

Abstract
Nuclear factor erythyroid factor 2 (Nrf2) transcribes genes in cultured endothelial cells that reduce reactive oxygen species (ROS) and generate nitric oxide (NO) or metabolize asymmetric dimethylarginine (ADMA), which inhibits NO synthase (NOS). Therefore, we undertook a functional study to test the hypothesis that activation of Nrf2 by tert-butylhydroquinone (tBHQ) preserves microvascular endothelial function during oxidative stress. Wild-type CB57BL/6 (wt), Nrf2 wt (+/+), or knockout (−/−) mice received vehicle (Veh) or tBHQ (0.1%; activator of Nrf2) during 14-day infusions of ANG II (to induce oxidative stress) or sham. MAP was recorded by telemetry. Mesenteric resistance arterioles were studied on isometric myographs and vascular NO and ROS by fluorescence microscopy. ANG II increased the mean arterial pressure (112 ± 5 vs. 145 ± 5 mmHg; P < 0.01) and excretion of 8-isoprostane F2α (2.8 ± 0.3 vs. 3.8 ± 0.3 ng/mg creatinine; P < 0.05) at 12–14 days. However, 12 days of ANG II reduced endothelium-derived relaxation (27 ± 5 vs. 17 ± 3%; P < 0.01) and NO (0.38 ± 0.07 vs. 0.18 ± 0.03 units; P < 0.01) but increased microvascular remodeling, endothelium-derived contractions (7.5 ± 0.5 vs. 13.0 ± 1.7%; P < 0.01), superoxide (0.09 ± 0.03 vs. 0.29 ± 0.08 units; P < 0.05), and contractions to U-46,619 (87 ± 6 vs. 118 ± 3%; P < 0.05), and endothelin-1(89 ± 4 vs. 123 ± 12%; P < 0.05). tBHQ prevented all of these effects of ANG II at 12–14 days in Nrf2+/+ mice but not in Nrf2−/− mice. In conclusion, tBHQ activates Nrf2 to prevent microvascular endothelial dysfunction, remodeling, and contractility, and moderate ADMA and hypertension at 12–14 days of ANG II infusion, thereby preserving endothelial function and preventing hypertension.
Keywords: endothelin-1, nitric oxide, oxygen species, tBHQ, thromboxane, reactive
INTRODUCTION
Nuclear factor erythyroid factor 2 (Nrf2) binds to antioxidant response elements that transcribe many genes encoding antioxidant proteins (6, 14), including hemoxygenase-1 (HO-1) (14, 31), superoxide dismutase-2 (14), and glutathione peroxidase (14). We have reported recently that tert-butylhydroquinone (tBHQ) activated Nrf2 in human cultured glomerular endothelial cells, where it enhanced the expression of the genes for dimethylarginine dimethylaminohydrolase (DDAH)-1 and -2 and, thereby, enhanced the activity of DDAH, which metabolizes asymmetric dimethylarginine (ADMA) (14). ADMA is an endogenous inhibitor of nitric oxide synthase (NOS) (32). tBHQ also transcribed the genes for peroxisome proliferator activator receptor gamma (PPARγ) and, thereby, enhanced the transcription of the genes for endothelial NOS (eNOS) and increased its activation by phosphorylation at Ser-1174 (p-eNOSser1174) (14). Incubation of human glomerular endothelial cells with ANG II increased the generation of superoxide (O2·−) and ADMA and reduced the generation of NO, but these effects were prevented by coincubation with tBHQ (14). This suggests that tBHQ may preserve ACh-induced endothelium-derived relaxation factor (EDRF) and nitric oxide (NO) during infusion of ANG II. However, it is unclear whether these cellular effects are translated in vivo into changes in blood pressure (BP) and microvascular function. Therefore, the present functional study in mice tested the hypothesis that tBHQ reduces the hypertension, microvascular remodeling, reactive oxygen species (ROS), and ADMA and restores NO and EDRF of mesenteric resistance arterioles (MRAs) from mice with oxidative stress caused by a 2-wk infusion of ANG II at a slow pressor rate. It tests further whether these effects are mediated in vivo by activation of Nrf2, by comparing the effects of tBHQ given to Nrf2 wild-type (+/+) and knockout (−/−) mice.
Hypertension, oxidative stress (1, 12, 20, 23, 30, 33, 34), and increased ADMA (2, 21, 27, 29, 30) are implicated in vascular and organ dysfunction, yet there are no proven strategies to prevent these deleterious conditions. The vascular dysfunction and oxidative stress in the spontaneously hypertensive rat have been related to defective Nrf2 function because of excessive vascular accumulation of Bach-1, which blocks the activation of antioxidant response elements by Nrf2 (13). A number of natural compounds and some drugs can activate Nrf2 and might, thereby, be beneficial in the prevention of CVD. For example, bardoxolone methyl improved the glomerular filtration rate of patients with diabetic nephropathy (16). However, a subsequent large-scale trial in patients with diabetic nephropathy was halted prematurely because of a paradoxical excess of cardiovascular disease (CVD) events (4). Thus, Nrf2 activators may be unique in simultaneously reducing the vascular ROS and ADMA and enhancing PPARγ and NO and, thereby, could have widespread application to prevent the complications of hypertension, but this requires testing in vivo because of the potential for adverse actions on the cardiovascular system (4). We selected tBHQ to activate Nrf2, since it is orally active and apparently specific for Nrf2 (14). It also has been given to human subjects as a food preservative (5).
The effects of Nrf2 become apparent during oxidative stress accompanying a slow pressor infusion of ANG II. ANG II activates vascular nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and increases ADMA and signaling, via the thromboxane A2-prostaglandin receptor (TPR) and endothelin-1 (ET-1) (26, 33). The generation of O2·− in microvessels bioinactivates NO (1) and, thereby, impairs ACh-induced EDRF. This endothelial dysfunction is worsened by inhibition of NOS by ADMA and by the generation of O2·− and ligands for the TPR that generate endothelium-derived contraction factor (EDCF) and enhance microvascular contractility to thromboxane A2 (TxA2) and ET-1 (26). Thus, we assessed the effects of tBHQ on these responses in mice infused with ANG II to test the effects of Nrf2 during oxidative stress. Key studies were undertaken in Nrf2−/− mice to determine whether any effects of tBHQ depended on Nrf2, and, thereby, whether tBHQ is a specific activator of this pathway in vivo.
