provided little direct evidence for this action (80).
More recently, we found that Relm-β is strongly expressed in the intestines of conventionally housed Muc2−/− mice, contributing to their development of spontaneous colitis, as characterized by the clinical symptoms of body-weight loss, rectal prolapse, and diarrhea (81). Notably, in aged mice (>4 mo old), their spontaneous colitis was also associated with elevated expression of the antimicrobial lectins RegIIIβ and RegIIIγ and commensal microbial dysbiosis, including a dramatic depletion in the Lactobacilli species. To test whether Relm-β played any role in this model, Muc2−/−/Retnlb−/− mice were generated and directly compared with Muc2−/− mice. The Muc2−/−/Retnlb−/− mice exhibited a dramatic reduction in RegIII lectin production, along with increased numbers of commensal Lactobacilli spp. and reduced clinical symptoms of colitis (81). We speculated that in the absence of a mucus layer, the resulting increase in stimulation by luminal microbes leads GCs to produce more Relm-β, thereby increasing antimicrobial defenses at the mucosal surface by upregulating expression of the RegIIIβ and RegIIIγ lectins (Fig. 1D). Whereas this response would likely be protective against acute microbial infections, over several months, the increased antimicrobial activity shifts the composition of the intestinal microbiota, leading to the loss of Lactobacilli spp and promoting the development of spontaneous inflammation.
These results point out that GC responses during experimental colitis, as well as in IBD, are similar to those described during long-lasting infections. GC depletion is a key feature found in IBD patients, as well as a common characteristic of enteric infections. In both cases, although the basis for GC depletion remains unclear, the pathology results in a thinner mucus layer that is more penetrable by bacteria contributing to the pathogenesis of the disease. Moreover, ER stress and its dramatic effects on GC secretion are also seen in long-lasting infections. Recently, mutations in various proteins related to the ER stress response, such as AGR2 and X-box-binding protein 1, have been shown to increase the risk of developing IBD (71, 132). Furthermore, in mouse models, both of these molecules have been associated with protection against enteric pathogen infection by promoting normal GC responses. The fact that GC dysfunction is a common feature of colitis/IBD and chronic infection highlights the importance of these secretory cells in regulating host protection vs. noxious luminal stimuli.
NEW APPROACHES TO STUDY GC-MICROBE INTERACTIONS
As outlined above, it is clear that GCs will be an important focus for future studies regarding many aspects of GI health and disease. With the emergence of new techniques, as well as new mouse models, their application will offer significant opportunities for advancing research in the field. First, the development of key transgenic mice, including Muc2−/−, Winnie, as well as IEC-C1galt1−/− and B3gnt6−/−, has helped illustrate the key role played by Muc2 and its glycosylation in promoting intestinal homeostasis (3, 13, 50, 94, 118). Moreover, these mice have helped demonstrate that intestinal mucus composition and structure are essential to the generation and maintenance of a healthy gut microbiome. Whereas there are, as yet, no GC-specific Cre-loxP systems, there are tissue-specific deletion mouse models available to target IECs specifically, including GCs (72). As mentioned above, Bhinder et al. (16) used the Villin-cre system to show that the loss of MyD88 signaling within the murine intestinal epithelium leads to impaired, GC-specific responses at the very early stages of enteric infections (72). Recently, the Hansson group (19) developed a new mouse model, allowing the visualization of specific GCs by labeling MUC2. The resulting mCherryMUC2 transgenic mouse model expresses the human MUC2 protein with a mCherry tag, thereby providing an easy means to visualize GCs fluorescently, without the need for immunostaining (19). So far, this model has been used to study the dynamics of mucin secretion by its direct visualization in ex vivo explant cultures; however, it is possible that the mCherryMUC2 mouse could be used to isolate GCs via fluorescent-activated cell sorting, a specialized form of flow cytometry. A table summarizing the array of mouse models and in vitro techniques suitable for the study of intestinal GC is presented in Table 2.
Table 2.
Mouse models available to study goblet cell functions
| Models | Type of Models | Phenotype | Useful for Studying | Reference |
|---|---|---|---|---|
| Muc2−/− mice | KO mouse | Lack of Muc2 protein, mucus barrier loss, spontaneous colitis | Impact of mucus barrier on intestinal homeostasis, host defense, and colitis | (14, 118) |
| C1galt1−/− mice | KO mouse | Lack of core 1 glycosylation on Muc2 associated with thin mucus layer | Role of mucus structure and glycosylation on intestinal homeostasis, host defense, and colitis | (13, 115) |
| C3Gnt−/− mice | KO mouse | Lack of core 3 glycosylation on Muc2 associated with modest effects on mucus layer | Role of mucus structure and glycosylation on intestinal homeostasis, host defense, and colitis | (13, 115) |
| Tff3−/− mice | KO mouse | Lack of Tff3, impaired mucosal healing, and susceptibility to intestinal inflammation | Impact of this goblet cell-secreted factor in promoting tissue repair and antimicrobial functions in gut | (30) |
| Relmb−/− mice | KO mouse | Lack of Relmb, decreased susceptibility to colitis, increased susceptibility to infection | Role of this goblet cell-secreted factor in modulating immune and antimicrobial functions in gut | (15, 81) |
| Muc2/Relmb−/− mice | KO mouse | Lack of Muc2 and Relmb, mucus barrier loss, but delayed spontaneous colitis | The combined effects of these goblet cell-secreted factors in intestinal homeostasis/colitis | (81) |
| Winnie mice | Transgenic mouse (nonsense mutation) | ER stress, goblet cell depletion, spontaneous colitis | Impact of ER stress and mucus composition/formation on intestinal homeostasis/colitis | (50, 94) |
| Villin-cre mice | Tissue-specific KO mouse | Allows gene-targeting/deletion in intestinal epithelial cells, including goblet cells | Impact of specific deletion of genes in intestinal epithelial cells, including goblet cells | (16, 72) |
| mCherryMUC2 mice | Transgenic mouse (exogenous protein expression) | Expression of human MUC2 protein tagged to mCHERRY-Flag in the mouse intestine | The location and function of human exogenous Muc2 in mice | (19) |
| Ex vivo colonic explant | In vitro model | Colonic tissue explant cultured in vitro | Mucus dynamics and secretion studies | (45) |
| LS174T cells | In vitro model | Intestinal epithelial cell lines expressing Muc2 | In vitro goblet cell-like cell lines | (8) |
| HT29 or HT29-MTX cells | In vitro model | Intestinal epithelial cell lines expressing Muc2 | In vitro goblet cell-like cell lines | (22, 39) |
| HT29 organotypic | In vitro model | Organotypic culture derived from transformed intestinal epithelial cell lines | In vitro goblet cell-like cell lines in an organotypic model | (54) |
| Organoids | In vitro model | Intestinal stem cells grown in 3D culture containing differentiated cells, such as goblet cells | In vitro goblet cell response to various stimuli (infection, cytokines, metabolites) | (55, 119) |
KO, knockout; MTX, methotrexate.
