

Abstract
We previously reported that microinjection of the proinflammatory cytokine interleukin-1β (IL-1β) into the subfornical organ (SFO) elicits a pressor response accompanied by increases in inflammation and renin-angiotensin system (RAS) activity in the SFO and hypothalamic paraventricular nucleus (PVN). The present study sought to determine whether blood-borne IL-1β induces similar neurochemical changes in the SFO and PVN and, if so, whether increased inflammation and RAS activity at the SFO level orchestrate the sympathoexcitatory response to circulating IL-1β. In urethane-anesthetized male Sprague-Dawley rats, intravenous injection of IL-1β (500 ng) increased blood pressure, heart rate, renal sympathetic nerve activity, and mRNA for angiotensin-converting enzyme, angiotensin II type 1a receptor, cyclooxygenase-2, tumor necrosis factor-α, and IL-1β, as well as the tumor necrosis factor-α p55 receptor and the IL-1 receptor, in the SFO and PVN. Pretreatment with SFO microinjections of the angiotensin II type 1a receptor blocker losartan (1 µg), the angiotensin-converting enzyme inhibitor captopril (1 µg), or the cyclooxygenase-2 inhibitor NS-398 (2 µg) attenuated expression of these excitatory mediators in the SFO and downstream in the PVN and the IL-1β-induced pressor responses. An SFO lesion minimized the IL-1β-induced expression of inflammatory and RAS components as well as c-Fos, an indicator of neuronal excitation, in the PVN. These studies demonstrate that circulating IL-1β, which increases in cardiovascular disorders such as hypertension and heart failure, acts on the SFO to increase inflammation and RAS activity in the SFO and PVN and that intervening in these neurochemical processes in the SFO can significantly reduce the sympathetic response.
Keywords: brain, cyclooxygenase-2, proinflammatory cytokines, renin-angiotensin system, sympathetic nervous system
INTRODUCTION
Peripheral and central actions of blood-borne proinflammatory cytokines (PICs) have been implicated in the pathogenesis of heart failure (HF) and hypertension (HTN) (22, 27, 31, 37, 47, 54). Circulating PICs act on the brain to increase sympathetic drive (15, 39, 75), but how they do so remains a matter for conjecture. Recent work from our laboratory has demonstrated that the sympathetically mediated pressor response to intravenous or intracarotid administration of the prototypical PICs tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) is substantially reduced by lesioning the subfornical organ (SFO) (75), a forebrain circumventricular organ that lacks an effective blood-brain barrier and senses circulating humoral factors (16, 49). Neurons in the SFO project directly to the hypothalamic paraventricular nucleus (PVN) (38), an integrative forebrain nucleus (17) that regulates sympathetic outflow (45) and has been identified as a source of increased sympathetic activity in neurogenic HTN (6, 11) and HF (53). Direct microinjection of TNF-α or IL-1β into the SFO, in doses that elicit a sympathetically mediated pressor response, stimulates inflammation and the expression of renin-angiotensin system (RAS) components not only in the SFO but also downstream in the PVN (73), where inflammation and RAS activity are thought to contribute to the sympathetic overactivity in neurogenic HTN and HF (8, 55, 80). Pretreatment of the SFO with inhibitors of RAS activity and cyclooxygenase (COX)-2, a mediator of the inflammatory response to PICs, reduces the sympathetic response to SFO microinjection of TNF-α or IL-1β (73). These studies have demonstrated that the SFO is essential for the full manifestation of the pressor response to systemically administered PICs and that PICs can act within the SFO to increase the sympathoexcitatory neurochemical milieu downstream in the PVN.
The present study sought to determine whether circulating PICs act via the SFO to upregulate RAS activity and inflammation in the PVN and increase sympathetic drive. We examined the effects of intravenously administered IL-1β on molecular mechanisms in the SFO and PVN that drive sympathetic activity. The findings have important implications for cardiovascular disorders such as HTN and HF, in which the central nervous system effects of circulating PICs may contribute to the progression of disease.
METHODS
Animals
Adult male Sprague-Dawley rats (275–325 g; Envigo/Harlan, Indianapolis, IN) were maintained in a temperature-controlled (23 ± 2°C) and light-controlled animal care facility at the University of Iowa; rat chow was provided ad libitum. All experimental procedures were approved by the University of Iowa Institutional Animal Care and Use Committee. The experimental protocols were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Surgical Preparations
Hemodynamic and sympathetic recording.
The surgical preparation for electrophysiological recording of blood pressure (BP), heart rate (HR), and renal sympathetic nerve activity (RSNA) has been described in detail previously (71, 74). Briefly, rats were anesthetized with urethane (1.5 g/kg ip), supplemented as needed (0.1 g/kg iv). PE-50 catheters were inserted into the left femoral artery for BP recording and into the left femoral vein for intravenous injection. The left kidney was exposed via a flank incision, and a renal nerve was isolated from surrounding tissue. The renal nerve was placed on bipolar silver wire recording electrodes to record RSNA. The nerve and electrodes were covered with Kwik-Cast silicone sealant (WPI, Sarasota, FL) before the incisions were sutured. Body temperature was maintained at 37 ± 1°C with a heating pad and heat lamp. Baseline RSNA and BP were stable for ≥30 min before the recording was initiated.
SFO microinjection.
The method for SFO microinjection is described elsewhere (73). Briefly, while still under urethane anesthesia, rats were fixed in a stereotaxic frame, and the skull was exposed by a midline incision and leveled between the bregma and lambda. A 30-gauge guide cannula was placed 0.9 mm posterior to bregma, along the midline, for SFO microinjection. The cannula tip was advanced to a final position 2.7 mm ventral to the cranial surface and 2 mm above the SFO. A 35-gauge injection cannula was inserted into the guide cannula and extended 2 mm beyond the tip of the guide cannula. Microinjection of SFO (0.2 μl over 10 s) was carried out using a 1-μl Hamilton microsyringe connected to the injection cannula.
Intracerebroventricular injection.
While still under urethane anesthesia, rats were fixed in a stereotaxic frame, and the skull was exposed as described above. A 29-gauge stainless steel guide cannula was implanted, with the cannula tip 2 mm above the left lateral cerebral ventricle, using the following coordinates: anteroposterior, −1.0 mm; dorsoventral, −2.5 mm; and mediolateral, 1.5 mm (left), with bregma as a reference. A 33-gauge injection cannula connected to a 1-µl Hamilton microsyringe was inserted into the guide cannula and extended 2 mm past the tip of the guide cannula for intracerebroventricular (ICV) injection (0.2 µl).
SFO lesions.
The detailed methods for SFO lesions (SFO-x) are described elsewhere (63, 75). Rats were anesthetized with ketamine (90 mg/kg ip)-xylazine (10 mg/kg ip) and secured in a stereotaxic apparatus (Kopf Instruments, Tujunga, CA). Under sterile conditions, a midline incision was made to visualize the skull. The skull was leveled between bregma and lambda. Lesions of the SFO were made by means of three penetrations along the midline through a 3.0-mm trephine hole that was made at bregma. The stereotaxic coordinates (in mm) of the three lesion sites, referred to bregma and the ventral surface of the midsagittal sinus, were −0.4, −4.3; −0.7, −4.5; and −1.0, −4.7. A monopolar electrode made of tungsten wire (0.008-inch diameter; A-M Systems, Sequim, WA), insulated except at the tip, was lowered to the points described. The lesions were made by a 1-mA anodal current that was passed through the bare tip for 8 s per penetration. The reference electrode was placed in the rectum. In sham SFO lesion (SFO-s) surgery, the monopolar electrode was placed 1.0 mm above the target coordinates, but no current was applied. The animals were allowed to recover and resume their normal chow diet. Postoperative pain was controlled with buprenorphine (0.03 mg/kg sc, every 12 h for 2 days, then as needed).
Experimental Protocols
Urethane-anesthetized intact rats (n = 30) underwent electrophysiological and hemodynamic recordings to determine sympathetic nerve activity and hemodynamic responses to intravenous IL-1β (500 ng in 10 µl) 10–15 min after SFO microinjection of the vehicle (Veh) artificial cerebrospinal fluid (aCSF, n = 6), the COX-2 inhibitor NS-398 (2 µg, n = 6), the angiotensin II type 1a receptor (AT1aR) blocker losartan (1 µg, n = 6), or the angiotensin-converting enzyme (ACE) inhibitor captopril (1 µg, n = 6). Intravenous injection of 0.9% saline in SFO Veh-pretreated rats (n = 6) served as the control. A small volume (0.05 µl) of 2% pontamine sky blue was microinjected to verify the SFO location of injections. At the conclusion of experiments (5–6 h after the intravenous IL-1β), rats were euthanized to collect SFO, PVN, and cortex tissues to determine mRNA expression of ACE, AT1aR, COX-2, COX-1, TNF-α, IL-1β, TNF-α receptor 1 (TNFR1), and IL-1 receptor (IL-1R).