These studies confirmed widespread effects of ANG II infusion at 12–14 days to increase the BP, parameters of oxidative stress, TxA2 generation, TxA2 and ET-1 signaling, EDCF, and ADMA and reduce EDRF and NO at 12–14 days of infusion. All of these effects were prevented by tBHQ, but the actions of tBHQ were lost in Nrf2−/− mice.
METHODS
Animal preparation and ANG II infusion.
Male C57BL/6 mice weighing 25–30 g (Charles River Laboratory, Germantown, MD) and Nrf2−/− and Nrf2+/+ mice (C57BL/6 background; Jackson Laboratories, Bar Harbor, ME) (3) were maintained on tap water and standard chow (Na+ content 0.4 g/100 g; Harlan, Teklad). All procedures conformed to the “Guide for Care and Use of Laboratory Animals” by the National Institutes of Health, Institute for Laboratory Animal Research and were submitted to and approved by the Georgetown University Animal Care and Use Committee.
Mice were anesthetized with 2% isoflurane and prepared for infusion with ANG II (Peninsula Laboratory, San Carlos, CA; 400 ng·kg–1·min–1) subcutaneously for 12–14 days via osmotic minipumps (model 1002; Alet, Palo Alto, CA) (8) or received sham procedures (sham). All mice received vehicle or 0.1% tBHQ in their drinking water for 12–14 days. Groups of mice (n = 6–8 per group) were anesthetized for insertion of telemeters to measure 24-h mean arterial pressure (MAP), as described previously (7).
Measurement of urinary 8-isoprostane F2α, thromboxane B2, malondialdehyde, albumin, creatinine, and plasma ADMA, and l-arginine.
Mice were housed in metabolic cages (Nalgene Nunc International, Rochester, NY) to collect urine for 24 h into tubes containing 0.1 ml of antibiotics (ampicillin: 0.4 g; streptomycin: 1–3 g; amphotericin B: 2.25 g), as described previously (8, 20). 8-isoprostane F2α and thromboxane B2 (TxB2) were quantified by ELISA (Enzo Life Science, Farmingdale, NY) after purification and extraction with individual recovery assessed by the addition of [3H]prostaglandin E2, as described and validated previously against gas chromatography mass spectrometry (20). Malondialdehyde was measured by an assay kit (Cayman Chemical, Ann Arbor, MI), and albumin was measured by an ELISA kit (Exocell, Philadelphia, PA). Values were normalized with creatinine, which was measured by a urinary creatinine assay kit (Exocell).
Plasma ADMA and l-arginine were measured in an Agilent 6400 series triple quadrupole liquid chromatography-tandem mass spectrometry system, as described previously (22).
Preparation and study of mesenteric resistance arterioles.
Vessels (mean luminal diameter: 145 ± 10 μm and length: ~2 mm) were separated from the superior mesenteric bed, mounted on a wire myograph (M610, Danish Myotechnology; Aarhus, Denmark), and studied as described previously (26).
MAP, mesenteric resistance arteriolar remodeling, ACh-induced endothelial responses, NO and ROS activities, and contractions.
For the first series, mice were fitted with telemeters (Transonic), and MAP was recorded continuously for 24 h/day for 3 days before (baseline) and for 13 days of vehicle or ANG II infusion, and the daily average values were calculated, with or without tBHQ (8).
For the second series, the cross-sectional areas of the media and lumen of MRAs were measured, as described previously (12). Other arterioles were used to obtain ACh-induced endothelium dependent relaxation (EDR), EDRF, endothelium independent relaxation, and EDCF, as described previously (29). For EDR and EDRF, arterioles were preconstricted with 10−5 mol/l norepinephrine to study EDRF from change in relaxation to ACh after blockade of NOS with 10−5 mol/l of NG-nitroarginine methylester (l-NAME). Other preconstricted MRAs were loaded with 5×10−5 mol/l 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA) to study ACh-induced NO activity. The change of fluorescence (ΔF1/F0) with 10−5 mol/l ACh was quantitated by PTI RatioMaster (Photon Technology International, London, ON, Canada), as described previously (29).
For the third series, EDCF was measured in vessels under spontaneous tone with relaxation pathways blocked by l-NAME plus 10−6 mol/l apamin plus 10−5 mol/l charybdotoxin (blockers of big, intermediate, and small calcium-activated potassium channels) and contracted with ACh. Other vessels were prepared similarly, loaded with 5×10−5 mol/l dihydroethidium (DHE), and the change of vessel fluorescence of ethidium (E) relative to DHE (E: DHE) with 10−4 mol/l ACh was determined to quantify vascular ROS, as described previously (29).
For the fourth series, dose-response relationships were obtained for contractions to phenylephrine (10−9 to 10−5 mol/l), U-46,619 (10−10 to 10−6 mol/l), and ET-1 (10−11 to 10−7 mol/l). Additionally, vascular ET-1 (10−7 mol/l)-induced cellular ROS production was determined in vessels loaded with 5 × 10−5 mol/l of DHE.
Chemicals and solutions.
Agents were purchased from Sigma (St. Louis, MO) and dissolved in physiological salt solution.
Statistical analysis.
The number of mice used for each protocol was selected after a power analysis based on our prior experiences of ANG II-induced changes in mice (25) and rats (29). Data on MAP were recorded daily, but all other results refer to studies before, and at 12–14 days of infusion of ANG II or vehicle. Data are presented as means ± SE. Cumulative concentration-response experiments were analyzed by nonlinear regression (curve fit), and differences were assessed by two-way, repeated-measures ANOVA to assess the effects of ANG II, tBHQ, and their interaction (effects of tBHQ on the response to ANG II). In other studies, a similar ANOVA was undertaken in Nrf2+/+ and Nrf2−/− mice infused with ANG II to assess the effect of tBHQ, Nrf2 genotype, and their interaction (effect of Nrf2 genotype on response to tBHQ). This was followed, if appropriate, with Bonferroni post hoc t-tests for multiple comparisons. A probability value <0.05 was considered statistically significant.
