

Abstract
Protein tyrosine phosphatase 1B (PTP1B) is a negative regulator of leptin receptor signaling and may contribute to leptin resistance in diet-induced obesity. Although PTP1B inhibition has been suggested as a potential weight loss therapy, the role of specific neuronal PTP1B signaling in cardiovascular and metabolic regulation and the importance of sex differences in this regulation are still unclear. In this study, we investigated the impact of proopiomelanocortin (POMC) neuronal PTP1B deficiency in cardiometabolic regulation in male and female mice fed a high-fat diet (HFD). When compared with control mice (PTP1Bflox/flox), male and female mice deficient in POMC neuronal PTP1B (PTP1Bflox/flox/POMC-Cre) had attenuated body weight gain (males: −18%; females: −16%) and fat mass (males: −33%; female: −29%) in response to HFD. Glucose tolerance was improved by 40%, and liver lipid accumulation was reduced by 40% in PTP1B/POMC-Cre males but not in females. When compared with control mice, deficiency of POMC neuronal PTP1B did not alter mean arterial pressure (MAP) in male or female mice (males: 112 ± 1 vs. 112 ± 1 mmHg in controls; females: 106 ± 3 vs. 109 ± 3 mmHg in controls). Deficiency of POMC neuronal PTP1B also did not alter MAP response to acute stress in males or females compared with control mice (males: Δ32 ± 0 vs. Δ29 ± 4 mmHg; females: Δ22 ± 2 vs. Δ27 ± 4 mmHg). These data demonstrate that POMC-specific PTP1B deficiency improved glucose tolerance and attenuated diet-induced fatty liver only in male mice and attenuated weight gain in males and females but did not enhance the MAP and HR responses to a HFD or to acute stress.
Keywords: blood pressure, glucose, leptin, lipid, liver, obesity
INTRODUCTION
Protein tyrosine phosphatase 1B (PTP1B) is a nontransmembrane protein anchored to the cytosolic face of the endoplasmic reticulum (ER) (17). It serves as an enzyme with multiple functions, including inhibition of leptin and insulin signaling (31). Obesity and proinflammatory proteins such as nuclear factor-kB (NF-κB) and increased ER stress (27) have been reported to activate PTP1B. When activated, PTP1B translocates through the cytosol to dephosphorylate plasma membrane-bound Janus kinase 2 (JAK2), which is attached to the leptin receptor (LR) and initiates LR signaling. Therefore, this effect of PTP1B may negatively influence LR signaling.
PTP1B is found in many tissues, including skeletal muscle, adipose tissue, liver, and the brain (29, 31). PTP1B has been suggested as a potential weight loss and appetite-suppressing target due to the reductions in body weight (BW) and food intake observed in mice with whole body PTP1B deficiency (20). Some of these metabolic effects of PTP1B have been attributed to central nervous system (CNS) actions (5). Although whole body and total CNS deletion of PTP1B have been reported to have beneficial metabolic effects (3, 5, 20), Chiappini et al. (8) reported that ventromedial hypothalamic (VMH) deletion of Ptpn1, the gene encoding PTP1B expression, resulted in increased age-related weight gain in high-fat diet (HFD)-fed female mice due to reductions in spontaneous motor activity and energy expenditure. These observations suggest that PTP1B may have heterogeneous metabolic effects, depending on the neuronal population in which PTP1B is expressed.
Because increased PTP1B attenuates LR and insulin signaling, which in turn have been suggested to play a role in regulating sympathetic nervous system (SNS) activity and blood pressure (BP) in obesity (12, 23), there has also been interest in possible cardiovascular actions of PTP1B. Whole body PTP1B deficiency was reported to increase mean arterial pressure (MAP) in response to leptin infusion in mice fed a standard chow diet (4). However, it is not clear whether these effects on BP are due to CNS actions or to peripheral vascular effects. Thus, the potential role of neuronal-specific PTP1B in cardiovascular regulation and in modulating the chronic BP effects of CNS LR signaling are unclear, especially in conditions in which PTP1B may be activated, such as in diet-induced obesity. Also, the specific neuronal populations responsible for mediating chronic cardiometabolic effects of PTP1B in obesity are unknown.