To control for potential leakage of drug from the SFO microinjections into the third ventricle, the hemodynamic and sympathetic responses to intravenous IL-1β (500 ng) were assessed 10–15 min after ICV injections of Veh (aCSF, n = 4), NS-398 (2 µg, n = 4), losartan (1 µg, n = 4), or captopril (1 µg, n = 4).
Urethane-anesthetized rats that had undergone SFO-x (n = 12) or SFO-s (n = 12) 2–3 wk earlier were treated with intravenous IL-1β (500 ng) to determine its effects on neurochemical mediators in the PVN. A group of SFO-s (n = 12) rats that received an intravenous injection of 0.9% saline Veh served as control. After 5-6 h, some rats (n = 6 in each group) were euthanized for collection of PVN tissue to determine mRNA expression of ACE, AT1aR, COX-2 and COX-1, TNF-α, IL-1β, TNFR1, and IL-1R. Others (n = 6 in each group) were euthanized for collection of PVN tissue for Western blot analysis of nuclear factor-κB (NF-κB) activity and c-Fos expression. Frozen serial sagittal sections (30 µm) through the SFO region were examined with light microscopy to verify the SFO lesions.
Acquisition of Hemodynamic and Sympathetic Recording Data
Data were collected with a laboratory interface [model 1401, Cambridge Electronics Design (CED), Cambridge, UK] connected to a personal computer. A pressure transducer was connected to the femoral artery catheter to transfer the blood pressure signals to a chart recorder (model TA240S, Gould Instruments, Valley View, OH) linked to the laboratory interface. HR was derived from the arterial pressure waveform. The raw RSNA was amplified by a preamplifier (Grass Instrument, Quincy, MA) and then rectified and integrated by a Paynter filter (20-ms time constant; BAK Electronics, Germantown, MD) before being passed to the laboratory interface. Digitized data were stored for subsequent off-line analysis with Spike2 software (CED). Mean BP (MBP, mmHg), HR (beats/min), and integrated RSNA (mV) were averaged over 5-min intervals after intravenous IL-1β or SFO microinjection and compared with baseline values averaged over 5-min intervals immediately before each intervention. Changes in integrated RSNA are reported as percentage of baseline control.
Molecular Studies
Tissue preparations.
To obtain tissues for real-time PCR and Western blot measurements, rats were anesthetized with urethane (1.5 g/kg ip) and decapitated. The brains were immediately removed, frozen in liquid nitrogen, and stored at − 80°C. The frozen brain was subsequently cut into 300-μm coronal sections, and the target tissues, including the SFO, PVN, and cortex, were obtained using a punch (1.5 mm inner diameter) device (Stoelting, Wood Dale, IL). Tissue was collected from both sides of the PVN in two sections for each rat. Tissue was collected from two or three sections of the SFO for each rat, depending on whether the third section contained SFO tissue. Some immediately surrounding tissue was usually included in the punch biopsies. For SFO-x rats, only PVN tissue was harvested.
Real-time PCR.
Total RNA was extracted using the RNeasy Plus Mini Kit according to the manufacturer’s instructions (Qiagen). The RNA samples were treated with DNaseI (Qiagen) to eliminate possible genomic DNA contamination. The concentration of RNA was determined by measurement of absorbance at 260 nm and the ratio of 260 nm to 280 nm in a spectrophotometer. RNA (400 ng) was used for the reverse-transcription reaction to make a final volume of 20 μl of cDNA using the TaqMan Gold reverse-transcription PCR kit (Applied Biosystems) with random hexamers. The reaction mixture was incubated at 25°C for 10 min, 48°C for 30 min, and 95°C for 5 min and subsequently stored at −80°C until use.
The mRNA levels of IL-1R and ACE were measured with TaqMan PCR from 2 µl of cDNA using TaqMan universal master mix in a final 25-µl mixture volume. The sequences for the primers and probe are shown in Table 1. Primer and probe of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) for TaqMan PCR were obtained from Applied Biosystems. The mRNA levels of AT1aR, COX-2, COX-1, TNF-α, IL-1β, TNFR1, and GAPDH (internal control for SYBR) were analyzed with SYBR Green real-time PCR using SYBR universal PCR master mix. The sequences for the primers are indicated in Table 1. Real-time PCR was performed using the ABI Prism 7300 sequence detection system (Applied Biosystems, Carlsbad, CA). Cycling conditions were as follows: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of amplification (95°C for 15 s, 60°C for 1 min). Each sample was assayed in duplicate. To ensure the absence of genomic DNA contamination, a control sample of non-reverse-transcribed RNA was run for each set of RNA extractions. The values of each gene of interest were corrected by the housekeeping gene GAPDH (TaqMan or SYBR), and relative quantification was calculated using the cycle threshold [2−(ΔΔCt)] method (43). Quantitative mRNA data are expressed as fold change relative to Veh control.
Table 1.
Sequences for primers and probes
| Gene | Primers and Probes | Ref. No. |
|---|---|---|
| IL-1β (SYBR) | 68 | |
| Forward primer | 5′-CGACAGAATCTAGTTGTCC-3′ | |
| Reverse primer | 5′-TCATAAACACTCTCATCCACAC-3′ | |
| TNF-α (SYBR) | 68 | |
| Forward primer | 5′-CCTTATCTACTCCCAGGTTCTC-3′ | |
| Reverse primer | 5′-TTTCTCCTGGTATGAATGGC-3′ | |
| COX-1 (SYBR) | 76 | |
| Forward primer | 5′-AGAGATCACCAATGCCAGCT-3′ | |
| Reverse primer | 5′-ACTGGATGGTACGCTTGGTC-3′ | |
| COX-2 (SYBR) | 46 | |
| Forward primer | 5′-AAGGGAGTCTGGAACATTGTGAAC-3′ | |
| Reverse primer | 5′-CAAATGTGATCTGGACGTCAACA-3′ | |
| TNFR1 (SYBR) | 41 | |
| Forward primer | 5′-GGTTCCTTTGTGGCACTTGGT-3′ | |
| Reverse primer | 5′-CTCTTGGTGACCGGGAGAAG-3′ | |
| AT1aR (SYBR) | 60 | |
| Forward primer | 5′-GGATGGTTCTCAGAGAGAGTACAT-3′ | |
| Reverse primer | 5′-CCTGCCCTCTTGTACCTGTTG-3′ | |
| GAPDH (SYBR) | 77 | |
| Forward primer | 5′-AAGGTCATCCCAGAGCTGAA-3′ | |
| Reverse primer | 5′-ATGTAGGCCATGAGGTCCAC-3′ | |
| IL-1R (TaqMan) | ||
| Forward primer | 5′-GCCCACGGAATGAGACGAT-3′ | 14 |
| Reverse primer | 5′-GCCCGTGACGTTGCAGA-3′ | |
| Probe | 5′-AGCTGACCCAGGATCCACGATACAACTG-3′ | |
| ACE (TaqMan) | 29 | |
| Forward primer | 5′-GGAGACGACTTACAGTGTAGCC-3′ | |
| Reverse primer | 5′-CACACCCAAAGCAATTCTTC-3′ | |
| Probe | 5′-AATGGCCACGTCCCGGAAAT-3′ |
IL-1β, interleukin (IL)-1β; TNF-α, tumor necrosis factor-α; COX, cyclooxygenase; TNFR1, TNF-α receptor 1; AT1aR, angiotensin II type 1a receptor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL-1R, IL-1 receptor; ACE, angiotensin-converting enzyme.
Western blot analysis.