RESULTS
MAP, mesenteric resistance arteriolar remodeling, body and organ weights, and excretion of markers of nitric oxide, thromboxane, and oxidative stress.
MAP was unchanged in mice receiving vehicle, but ANG II alone increased MAP after a delay of 4 days (Fig. 1). tBHQ alone did not change MAP but, when given with ANG II, it increased the MAP above levels of ANG II alone over the first 3 days, yet reduced MAP below levels of ANG II over days 8–13.
Fig. 1.
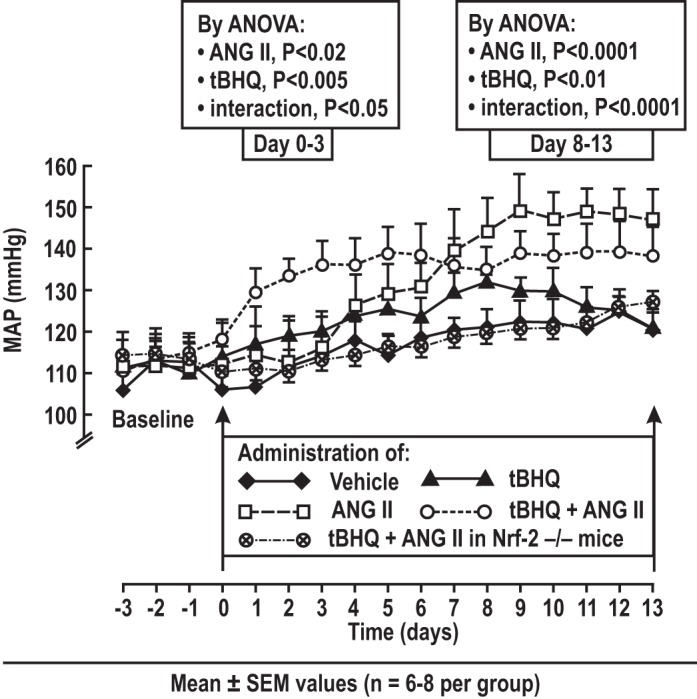
Daily mean arterial pressure (MAP) in conscious mice recorded by telemetry, showing effects of tert-butylhydroquinone (tBHQ). Daily values (means ± SE) in groups of mice (6–8 per group) infused with vehicle, ANG II (400 ng·kg−1·min−1 sc × 13 days) with or without tBHQ (0.1% in the drinking water).
ANG II did not change body, heart, or aorta weights (Table 1). ANG II caused microvascular remodeling (Table 3) and increased the excretion of TxB2, malondialdehyde, and 8-isoprostane F2α and reduced the excretion of nitrite. All of these were prevented by tBHQ (Table 2). ANG II did not change urinary albumin, but this was increased by tBHQ. All of the effects of tBHQ during infusion of ANG II were lost in Nrf2−/− mice.
Table 1.
Body weight and heart and aorta weight relative to body weight, urinary albumin, thromboxane B2, 8-isoprostane F2α, malondialdehyde, and nitrate/nitrite: effects of ANG II infusion for 12–14 days and oral tBHQ in C57BL/6 mice
| Vehicle |
tBHQ |
Effects via ANOVA |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | Sham | ANG II | Sham | ANG II | ANG II | tBHQ | Interaction |
| Body weight, g | 27.7 ± 0.9 | 27.7 ± 0.5 | 26.0 ± 0.8 | 28.4 ± 0.4 | NS | NS | NS |
| Heart/body weight, mg/g | 5.4 ± 0.3 | 4.7 ± 0.2 | 5.1 ± 0.3 | 4.8 ± 0.2 | NS | NS | NS |
| Aorta/heart, % | 33 ± 2 | 30 ± 2 | 32 ± 1 | 33 ± 3 | NS | NS | NS |
| Urinary albumin, µg/mg creatinine | 24.7 ± 2.4 | 26.1 ± 5.7 | 33.6 ± 3.3 | 51.6 ± 7.9† | NS | P < 0.05 | NS |
| Urinary TxB2, ng/mg creatinine | 2.06 ± 0.16 | 2.76 ± 0.19* | 1.96 ± 0.06 | 2.13 ± 0.14† | P < 0.05 | NS | P < 0.05 |
| Urinary 8-Iso F2α, ng/mg creatinine | 1.41 ± 0.14 | 1.89 ± 0.15* | 1.39 ± 0.07 | 1.43 ± 0.09† | P < 0.05 | NS | P < 0.05 |
| Urinary MDA, mmol/mg creatinine | 49.5 ± 5.1 | 82.5 ± 11.6* | 55.4 ± 8.0 | 53.7 ± 3.9† | P < 0.05 | NS | P < 0.05 |
| Urinary NOx, nmol/mg creatinine | 246 ± 18 | 140 ± 11* | 226 ± 18 | 214 ± 24† | P < 0.05 | NS | P < 0.05 |
Values are expressed as means ± SE values (n = 6 per group) for results obtained at days 12–14 of infusion of ANG II or vehicle. MDA, malondialdehyde; tBHQ, tert- butylhydroquinone; NOx, nitrite/nitrate; NS, not significant.
P < 0.05, compared with sham.
P < 0.05, compared with vehicle.
Table 3.