Proopiomelanocortin (POMC) neurons located within the arcuate nucleus (ARC) of the hypothalamus and in the nucleus tractus solitarius (NTS) of the brain stem are thought to be important targets for the effects of leptin on sympathetic activity, BP, appetite, and glucose regulation (18, 19). We previously showed that POMC neuronal-specific LR deletion abolished the chronic effects of leptin to increase BP and to reduce insulin and glucose levels (11). POMC neurons, however, appear to play a lesser role in mediating leptin’s effects to reduce appetite and increase energy expenditure (11). Banno et al. (3) showed that POMC neuronal-specific PTP1B deletion caused only small reductions in body weight (BW) as well as improvements in glucose regulation in male mice fed a HFD. However, the role of POMC neuronal PTP1B in cardiovascular regulation in dietary-induced obesity is still unclear. Importantly, there have been no previous studies to our knowledge that have investigated potential sex differences in cardiometabolic regulation by PTP1B in POMC neurons.
Although deficiency of melanocortin 4 receptors (MC4R) is known to be associated with obesity and liver steatosis, whether POMC neurons regulate liver lipids independent of effects on overall adiposity is unclear. Also, there have been no previous studies, to our knowledge, that have investigated the possible role of POMC neuronal PTP1B signaling in protecting against liver steatosis.
The main goal of the current study was to test the hypothesis that POMC neuronal-specific PTP1B deficiency protects against the adverse metabolic effects of a chronic HFD, including weight gain, impaired glucose tolerance, and increased liver lipids while increasing BP and heart rate (HR). We also investigated potential sex differences in POMC neuronal PTP1B regulation of cardiovascular and metabolic function.
MATERIALS AND METHODS
All experimental protocols and procedures were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center, Jackson, MS. Mice were placed in a 12-h dark (6 PM to 6 AM) and light (6 AM to 6 PM) cycle and given free access to food and water throughout the study.
Animals.
Male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice were used in these studies. PTP1Bflox/flox/POMC-Cre mice were generated by crossing POMC-Cre mice that express Cre recombinase specifically in POMC neurons on a Friend Virus B (B6.FVB) background (generously provided by Dr. Joel Elmquist; University of Texas Southwestern Medical School, Dallas, TX) with PTP1Bflox/flox mice on a mixed 129Sv/J × C57BL/6 background (generously provided by Dr. Kendra Bence; Pfizer, Cambridge, MA). The PTP1Bflox/flox mice have LoxP sites inserted into the intronic sequence surrounding exons 6–8, which encode the PTP1B active site and surrounding parts of the catalytic domain. Therefore, crossing POMC-Cre mice with PTP1Bflox/flox mice led to the generation of mice with PTP1B deficiency only in POMC neurons. Homozygous PTP1Bflox/flox mice from our colony were used as controls. Specificity of Cre expression in POMC neurons and selective deletion of PTP1B in POMC neurons have been reported previously (2, 5). To visualize Cre recombinase expression in POMC neurons, we also bred in the tomato reporter gene using B6.Cg-Gt (Rosa)26Sor/J on a C57BL/6J background purchased from Jackson Laboratories in a subset of mice.
Body weight, body composition, and glucose tolerance analysis: control diet.
Control PTP1Bflox/flox (n = 15) and PTP1Bflox/flox/POMC-Cre (n = 9) mice were individually housed and fed a control diet (Harlan Teklad/ENVIGO, CA 170955, 4 kcal/g, 13% fat) starting at 6 wk of age and continuing until the experiments were completed at 29 wk of age. Body weights were measured twice/wk from 6 to 20 wk of age. Weekly changes in body composition were analyzed using magnetic resonance imaging (4in1 EchoMRI-900TM; Echo Medical System, Houston, TX). Glucose tolerance tests were completed at 20 wk of age (PTP1Bflox/flox, n = 15; and PTP1Bflox/flox/POMC-Cre, n = 6). Animals were euthanized at 29 wk of age for liver lipid analysis (PTP1Bflox/flox; n = 5) and PTP1Bflox/flox/POMC-Cre; n = 3).