The punched tissues were homogenized in cell lysis buffer (Cell Signaling Technology, Beverly, MA) with the addition of cOmplete protease inhibitor cocktail tablets and then centrifuged at 12,000 rpm at 4°C for 20 min. The supernatant was collected, and protein concentration was measured using the Bradford assay. Expression of the NF-κB transcription factor p65 subunit and its inhibitor of κB (IκB-α), as well as c-Fos expression, in the PVN was assessed by Western blotting. Briefly, protein samples (30 μg) were separated by 10% SDS-polyacrylamide gel and then transferred to a PVDF membrane. One protein standard (catalog no. 161-0394, Bio-Rad, Hercules, CA) was also run with the protein sample to determine the size of the protein. Nonspecific binding was blocked by incubation with 5% nonfat dry milk for 1 h at room temperature. Membranes were then incubated overnight at 4°C with primary polyclonal rabbit antibodies against NF-κB p65 and IκB-α (1:500 dilution; catalog nos. 8242 and 9242, respectively; Cell Signaling Technology), rabbit monoclonal antibody against c-Fos (1:500 dilution; catalog no. 2250, Cell Signaling Technology), or rabbit anti-rat β-actin monoclonal antibody (1:1,000 dilution; catalog no. 4970, Cell Signaling Technology), respectively. The secondary antibody was goat anti-rabbit IgG-horseradish peroxidase (1:5,000 dilution; catalog no. SC-2004, Santa Cruz Biotechnology). Immunoblots were visualized with an enhanced chemiluminescence reagent. Band intensities were quantified with Image Lab analysis software (Bio-Rad). The protein levels of NF-κB p65, IκB-α, and c-Fos were normalized by the total β-actin.
Drug Administered
IL-1β was purchased from Millipore (Billerica, MA) and dissolved in 0.9% saline. Losartan and captopril were purchased from Sigma (St. Louis, MO) and dissolved in aCSF. NS-398 was purchased from Tocris (Ellisville, MO) and was first dissolved in dimethyl sulfoxide (DMSO) and then diluted in aCSF to make a 5% final DMSO concentration. The dose of intravenous IL-1β (500 ng) was based on our previous observations showing that it elicited a robust sympathetic response and a significant change in expression of excitatory and inflammatory mediators in the brain (75). The doses of NS-398, losartan, and captopril for SFO microinjection were previously shown to reduce the sympathetic and hemodynamic response to SFO microinjections of IL-1β (73).
Statistical Analysis
Values are means ± SE. The significance of differences among groups was analyzed by one-way repeated-measure ANOVA followed by post hoc Fisher’s test. Student's t-test was used to determine statistical significance between paired data for a single comparison. P < 0.05 was considered to indicate statistical significance.
RESULTS
Effects of SFO Microinjection of AT1R Blocker, ACE Inhibitor, and COX-2 Inhibitor on Intravenous IL-1β-Induced Responses
In the rats pretreated with SFO microinjections of Veh (aCSF), intravenous injections of 0.9% saline (10 µl) had no significant effect on baseline MBP (95.3 ± 2.3 mmHg), HR (332 ± 15 beats/min), or integrated RSNA (10.3 ± 2.4 mV). In the rats pretreated with SFO microinjections of Veh, intravenous injection of an equal volume of IL-1β (500 ng; Fig. 1, A and E) induced dramatic and persistent increases in MBP, HR, and RSNA. These excitatory responses began within 20–30 min after intravenous injection of IL-1β. The peak responses of MBP (19.7 ± 2.5 mmHg), HR (83.4 ± 9.1 beats/min), and RSNA (87.1 ± 10.1% change) occurred 2–3 h after the IL-1β injection and remained at higher-than-baseline levels for ≥5–6 h. Pretreatment with SFO microinjection of losartan (Fig. 1, B and E), captopril (Fig. 1, C and E), or NS-398 (Fig. 1, D and E) significantly reduced MBP [10.9 ± 2.2, 12.5 ± 2.5, and 11.2 ± 2.4 mmHg, respectively; time effect: F(9, 230) = 28.59, P < 0.0001; treatment effect: F(3, 236) = 30.67, P < 0.0001], HR [45.0 ± 9.3, 46.5 ± 7.1, and 43.4 ± 10.8 beats/min, respectively; time effect: F(9, 230) = 34.47, P < 0.0001; treatment effect: F(3, 236) = 27.94, P < 0.0001], and RSNA [ 46.4 ± 6.7, 52.1 ± 7.7, and 52.2 ± 9.0% change, respectively; time effect: F(9, 230) = 37.73, P < 0.0001; treatment effect: F(3, 236) = 24.12, P < 0.0001] responses to intravenous injection of IL-1β.
Fig. 1.

Representative traces (A–D) and grouped data (E) showing effects of intravenous (IV) injection of interleukin (IL)-1β (500 ng) on blood pressure (BP), heart rate (HR, beats/min), and renal sympathetic nerve activity (RSNA), windowed (spikes/s) and integrated (mV), in rats pretreated with subfornical organ (SFO) microinjections of vehicle (Veh), losartan, captopril, and NS-398. Arrows indicate timing of injections. MBP, mean blood pressure; ΔRSNA, change from baseline in integrated RSNA. Values are means ± SE. *P < 0.05 vs. baseline; †P < 0.05 vs. SFO Veh + IV IL-1β.
In rats pretreated with ICV Veh (aCSF), intravenous injection of IL-1β elicited excitatory responses in MBP (20.0 ± 2.9 mmHg), HR (85.2 ± 10.9 beats/min), and RSNA (88.6 ± 13.2% change) that were similar with those observed in rats pretreated with SFO microinjection of Veh. ICV injections of losartan, captopril, or NS-398, in the same dose and volume that were used for the SFO microinjections, did not significantly attenuate the increases in MBP (16.1 ± 2.4, 16.8 ± 2.2, 15.8 ± 2.8 mmHg, respectively), HR (73.1 ± 7.6, 71.4 ± 8.1, and 68.9 ± 8.6 beats/min, respectively), and RSNA (73.9 ± 8.8, 71.5 ± 8.7, and 70.2 ± 9.5% change, respectively) induced by intravenous injection of IL-1β.
Effects of SFO Microinjection of AT1R Blocker, ACE Inhibitor, and COX-2 Inhibitor on Intravenous IL-1β-Induced Expression of Excitatory Mediators in SFO and PVN
In rats pretreated with SFO microinjections of Veh, real-time PCR analysis revealed that ACE mRNA and AT1aR mRNA were significantly elevated in the SFO (ACE: 2.96 ± 0.21 vs. 1.03 ± 0.13-fold; AT1aR: 2.45 ± 0.19 vs. 1.05 ± 0.13-fold) and downstream in the PVN (ACE: 2.50 ± 0.16 vs. 1.02 ± 0.11-fold; AT1aR: 2.04 ± 0.14 vs. 1.03 ± 0.10-fold) (Fig. 2) 5–6 h after the intravenous injection of IL-1β compared with intravenous injection of Veh. COX-2 mRNA was also increased in the SFO (3.26 ± 0.31 vs. 1.05 ± 0.2-fold) and PVN (2.69 ± 0.19 vs. 1.02 ± 0.08-fold) of the rats treated with intravenous IL-1β. There was no significant change in COX-1 mRNA in the SFO (1.34 ± 0.18 vs. 1.03 ± 0.10-fold) or in the PVN (1.29 ± 0.13 vs. 1.02 ± 0.09-fold) (Fig. 2). In the rats pretreated with SFO microinjection of losartan, captopril, and NS-398, intravenous IL-1β-induced expression of AT1aR, ACE, and COX-2 mRNA in the SFO [AT1aR: F(4, 25) = 8.119, P = 0.0002; ACE: F(4, 25) = 11.22, P < 0.0001; COX-2: F(4, 25) = 10.08, P < 0.0001] and PVN [AT1aR: F(4, 25) = 10.32, P < 0.0001; ACE: F(4, 25) = 15.68, P < 0.0001; COX-2: F(4, 25) = 12, P < 0.0001] was significantly lower than in the rats pretreated with SFO microinjection of Veh (Fig. 2).
Fig. 2.

Quantitative analysis by real-time PCR showing effects of intravenous (IV) injection of interleukin (IL)-1β or 0.9% saline on mRNA expression of angiotensin-converting enzyme (ACE), angiotensin II type 1a receptor (AT1aR), cyclooxygenase (COX)-2, and COX-1 in the cortex, subfornical organ (SFO), and hypothalamic paraventricular nucleus (PVN) of rats pretreated with SFO microinjection of vehicle (Veh), losartan, captopril, and NS-398. Values are means ± SE (n = 6 for each group) and expressed as fold change relative to Veh control. *P < 0.05 vs. SFO Veh + IV saline; †P < 0.05 vs. SFO Veh + IV IL-1β.