Arginine, ADMA, and l-arginine-to-ADMA ratio in mesenteric resistance arterioles, heart, and plasma: effects of ANG II infusion for 12–14 days and oral tBHQ in C57BL/6 mice
| Vehicle |
tBHQ |
Effects via ANOVA |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | Sham | ANG II | Sham | ANG II | ANG II | tBHQ | Interaction |
| MRA l-arginine, µmol/mg protein | 21.1 ± 5.0 | 14.9 ± 2.6 | 20.7 ± 2.7 | 21.2 ± 3.6† | P < 0.05 | NS | P < 0.05 |
| MRA ADMA, nmol/mg protein | 249 ± 15 | 339 ± 22* | 251 ± 12 | 277 ± 13† | P < 0.05 | NS | P < 0.05 |
| MRA l-arginine/ADMA | 83 ± 18 | 40 ± 7* | 85 ± 12 | 103 ± 28† | P < 0.05 | NS | P < 0.05 |
| Heart l-arginine, µmol/mg protein | 2.1 ± 0.4 | 1.4 ± 0.3* | 1.4 ± 0.3 | 2.1 ± 0.2† | P < 0.05 | NS | P < 0.05 |
| Heart ADMA, nmol/mg protein | 40 ± 2 | 57 ± 3* | 57 ± 2 | 45 ± 4† | P < 0.05 | NS | P < 0.05 |
| Heart l-arginine/ADMA | 53 ± 10 | 25 ± 6* | 37 ± 5 | 48 ± 7† | P < 0.05 | NS | P < 0.05 |
| Plasma l-arginine, µmol/l | 117 ± 10 | 83 ± 12 | 131 ± 21 | 123 ± 14 | NS | NS | NS |
| Plasma ADMA, µmol/l | 2.2 ± 0.2 | 3.0 ± 0.4 | 2.1 ± 0.1 | 2.2 ± 0.2 | NS | NS | NS |
| Plasma l-arginine/ADMA | 63 ± 7 | 30 ± 2* | 54 ± 11 | 58 ± 10 | P < 0.05 | NS | P < 0.05 |
Values are expressed as means ± SE values (n = 6 per group) for results obtained at days 12–14 of infusion of ANG II or vehicle. ADMA, asymmetric dimethylarginine; MRA, mesenteric resistance arterioles; NS, not significant.
P < 0.05, compared with sham.
P < 0.05, compared with vehicle.
Table 2.
Body weight and heart and aorta weight relative to body weight, urinary albumin, thromboxane B2, 8-isoprostane F2α, malondialdehyde, and nitrate/nitrite during ANG II infusion for 12–14 days: effects of oral tBHQ in Nrf2 mice infused with ANG II for 12–14 days
| Vehicle |
tBHQ |
Effects via ANOVA |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | Nrf2+/+ | Nrf2−/− | Nrf2+/+ | Nrf2−/− | Nrf2 | tBHQ | Interaction |
| Body weight, g | 32.7 ± 1.6 | 31.6 ± 1.2 | 28.5 ± 1.3 | 28.8 ± 0.8 | NS | P < 0.05 | NS |
| Heart/body weight, mg/g | 4.7 ± 0.1 | 4.6 ± 0.1 | 4.4 ± 0.1 | 4.3 ± 0.3 | NS | NS | NS |
| Aorta/heart, % | 32 ± 1 | 32 ± 2 | 34 ± 1 | 33 ± 1 | NS | NS | NS |
| Urinary albumin, µg/mg creatinine | 54.7 ± 3.8 | 56.1 ± 4.9 | 35.6 ± 3.3† | 52.5 ± 8.9 | NS | P < 0.05 | P < 0.05 |
| Urinary TxB2, ng/mg creatinine | 1.59 ± 0.0.07 | 1.54 ± 0.08 | 1.10 ± 0.04† | 1.41 ± 0.08 | NS | P < 0.05 | P < 0.05 |
| Urinary 8-Iso F2α, ng/mg creatinine | 2.86 ± 0.21 | 2.65 ± 0.31 | 1.40 ± 0.06† | 2.19 ± 0.21 | NS | P < 0.05 | P < 0.05 |
| Urinary MDA, mmol/mg creatinine | 81.1 ± 10.3 | 84.4 ± 8.1 | 48.4 ± 2.3† | 88.3 ± 9.1 | NS | P < 0.05 | P < 0.05 |
| Urinary NOx, nmol/mg creatinine | 153 ± 13 | 148 ± 7 | 208 ± 13† | 134 ± 11 | NS | P < 0.05 | P < 0.05 |
Values are expressed as means ± SE values (n = 6 per group) for results obtained at days 12–14 of infusion of ANG II. MDA, malondialdehyde; tBHQ, tert- butylhydroquinone; NOx, nitrite/nitrate; NS, not significant.
P < 0.05, compared with vehicle.
l-arginine, ADMA, and l-arginine: ADMA ratio.
ANG II infusion reduced l-arginine but increased ADMA in mesenteric resistance arterioles, heart, and plasma (Table 3), resulting in a reduced l-arginine-to-ADMA ratio. All of the effects of ANG II were prevented by tBHQ.
ACh-induced EDR, EDRF and NO generation and ACh-induced EDCF and O2·− generation in mesenteric arterioles.
ANG II infusion reduced ACh-induced EDR, EDRF, and NO generation (Fig. 2, Table 4) and increased ACh-induced EDCF and ROS generation (Fig. 2, Table 4). All of these effects of ANG II were prevented by tBHQ, but these effects of tBHQ were lost in Nrf2−/− mice (Table 5).
Fig. 2.

ACh-induced endothelium-dependent relaxation (EDR), endothelium-derived relaxation factor(EDRF)/nitric oxide (NO), and contraction factor (EDCF) in mesenteric resistance vessels of C57BL/6 mice, showing effects of ANG II infusion and/or tert-butylhydroquinone (tBHQ) (n = 6/groups). The dose response from mice drinking vehicle (sham) or 0.1% tBHQ with ANG II (400 ng·kg−1·min−1) or vehicle infused subcutaneously for 12–14 days. *P < 0.05, compared with ANG II, tBHQ with ANG II, and vehicle.
Table 4.