Body weight and body composition analysis: high-fat diet.
Control male (n = 10) and female (n = 9) PTP1Bflox/flox and male (n = 7) and female (n = 7) PTP1Bflox/flox/POMC-Cre mice were individually housed and fed a HFD (TD-0881, 4.7 kcal/g, 45% fat; Harlan Teklad/ENVIGO) starting at 6 wk of age and continuing until the experiments were completed at 29 wk of age. Food intake and body weight were measured twice/wk from 6 to 20 wk of age. Weekly changes in body composition were analyzed using magnetic resonance imaging. Animals were euthanized at 29 wk of age for liver lipid analysis.
Food intake response to acute leptin injections.
In nonfasted, noninstrumented PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice (20 ± 2 wk of age) fed a HFD, leptin (5 µg/g) or saline vehicle (0.2 ml) was injected intraperitoneally at 5 PM, and food intake was measured 2, 4, 15, and 24 h later. Food intake response to saline injection was subtracted from food intake response after leptin injection and the difference plotted as change (Δ) in food intake over 24 h. Each animal served as its own control.
Immunohistochemistry.
To provide additional confirmation of selective deletion of PTP1B in POMC neurons, we used immunohistochemistry to detect expression of p-STAT3 in sections of the ARC from PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice. Mice were injected intraperitoneally with recombinant mouse leptin (5 mg/kg). After 45 min, mice were euthanized and perfused via a cannula inserted into the left ventricle with phosphate-buffered saline (PBS) containing phosphatase inhibitor (Roche); tissues were collected and kept overnight in formalin, after which the solution was switched to 30% sucrose, and tissues were kept overnight at 4°C. Frozen brain coronal sections, 30 μm thick, were cut and processed for immunofluorescence to verify the presence of p-STAT3 immunoreactivity. Sections were rinsed in PBS and then incubated in blocking solution (PBS, 0.3% Triton X-100) for 24 h and preincubated with 5% normal horse serum in PBS for 1 h at room temperature. After rinses with PBS, sections were incubated with rabbit anti-p-STAT3 (Cell Signaling) at a dilution 1:100 for 48 h at 4°C. After rinses (3 times) with PBS, sections were incubated with biotin-conjugated anti-rabbit IgG at a dilution of 1:100 for 1 h at room temperature. After rinses with PBS, sections were incubated with Avidin-conjugated DyLight 549 at a dilution of 1:200 for 1 h at room temperature in a dark environment. Sections were rinsed, mounted on slides, and examined in a fluorescence microscope at a 556-nm wavelength.
Oral glucose tolerance test.
d-Glucose (3 mg/kg lean body mass + 1 mg/kg fat mass) was administered by gavage after a 5-h fast in 20 ± 2-wk-old male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice fed a HFD. Blood samples were collected by tail snip, and blood glucose was measured using glucose strips (ReliOn) at baseline and 15, 30, 60, 90, and 120 min after glucose administration.
Liver composition.
Whole livers from male and female PTP1Bflox/flox mice and PTP1Bflox/flox/POMC-Cre fed a HFD were harvested and analyzed for fat and lean mass composition using EchoMRI.
We also performed Oil Red O staining in frozen liver sections from PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice to assess liver lipids. Sections (10 µm thick) were fixed in 10% buffered formalin for 5 min and stained for 10 min with 0.5% Oil Red O in 60% isopropyl alcohol. The slides were washed several times in water and counterstained in Mayer’s hematoxylin for 30 s and mounted in aqueous mounting media.
Liver triglyceride was analyzed from male and female PTP1Bflox/flox and male and female PTP1Bflox/flox/POMC-Cre mice using a colorimetric assay (BioVision K622, Milpitas, CA) according to the manufacturer’s instructions. Briefly, 100-mg liver samples were homogenized in a Douncer homogenizer in 5% NP-40. Samples were centrifuged, and the supernatant was isolated and diluted (1:1,000). Samples were incubated with lipase at room temperature for 20 min to convert triglyceride to glycerol. Reaction mix was then added, and the samples were incubated for a further 60 min at room temperature, protected from light. The samples were read at absorbance 540 nm in a microplate reader.