Similarly, in the rats pretreated with SFO microinjections of Veh, intravenous IL-1β markedly increased expression of TNF-α and IL-1β mRNA in the SFO (2.36 ± 0.21 vs. 1.01 ± 0.07-fold and 3.33 ± 0.32 vs. 1.02 ± 0.08-fold, respectively) and PVN (2.01 ± 0.15 vs. 1.03 ± 0.10-fold and 2.47 ± 0.27 vs. 1.02 ± 0.09-fold, respectively) 5–6 h after the injection compared with intravenous injection of Veh (Fig. 3). Intravenous IL-1β also significantly increased mRNA levels of the cytokine receptors TNFR1 and IL-1R in the SFO (2.12 ± 0.24 vs. 1.00 ± 0.09-fold and 2.30 ± 0.21 vs. 1.02 ± 0.10-fold, respectively) and PVN (1.98 ± 0.19 vs. 1.02 ± 0.10-fold and 2.10 ± 0.20 vs. 1.02 ± 0.08-fold, respectively) compared with intravenous injection of Veh (Fig. 3). In the rats pretreated with SFO microinjection of losartan, captopril, and NS-398, intravenous IL-1β-induced expression of TNF-α, IL-1β, TNFR1, and IL-1R was significantly lower in the SFO [TNF-α: F(4, 25) = 7.722, P = 0.0003; IL-1β: F(4, 25) = 14.19, P < 0.0001; TNFR1: F(4, 25) = 7.564, P = 0.0004; IL-1R: F(4, 25) = 7.577, P = 0.0004] and PVN [TNF-α: F(4, 25) = 9.593, P < 0.0001; IL-1β: F(4, 25) = 17.73, P < 0.0001; TNFR1: F(4, 25) = 9.485, P < 0.0001; IL-1R: F(4, 25) = 6.574, P = 0.0009] than in the rats pretreated with SFO microinjection of Veh (Fig. 3).
Fig. 3.

Quantitative analysis by real-time PCR showing effects of intravenous (IV) injection of interleukin (IL)-1β or 0.9% saline on mRNA expression of tumor necrosis factor (TNF)-α, IL-1β, TNF-α receptor 1 (TNFR1), and IL-1 receptor (IL-1R) in the cortex, subfornical organ (SFO), and hypothalamic paraventricular nucleus (PVN) of rats pretreated with SFO microinjection of vehicle (Veh), losartan, captopril, and NS-398. Values are means ± SE (n = 6 for each group) and expressed as fold change relative to Veh control. *P < 0.05 vs. SFO Veh + IV saline; †P < 0.05 vs. SFO Veh + IV IL-1β.
Intravenous injection of IL-1β had no significant effects on the mRNA levels of any of these excitatory mediators and inflammatory elements in the cortex (Figs. 2 and 3).
Effects of SFO Lesions on Intravenous IL-1β-Induced Expression of Excitatory Mediators in the PVN
In SFO-s rats, mRNA levels of the RAS components ACE and AT1aR and the inflammatory elements COX-2, TNF-α, IL-1β, TNFR1, and IL-1R were significantly increased in the PVN 5–6 h after administration of intravenous IL-1β compared with intravenous 0.9% saline (Fig. 4). In SFO-x rats, the intravenous IL-1β-induced PVN expression of these excitatory and inflammatory mediators was significantly lower [ACE: 1.45 ± 0.12 vs. 2.61 ± 0.22-fold, F(2, 15) = 20.31, P < 0.0001; AT1aR: 1.28 ± 0.11 vs. 2.10 ± 0.15-fold, F(2, 15) = 21.68, P < 0.0001; COX-2: 1.76 ± 0.10 vs. 2.77 ± 0.23-fold, F(2, 15) = 19.38, P < 0.0001; TNF-α: 1.35 ± 0.13 vs. 1.99 ± 0.15-fold, F(2, 15) = 10.56, P = 0.0014; IL-1β: 1.56 ± 0.13 vs. 2.35 ± 0.16-fold, F(2, 15) = 25.3, P < 0.0001; TNFR1: 1.36 ± 0.14 vs. 1.94 ± 0.18-fold, F(2, 15) = 9.98, P = 0.0018; IL-1R: 1.31 ± 0.15 vs. 2.03 ± 0.20-fold, F(2, 15) = 16.37, P = 0.0002] than in SFO-s rats (Fig. 4). COX-1 mRNA in PVN was unaffected by intravenous injection of IL-1β in SFO-s (1.33 ± 0.14-fold) or SFO-x (1.12 ± 0.13-fold) rats [F(2, 15) = 2.103, P = 0.1567].
Fig. 4.
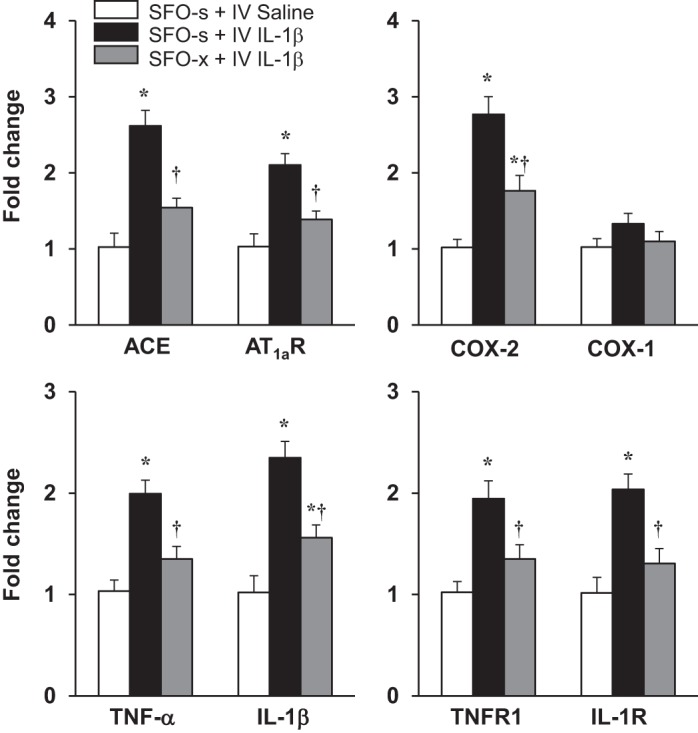
Quantitative analysis by real-time PCR showing effects of intravenous (IV) injection of interleukin (IL)-1β or 0.9% saline on mRNA expression of angiotensin-converting enzyme (ACE), angiotensin II type 1a receptor (AT1aR), cyclooxygenase (COX)-2, COX-1, tumor necrosis factor (TNF)-α, IL-1β, TNF-α receptor 1 (TNFR1), and IL-1 receptor (IL-1R) in the hypothalamic paraventricular nucleus (PVN) of rats with a subfornical organ (SFO) lesion (SFO-x) or a sham SFO lesion (SFO-s). Values are means ± SE (n = 6 for each group) and expressed as fold change relative to Veh control. *P < 0.05 vs. SFO-s + IV saline; †P < 0.05 vs. SFO-s + IV IL-1β.
Effects of SFO Lesions on Intravenous IL-1β-Induced NF-κB Activity and c-Fos Expression in the PVN
Western blot analysis revealed that intravenous injection of IL-1β upregulated NF-κB activity, with an increased level of NF-κB p65 subunit and a decreased level of IκB-α in the PVN in SFO-s rats. Expression of c-Fos, a classical indicator of neuronal excitation, was also increased in the PVN of SFO-s rats treated with intravenous IL-1β compared with intravenous 0.9% saline (Fig. 5). However, there were significant differences in expression of NF-κB p65 [F(2, 15) = 9.281, P = 0.0024], IκB-α [F(2, 15) = 4.463, P = 0.0301], and c-Fos [F(2, 15) = 4.913, P = 0.0229] in the PVN in response to intravenous IL-1β in the SFO-x rats compared with the SFO-s rats. NF-κB activity and c-Fos activity in the PVN, in response to intravenous IL-1β, were reduced in the SFO-x rats.
Fig. 5.