A major impediment impacting research of this cell type over the last several decades has been the lack of accurate in vitro cellular models for intestinal GCs. Currently, there are only a few “GC-like” cell lines, including the mucin-secreting LS174T and the HT29-methotrexate cell lines. Unfortunately, these cell lines do not secrete significant amounts of Muc2, have only a superficial structural resemblance to GCs, and express most key GC markers only at the gene transcript level (8, 39). Whereas the HT29 cell line has also been used to create a three-dimensional (3D) organotypic culture of IECs that showed localized Muc2 expression (54), this technique led to only a small number of mature GCs differentiating from the transformed cells (54). However, over the last several years, the Hansson group (45) has developed an exciting ex vivo technique adapted for the mouse, as well as human, small intestine and colon. Surgically resected intestinal tissues are mounted in a horizontal perfusion chamber to allow the analysis of GC and mucus properties. This explant method facilitates the study of the mechanisms by which GCs can be stimulated to secrete mucins. With the use of this approach, along with charcoal and variable-sized beads, they have been able to study the permeability of the intestinal mucus layer. Future studies undoubtedly will use specific gene-deficient mice to define the influence of key genes or signaling pathways on GC biology. Lastly, the rapidly moving field of in vitro enteroid/organoid culturing offers significant potential to facilitate GC studies. With the use of induced pluripotent stem cells, whole intestinal biopsies, or purified leucine-rich repeat-containing G-protein-coupled receptor 5-positive stem cells, enteroids can be grown in Matrigel, forming miniature 3D gut balls, consisting entirely of IECs, with their makeup matching that of the intestinal region from which the biopsy originated (68). Notably, of the epithelial cells that comprise colonic enteroids derived from either mouse or human biopsies, roughly 5–20% are GCs that appear to be functional when maintained under normal enteroid culture conditions (119). The increase or decrease of the differentiation of GC, as well as other IEC subtypes within enteroids, can be achieved by the alteration of the concentrations of R-spondin, Noggin, and Wnt3A within the culture medium (110). In addition, a pharmacological approach to inhibit Notch signaling by dibenzazepine (DBZ) dramatically increased GC numbers within enteroids (Fig. 4). Enteroids can be microinjected with different microbes to study the interactions of specific bacteria and IECs, such as GCs (Fig. 4). Enteroids have also been used to create a 2D monolayer that can be infected successfully by different pathogen strains, such as EHEC and S. Typhimurium, to recreate a valuable in vitro infection model (55, 64, 119, 130). Whereas these studies are just in their infancy, it is clear that the use of enteroids derived from patients with specific genetic mutations or gene-deficient mouse strains will rapidly facilitate our understanding of GC signaling and function.
Fig. 4.

Enteroids provide a useful model to study microbe interactions with goblet cells. Immunostaining of mouse colonic enteroids for E-cadherin (red), Muc2 (green), and DNA (DAPI; blue) under (A) baseline conditions or (B) following enteroid treatment with the Notch inhibitor dibenzazepine (DBZ) to increase goblet cell numbers. Muc2-producing goblet cells are seen under baseline conditions but are dramatically increased in number following DBZ treatment (white arrows). C: contrast-phase micrograph showing a mouse intestine enteroid microinjected with Salmonella enterica serovar Typhimurium. D: immunostaining of a mouse intestine enteroid showing the presence of luminal S. Typhimurium (green) in contact with epithelial cells stained with β-actin (red), whereas epithelial cell nuclei are stained with DAPI (blue). Original scale bars, 50 μm.
CONCLUSIONS
In conclusion, intestinal GCs play a key role in controlling host-microbe interactions within the GI tract. As the cell type responsible for the generation and structure of the mucus layer, GCs control the access of commensal microbes to the intestinal mucosal surface through Muc2 and other microbial aggregating factors. In addition, Muc2 glycosylation impacts the function of the mucus layer, as well as its ability to control microbe adherence and microbial nutrition at this key microbe-host interface. A key question to be addressed is whether impairment in mucus structure/function is a direct cause of IBD or solely a consequence. The acknowledgment of this will require well-designed longitudinal studies. Whereas Muc2 is the main constituent of the intestinal mucus barrier, recent studies have revealed that much of its protective nature may reflect myriad proteins and other host-derived factors that reside within intestinal mucus and which bind to or target gut microbes, limiting their ability to cross the mucus layers. Notably, GCs are now being recognized as active players in host defense by responding to their luminal environment through the compound exocytosis of their granules. The discovery that this response is inflammasome dependent raises the issue of whether it reflects GC intrinsic signaling and if there are other GC responses beyond mucin release that are triggered by inflammasome activation.
Aside from mucin release, it is also becoming clear that other GC-derived mediators, such as RELM-β, can play a key role in controlling gut microbes, as well as the host immune response, in the context of enteric infections and IBD. The potential that there are other GC mediators that have yet to be defined is an intriguing possibility. Moreover, the definition of the impact of GCs and their mediators in controlling susceptibility to IBD, as well as potentially driving the disease, will be a key area of study over the coming years. In concert with the increasing interest in GCs has been the development of new approaches to study their function. With new mouse strains lacking the expression of key genes solely in their intestinal epithelium, there is a growing potential to define the in vivo functions of GCs. New approaches to culture intestinal biopsies, as well as enteroids, also offer significant potential to define the functions of GCs and their mediators; however, at present, they are unable to differentiate between responses that are solely GC intrinsic and those that rely on communication among different epithelial cell types. Additional models that can specifically target GCs, whereas leaving other epithelial subtypes alone, would be a significant boon to this growing field.