Measurement of blood pressure and heart rate.
At 20 ± 2 wk old, male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice fed a HFD were anesthetized with 2% isoflurane, and under sterile conditions, a telemetry probe (TA11PA-C10; Data Science) was implanted in the left carotid artery and advanced into the aorta. Seven to 10 days after mice recovered from surgery, MAP and HR were measured by telemetry 24 h/day for 4 consecutive days using computerized methods for data collection, as previously described (11, 14). Daily MAP and HR were obtained from the average of 12:12-h light-dark recording using a sampling rate of 500 Hz with a duration of 10 s every 10-min period.
Acute air jet stress test.
To determine whether deleting PTP1B in POMC neurons alters MAP and HR responses to acute stress, male and female PTP1Bflox/flox and male and female PTP1Bflox/flox/POMC-Cre mice fed a HFD implanted with BP telemeters were placed in special cages used for air jet stress testing, as previously described (11). Mice were allowed to acclimate to the cages for ≥2 h and monitored until BP was stable for ≥10 min. The air jet stress was then administered in 5-s pulses every 10 s for 5 min while BP and HR were measured continuously. Changes in BP and HR to acute stress were measured by subtracting the average baseline measurement from the average measurement recorded during acute stress.
Statistical analyses.
Data are expressed as means ± SE. Significant differences between two groups were determined by Student’s t-test. Significant differences between two groups over time were determined by two-way ANOVA where possible, followed by the Sidak’s multiple-comparisons test. Differences between groups over time were determined by t-test following linear regression analysis. A P value of <0.05 indicates a significant difference.
RESULTS
POMC neuronal-specific PTP1B deficiency.
PCR data demonstrated that PTP1Bflox/flox/POMC-Cre animals were homozygous for PTP1Bflox/flox and expressed Cre recombination (Fig. 1A). We also confirmed positive expression of Cre-recombinase within POMC neurons of homozygous PTP1Bflox/flox/POMC-Cre mice. Whole brain sections from a subset of PTP1Bflox/flox/POMC-Cre mice inbred for the tomato red reporter gene showed tomato red fluorescence as an indicator of Cre-recombinase expression in the arcuate nucleus (ARC; −2.18 mm from bregma) of the hypothalamus and the nucleus tractus solitarius (NTS; −6.72 mm from bregma), where POMC neurons are known to be located (Fig. 1B). Furthermore, we used immunohistochemistry to detect expression of p-STAT3, a major leptin signaling protein, in sections of the ARC (−2.18 mm from bregma) in PTP1Bflox/flox/POMC-Cre and PTP1Bflox/flox mice following acute intraperitoneal leptin injection (Fig. 1C). Deletion of PTP1B specifically in POMC neurons resulted in a markedly greater p-STAT3 staining compared with PTP1Bflox/flox mice.
Fig. 1.

Genotype confirmation using PCR and immunofluorescence analysis. A: protein tyrosine phosphatase 1B (PTP1B; +/+), heterozygotes (+/−), and proopiomelanocortin (POMC)-Cre positive mice. Positive (+) and negative (−) DNA samples. B: tomato reporter gene expression in POMC-Cre positive neurons of a homozygous PTP1Bflox/flox/POMC-Cre mouse in arcuate nucleus (ARC) and nucleus tractus solitarius (NTS). C: immunohistochemistry of p-STAT3 signaling in the ARC of a PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mouse injected intraperitoneally with leptin.
Body weight, body composition, and glucose tolerance test of PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice fed a control diet.