Western blot analysis showing effects of intravenous (IV) injection of interleukin (IL)-1β or 0.9% saline on expression of the transcription factor nuclear factor NF-κB (NF-κB) p65 subunit (A), inhibitor of κB (IκB-α, B), and c-Fos (C) in the hypothalamic paraventricular nucleus (PVN) of rats with a subfornical organ lesion (SFO-x) or a sham SFO lesion (SFO-s). Values are means ± SE and normalized to β-actin (n = 6 in each group). *P < 0.05 vs. SFO-s + IV saline; †P < 0.05 vs. SFO-s + IV IL-1β. Representative Western blots are shown above each bar.
Confirmation of SFO Microinjections and SFO Lesions
Microinjection sites were confirmed by visualization of pontamine sky blue within the SFO during tissue processing for molecular studies. Twelve of 15 animals in which SFO lesions were made had ≥80% ablation of the SFO and constituted the SFO-x group. Representative images of an SFO lesion and microinjection site are shown in Fig. 6.
Fig. 6.

Histological images of the subfornical organ (SFO) from rats subjected to a sham lesion (SFO-s, A), an electrolytic lesion (SFO-x, B), and SFO microinjection (C). Arrow in C indicates a microinjection site labeled with pontamine sky blue.
DISCUSSION
By virtue of its exposure to the circulation and its connections with key cardiovascular-related nuclei inside the blood-brain barrier, the SFO is well positioned to sense peripheral stress signals and to orchestrate an appropriate central sympathoexcitatory response. This function of the SFO is well established for angiotensin II- and aldosterone-mediated HTN (52, 69). Recent studies have suggested a similar sensory and effector role for the SFO in immune activation of the sympathetic nervous system. Thus, in normal rats, the sympathoexcitatory response to blood-borne PICs is substantially reduced by an SFO lesion (75). Moreover, microinjection of PICs into the SFO of normal rats upregulates RAS activity and inflammation not only in the SFO, but also downstream in the PVN (73), where these are major factors contributing to sympathetic activation in HTN and HF (34, 59, 69). The present study was undertaken to determine whether a systemically (intravenously) administered PIC, mimicking the manner in which circulating PICs would normally access the SFO in HTN and HF, might act similarly to upregulate the sympathoexcitatory milieu in the PVN that drives sympathetic nerve activity and whether intervening in IL-1β-mediated molecular events in the SFO might prevent those downstream effects. Previous studies have shown that SFO activation of sympathetic activity is significantly reduced by interventions at the PVN level (2, 44, 69).
The present study confirmed our earlier observation that intravenous injection of IL-1β increases BP, HR, and RSNA (75) and extended that finding to show that 1) intravenous IL-1β upregulates mRNA for RAS (ACE and AT1aR mRNA) and inflammatory (TNF-α, IL-1β, TNFR1, IL-1R, and COX-2 mRNA) mediators in the SFO and the PVN; 2) inhibition of RAS activity and inflammation in the SFO significantly reduces intravenous IL-1β-induced RAS activity and inflammation downstream in the PVN and attenuates the sympathetic response; and 3) eliminating the SFO altogether normalizes intravenous IL-1β-mediated increases in RAS activity and significantly reduces the indicators of inflammation in the PVN, along with c-Fos protein as an indicator of PVN neuronal excitation. These findings demonstrate that the sympathetic response to circulating IL-1β is largely dependent on actions in the SFO that induce production of an excitatory neurochemical milieu of the PVN.
Previous studies examining the central effects of PICs on sympathetic drive in HTN and HF have focused entirely, and quite logically, on events in the PVN as a source of presympathetic neurons. They have demonstrated that RAS activity and inflammation are upregulated in the PVN in HTN (6, 57, 58) and HF (20, 25, 26, 35, 36, 61), facilitatory interactions between these two excitatory neurochemical systems occur in the PVN (26, 33, 34), and interventions that reduce the activity of either system in the PVN can substantially reduce sympathetic nerve activity (24, 34, 70), with beneficial effects on BP in HTN (69) and on hemodynamics and cardiac remodeling in HF (33, 36, 72). Those studies have not addressed the mechanism(s) responsible for upregulating RAS activity and inflammation in the PVN. The present study suggests that events in the SFO driven by blood-borne PICs, in this case, by IL-1β, contribute significantly to the neurochemical changes in the PVN that drive sympathetic excitation in those settings. We found that systemically administered IL-1β upregulates RAS activity and inflammation in the SFO and PVN and that pretreatment of the SFO with the inhibitors of RAS activity (losartan or captopril) or inflammation (NS-398) reduces the IL-1β-induced upregulation of ACE and AT1aR, TNF-α, IL-1β, and COX-2 mRNA downstream in the PVN and at least partially reduces the peripheral sympathetic response. Consistent with a prominent role for the SFO in activation of the PVN, rats with an SFO lesion had substantially lower levels of RAS activity and inflammatory indicators in the PVN, as well as c-Fos as an indicator of neuronal excitation, in response to intravenous IL-1β. These findings demonstrate that SFO neurons excited by IL-1β, either directly (18) or indirectly via its effects on RAS activity or COX-2 activity, upregulate RAS and inflammatory elements in the PVN that augment sympathetic nerve activity. The precise mechanisms, SFO cell types, and neurotransmitters/modulators released in the PVN remain to be determined. Nevertheless, these results strongly support a major role for the SFO as an interface between peripheral inflammation and central neural mechanisms driving the sympathetic response, similar to its role as a mediator of angiotensin II-dependent HTN.
Previous studies of the peripheral and central effects of PICs in experimental models of HTN and HF have focused on effects mediated by TNF-α (1, 3, 36, 67), perhaps because of the availability of specific TNF-α blockers (etanercept and infliximab) and a TNF-α synthesis inhibitor (pentoxifylline) that have been used in clinical studies (7, 48). Studies in experimental models have shown that administration of these agents systemically, intracerebroventricularly, or by direct microinjection into the PVN can substantially reduce sympathetic activity in HTN and HF (25, 32, 34–36). Less is known regarding the effects of IL-1β in those settings, even though levels of IL-1β, like levels of TNF-α (13), are increased in HF and are associated with adverse outcomes (27, 66). However, there is growing interest in the potential benefits of blocking the IL-1R in various HF settings using the IL-1R blocker anakinra (64, 65). The present study provides supportive experimental evidence suggesting that blood-borne IL-1β may have a significant independent influence on central mechanisms driving sympathetic activity in HF. In fact, it is conceivable that reductions in IL-1β may account, at least in part, for the beneficial effects of reducing TNF-α in experimental models of HF. Our previous studies have shown that, in rats with HF, treatment with etanercept or pentoxifylline lowers IL-1β, as well as TNF-α, levels in plasma and in brain tissue (35, 36), likely because of the propensity of PICs to produce more PICs via activation of NF-κB (21).
There are several potential explanations for the excitatory effects of blood-borne IL-1β on SFO neurons. In brain slice preparations, physiological levels of IL-1β were shown to have direct excitatory effects on the majority of SFO neurons (12), although the projection sites of those neurons were not identified. In addition, IL-1β induces expression of the COX-2 enzyme, which catalyzes the production of prostaglandin E2 (PGE2). IL-1β induction of COX-2 has been shown to excite parvocellular PVN neurons via a PGE2-mediated reduction of GABAergic inhibition (19). Whether a similar PGE2 mechanism might contribute to the excitability of SFO neurons has not been determined. However, in the present study, pretreatment with microinjection of the COX-2 inhibitor NS-398 significantly reduced intravenous IL-1β-induced COX-2 expression in the SFO, RAS, and inflammatory indicators in the PVN and sympathetic nerve activity. Yet another possibility is IL-1β-induced angiotensinergic activation of SFO neurons (56, 62). AT1Rs are densely distributed in the SFO, and our data demonstrate that blood-borne IL-1β upregulates ACE and AT1aR mRNA in the SFO. Moreover, pretreatment of the SFO with the AT1R blocker losartan or the ACE inhibitor captopril significantly reduced the effects of intravenous IL-1β on PVN neurochemistry and sympathetic drive. Finally, reactive oxygen species (ROS) in the SFO are known to contribute to sympathetic drive in HTN (30, 81) and HF (42). ROS can be generated by RAS activity (4, 23, 82) and PICs (36, 78) and, in turn, may facilitate RAS and PIC production (40, 51). ROS may be a key modulator of IL-1β-induced molecular mechanisms, via activation of NF-κB and mitogen-activated protein kinase signaling.