GRANTS
Research in the B. A. Vallance laboratory is supported by operating grants from the National Science and Engineering Research Council (NSERC; RGPIN 436233-13) and the Canadian Institutes of Health Research (MOP-126051 and PJT-148846; both to B. A. Vallance) and from Crohn’s and Colitis Canada (to B. A. Vallance and L. A. Knodler). Support for J. M. Allaire was provided by a fellowship from the Fond de Recherche en Santé du Québec (FRQS) and for M. Stahl, by a Mitacs Accelerate award. Support for K. Bhullar was provided by a Vanier Graduate Canada Scholarship (VGS). B. A. Vallance is the Children with Intestinal and Liver Disorders (Ch.I.L.D.) Foundation Research Chair in Pediatric Gastroenterology. K. Jacobson is a senior clinician scientist supported by the BC Children’s and Women’s Hospital Research Institute (BCCHRI) Clinician Scientists Award Program, University of British Columbia (UBC), and Ch.I.L.D. Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.M.A., V.M., and M.S. prepared figures; J.M.A., V.M., S.M.C., M.S., and B.A.V. drafted manuscript; J.M.A., V.M., S.M.C., H.Y., K.B., L.A.K., B.B., K.J., and B.A.V. edited and revised manuscript; J.M.A. and B.A.V. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Caixia Ma and Tina Huang for technical assistance.
Present address of V. Morampudi: enGene, Montreal, QC, Canada H4S 1Z9.
Present address of K. Bhullar: StemCell Technologies, Vancouver, BC, Canada V6A 1B6.
REFERENCES
- 1.Alipour M, Zaidi D, Valcheva R, Jovel J, Martínez I, Sergi C, Walter J, Mason AL, Wong GK, Dieleman LA, Carroll MW, Huynh HQ, Wine E. Mucosal barrier depletion and loss of bacterial diversity are primary abnormalities in paediatric ulcerative colitis. J Crohn’s Colitis 10: 462–471, 2016. doi: 10.1093/ecco-jcc/jjv223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ambort D, Johansson ME, Gustafsson JK, Ermund A, Hansson GC. Perspectives on mucus properties and formation—lessons from the biochemical world. Cold Spring Harb Perspect Med 2: 2, 2012. doi: 10.1101/cshperspect.a014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.An G, Wei B, Xia B, McDaniel JM, Ju T, Cummings RD, Braun J, Xia L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J Exp Med 204: 1417–1429, 2007. doi: 10.1084/jem.20061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arike L, Hansson GC. The densely O-glycosylated MUC2 mucin protects the intestine and provides food for the commensal bacteria. J Mol Biol 428: 3221–3229, 2016. doi: 10.1016/j.jmb.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arike L, Holmén-Larsson J, Hansson GC. Intestinal Muc2 mucin O-glycosylation is affected by microbiota and regulated by differential expression of glycosyltranferases. Glycobiology 27: 318–328, 2017. doi: 10.1093/glycob/cww134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Artis D, Wang ML, Keilbaugh SA, He W, Brenes M, Swain GP, Knight PA, Donaldson DD, Lazar MA, Miller HR, Schad GA, Scott P, Wu GD. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc Natl Acad Sci USA 101: 13596–13600, 2004. doi: 10.1073/pnas.0404034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asker N, Axelsson MA, Olofsson SO, Hansson GC. Dimerization of the human MUC2 mucin in the endoplasmic reticulum is followed by a N-glycosylation-dependent transfer of the mono- and dimers to the Golgi apparatus. J Biol Chem 273: 18857–18863, 1998. doi: 10.1074/jbc.273.30.18857. [DOI] [PubMed] [Google Scholar]
- 8.Becker S, Oelschlaeger TA, Wullaert A, Vlantis K, Pasparakis M, Wehkamp J, Stange EF, Gersemann M. Bacteria regulate intestinal epithelial cell differentiation factors both in vitro and in vivo. PLoS One 8: e55620, 2013. doi: 10.1371/journal.pone.0055620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bengtsson RJ, MacIntyre N, Guthrie J, Wilson AD, Finlayson H, Matika O, Pong-Wong R, Smith SH, Archibald AL, Ait-Ali T. Lawsonia intracellularis infection of intestinal crypt cells is associated with specific depletion of secreted MUC2 in goblet cells. Vet Immunol Immunopathol 168: 61–67, 2015. doi: 10.1016/j.vetimm.2015.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergström A, Kristensen MB, Bahl MI, Metzdorff SB, Fink LN, Frøkiaer H, Licht TR. Nature of bacterial colonization influences transcription of mucin genes in mice during the first week of life. BMC Res Notes 5: 402, 2012. doi: 10.1186/1756-0500-5-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergström JH, Berg KA, Rodríguez-Piñeiro AM, Stecher B, Johansson ME, Hansson GC. AGR2, an endoplasmic reticulum protein, is secreted into the gastrointestinal mucus. PLoS One 9: e104186, 2014. doi: 10.1371/journal.pone.0104186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergström JH, Birchenough GM, Katona G, Schroeder BO, Schütte A, Ermund A, Johansson ME, Hansson GC. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc Natl Acad Sci USA 113: 13833–13838, 2016. doi: 10.1073/pnas.1611400113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergstrom K, Fu J, Johansson ME, Liu X, Gao N, Wu Q, Song J, McDaniel JM, McGee S, Chen W, Braun J, Hansson GC, Xia L. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. Mucosal Immunol 10: 91–103, 2017. doi: 10.1038/mi.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bergstrom KS, Kissoon-Singh V, Gibson DL, Ma C, Montero M, Sham HP, Ryz N, Huang T, Velcich A, Finlay BB, Chadee K, Vallance BA. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog 6: e1000902, 2010. doi: 10.1371/journal.ppat.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bergstrom KS, Morampudi V, Chan JM, Bhinder G, Lau J, Yang H, Ma C, Huang T, Ryz N, Sham HP, Zarepour M, Zaph C, Artis D, Nair M, Vallance BA. Goblet cell derived RELM-β recruits CD4+ T cells during infectious colitis to promote protective intestinal epithelial cell proliferation. PLoS Pathog 11: e1005108, 2015. doi: 10.1371/journal.ppat.1005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, Vallance BA. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun 82: 3753–3763, 2014. doi: 10.1128/IAI.02045-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhullar K, Zarepour M, Yu H, Yang H, Croxen M, Stahl M, Finlay BB, Turvey SE, Vallance BA. The serine protease autotransporter Pic modulates Citrobacter rodentium pathogenesis and its innate recognition by the host. Infect Immun 83: 2636–2650, 2015. doi: 10.1128/IAI.00025-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birchenough GM, Johansson ME, Gustafsson JK, Bergström JH, Hansson GC. New developments in goblet cell mucus secretion and function. Mucosal Immunol 8: 712–719, 2015. doi: 10.1038/mi.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birchenough GM, Nyström EE, Johansson ME, Hansson GC. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352: 1535–1542, 2016. doi: 10.1126/science.aaf7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boltin D, Perets TT, Vilkin A, Niv Y. Mucin function in inflammatory bowel disease: an update. J Clin Gastroenterol 47: 106–111, 2013. doi: 10.1097/MCG.0b013e3182688e73. [DOI] [PubMed] [Google Scholar]