MRA media-to-lumen ratio, ACh-induced relaxation, endothelium-dependent relaxation and contraction, sodium nitroprusside-induced endothelium-independent relaxation, and ACh-induced NO and ROS generation: effects of ANG II infusion for 12–14 days and oral tBHQ in C57BL/6 mice
| Vehicle |
tBHQ |
Effects via ANOVA |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | Sham | ANG II | Sham | ANG II | ANG II | tBHQ | Interaction |
| Ratio of media to lumen of MRA | 0.28 ± 0.02 | 0.41 ± 0.06* | 0.29 ± 0.03 | 0.31 ± 0.05 | P < 0.05 | NS | P < 0.05 |
| ACh relaxation, % | 77.4 ± 2.8 | 53.0 ± 5.4* | 75.5 ± 2.7 | 75.3 ± 1.3† | P < 0.05 | NS | P < 0.05 |
| EDRF, % | 27.4 ± 4.6 | 16.8 ± 3.3* | 29.0 ± 3.4 | 23.0 ± 2.4† | P < 0.05 | NS | P < 0.05 |
| EDCF, % | 7.4 ± 0.4 | 13.0 ± 1.7* | 7.2 ± 0.2 | 7.6 ± 0.8† | P < 0.05 | NS | P < 0.05 |
| EIR, % | 86.4 ± 2.9 | 89.5 ± 3.6 | 89.1 ± 2.1 | 85.6 ± 2.0 | NS | NS | NS |
| ACh-NO, Δunit | 0.36 ± 0.07 | 0.18 ± 0.03* | 0.35 ± 0.07 | 0.28 ± 0.05† | P < 0.05 | NS | P < 0.05 |
| EDCF-ROS, Δunit | 0.09 ± 0.03 | 0.29 ± 0.08* | 0.09 ± 0.03 | 0.09 ± 0.03† | P < 0.05 | NS | P < 0.05 |
| PE contraction, % | 67.8 ± 3.8 | 61.1 ± 5.9 | 67.2 ± 11.8 | 61.1 ± 5.9 | NS | NS | NS |
| U-46,619 contraction, % | 87.1 ± 5.9 | 118.0 ± 2.6* | 92.5 ± 3.9 | 81.9 ± 8.5† | P < 0.05 | NS | P < 0.05 |
| ET-1 contraction, % | 89.1 ± 4.0 | 122.7.1 ± 12.3* | 92.8 ± 5.1 | 81.3 ± 8.9† | P < 0.05 | NS | P < 0.05 |
| ET-1-ROS, Δratio | 0.13 ± 0.05 | 0.43 ± 0.08* | 0.10 ± 0.03 | 0.11 ± 0.03† | P < 0.05 | P < 0.05 | P < 0.05 |
Values are expressed as means ± SE values (n = 6 per group) for results obtained at days 12–14 of infusion of ANG II or vehicle. EDCF, endothelium-derived contraction factor; EDRF, endothelium-derived relaxation factor; EIR, endothelium-independent relaxation; ET-1, endothelin-1; NO, nitric oxide; PE, phenylephrine; ROS, reactive oxygen species; NS, not significant.
P < 0.05, compared with sham.
P < 0.05, compared with vehicle.
Table 5.
MRA media-to-lumen ratio, ACh-induced relaxation, endothelium-dependent relaxation and contraction, sodium nitroprusside-induced endothelium-independent relaxation, and ACh-induced NO and ROS generation: effects of ANG II infusion for 12–14 days and oral tBHQ in Nrf2 mice infused with ANG II
| Vehicle |
tBHQ |
Effects via ANOVA |
|||||
|---|---|---|---|---|---|---|---|
| Parameter | Nrf2+/+ | Nrf2−/− | Nrf2+/+ | Nrf2−/− | Nrf2 | tBHQ | Interaction |
| Ratio of media to lumen of MRA | 0.39 ± 0.05 | 0.40 ± 0.04 | 0.29 ± 0.03† | 0.38 ± 0.06 | NS | P < 0.05 | P < 0.05 |
| ACh relaxation, % | 53.0 ± 5.4 | 51.3 ± 4.7 | 69.2 ± 5.5† | 45.8 ± 5.3 | NS | P < 0.05 | P < 0.05 |
| EDRF, % | 16.8 ± 3.3 | 13.3 ± 2.4 | 27.1 ± 2.7† | 14.7 ± 2.7 | P < 0.05 | P < 0.05 | P < 0.05 |
| EDCF, % | 13.0 ± 1.7 | 15.6 ± 0.8 | 7.7 ± 0.5† | 12.4 ± 0.9 | P < 0.05 | P < 0.05 | P < 0.05 |
| ACh-NO, Δunit | 0.18 ± 0.03 | 0.16 ± 0.05 | 0.34 ± 0.03† | 0.19 ± 0.03 | P < 0.05 | P < 0.05 | P < 0.05 |
| EDCF-ROS, Δunit | 0.29 ± 0.08 | 0.32 ± 0.03 | 0.12 ± 0.04† | 0.27 ± 0.06 | P < 0.05 | P < 0.05 | P < 0.05 |
| U-46,619 contraction, % | 118.0 ± 2.6 | 125.9 ± 8.5 | 79.4 ± 3.9† | 131.4 ± 5.9 | P < 0.05 | P < 0.05 | P < 0.05 |
| ET-1 contraction, % | 122.7.1 ± 7.3 | 133.3 ± 8.9 | 92.6 ± 6.5† | 137.9 ± 6.8 | P < 0.05 | NS | P < 0.05 |
| ET-1-ROS, Δratio | 0.43 ± 0.08 | 0.44 ± 0.03 | 0.16 ± 0.06† | 0.40 ± 0.07 | NS | P < 0.05 | P < 0.05 |
Values are expressed as means ± SE values (n = 6 per group) for results obtained at days 12–14 of infusion of ANG II or vehicle. EDCF, endothelium-derived contraction factor; EDRF, endothelium-derived relaxation factor; ET-1, endothelin-1; NO, nitric oxide; ROS, reactive oxygen species; NS, not significant.
P < 0.05, compared with vehicle.
Mesenteric resistance arteriolar contractions to phenylephrine, U-46,619, and endothelin, as well as endothelin 1-induced superoxide generation.
ANG II infusion did not change the contraction to phenylephrine (Fig. 3; Table 5), but increased the contractions to U-46,619 and ET-1, as well as ET-1-induced O2·− generation (Table 5). These effects of ANG II were prevented by tBHQ, but the effects of tBHQ were lost in Nrf2 −/− mice (Table 5).
Fig. 3.

Moderation by Nrf2 of enhanced contraction to phenylephrine (PE), U-46,619, and endothelin 1 (ET-1) in mesenteric resistance arteries of C57BL/6 mice, showing effects of ANG II infusion and/or tert-butylhydroquinone (tBHQ). The dose response from mice drinking vehicle (sham) or 0.1% tBHQ with or without ANG II (400 ng·kg−1·min−1) or vehicle infused subcutaneously for 12–14 days. *P < 0.05, compared with ANG II, tBHQ with ANG II, and vehicle.