Combined male and female data in mice fed a normal diet from 6 wk of age demonstrate that, compared with control PTP1Bflox/flox mice, deletion of PTP1B specifically in POMC neurons (PTP1Bflox/flox/POMC-Cre) had no significant effect on body weight, fat mass, or lean mass (Fig. 2, A–C). Glucose tolerance was not significantly altered in PTP1Bflox/flox/POMC-Cre mice compared with controls (Fig. 2D). Liver lean mass was slightly reduced in PTP1Bflox/flox/POMC-Cre compared with controls, and this was balanced by a small increase in fat mass (mg/g tissue), although the differences were not statistically significant (Fig. 2, E and F).
Fig. 2.

Body composition, glucose tolerance, and liver lipid analysis of mice fed a normal control diet. Body weights were measured twice weekly in PTP1Bflox/flox (n = 15) and PTP1Bflox/flox/POMC-Cre (n = 9) mice. EchoMRI for lean and fat mass were conducted once per week for the duration of the study. A: body weight (g) from 6 to 20 wk of age. B: fat mass [%body weight (%BW)] from 6 to 20 wk of age. C: lean mass (%BW) from 6 to 20 wk of age. D: after a 5-h fast, glucose tolerance was measured in PTP1Bflox/flox (n = 15) and PTP1Bflox/flox/POMC-Cre (n = 6) over 120 min postglucose gavage. E and F: whole liver lean mass (mg/g liver tissue wt) and lipid accumulation (mg/g liver tissue wt) analysis in PTP1Bflox/flox (n = 5) and PTP1Bflox/flox/POMC-Cre (n = 3) mice assessed with EchoMRI. Data are expressed as means ± SE. There were no statistically significant changes as assessed by 2-way ANOVA with post hoc Sidak’s multiple comparison, t-test following linear regression analysis (A–D), or unpaired Student’s t-test (E and F).
Body weight and body composition of PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice fed a HFD.
Combined male and female data shown in Fig. 3A demonstrate that, compared with control PTP1Bflox/flox mice, PTP1Bflox/flox/POMC-Cre mice had significant attenuations in weight gain from 14 wk onward (P < 0.05), with an 18% body weight reduction in males (P < 0.05) and a 16% reduction in females at 20 wk of age (P < 0.01) (Fig. 3B). This was accounted for mainly by reduced fat mass (Fig. 3C); in male and female PTP1Bflox/flox/POMC-Cre mice, fat mass (g) was reduced by 33 (P < 0.05) and 29% (P < 0.05), respectively, at 20 wk of age compared with control PTP1Bflox/flox mice. Fat mass, as percent body weight (%BW) are presented in Fig. 3D. Total lean body mass (g) was not significantly higher in PTP1Bflox/flox/POMC-Cre mice compared with PTP1Bflox/flox mice (data not shown). Lean mass (expressed as %BW) was significantly higher in male PTP1Bflox/flox/POMC-Cre than in male PTP1Bflox/flox mice (P < 0.05), but the increase in females was not quite statistically significant (P = 0.053) (Fig. 3, E and F).
Fig. 3.

Body composition and plasma analysis of male (n = 8) and female (n = 7) PTP1Bflox/flox and male (n = 5) and female (n = 7) PTP1Bflox/flox/POMC-Cre mice during high-fat diet (HFD) feeding. Body weight and food intake were measured twice weekly. EchoMRIs for lean and fat mass were conducted once per week for the duration of the study. A: body weight (g) from 6 to 20 wk of age. B. body weight in male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice at 20 wk of age. C: total fat mass (g) as measured by EchoMRI (g). D: total lean mass (g) as measured by EchoMRI. E: total fat mass in male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice at 20 wk of age. F: average daily food intake (g). G: cumulative food intake from 10 to 14 wk (g). H: plasma leptin (ng/ml). I: plasma insulin (ng/ml). Data are expressed as means ± SE. *P < 0.05. A, C, D, and F: 2-way ANOVA with post hoc Sidak’s multiple comparison; t-test following linear regression analysis. B, E, G, H, and I: unpaired Student’s t-test.