- 21.Chan JM, Bhinder G, Sham HP, Ryz N, Huang T, Bergstrom KS, Vallance BA. CD4+ T cells drive goblet cell depletion during Citrobacter rodentium infection. Infect Immun 81: 4649–4658, 2013. doi: 10.1128/IAI.00655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cobo ER, Kissoon-Singh V, Moreau F, Holani R, Chadee K. MUC2 mucin and butyrate contribute to the synthesis of the antimicrobial peptide cathelicidin in response to Entamoeba histolytica- and dextran sodium sulfate-induced colitis. Infect Immun 85: 85, 2017. doi: 10.1128/IAI.00905-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comstock LE. Importance of glycans to the host-bacteroides mutualism in the mammalian intestine. Cell Host Microbe 5: 522–526, 2009. doi: 10.1016/j.chom.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev 61: 75–85, 2009. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Corfield AP, Myerscough N, Warren BF, Durdey P, Paraskeva C, Schauer R. Reduction of sialic acid O-acetylation in human colonic mucins in the adenoma-carcinoma sequence. Glycoconj J 16: 307–317, 1999. doi: 10.1023/A:1007026314792. [DOI] [PubMed] [Google Scholar]
- 26.Coulombe G, Langlois A, De Palma G, Langlois MJ, McCarville JL, Gagné-Sanfaçon J, Perreault N, Feng GS, Bercik P, Boudreau F, Verdu EF, Rivard N. SHP-2 phosphatase prevents colonic inflammation by controlling secretory cell differentiation and maintaining host-microbiota homeostasis. J Cell Physiol 231: 2529–2540, 2016. doi: 10.1002/jcp.25407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Croix JA, Carbonero F, Nava GM, Russell M, Greenberg E, Gaskins HR. On the relationship between sialomucin and sulfomucin expression and hydrogenotrophic microbes in the human colonic mucosa. PLoS One 6: e24447, 2011. doi: 10.1371/journal.pone.0024447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 7: 349–359, 2006. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- 29.Dann SM, Eckmann L. Innate immune defenses in the intestinal tract. Curr Opin Gastroenterol 23: 115–120, 2007. doi: 10.1097/MOG.0b013e32803cadf4. [DOI] [PubMed] [Google Scholar]
- 30.Dehlawi MS, Mahida YR, Hughes K, Wakelin D. Effects of Trichinella spiralis infection on intestinal pathology in mice lacking interleukin-4 (IL-4) or intestinal trefoil factor (ITF/TFF3). Parasitol Int 55: 207–211, 2006. doi: 10.1016/j.parint.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 31.Deplancke B, Gaskins HR. Microbial modulation of innate defense: goblet cells and the intestinal mucus layer. Am J Clin Nutr 73: 1131S–1141S, 2001. [DOI] [PubMed] [Google Scholar]
- 32.Dwivedi R, Nothaft H, Garber J, Xin Kin L, Stahl M, Flint A, van Vliet AH, Stintzi A, Szymanski CM. L-Fucose influences chemotaxis and biofilm formation in Campylobacter jejuni. Mol Microbiol 101: 575–589, 2016. doi: 10.1111/mmi.13409. [DOI] [PubMed] [Google Scholar]
- 33.Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol 308: G510–G524, 2015. doi: 10.1152/ajpgi.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erdem AL, Avelino F, Xicohtencatl-Cortes J, Girón JA. Host protein binding and adhesive properties of H6 and H7 flagella of attaching and effacing Escherichia coli. J Bacteriol 189: 7426–7435, 2007. doi: 10.1128/JB.00464-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabich AJ, Jones SA, Chowdhury FZ, Cernosek A, Anderson A, Smalley D, McHargue JW, Hightower GA, Smith JT, Autieri SM, Leatham MP, Lins JJ, Allen RL, Laux DC, Cohen PS, Conway T. Comparison of carbon nutrition for pathogenic and commensal Escherichia coli strains in the mouse intestine. Infect Immun 76: 1143–1152, 2008. doi: 10.1128/IAI.01386-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freitas M, Cayuela C, Antoine JM, Piller F, Sapin C, Trugnan G. A heat labile soluble factor from Bacteroides thetaiotaomicron VPI-5482 specifically increases the galactosylation pattern of HT29-MTX cells. Cell Microbiol 3: 289–300, 2001. doi: 10.1046/j.1462-5822.2001.00113.x. [DOI] [PubMed] [Google Scholar]
- 37.Fu T, Znalesniak EB, Kalinski T, Möhle L, Biswas A, Salm F, Dunay IR, Hoffmann W. TFF peptides play a role in the immune response following oral infection of mice with Toxoplasma gondii. Eur J Microbiol Immunol (Bp) 5: 221–231, 2015. doi: 10.1556/1886.2015.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gagné-Sansfaçon J, Allaire JM, Jones C, Boudreau F, Perreault N. Loss of sonic hedgehog leads to alterations in intestinal secretory cell maturation and autophagy. PLoS One 9: e98751, 2014. doi: 10.1371/journal.pone.0098751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gagnon M, Zihler Berner A, Chervet N, Chassard C, Lacroix C. Comparison of the Caco-2, HT-29 and the mucus-secreting HT29-MTX intestinal cell models to investigate Salmonella adhesion and invasion. J Microbiol Methods 94: 274–279, 2013. doi: 10.1016/j.mimet.2013.06.027. [DOI] [PubMed] [Google Scholar]
- 40.Gerbe F, Jay P. Intestinal tuft cells: epithelial sentinels linking luminal cues to the immune system. Mucosal Immunol 9: 1353–1359, 2016. doi: 10.1038/mi.2016.68. [DOI] [PubMed] [Google Scholar]