DISCUSSION
We confirm that a slow pressor infusion of ANG II causes oxidative stress (13) with increased generation of TxA2 (8) and increased responsiveness to thromboxane and ET-1 (26). This was accompanied by a delayed increase in MAP and by microvascular remodeling (12), endothelial dysfunction with reduced ACh-induced EDR, EDRF, and NO, but enhanced ACh-induced ROS and EDCF (26, 29). ANG II reduced l-arginine, but increased ADMA and reduced l-arginine-to-ADMA ratio in microvessels, heart, and plasma (29, 32). The main new findings are that tBHQ given to sham mice had no effects on these parameters but when given to wild-type or Nrf2+/+ mice infused with ANG II, tBHQ prevented all these widespread and potentially adverse effects of ANG II. All of the effects of tBHQ were lost in Nrf2−/− mice infused with ANG II.
tBHQ had time-dependent effects on telemetric measurements of MAP during ANG II. Over the first 3 days of ANG II alone, the MAP did not change, confirming that this is a slow pressor dose, but by day 8, the MAP increased to a plateau of ~145 mmHg. tBHQ alone did not change MAP. However, when given with ANG II, tBHQ increased the MAP by ~15–20 mmHg over the first 3 days, yet reduced the MAP below levels of ANG II by 10–15 mmHg over days 8–13. The early rise in MAP is reminiscent of the report of an early increase in CVD events and BP in the trial of bardoxolone for diabetic nephropathy (4) and requires further investigation. The reduction in ANG II-induced hypertension over 8–13 days is consistent with the increase in NO and reduction in contractility to thromboxane and ET-1 and the reduction in oxidative stress at this time.
The oxidative stress that developed during ANG II infusion likely derived from activation of NADPH oxidase, since silencing of p22phox prevents oxidative stress and hypertension in rats infused with ANG II (15). The endothelial dysfunction caused by ANG II was likely multifactorial and derived from a reduced ratio of eNOS substrate (l-arginine) to inhibitor (ADMA), and increased microvascular O2·− that oxidizes the eNOS cofactor tetrahydrobiopterin to dihydrobiopterin and uncouples the enzyme (10). Moreover, O2·− bioinactivates NO (32). Remarkably, these widespread systemic and microvascular effects of ANG II to cause slowly developing hypertension, oxidative stress, endothelial dysfunction, and NO deficiency were all prevented by tBHQ. tBHQ also prevented the reduction in l-arginine-to-ADMA ratio, increased TxB2, and increased contractility to U-46,619 and ETI. All of these effects of ANG II depend on ROS (26, 32). Therefore, the effects of tBHQ to prevent these actions of ANG II likely derived largely from transcriptional generation of the genes for systems that metabolize ROS, such as HO-1, and for PPARγ, which upregulates eNOS and the active form of p-eNOSser1177 (32). In endothelial cells, tBHQ also upregulates DDAH1 and DDAH2 (14). DDAH enhances the metabolism of ADMA in microvessels. This could account for the reduced plasma and tissue concentration of ADMA in ANG II-infused mice given tBHQ.
There were no effects of tBHQ without oxidative stress induced by ANG II infusion, except for an increase in albumin excretion. Activation of Nrf2 with bardoxolone methyl increased albumin excretion, but this was related to decreased tubular albumin reabsorption because of decreased expression of megalin and tubulin (18). Thus, the increased albumin excretion with tBHQ likely does not represent glomerular damage. Cellular silencing of Nrf2 in vitro (14), or Nrf2 gene deletion in vivo in this study, both prevented the effects of tBHQ, thereby demonstrating that tBHQ is a specific activator of Nrf2. tBHQ translocated Nrf2 to the nucleus of endothelial cells, where it bound to antioxidant response elements on many genes (14). Thus, tBHQ is a potent, orally active, inexpensive, and specific activator of Nrf2 both in vitro and in vivo and should, thereby, be a useful tool to probe the functions of Nrf2.
Microvascular remodeling can be induced by O2·− (12) and H2O2 (34). Thus, the prevention of remodeling of vessels in ANG II-infused mice by tBHQ likely reflects reduced microvascular ROS. Microvascular endothelial dysfunction and remodeling are strong predictors of future cardiovascular events (19, 23).
ADMA is generated by a two-step methylation of l-arginine epitopes by specific isoforms of protein arginine methyltransferase (21). Tissue levels of ADMA are largely regulated by its metabolism by DDAH-1 and DDAH-2 (2), which are upregulated by tBHQ in endothelial cells (14). We reported that DDAH2 in MRAs reduced vascular ADMA and preserved endothelial function (27). ANG II infusion increased ADMA in mesenteric arterioles, heart, and plasma, but this was prevented by tBHQ, consistent with activation of DDAH (14). ANG II infusion also reduced the tissue and plasma levels of l-arginine that, in the presence of excess ADMA, may limit NO generation further (27). This reduction in l-arginine may be related to effects of ROS and ANG II to increase the expression of the hepatic cationic amino acid transferase (2A) that transports l-arginine into liver cells, where it is metabolized by arginase (9). The reduction in NOS substrate-to-inhibitor ratio by ANG II likely contributed to the endothelial dysfunction and reduced vascular NO.
We confirmed that infusion of ANG II enhanced the microvascular ROS generation (26) that is blocked by tBHQ. Previous studies have reported that ROS increased the microvascular expression and responsiveness of TPRs and endothelin type A receptors (ETAs) (24, 25), whereas norepinephrine does not generate ROS, and its responses are unchanged by ANG II infusion (28), similar to the findings in this study. Thus, the antioxidant actions of tBHQ likely contributed to its moderation of microvascular contractility to U-46,619 and ET-1, while maintaining contractility to phenylephrine. Infusion of ANG II increased TxB2 excretion (7), but this was prevented by tBHQ. A reduction in TxA2 could be an important effect of Nrf2, since we have shown that TPRs in mice are required for ANG II to cause oxidative stress, hypertension, and increased renal vascular resistance (7). Thus, the reduction by tBHQ of the responsiveness of TPRs, coupled with a reduced generation of TxA2, provides a robust pathway to counter hypertension and oxidative stress.