Average daily food intakes for male and female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice were not significantly different from 6 to 20 wk of age (Fig. 3G). However, cumulative food intake over a 5-wk period (weeks 10–14 inclusive) was lower in PTP1Bflox/flox/POMC-Cre mice compared with PTP1Bflox/flox (P < 0.05; Fig. 3H). Fasting plasma leptin (Fig. 3I) and insulin (Fig. 3J) levels were also slightly lower in PTP1Bflox/flox/POMC-Cre mice compared with PTP1Bflox/flox mice at 20 wk of age, but the differences were not statistically significant in male or female mice.
Impact of POMC neuronal-specific PTP1B deficiency on food intake responses to leptin.
With the use of intraperitoneal saline injection as a baseline control, leptin injection resulted in similar 24-h reductions in food intake in PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre male and female mice fed a HFD (Fig. 4). There were no significant sex differences in the anorexic effect of acute leptin injections (data not shown).
Fig. 4.
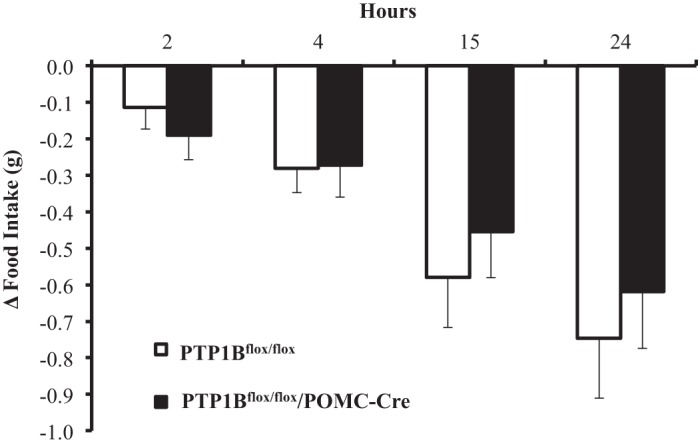
Food intake response to acute leptin administration. The change (Δ) in 24-h food intake after a saline injection subtracted from food intake response after leptin injection (5 mg/kg ip) in PTP1Bflox/flox (n = 15) and PTP1Bflox/flox/POMC-Cre (n = 11) mice. Data are expressed as means ± SE. There were no statistically significant changes as assessed by 2-way ANOVA with post hoc Sidak’s multiple comparison.
Impact of POMC neuronal-specific PTP1B deficiency on glucose tolerance.
Sex differences in the responses to a glucose tolerance test (GTT) were noted at 20 wk of age, and therefore, data for male and female mice were analyzed separately. Male PTP1Bflox/flox/POMC-Cre mice had significantly improved glucose tolerance compared with male PTP1Bflox/flox mice (P < 0.05), as evidenced by a 40% reduction in area under the curve (AUC) (P < 0.05) (Fig. 5, A and B). Female PTP1Bflox/flox control mice had substantially better glucose tolerance and lower AUC compared with male PTP1Bflox/flox mice. However, female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice had similar glucose tolerances, and there were no significant differences in AUC (Fig. 5, C and D).
Fig. 5.

Glucose tolerance measured over 120 min in PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice fed a HFD. Glucose tolerance in male (n = 10) and female (n = 7) PTP1Bflox/flox and male (n = 7) and female (n = 7) PTP1Bflox/flox/POMC-Cre mice measured after a 5-h fast. A: male PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre blood glucose measured over 120 min postglucose gavage. B: blood glucose area under the curve (AUC) for male PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre. C: female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre blood glucose measured over 120 min postglucose gavage. D: blood glucose AUC for female PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre. Data are expressed as means ± SE. *P < 0.05. A and C: 2-way ANOVA with post hoc Sidak’s multiple comparison; n = 7–10/group. B and D: unpaired Student’s t-test.
Impact of POMC neuronal-specific PTP1B deficiency on liver lipid accumulation.