Limitations of the Study
Neither RAS inhibition nor COX-2 inhibition in the SFO completely blocked sympathetic activation by intravenous IL-1β. There are several potential explanations for this incomplete response. It may indicate the involvement of both systems, since simultaneous inhibition was not attempted in this study, or it may simply reflect incomplete inhibition by the drug doses used. Alternatively, it is quite possible that blood-borne IL-1β acts on other central nuclei that express IL-1R, including other circumventricular organs, such as the organum vasculosum of the lamina terminalis that also projects to the PVN, and the residual sympathetic excitation may represent the influences of these other neural pathways.
It is possible that some portion of the effects attributed to inhibiting RAS and COX-2 activity at the SFO level may have resulted from effects on other nuclei in the forebrain region via leakage of drug into the third ventricle. Notably, however, ICV administration of the same doses of these drugs that were microinjected into the SFO failed to significantly alter the hemodynamic and sympathetic responses to intravenous IL-1β.
Finally, and importantly, the specific mechanisms and cell types that mediate the IL-1β-induced increases in RAS activity and inflammation within the SFO and the nature of the SFO neurons that signal the PVN to alter its neurochemical milieu and how they accomplish these outcomes remain to be addressed.
Perspectives and Significance
The present study reveals that blood-borne IL-1β is capable of initiating molecular events in the SFO that result in an increase in sympathoexcitatory mediators in the PVN and an increase in sympathetic nerve activity, affecting BP and HR. In this scenario, in which it was administered intravenously to normal animals in a dose known to elicit a sympathoexcitatory response, IL-1β acted on the SFO to upregulate the expression of PIC receptors, brain RAS activity, and COX-2, key elements of three systems known to increase sympathetic excitation, in the SFO and PVN. An interesting implication of these findings is that PICs must lie upstream of the brain RAS in the sequence of molecular events leading to sympathetic excitation. There is evidence of PIC upregulation of RAS expression in the periphery (9, 28) and in the brain (34, 73, 79). However, pathophysiological states, such as HF and many forms of HTN, in which RAS activation coexists with the inflammatory cytokine response, present a more complex scenario. The RAS is proinflammatory in other settings (5, 10, 50), and previous studies have demonstrated cross talk between PICs and the RAS in the PVN (33, 57). It seems likely that similar interactions occur in the SFO. Thus, in settings in which the RAS and PICs are both active, a degree of redundancy exists, and the relative contribution of each is uncertain. Nevertheless, the present findings support the concept that central nervous system actions of blood-borne IL-1β may contribute to the progression of HTN and HF and should be considered in the development of new therapeutic approaches.
GRANTS
This material is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development and by National Heart, Lung, and Blood Institute Grant R01 HL-073986 (to R. B. Felder).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.-G.W., Y.Y., and R.B.F. conceived and designed research; S.-G.W. performed experiments; S.-G.W. analyzed data; S.-G.W., Y.Y., and R.B.F. interpreted results of experiments; S.-G.W. prepared figures; S.-G.W. and R.B.F. drafted manuscript; S.-G.W. and R.B.F. edited and revised manuscript; S.-G.W., Y.Y., and R.B.F. approved final version of manuscript.
REFERENCES
- 1.Azhar A, El-Bassossy HM. Pentoxifylline alleviates hypertension in metabolic syndrome: effect on low-grade inflammation and angiotensin system. J Endocrinol Invest 38: 437–445, 2015. doi: 10.1007/s40618-014-0209-z. [DOI] [PubMed] [Google Scholar]
- 2.Bains JS, Ferguson AV. Paraventricular nucleus neurons projecting to the spinal cord receive excitatory input from the subfornical organ. Am J Physiol Regul Integr Comp Physiol 268: R625–R633, 1995. doi: 10.1152/ajpregu.1995.268.3.R625. [DOI] [PubMed] [Google Scholar]
- 3.Bergman MR, Holycross BJ. Pharmacological modulation of myocardial tumor necrosis factor-α production by phosphodiesterase inhibitors. J Pharmacol Exp Ther 279: 247–254, 1996. [PubMed] [Google Scholar]
- 4.Braga VA, Medeiros IA, Ribeiro TP, França-Silva MS, Botelho-Ono MS, Guimarães DD. Angiotensin-II-induced reactive oxygen species along the SFO-PVN-RVLM pathway: implications in neurogenic hypertension. Braz J Med Biol Res 44: 871–876, 2011. doi: 10.1590/S0100-879X2011007500088. [DOI] [PubMed] [Google Scholar]
- 5.Brasier AR, Recinos A 3rd, Eledrisi MS. Vascular inflammation and the renin-angiotensin system. Arterioscler Thromb Vasc Biol 22: 1257–1266, 2002. doi: 10.1161/01.ATV.0000021412.56621.A2. [DOI] [PubMed] [Google Scholar]
- 6.Cardinale JP, Sriramula S, Mariappan N, Agarwal D, Francis J. Angiotensin II-induced hypertension is modulated by nuclear factor-κB in the paraventricular nucleus. Hypertension 59: 113–121, 2012. doi: 10.1161/HYPERTENSIONAHA.111.182154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Champion S, Lapidus N, Cherié G, Spagnoli V, Oliary J, Solal AC. Pentoxifylline in heart failure: a meta-analysis of clinical trials. Cardiovasc Ther 32: 159–162, 2014. doi: 10.1111/1755-5922.12076. [DOI] [PubMed] [Google Scholar]
- 8.Chen A, Huang BS, Wang HW, Ahmad M, Leenen FH. Knockdown of mineralocorticoid or angiotensin II type 1 receptor gene expression in the paraventricular nucleus prevents angiotensin II hypertension in rats. J Physiol 592: 3523–3536, 2014. doi: 10.1113/jphysiol.2014.275560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowling RT, Zhang X, Reese VC, Iwata M, Gurantz D, Dillmann WH, Greenberg BH. Effects of cytokine treatment on angiotensin II type 1A receptor transcription and splicing in rat cardiac fibroblasts. Am J Physiol Heart Circ Physiol 289: H1176–H1183, 2005. doi: 10.1152/ajpheart.00088.2005. [DOI] [PubMed] [Google Scholar]
- 10.Dandona P, Dhindsa S, Ghanim H, Chaudhuri A. Angiotensin II and inflammation: the effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockade. J Hum Hypertens 21: 20–27, 2007. doi: 10.1038/sj.jhh.1002101. [DOI] [PubMed] [Google Scholar]
- 11.Dange RB, Agarwal D, Teruyama R, Francis J. Toll-like receptor 4 inhibition within the paraventricular nucleus attenuates blood pressure and inflammatory response in a genetic model of hypertension. J Neuroinflammation 12: 31, 2015. doi: 10.1186/s12974-015-0242-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desson SE, Ferguson AV. Interleukin 1β modulates rat subfornical organ neurons as a result of activation of a non-selective cationic conductance. J Physiol 550: 113–122, 2003. doi: 10.1113/jphysiol.2003.041210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 103: 2055–2059, 2001. doi: 10.1161/01.CIR.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 14.Docagne F, Campbell SJ, Bristow AF, Poole S, Vigues S, Guaza C, Perry VH, Anthony DC. Differential regulation of type I and type II interleukin-1 receptors in focal brain inflammation. Eur J Neurosci 21: 1205–1214, 2005. doi: 10.1111/j.1460-9568.2005.03965.x. [DOI] [PubMed] [Google Scholar]
- 15.Felder RB, Francis J, Zhang ZH, Wei SG, Weiss RM, Johnson AK. Heart failure and the brain: new perspectives. Am J Physiol Regul Integr Comp Physiol 284: R259–R276, 2003. doi: 10.1152/ajpregu.00317.2002. [DOI] [PubMed] [Google Scholar]
- 16.Ferguson AV. Circumventricular organs: integrators of circulating signals controlling hydration, energy balance, and immune function. In: Neurobiology of Body Fluid Homeostasis: Transduction and Integration, edited by De Luca LA Jr, Menani JV, Johnson AK. Boca Raton, FL: CRC, 2014. [PubMed] [Google Scholar]