We acknowledge some limitations of this study. First, the molecular mechanisms of Nrf2 activation were not studied, but these were the subject of an extensive prior investigation in isolated cells (14). Second, the hypertensive actions of ANG II, and its prevention by tBHQ, may have contributed to the results. However, oxidative stress with ANG II is specific and not seen with equipressor infusions of norepinephrine (17). Moreover, a similar profile of effects of ANG II, and their blockade by tBHQ, was observed in isolated endothelial cells (14). Third, the study is relatively short, and the intervention with tBHQ was given coincident with starting the ANG II infusion, which limits its clinical applicability. Fourth, tBHQ unexpectedly increased MAP when given over the first 3 days of ANG II infusion. This requires further study. Fifth, the effects of tBHQ on endothelium-dependent hyperpolarization factor were not studied. However, the aim of this in vivo study was to test hypotheses relating to the generation of NO and ROS in endothelial cells by tBHQ, where the hyperpolarization factor was not studied (14).
In conclusion, the increased oxidative stress, microvascular ROS, endothelial dysfunction, reduced l-arginine-to-ADMA ratio, as well as enhanced microvascular remodeling, contractility, and hypertension that develop after 12–14 days of ANG II infusion can be prevented by activation of Nrf2 with tBHQ. Thus, Nrf2 emerges as a major factor whose activation can prevent many adverse systemic microvascular effects of ANG II.
Perspectives and Significance
Endothelial dysfunction and vascular remodeling mediated by ROS, ADMA, and NO deficiency are apparent early in human hypertension (30) and precede clinical signs of cardiovascular disease (17, 19), which is, itself, the principal identified cause of death and disability worldwide (11). Therefore, the prevention of these adverse events by Nrf2 suggests potentially important new approaches to the prevention of hypertensive CVD. However, the paradoxical adverse cardiovascular effects of bardoxolone methyl reported in a recent clinical trial (4) and the early rise in MAP with tBHQ over the first 3 days of ANG II infusion in this study, must be explained and prevented before this can be recommended as a therapeutic strategy.
GRANTS
This study was supported by grants from the National Institutes of Health to C. S. Wilcox from the National Institute of Diabetes and Digestive and Kidney Diseases (DK-036079 and DK-049870) and from the National Heart, Lung, and Blood Institute (HL-068686), and by funds from the George E. Schreiner Chair of Nephrology and the Georgetown University Hypertension Center. Also, a grant from the National Heart Lung and Blood Institute (HL-134511) was given to C. S. Wilcox and D. Wang.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.W., Z.L., W.J.W., C.S.W., and D.W. conceived and designed research; C.W., J.T., J.L., and D.W. performed experiments; C.W., Z.L., G.C., A.W., J.T., C.S.W., and D.W. analyzed data; C.W., Z.L., A.W., P.A.J., J.L., W.J.W., C.S.W., and D.W. interpreted results of experiments; C.W., Z.L., G.C., A.W., P.A.J., J.T., J.L., W.J.W., C.S.W., and D.W. approved final version of manuscript; Z.L., C.S.W., and D.W. drafted manuscript; J.T., J.L., W.J.W., C.S.W., and D.W. edited and revised manuscript; C.S.W. and D.W. prepared figures.
REFERENCES
- 1.Araujo M, Wilcox CS. Oxidative stress in hypertension: role of the kidney. Antioxid Redox Signal 20: 74–101, 2014. doi: 10.1089/ars.2013.5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arrigoni F, Ahmetaj B, Leiper J. The biology and therapeutic potential of the DDAH/ADMA pathway. Curr Pharm Des 16: 4089–4102, 2010. doi: 10.2174/138161210794519246. [DOI] [PubMed] [Google Scholar]
- 3.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 26: 175–182, 2002. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 4.de Zeeuw D, Akizawa T, Audhya P, Bakris GL, Chin M, Christ-Schmidt H, Goldsberry A, Houser M, Krauth M, Lambers Heerspink HJ, McMurray JJ, Meyer CJ, Parving HH, Remuzzi G, Toto RD, Vaziri ND, Wanner C, Wittes J, Wrolstad D, Chertow GM; BEACON Trial Investigators . Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N Engl J Med 369: 2492–2503, 2013. doi: 10.1056/NEJMoa1306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gharavi N, Haggarty S, El-Kadi AO. Chemoprotective and carcinogenic effects of tert-butylhydroquinone and its metabolites. Curr Drug Metab 8: 1–7, 2007. doi: 10.2174/138920007779315035. [DOI] [PubMed] [Google Scholar]
- 6.Jiang S, Yang Y, Li T, Ma Z, Hu W, Deng C, Fan C, Lv J, Sun Y, Yi W. An overview of the mechanisms and novel roles of Nrf2 in cardiovascular diseases. Expert Opin Ther Targets 20: 1413–1424, 2016. doi: 10.1080/14728222.2016.1250887. [DOI] [PubMed] [Google Scholar]
- 7.Kawada N, Dennehy K, Solis G, Modlinger P, Hamel R, Kawada JT, Aslam S, Moriyama T, Imai E, Welch WJ, Wilcox CS. TP receptors regulate renal hemodynamics during angiotensin II slow pressor response. Am J Physiol Renal Physiol 287: F753–F759, 2004. doi: 10.1152/ajprenal.00423.2003. [DOI] [PubMed] [Google Scholar]
- 8.Kawada N, Imai E, Karber A, Welch WJ, Wilcox CS. A mouse model of angiotensin II slow pressor response: role of oxidative stress. J Am Soc Nephrol 13: 2860–2868, 2002. doi: 10.1097/01.ASN.0000035087.11758.ED. [DOI] [PubMed] [Google Scholar]
- 9.Kitiyakara C, Chabrashvili T, Jose P, Welch WJ, Wilcox CS. Effects of dietary salt intake on plasma arginine. Am J Physiol Regul Integr Comp Physiol 280: R1069–R1075, 2001. doi: 10.1152/ajpregu.2001.280.4.R1069. [DOI] [PubMed] [Google Scholar]