- 41.Gonçalves NS, Ghaem-Maghami M, Monteleone G, Frankel G, Dougan G, Lewis DJ, Simmons CP, MacDonald TT. Critical role for tumor necrosis factor alpha in controlling the number of lumenal pathogenic bacteria and immunopathology in infectious colitis. Infect Immun 69: 6651–6659, 2001. doi: 10.1128/IAI.69.11.6651-6659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graziani F, Pujol A, Nicoletti C, Dou S, Maresca M, Giardina T, Fons M, Perrier J. Ruminococcus gnavus E1 modulates mucin expression and intestinal glycosylation. J Appl Microbiol 120: 1403–1417, 2016. doi: 10.1111/jam.13095. [DOI] [PubMed] [Google Scholar]
- 43.Grootjans J, Hundscheid IH, Buurman WA. Goblet cell compound exocytosis in the defense against bacterial invasion in the colon exposed to ischemia-reperfusion. Gut Microbes 4: 232–235, 2013. doi: 10.4161/gmic.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gulhane M, Murray L, Lourie R, Tong H, Sheng YH, Wang R, Kang A, Schreiber V, Wong KY, Magor G, Denman S, Begun J, Florin TH, Perkins A, Cuív PO, McGuckin MA, Hasnain SZ. High fat diets induce colonic epithelial cell stress and inflammation that is reversed by IL-22. Sci Rep 6: 28990, 2016. doi: 10.1038/srep28990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gustafsson JK, Ermund A, Johansson ME, Schütte A, Hansson GC, Sjövall H. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am J Physiol Gastrointest Liver Physiol 302: G430–G438, 2012. doi: 10.1152/ajpgi.00405.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halm DR, Halm ST. Secretagogue response of goblet cells and columnar cells in human colonic crypts. Am J Physiol Cell Physiol 278: C212–C233, 2000. doi: 10.1152/ajpcell.2000.278.1.C212. [DOI] [PubMed] [Google Scholar]
- 47.Hansson GC, Johansson ME. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 1: 51–54, 2010. doi: 10.4161/gmic.1.1.10470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harrington SM, Sheikh J, Henderson IR, Ruiz-Perez F, Cohen PS, Nataro JP. The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect Immun 77: 2465–2473, 2009. doi: 10.1128/IAI.01494-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He W, Wang ML, Jiang HQ, Steppan CM, Shin ME, Thurnheer MC, Cebra JJ, Lazar MA, Wu GD. Bacterial colonization leads to the colonic secretion of RELMbeta/FIZZ2, a novel goblet cell-specific protein. Gastroenterology 125: 1388–1397, 2003. doi: 10.1016/j.gastro.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 5: e54, 2008. doi: 10.1371/journal.pmed.0050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hews CL, Tran SL, Wegmann U, Brett B, Walsham AD, Kavanaugh D, Ward NJ, Juge N, Schüller S. The StcE metalloprotease of enterohaemorrhagic Escherichia coli reduces the inner mucus layer and promotes adherence to human colonic epithelium ex vivo. Cell Microbiol 19: e12717, 2017. doi: 10.1111/cmi.12717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hogan SP, Seidu L, Blanchard C, Groschwitz K, Mishra A, Karow ML, Ahrens R, Artis D, Murphy AJ, Valenzuela DM, Yancopoulos GD, Rothenberg ME. Resistin-like molecule beta regulates innate colonic function: barrier integrity and inflammation susceptibility. J Allergy Clin Immunol 118: 257–268, 2006. doi: 10.1016/j.jaci.2006.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmén Larsson JM, Thomsson KA, Rodríguez-Piñeiro AM, Karlsson H, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. III. Gastrointestinal Muc5ac and Muc2 mucin O-glycan patterns reveal a regiospecific distribution. Am J Physiol Gastrointest Liver Physiol 305: G357–G363, 2013. doi: 10.1152/ajpgi.00048.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Höner zu Bentrup K, Ramamurthy R, Ott CM, Emami K, Nelman-Gonzalez M, Wilson JW, Richter EG, Goodwin TJ, Alexander JS, Pierson DL, Pellis N, Buchanan KL, Nickerson CA. Three-dimensional organotypic models of human colonic epithelium to study the early stages of enteric salmonellosis. Microbes Infect 8: 1813–1825, 2006. doi: 10.1016/j.micinf.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 55.In J, Foulke-Abel J, Zachos NC, Hansen AM, Kaper JB, Bernstein HD, Halushka M, Blutt S, Estes MK, Donowitz M, Kovbasnjuk O. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol 2: 48–62, 2016. doi: 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johansson ME, Ambort D, Pelaseyed T, Schütte A, Gustafsson JK, Ermund A, Subramani DB, Holmén-Larsson JM, Thomsson KA, Bergström JH, van der Post S, Rodriguez-Piñeiro AM, Sjövall H, Bäckström M, Hansson GC. Composition and functional role of the mucus layers in the intestine. Cell Mol Life Sci 68: 3635–3641, 2011. doi: 10.1007/s00018-011-0822-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johansson ME, Gustafsson JK, Sjöberg KE, Petersson J, Holm L, Sjövall H, Hansson GC. Bacteria penetrate the inner mucus layer before inflammation in the dextran sulfate colitis model. PLoS One 5: e12238, 2010. doi: 10.1371/journal.pone.0012238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johansson ME, Jakobsson HE, Holmén-Larsson J, Schütte A, Ermund A, Rodríguez-Piñeiro AM, Arike L, Wising C, Svensson F, Bäckhed F, Hansson GC. Normalization of host intestinal mucus layers requires long-term microbial colonization. Cell Host Microbe 18: 582–592, 2015. doi: 10.1016/j.chom.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA 108, Suppl 1: 4659–4665, 2011. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci USA 105: 15064–15069, 2008. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Johansson ME, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol 10: 352–361, 2013. doi: 10.1038/nrgastro.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johansson ME, Thomsson KA, Hansson GC. Proteomic analyses of the two mucus layers of the colon barrier reveal that their main component, the Muc2 mucin, is strongly bound to the Fcgbp protein. J Proteome Res 8: 3549–3557, 2009. doi: 10.1021/pr9002504. [DOI] [PubMed] [Google Scholar]