- 17.Ferguson AV, Latchford KJ, Samson WK. The paraventricular nucleus of the hypothalamus—a potential target for integrative treatment of autonomic dysfunction. Expert Opin Ther Targets 12: 717–727, 2008. doi: 10.1517/14728222.12.6.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferri CC, Ferguson AV. Interleukin-1β depolarizes paraventricular nucleus parvocellular neurones. J Neuroendocrinol 15: 126–133, 2003. doi: 10.1046/j.1365-2826.2003.00870.x. [DOI] [PubMed] [Google Scholar]
- 19.Ferri CC, Ferguson AV. Prostaglandin E2 mediates cellular effects of interleukin-1β on parvocellular neurones in the paraventricular nucleus of the hypothalamus. J Neuroendocrinol 17: 498–508, 2005. doi: 10.1111/j.1365-2826.2005.01336.x. [DOI] [PubMed] [Google Scholar]
- 20.Francis J, Chu Y, Johnson AK, Weiss RM, Felder RB. Acute myocardial infarction induces hypothalamic cytokine synthesis. Am J Physiol Heart Circ Physiol 286: H2264–H2271, 2004. doi: 10.1152/ajpheart.01072.2003. [DOI] [PubMed] [Google Scholar]
- 21.Gosselin D, Rivest S. Role of IL-1 and TNF in the brain: twenty years of progress on a Dr. Jekyll/Mr. Hyde duality of the innate immune system. Brain Behav Immun 21: 281–289, 2007. doi: 10.1016/j.bbi.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Granger JP. Inflammatory cytokines, vascular function, and hypertension. Am J Physiol Regul Integr Comp Physiol 286: R989–R990, 2004. doi: 10.1152/ajpregu.00157.2004. [DOI] [PubMed] [Google Scholar]
- 23.Griendling KK, Ushio-Fukai M. Reactive oxygen species as mediators of angiotensin II signaling. Regul Pept 91: 21–27, 2000. doi: 10.1016/S0167-0115(00)00136-1. [DOI] [PubMed] [Google Scholar]
- 24.Guggilam A, Cardinale JP, Mariappan N, Sriramula S, Haque M, Francis J. Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: a role for superoxide and nitric oxide. Basic Res Cardiol 106: 273–286, 2011. doi: 10.1007/s00395-010-0146-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guggilam A, Haque M, Kerut EK, McIlwain E, Lucchesi P, Seghal I, Francis J. TNF-α blockade decreases oxidative stress in the paraventricular nucleus and attenuates sympathoexcitation in heart failure rats. Am J Physiol Heart Circ Physiol 293: H599–H609, 2007. doi: 10.1152/ajpheart.00286.2007. [DOI] [PubMed] [Google Scholar]
- 26.Guggilam A, Patel KP, Haque M, Ebenezer PJ, Kapusta DR, Francis J. Cytokine blockade attenuates sympathoexcitation in heart failure: cross-talk between nNOS, AT-1R and cytokines in the hypothalamic paraventricular nucleus. Eur J Heart Fail 10: 625–634, 2008. doi: 10.1016/j.ejheart.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 122: 23–35, 2012. doi: 10.1159/000338166. [DOI] [PubMed] [Google Scholar]
- 28.Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH. IL-1β and TNF-α upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J Mol Cell Cardiol 38: 505–515, 2005. doi: 10.1016/j.yjmcc.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 29.Harada E, Yoshimura M, Yasue H, Nakagawa O, Nakagawa M, Harada M, Mizuno Y, Nakayama M, Shimasaki Y, Ito T, Nakamura S, Kuwahara K, Saito Y, Nakao K, Ogawa H. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation 104: 137–139, 2001. doi: 10.1161/01.CIR.104.2.137. [DOI] [PubMed] [Google Scholar]
- 30.Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am 93: 621–635, 2009. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev 15: 543–562, 2010. doi: 10.1007/s10741-010-9168-4. [DOI] [PubMed] [Google Scholar]
- 32.Kang YM, He RL, Yang LM, Qin DN, Guggilam A, Elks C, Yan N, Guo Z, Francis J. Brain tumour necrosis factor-α modulates neurotransmitters in hypothalamic paraventricular nucleus in heart failure. Cardiovasc Res 83: 737–746, 2009. doi: 10.1093/cvr/cvp160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang YM, Ma Y, Elks C, Zheng JP, Yang ZM, Francis J. Cross-talk between cytokines and renin-angiotensin in hypothalamic paraventricular nucleus in heart failure: role of nuclear factor-κB. Cardiovasc Res 79: 671–678, 2008. doi: 10.1093/cvr/cvn119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang YM, Wang Y, Yang LM, Elks C, Cardinale J, Yu XJ, Zhao XF, Zhang J, Zhang LH, Yang ZM, Francis J. TNF-α in hypothalamic paraventricular nucleus contributes to sympathoexcitation in heart failure by modulating AT1 receptor and neurotransmitters. Tohoku J Exp Med 222: 251–263, 2010. doi: 10.1620/tjem.222.251. [DOI] [PubMed] [Google Scholar]
- 35.Kang YM, Zhang ZH, Johnson RF, Yu Y, Beltz T, Johnson AK, Weiss RM, Felder RB. Novel effect of mineralocorticoid receptor antagonism to reduce proinflammatory cytokines and hypothalamic activation in rats with ischemia-induced heart failure. Circ Res 99: 758–766, 2006. doi: 10.1161/01.RES.0000244092.95152.86. [DOI] [PubMed] [Google Scholar]
- 36.Kang YM, Zhang ZH, Xue B, Weiss RM, Felder RB. Inhibition of brain proinflammatory cytokine synthesis reduces hypothalamic excitation in rats with ischemia-induced heart failure. Am J Physiol Heart Circ Physiol 295: H227–H236, 2008. doi: 10.1152/ajpheart.01157.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kapadia SR. Cytokines and heart failure. Cardiol Rev 7: 196–206, 1999. doi: 10.1097/00045415-199907000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Kawano H, Masuko S. Region-specific projections from the subfornical organ to the paraventricular hypothalamic nucleus in the rat. Neuroscience 169: 1227–1234, 2010. doi: 10.1016/j.neuroscience.2010.05.065. [DOI] [PubMed] [Google Scholar]
- 39.Kenney MJ, Ganta CK. Autonomic nervous system and immune system interactions. Compr Physiol 4: 1177–1200, 2014. doi: 10.1002/cphy.c130051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kobori H, Nishiyama A. Effects of tempol on renal angiotensinogen production in Dahl salt-sensitive rats. Biochem Biophys Res Commun 315: 746–750, 2004. doi: 10.1016/j.bbrc.2004.01.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, Zhou B, Liu J, Li F, Li Y, Kang X, Sun H, Wu S. Administration of progranulin (PGRN) triggers ER stress and impairs insulin sensitivity via PERK-eIF2α-dependent manner. Cell Cycle 14: 1893–1907, 2015. doi: 10.1080/15384101.2015.1041686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lindley TE, Infanger DW, Rishniw M, Zhou Y, Doobay MF, Sharma RV, Davisson RL. Scavenging superoxide selectively in mouse forebrain is associated with improved cardiac function and survival following myocardial infarction. Am J Physiol Regul Integr Comp Physiol 296: R1–R8, 2009. doi: 10.1152/ajpregu.00078.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods 25: 402–408, 2001. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 44.Llewellyn T, Zheng H, Liu X, Xu B, Patel KP. Median preoptic nucleus and subfornical organ drive renal sympathetic nerve activity via a glutamatergic mechanism within the paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol 302: R424–R432, 2012. doi: 10.1152/ajpregu.00403.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loewy AD. Descending pathways to the sympathetic preganglionic neurons. Prog Brain Res 57: 267–277, 1982. doi: 10.1016/S0079-6123(08)64133-3. [DOI] [PubMed] [Google Scholar]
- 46.Luyendyk JP, Lehman-McKeeman LD, Nelson DM, Bhaskaran VM, Reilly TP, Car BD, Cantor GH, Deng X, Maddox JF, Ganey PE, Roth RA. Coagulation-dependent gene expression and liver injury in rats given lipopolysaccharide with ranitidine but not with famotidine. J Pharmacol Exp Ther 317: 635–643, 2006. doi: 10.1124/jpet.105.096305. [DOI] [PubMed] [Google Scholar]
- 47.Mann DL. Stress-activated cytokines and the heart: from adaptation to maladaptation. Annu Rev Physiol 65: 81–101, 2003. doi: 10.1146/annurev.physiol.65.092101.142249. [DOI] [PubMed] [Google Scholar]