- 10.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 111: 1201–1209, 2003. doi: 10.1172/JCI200314172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawes CM, Vander Hoorn S, Rodgers A; International Society of Hypertension . Global burden of blood-pressure-related disease, 2001. Lancet 371: 1513–1518, 2008. doi: 10.1016/S0140-6736(08)60655-8. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Feng D, Luo Z, Welch WJ, Wilcox CS, Lai EY. Remodeling of afferent arterioles from mice with oxidative stress does not account for increased contractility but does limit excessive wall stress. Hypertension 66: 550–556, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM. Downregulation of nuclear factor erythroid 2-related factor and associated antioxidant genes contributes to redox-sensitive vascular dysfunction in hypertension. Hypertension 66: 1240–1250, 2015. doi: 10.1161/HYPERTENSIONAHA.115.06163. [DOI] [PubMed] [Google Scholar]
- 14.Luo Z, Aslam S, Welch WJ, Wilcox CS. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-γ/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension 65: 896–902, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modlinger P, Chabrashvili T, Gill PS, Mendonca M, Harrison DG, Griendling KK, Li M, Raggio J, Wellstein A, Chen Y, Welch WJ, Wilcox CS. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension 47: 238–244, 2006. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- 16.Pergola PE, Raskin P, Toto RD, Meyer CJ, Huff JW, Grossman EB, Krauth M, Ruiz S, Audhya P, Christ-Schmidt H, Wittes J, Warnock DG; BEAM Study Investigators . Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N Engl J Med 365: 327–336, 2011. doi: 10.1056/NEJMoa1105351. [DOI] [PubMed] [Google Scholar]
- 17.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 97: 1916–1923, 1996. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reisman SA, Chertow GM, Hebbar S, Vaziri ND, Ward KW, Meyer CJ. Bardoxolone methyl decreases megalin and activates Nrf2 in the kidney. J Am Soc Nephrol 23: 1663–1673, 2012. doi: 10.1681/ASN.2012050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rizzoni D, Porteri E, Boari GE, De Ciuceis C, Sleiman I, Muiesan ML, Castellano M, Miclini M, Agabiti-Rosei E. Prognostic significance of small-artery structure in hypertension. Circulation 108: 2230–2235, 2003. doi: 10.1161/01.CIR.0000095031.51492.C5. [DOI] [PubMed] [Google Scholar]
- 20.Schnackenberg CG, Wilcox CS. Two-week administration of tempol attenuates both hypertension and renal excretion of 8-Iso prostaglandin F2α. Hypertension 33: 424–428, 1999. doi: 10.1161/01.HYP.33.1.424. [DOI] [PubMed] [Google Scholar]
- 21.Teerlink T, Luo Z, Palm F, Wilcox CS. Cellular ADMA: regulation and action. Pharmacol Res 60: 448–460, 2009. doi: 10.1016/j.phrs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomlinson JA, Caplin B, Boruc O, Bruce-Cobbold C, Cutillas P, Dormann D, Faull P, Grossman RC, Khadayate S, Mas VR, Nitsch DD, Wang Z, Norman JT, Wilcox CS, Wheeler DC, Leiper J. Reduced renal methylarginine metabolism protects against progressive kidney damage. J Am Soc Nephrol 26: 3045–3059, 2015. doi: 10.1681/ASN.2014030280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Touyz RM, Tabet F, Schiffrin EL. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin Exp Pharmacol Physiol 30: 860–866, 2003. doi: 10.1046/j.1440-1681.2003.03930.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang C, Luo Z, Kohan D, Wellstein A, Jose PA, Welch WJ, Wilcox CS, Wang D. Thromboxane prostanoid receptors enhance contractions, endothelin-1, and oxidative stress in microvessels from mice with chronic kidney disease. Hypertension 65: 1055–1063, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D, Chabrashvili T, Borrego L, Aslam S, Umans JG. Angiotensin II infusion alters vascular function in mouse resistance vessels: roles of O and endothelium. J Vasc Res 43: 109–119, 2006. doi: 10.1159/000089969. [DOI] [PubMed] [Google Scholar]
- 26.Wang D, Chabrashvili T, Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane prostanoid receptors, and endothelium. Circ Res 94: 1436–1442, 2004. doi: 10.1161/01.RES.0000129578.76799.75. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Gill PS, Chabrashvili T, Onozato ML, Raggio J, Mendonca M, Dennehy K, Li M, Modlinger P, Leiper J, Vallance P, Adler O, Leone A, Tojo A, Welch WJ, Wilcox CS. Isoform-specific regulation by N(G),N(G)-dimethylarginine dimethylaminohydrolase of rat serum asymmetric dimethylarginine and vascular endothelium-derived relaxing factor/NO. Circ Res 101: 627–635, 2007. doi: 10.1161/CIRCRESAHA.107.158915. [DOI] [PubMed] [Google Scholar]
- 28.Wang D, Jose P, Wilcox CS. β1 Receptors protect the renal afferent arteriole of angiotensin-infused rabbits from norepinephrine-induced oxidative stress. J Am Soc Nephrol 17: 3347–3354, 2006. doi: 10.1681/ASN.2006030212. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, Luo Z, Wang X, Jose PA, Falck JR, Welch WJ, Aslam S, Teerlink T, Wilcox CS. Impaired endothelial function and microvascular asymmetrical dimethylarginine in angiotensin II-infused rats: effects of tempol. Hypertension 56: 950–955, 2010. doi: 10.1161/HYPERTENSIONAHA.110.157115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang D, Strandgaard S, Iversen J, Wilcox CS. Asymmetric dimethylarginine, oxidative stress, and vascular nitric oxide synthase in essential hypertension. Am J Physiol Regul Integr Comp Physiol 296: R195–R200, 2009. doi: 10.1152/ajpregu.90506.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wenzel P, Rossmann H, Müller C, Kossmann S, Oelze M, Schulz A, Arnold N, Simsek C, Lagrange J, Klemz R, Schönfelder T, Brandt M, Karbach SH, Knorr M, Finger S, Neukirch C, Häuser F, Beutel ME, Kröller-Schön S, Schulz E, Schnabel RB, Lackner K, Wild PS, Zeller T, Daiber A, Blankenberg S, Münzel T. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J 36: 3437–3446, 2015. doi: 10.1093/eurheartj/ehv544. [DOI] [PubMed] [Google Scholar]
- 32.Wilcox CS. Asymmetric dimethylarginine and reactive oxygen species: unwelcome twin visitors to the cardiovascular and kidney disease tables. Hypertension 59: 375–381, 2012. doi: 10.1161/HYPERTENSIONAHA.111.187310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilcox CS. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol Ther 126: 119–145, 2010. doi: 10.1016/j.pharmthera.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension 46: 732–737, 2005. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]