- 63.Jung K, Saif LJ. Goblet cell depletion in small intestinal villous and crypt epithelium of conventional nursing and weaned pigs infected with porcine epidemic diarrhea virus. Res Vet Sci 110: 12–15, 2017. doi: 10.1016/j.rvsc.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 64.Karve SS, Pradhan S, Ward DV, Weiss AA. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS One 12: e0178966, 2017. doi: 10.1371/journal.pone.0178966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Khan MA, Bouzari S, Ma C, Rosenberger CM, Bergstrom KS, Gibson DL, Steiner TS, Vallance BA. Flagellin-dependent and -independent inflammatory responses following infection by enteropathogenic Escherichia coli and Citrobacter rodentium. Infect Immun 76: 1410–1422, 2008. doi: 10.1128/IAI.01141-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim JC, Yoon JW, Kim CH, Park MS, Cho SH. Repression of flagella motility in enterohemorrhagic Escherichia coli O157:H7 by mucin components. Biochem Biophys Res Commun 423: 789–792, 2012. doi: 10.1016/j.bbrc.2012.06.041. [DOI] [PubMed] [Google Scholar]
- 67.Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16: 249–256, 2014. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kretzschmar K, Clevers H. Organoids: modeling development and the stem cell niche in a dish. Dev Cell 38: 590–600, 2016. doi: 10.1016/j.devcel.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 69.Laohachai KN, Bahadi R, Hardo MB, Hardo PG, Kourie JI. The role of bacterial and non-bacterial toxins in the induction of changes in membrane transport: implications for diarrhea. Toxicon 42: 687–707, 2003. doi: 10.1016/j.toxicon.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 70.Li H, Limenitakis JP, Fuhrer T, Geuking MB, Lawson MA, Wyss M, Brugiroux S, Keller I, Macpherson JA, Rupp S, Stolp B, Stein JV, Stecher B, Sauer U, McCoy KD, Macpherson AJ. The outer mucus layer hosts a distinct intestinal microbial niche. Nat Commun 6: 8292, 2015. doi: 10.1038/ncomms9292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Luo K, Cao SS. Endoplasmic reticulum stress in intestinal epithelial cell function and inflammatory bowel disease. Gastroenterol Res Pract 2015: 328791, 2015. doi: 10.1155/2015/328791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Madison BB, Dunbar L, Qiao XT, Braunstein K, Braunstein E, Gumucio DL. Cis elements of the Villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 277: 33275–33283, 2002. doi: 10.1074/jbc.M204935200. [DOI] [PubMed] [Google Scholar]
- 73.Manko A, Motta JP, Cotton JA, Feener T, Oyeyemi A, Vallance BA, Wallace JL, Buret AG. Giardia co-infection promotes the secretion of antimicrobial peptides beta-defensin 2 and trefoil factor 3 and attenuates attaching and effacing bacteria-induced intestinal disease. PLoS One 12: e0178647, 2017. doi: 10.1371/journal.pone.0178647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Martens EC, Roth R, Heuser JE, Gordon JI. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J Biol Chem 284: 18445–18457, 2009. doi: 10.1074/jbc.M109.008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mashimo H, Wu DC, Podolsky DK, Fishman MC. Impaired defense of intestinal mucosa in mice lacking intestinal trefoil factor. Science 274: 262–265, 1996. doi: 10.1126/science.274.5285.262. [DOI] [PubMed] [Google Scholar]
- 76.Matsuo K, Ota H, Akamatsu T, Sugiyama A, Katsuyama T. Histochemistry of the surface mucous gel layer of the human colon. Gut 40: 782–789, 1997. doi: 10.1136/gut.40.6.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCormick BA, Stocker BA, Laux DC, Cohen PS. Roles of motility, chemotaxis, and penetration through and growth in intestinal mucus in the ability of an avirulent strain of Salmonella typhimurium to colonize the large intestine of streptomycin-treated mice. Infect Immun 56: 2209–2217, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McGuckin MA, Lindén SK, Sutton P, Florin TH. Mucin dynamics and enteric pathogens. Nat Rev Microbiol 9: 265–278, 2011. doi: 10.1038/nrmicro2538. [DOI] [PubMed] [Google Scholar]
- 79.McLoughlin K, Schluter J, Rakoff-Nahoum S, Smith AL, Foster KR. Host selection of microbiota via differential adhesion. Cell Host Microbe 19: 550–559, 2016. doi: 10.1016/j.chom.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 80.McVay LD, Keilbaugh SA, Wong TM, Kierstein S, Shin ME, Lehrke M, Lefterova MI, Shifflett DE, Barnes SL, Cominelli F, Cohn SM, Hecht G, Lazar MA, Haczku A, Wu GD. Absence of bacterially induced RELMbeta reduces injury in the dextran sodium sulfate model of colitis. J Clin Invest 116: 2914–2923, 2006. doi: 10.1172/JCI28121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Morampudi V, Dalwadi U, Bhinder G, Sham HP, Gill SK, Chan J, Bergstrom KS, Huang T, Ma C, Jacobson K, Gibson DL, Vallance BA. The goblet cell-derived mediator RELM-β drives spontaneous colitis in Muc2-deficient mice by promoting commensal microbial dysbiosis. Mucosal Immunol 9: 1218–1233, 2016. doi: 10.1038/mi.2015.140. [DOI] [PubMed] [Google Scholar]
- 82.Nakamura K, Sakuragi N, Takakuwa A, Ayabe T. Paneth cell α-defensins and enteric microbiota in health and disease. Biosci Microbiota Food Health 35: 57–67, 2016. doi: 10.12938/bmfh.2015-019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nevola JJ, Stocker BA, Laux DC, Cohen PS. Colonization of the mouse intestine by an avirulent Salmonella typhimurium strain and its lipopolysaccharide-defective mutants. Infect Immun 50: 152–159, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Noah TK, Kazanjian A, Whitsett J, Shroyer NF. SAM pointed domain ETS factor (SPDEF) regulates terminal differentiation and maturation of intestinal goblet cells. Exp Cell Res 316: 452–465, 2010. doi: 10.1016/j.yexcr.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Okumura R, Kurakawa T, Nakano T, Kayama H, Kinoshita M, Motooka D, Gotoh K, Kimura T, Kamiyama N, Kusu T, Ueda Y, Wu H, Iijima H, Barman S, Osawa H, Matsuno H, Nishimura J, Ohba Y, Nakamura S, Iida T, Yamamoto M, Umemoto E, Sano K, Takeda K. Lypd8 promotes the segregation of flagellated microbiota and colonic epithelia. Nature 532: 117–121, 2016. doi: 10.1038/nature17406. [DOI] [PubMed] [Google Scholar]
- 86.Pácha J. Development of intestinal transport function in mammals. Physiol Rev 80: 1633–1667, 2000. doi: 10.1152/physrev.2000.80.4.1633. [DOI] [PubMed] [Google Scholar]
- 87.Pacheco AR, Curtis MM, Ritchie JM, Munera D, Waldor MK, Moreira CG, Sperandio V. Fucose sensing regulates bacterial intestinal colonization. Nature 492: 113–117, 2012. doi: 10.1038/nature11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Parham NJ, Srinivasan U, Desvaux M, Foxman B, Marrs CF, Henderson IR. PicU, a second serine protease autotransporter of uropathogenic Escherichia coli. FEMS Microbiol Lett 230: 73–83, 2004. doi: 10.1016/S0378-1097(03)00862-0. [DOI] [PubMed] [Google Scholar]