- 48.Mann DL. Targeted anticytokine therapy and the failing heart. Am J Cardiol 95: 9–16, 2005. doi: 10.1016/j.amjcard.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 49.McKinley MJ, McAllen RM, Davern P, Giles ME, Penschow J, Sunn N, Uschakov A, Oldfield BJ. The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol 172: III–XII, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol 292: C82–C97, 2007. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 51.Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med 208: 417–420, 2011. doi: 10.1084/jem.20110367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Osborn JW, Hendel MD, Collister JP, Ariza-Guzman PA, Fink GD. The role of the subfornical organ in angiotensin II-salt hypertension in the rat. Exp Physiol 97: 80–88, 2012. doi: 10.1113/expphysiol.2011.060491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel KP. Role of paraventricular nucleus in mediating sympathetic outflow in heart failure. Heart Fail Rev 5: 73–86, 2000. doi: 10.1023/A:1009850224802. [DOI] [PubMed] [Google Scholar]
- 54.Peeters AC, Netea MG, Janssen MC, Kullberg BJ, Van der Meer JW, Thien T. Pro-inflammatory cytokines in patients with essential hypertension. Eur J Clin Invest 31: 31–36, 2001. doi: 10.1046/j.1365-2362.2001.00743.x. [DOI] [PubMed] [Google Scholar]
- 55.Shi P, Diez-Freire C, Jun JY, Qi Y, Katovich MJ, Li Q, Sriramula S, Francis J, Sumners C, Raizada MK. Brain microglial cytokines in neurogenic hypertension. Hypertension 56: 297–303, 2010. doi: 10.1161/HYPERTENSIONAHA.110.150409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson JB, Routtenberg A. Subfornical organ: site of drinking elicitation by angiotensin II. Science 181: 1172–1175, 1973. doi: 10.1126/science.181.4105.1172. [DOI] [PubMed] [Google Scholar]
- 57.Sriramula S, Cardinale JP, Francis J. Inhibition of TNF in the brain reverses alterations in RAS components and attenuates angiotensin II-induced hypertension. PLoS One 8: e63847, 2013. doi: 10.1371/journal.pone.0063847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II-induced hypertension. Cardiovasc Res 92: 401–408, 2011. doi: 10.1093/cvr/cvr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-α in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension 51: 1345–1351, 2008. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stenman E, Edvinsson L. Cerebral ischemia enhances vascular angiotensin AT1 receptor-mediated contraction in rats. Stroke 35: 970–974, 2004. doi: 10.1161/01.STR.0000121642.53822.58. [DOI] [PubMed] [Google Scholar]
- 61.Tan J, Wang H, Leenen FH. Increases in brain and cardiac AT1 receptor and ACE densities after myocardial infarct in rats. Am J Physiol Heart Circ Physiol 286: H1665–H1671, 2004. doi: 10.1152/ajpheart.00858.2003. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka J, Saito H, Kaba H, Seto K. Subfornical organ neurons act to enhance the activity of paraventricular vasopressin neurons in response to intravenous angiotensin II. Neurosci Res 4: 424–427, 1987. doi: 10.1016/0168-0102(87)90008-3. [DOI] [PubMed] [Google Scholar]
- 63.Thunhorst RL, Beltz TG, Johnson AK. Effects of subfornical organ lesions on acutely induced thirst and salt appetite. Am J Physiol Regul Integr Comp Physiol 277: R56–R65, 1999. [DOI] [PubMed] [Google Scholar]
- 64.Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, Turlington JS, Thurber CJ, Christopher S, Dixon DL, Fronk DT, Thomas CS, Rose SW, Buckley LF, Dinarello CA, Biondi-Zoccai G, Abbate A. Interleukin-1 blockade in acute decompensated heart failure: a randomized, double-blinded, placebo-controlled pilot study. J Cardiovasc Pharmacol 67: 544–551, 2016. doi: 10.1097/FJC.0000000000000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Tassell BW, Buckley LF, Carbone S, Trankle CR, Canada JM, Dixon DL, Abouzaki N, Oddi-Erdle C, Biondi-Zoccai G, Arena R, Abbate A. Interleukin-1 blockade in heart failure with preserved ejection fraction: rationale and design of the diastolic heart failure anakinra response trial 2 (D-HART2). Clin Cardiol 40: 626–632, 2017. doi: 10.1002/clc.22719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Tassell BW, Raleigh JM, Abbate A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep 12: 33–41, 2015. doi: 10.1007/s11897-014-0231-7. [DOI] [PubMed] [Google Scholar]
- 67.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-α antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649, 2010. doi: 10.1161/HYPERTENSIONAHA.110.157685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Walker RJ, Steinle JJ. Role of β-adrenergic receptors in inflammatory marker expression in Müller cells. Invest Ophthalmol Vis Sci 48: 5276–5281, 2007. doi: 10.1167/iovs.07-0129. [DOI] [PubMed] [Google Scholar]
- 69.Wang HW, Huang BS, Chen A, Ahmad M, White RA, Leenen FH. Role of brain aldosterone and mineralocorticoid receptors in aldosterone-salt hypertension in rats. Neuroscience 314: 90–105, 2016. doi: 10.1016/j.neuroscience.2015.11.055. [DOI] [PubMed] [Google Scholar]
- 70.Wang L, Hiller H, Smith JA, de Kloet AD, Krause EG. Angiotensin type 1a receptors in the paraventricular nucleus of the hypothalamus control cardiovascular reactivity and anxiety-like behavior in male mice. Physiol Genomics 48: 667–676, 2016. doi: 10.1152/physiolgenomics.00029.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wei SG, Felder RB. Forebrain renin-angiotensin system has a tonic excitatory influence on renal sympathetic nerve activity. Am J Physiol Heart Circ Physiol 282: H890–H895, 2002. doi: 10.1152/ajpheart.2002.282.3.H890. [DOI] [PubMed] [Google Scholar]
- 72.Wei SG, Yu Y, Weiss RM, Felder RB. Endoplasmic reticulum stress increases brain MAPK signaling, inflammation and renin-angiotensin system activity and sympathetic nerve activity in heart failure. Am J Physiol Heart Circ Physiol 311: H871–H880, 2016. doi: 10.1152/ajpheart.00362.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei SG, Yu Y, Zhang ZH, Felder RB. Proinflammatory cytokines upregulate sympathoexcitatory mechanisms in the subfornical organ of the rat. Hypertension 65: 1126–1133, 2015. doi: 10.1161/HYPERTENSIONAHA.114.05112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wei SG, Yu Y, Zhang ZH, Weiss RM, Felder RB. Angiotensin II-triggered p44/42 mitogen-activated protein kinase mediates sympathetic excitation in heart failure rats. Hypertension 52: 342–350, 2008. doi: 10.1161/HYPERTENSIONAHA.108.110445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wei SG, Zhang ZH, Beltz TG, Yu Y, Johnson AK, Felder RB. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62: 118–125, 2013. doi: 10.1161/HYPERTENSIONAHA.113.01404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E, Zhang J, Peng Q, Duan W, Reeves SA, Cattaneo E, Krainc D. Peroxisome-proliferator-activated receptor-γ coactivator 1α contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci 31: 9544–9553, 2011. doi: 10.1523/JNEUROSCI.1291-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yalcin A. Quantification of thioredoxin mRNA expression in the rat hippocampus by real-time PCR following oxidative stress. Acta Biochim Pol 51: 1059–1065, 2004. [PubMed] [Google Scholar]
- 78.Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res 85: 462–472, 2007. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yu Y, Wei SG, Weiss RM, Felder RB. TNF-α receptor 1 knockdown in the subfornical organ ameliorates sympathetic excitation and cardiac hemodynamics in heart failure rats. Am J Physiol Heart Circ Physiol 313: H744–H756, 2017. doi: 10.1152/ajpheart.00280.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yu Y, Zhang ZH, Wei SG, Weiss RM, Felder RB. Peroxisome proliferator-activated receptor-γ regulates inflammation and renin-angiotensin system activity in the hypothalamic paraventricular nucleus and ameliorates peripheral manifestations of heart failure. Hypertension 59: 477–484, 2012. doi: 10.1161/HYPERTENSIONAHA.111.182345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95: 210–216, 2004. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 82.Zucker IH, Gao L. The regulation of sympathetic nerve activity by angiotensin II involves reactive oxygen species and MAPK. Circ Res 97: 737–739, 2005. doi: 10.1161/01.RES.0000188261.94569.1f. [DOI] [PubMed] [Google Scholar]