- 89.Park SW, Zhen G, Verhaeghe C, Nakagami Y, Nguyenvu LT, Barczak AJ, Killeen N, Erle DJ. The protein disulfide isomerase AGR2 is essential for production of intestinal mucus. Proc Natl Acad Sci USA 106: 6950–6955, 2009. doi: 10.1073/pnas.0808722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pelaseyed T, Bergström JH, Gustafsson JK, Ermund A, Birchenough GM, Schütte A, van der Post S, Svensson F, Rodríguez-Piñeiro AM, Nyström EE, Wising C, Johansson ME, Hansson GC. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev 260: 8–20, 2014. doi: 10.1111/imr.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Muñoz ME, Arrieta MC, Ramer-Tait AE, Walter J. A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 5: 48, 2017. doi: 10.1186/s40168-017-0268-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Propheter DC, Chara AL, Harris TA, Ruhn KA, Hooper LV. Resistin-like molecule β is a bactericidal protein that promotes spatial segregation of the microbiota and the colonic epithelium. Proc Natl Acad Sci USA 114: 11027–11033, 2017. doi: 10.1073/pnas.1711395114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Raja SB, Murali MR, Devaraj H, Devaraj SN. Differential expression of gastric MUC5AC in colonic epithelial cells: TFF3-wired IL1 β/Akt crosstalk-induced mucosal immune response against Shigella dysenteriae infection. J Cell Sci 125: 703–713, 2012. doi: 10.1242/jcs.092148. [DOI] [PubMed] [Google Scholar]
- 94.Robinson AM, Rahman AA, Carbone SE, Randall-Demllo S, Filippone R, Bornstein JC, Eri R, Nurgali K. Alterations of colonic function in the Winnie mouse model of spontaneous chronic colitis. Am J Physiol Gastrointest Liver Physiol 312: G85–G102, 2017. doi: 10.1152/ajpgi.00210.2016. [DOI] [PubMed] [Google Scholar]
- 95.Rodrigues S, Van Aken E, Van Bocxlaer S, Attoub S, Nguyen QD, Bruyneel E, Westley BR, May FE, Thim L, Mareel M, Gespach C, Emami S. Trefoil peptides as proangiogenic factors in vivo and in vitro: implication of cyclooxygenase-2 and EGF receptor signaling. FASEB J 17: 7–16, 2003. doi: 10.1096/fj.02-0201com. [DOI] [PubMed] [Google Scholar]
- 96.Rodríguez-Piñeiro AM, Bergström JH, Ermund A, Gustafsson JK, Schütte A, Johansson ME, Hansson GC. Studies of mucus in mouse stomach, small intestine, and colon. II. Gastrointestinal mucus proteome reveals Muc2 and Muc5ac accompanied by a set of core proteins. Am J Physiol Gastrointest Liver Physiol 305: G348–G356, 2013. doi: 10.1152/ajpgi.00047.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rodríguez-Piñeiro AM, van der Post S, Johansson ME, Thomsson KA, Nesvizhskii AI, Hansson GC. Proteomic study of the mucin granulae in an intestinal goblet cell model. J Proteome Res 11: 1879–1890, 2012. doi: 10.1021/pr2010988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rogier EW, Frantz AL, Bruno ME, Kaetzel CS. Secretory IgA is concentrated in the outer layer of colonic mucus along with gut bacteria. Pathogens 3: 390–403, 2014. doi: 10.3390/pathogens3020390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rokhsefat S, Lin A, Comelli EM. Mucin-microbiota interaction during postnatal maturation of the intestinal ecosystem: clinical implications. Dig Dis Sci 61: 1473–1486, 2016. doi: 10.1007/s10620-016-4032-6. [DOI] [PubMed] [Google Scholar]
- 100.Sancho R, Cremona CA, Behrens A. Stem cell and progenitor fate in the mammalian intestine: Notch and lateral inhibition in homeostasis and disease. EMBO Rep 16: 571–581, 2015. doi: 10.15252/embr.201540188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, Maslowski KM, Hardt WD. Epithelium-intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe 16: 237–248, 2014. doi: 10.1016/j.chom.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 102.Shirazi T, Longman RJ, Corfield AP, Probert CS. Mucins and inflammatory bowel disease. Postgrad Med J 76: 473–478, 2000. doi: 10.1136/pmj.76.898.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Smirnov A, Tako E, Ferket PR, Uni Z. Mucin gene expression and mucin content in the chicken intestinal goblet cells are affected by in ovo feeding of carbohydrates. Poult Sci 85: 669–673, 2006. doi: 10.1093/ps/85.4.669. [DOI] [PubMed] [Google Scholar]
- 104.Songhet P, Barthel M, Stecher B, Müller AJ, Kremer M, Hansson GC, Hardt WD. Stromal IFN-γR-signaling modulates goblet cell function during Salmonella typhimurium infection. PLoS One 6: e22459, 2011. doi: 10.1371/journal.pone.0022459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sonnenburg JL, Xu J, Leip DD, Chen C-H, Westover BP, Weatherford J, Buhler JD, Gordon JI. Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307: 1955–1959, 2005. doi: 10.1126/science.1109051. [DOI] [PubMed] [Google Scholar]
- 106.Spence JR, Lauf R, Shroyer NF. Vertebrate intestinal endoderm development. Dev Dyn 240: 501–520, 2011. doi: 10.1002/dvdy.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stahl M, Friis LM, Nothaft H, Liu X, Li J, Szymanski CM, Stintzi A. L-Fucose utilization provides Campylobacter jejuni with a competitive advantage. Proc Natl Acad Sci USA 108: 7194–7199, 2011. doi: 10.1073/pnas.1014125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stahl M, Frirdich E, Vermeulen J, Badayeva Y, Li X, Vallance BA, Gaynor EC. The helical shape of Campylobacter jejuni promotes in vivo pathogenesis by aiding transit through intestinal mucus and colonization of crypts. Infect Immun 84: 3399–3407, 2016. doi: 10.1128/IAI.00751-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, Enders GH, Silberg DG, Wen X, Wu GD, Lazar MA. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA 98: 502–506, 2001. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sun M, Wu W, Liu Z, Cong Y. Microbiota metabolite short chain fatty acids, GPCR, and inflammatory bowel diseases. J Gastroenterol 52: 1–8, 2017. doi: 10.1007/s00535-016-1242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tailford LE, Crost EH, Kavanaugh D, Juge N. Mucin glycan foraging in the human gut microbiome. Front Genet 6: 81, 2015. doi: 10.3389/fgene.2015.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Taupin DR, Kinoshita K, Podolsky DK. Intestinal trefoil factor confers colonic epithelial resistance to apoptosis. Proc Natl Acad Sci USA 97: 799–804, 2000. doi: 10.1073/pnas.97.2.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Teng X, Xu LF, Zhou P, Sun HW, Sun M. Effects of trefoil peptide 3 on expression of TNF-alpha, TLR4, and NF-kappaB in trinitrobenzene sulphonic acid induced colitis mice. Inflammation 32: 120–129, 2009. doi: 10.1007/s10753-009-9110-x. [DOI] [PubMed] [Google Scholar]
- 114.Theodoratou E, Campbell H, Ventham NT, Kolarich D, Pučić-Baković M, Zoldoš V, Fernandes D, Pemberton IK, Rudan I, Kennedy NA, Wuhrer M, Nimmo E, Annese V, McGovern DP, Satsangi J, Lauc G. The role of glycosylation in IBD. Nat Rev Gastroenterol Hepatol 11: 588–600, 2014. doi: 10.1038/nrgastro.2014.78. [DOI] [PubMed] [Google Scholar]
- 115.Thomsson KA, Holmén-Larsson JM, Angström J, Johansson ME, Xia L, Hansson GC. Detailed O-glycomics of the Muc2 mucin from colon of wild-type, core 1- and core 3-transferase-deficient mice highlights differences compared with human MUC2. Glycobiology 22: 1128–1139, 2012. doi: 10.1093/glycob/cws083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Turner JE, Stockinger B, Helmby H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog 9: e1003698, 2013. doi: 10.1371/journal.ppat.1003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van der Post S, Hansson GC. Membrane protein profiling of human colon reveals distinct regional differences. Mol Cell Proteomics 13: 2277–2287, 2014. doi: 10.1074/mcp.M114.040204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Van der Sluis M, De Koning BA, De Bruijn AC, Velcich A, Meijerink JP, Van Goudoever JB, Büller HA, Dekker J, Van Seuningen I, Renes IB, Einerhand AW. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 131: 117–129, 2006. doi: 10.1053/j.gastro.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 119.VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, Stappenbeck TS. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64: 911–920, 2015. doi: 10.1136/gutjnl-2013-306651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.VanDussen KL, Samuelson LC. Mouse atonal homolog 1 directs intestinal progenitors to secretory cell rather than absorptive cell fate. Dev Biol 346: 215–223, 2010. doi: 10.1016/j.ydbio.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Verdugo P. Goblet cells secretion and mucogenesis. Annu Rev Physiol 52: 157–176, 1990. doi: 10.1146/annurev.ph.52.030190.001105. [DOI] [PubMed] [Google Scholar]
- 122.Wang H, Kim JJ, Denou E, Gallagher A, Thornton DJ, Shajib MS, Xia L, Schertzer JD, Grencis RK, Philpott DJ, Khan WI. New role of Nod proteins in regulation of intestinal goblet cell response in the context of innate host defense in an enteric parasite infection. Infect Immun 84: 275–285, 2015. doi: 10.1128/IAI.01187-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wlodarska M, Thaiss CA, Nowarski R, Henao-Mejia J, Zhang JP, Brown EM, Frankel G, Levy M, Katz MN, Philbrick WM, Elinav E, Finlay BB, Flavell RA. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156: 1045–1059, 2014. doi: 10.1016/j.cell.2014.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wlodarska M, Willing BP, Bravo DM, Finlay BB. Phytonutrient diet supplementation promotes beneficial Clostridia species and intestinal mucus secretion resulting in protection against enteric infection. Sci Rep 5: 9253, 2015. doi: 10.1038/srep09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Worthington JJ. The intestinal immunoendocrine axis: novel cross-talk between enteroendocrine cells and the immune system during infection and inflammatory disease. Biochem Soc Trans 43: 727–733, 2015. doi: 10.1042/BST20150090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wrzosek L, Miquel S, Noordine ML, Bouet S, Joncquel Chevalier-Curt M, Robert V, Philippe C, Bridonneau C, Cherbuy C, Robbe-Masselot C, Langella P, Thomas M. Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol 11: 61, 2013. doi: 10.1186/1741-7007-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xu LF, Teng X, Guo J, Sun M. Protective effect of intestinal trefoil factor on injury of intestinal epithelial tight junction induced by platelet activating factor. Inflammation 35: 308–315, 2012. doi: 10.1007/s10753-011-9320-x. [DOI] [PubMed] [Google Scholar]
- 128.Xue Y, Zhang H, Wang H, Hu J, Du M, Zhu MJ. Host inflammatory response inhibits Escherichia coli O157:H7 adhesion to gut epithelium through augmentation of mucin expression. Infect Immun 82: 1921–1930, 2014. doi: 10.1128/IAI.01589-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zarepour M, Bhullar K, Montero M, Ma C, Huang T, Velcich A, Xia B, Vallance BA. Muc2 limits pathogen burdens and epithelial barrier dysfunction during Salmonella colitis. Infect Immun 81: 3672–3683, 2013. doi: 10.1128/IAI.00854-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang YG, Wu S, Xia Y, Sun J. Salmonella-infected crypt-derived intestinal organoid culture system for host-bacterial interactions. Physiol Rep 2: e12147, 2014. doi: 10.14814/phy2.12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhao F, Edwards R, Dizon D, Afrasiabi K, Mastroianni JR, Geyfman M, Ouellette AJ, Andersen B, Lipkin SM. Disruption of Paneth and goblet cell homeostasis and increased endoplasmic reticulum stress in Agr2−/− mice. Dev Biol 338: 270–279, 2010. doi: 10.1016/j.ydbio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zheng W, Rosenstiel P, Huse K, Sina C, Valentonyte R, Mah N, Zeitlmann L, Grosse J, Ruf N, Nürnberg P, Costello CM, Onnie C, Mathew C, Platzer M, Schreiber S, Hampe J. Evaluation of AGR2 and AGR3 as candidate genes for inflammatory bowel disease. Genes Immun 7: 11–18, 2006. doi: 10.1038/sj.gene.6364263. [DOI] [PubMed] [Google Scholar]
- 133.Zhou Y, Rychahou P, Wang Q, Weiss HL, Evers BM. TSC2/mTORC1 signaling controls Paneth and goblet cell differentiation in the intestinal epithelium. Cell Death Dis 6: e1631, 2015. doi: 10.1038/cddis.2014.588. [DOI] [PMC free article] [PubMed] [Google Scholar]