Mice were euthanized at 29 ± 1 wk of age, and livers harvested from PTP1Bflox/flox/POMC-Cre mice weighed significantly less than livers from PTP1Bflox/flox mice (P < 0.05; Fig. 6A). However, when liver weight was normalized as percentage of total body weight, only male PTP1Bflox/flox/POMC-Cre mice livers were significantly protected from the effects of a HFD on fat accumulation compared with control PTP1Bflox/flox mice (P < 0.05; Fig. 6B). When compared with controls, only male PTP1Bflox/flox/POMC-Cre mice had significantly reduced liver fat accumulation, as measured by EchoMRI (P < 0.05; Fig. 6C). Lean liver mass was significantly increased in male PTP1Bflox/flox/POMC-Cre mice compared with male flox/flox controls (P < 0.05; Fig. 6D). There were no significant differences in liver lipid accumulation between female PTP1Bflox/flox mice and female PTP1Bflox/flox/POMC-Cre mice.
Fig. 6.

Whole liver lipid accumulation analysis in male (n = 10) and female (n = 9) PTP1Bflox/flox and male (n = 6) and female (n = 7) PTP1Bflox/flox/POMC-Cre mice fed a HFD. Liver fat and lean mass were assessed using EchoMRI. A: whole liver weight (g). B: liver weight as percentage of total body weight (%TBW). C: fat mass (mg/g liver wt). D: lean mass (mg/g liver wt). Data are expressed as means ± SE. *P < 0.05. A–D: unpaired Student’s t-test.
Liver sections from male PTP1Bflox/flox/POMC-Cre mice had reduced lipid content compared with PTP1Bflox/flox mice, as shown by a reduction in Oil Red O staining (representative images; Fig. 7A). Significant reductions in liver triacylglycerol were also observed only in male PTP1Bflox/flox/POMC-Cre mice compared with PTP1Bflox/flox mice (P < 0.05; Fig. 7B).
Fig. 7.

Liver triacylglycerol content in male (n = 6) and female (n = 4) PTP1Bflox/flox and male (n = 4) and female (n = 6) PTP1Bflox/flox/POMC-Cre mice fed a HFD. A: PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre liver sections stained for Oil Red O and visualized at ×200 magnification. The nucleus was counterstained with Mayer’s hematoxylin. B: PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre liver triacylglycerol content (mg/dl). Data are expressed as means ± SE. *P < 0.05. B: unpaired Student’s t-test.
Impact of POMC neuronal-specific PTP1B deficiency on blood pressure and heart rate.
When compared with control mice, deficiency of PTP1B specifically in POMC neurons did not significantly alter MAP in male or female HFD-fed mice. Therefore, the BP data for males and females were combined in Fig. 8A. There were also no significant differences observed in systolic or diastolic pressures in mice with POMC-specific PTP1B deficiency compared with control mice fed a chronic HFD (Fig. 8, B and C). HR was similar in PTP1Bflox/flox/POMC-Cre mice compared with PTP1Bflox/flox mice (Fig. 8D) fed a HFD.
Fig. 8.

Blood pressure (BP) and heart rate (HR) in PTP1Bflox/flox (n = 4) and PTP1Bflox/flox/POMC-Cre (n = 7) mice fed a HFD. BP and heart rate in PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice were measured for 12/12 h day/night for 4 consecutive days. A: mean arterial pressure (MAP). B: systolic BP. C: diastolic BP. D: HR. Data are expressed as means ± SE. *P < 0.05. A–D: unpaired Student’s t-test.
Impact of POMC neuronal-specific PTP1B deficiency on blood pressure and heart rate responses to acute stress.
Prestress resting measurements of MAP were not significantly different in PTP1Bflox/flox compared with PTP1Bflox/flox/POMC-Cre mice (Fig. 9A). In response to acute stress, MAP of PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice increased by 29 ± 4 and 32 ± 0 mmHg in males and 27 ± 4 and 22 ± 2 in females, respectively (Fig. 9B). At baseline, HR was not significantly different in PTP1Bflox/flox compared with PTP1Bflox/flox/POMC-Cre mice (Fig. 9C). Acute stress raised HR equally in PTP1Bflox/flox and PTP1Bflox/flox/POMC-Cre mice (Fig. 9D). Female PTP1Bflox/flox/POMC-Cre mice had an attenuated MAP response to acute stress compared with male PTP1Bflox/flox/POMC-Cre, as shown in Fig. 9B. No other sex differences were observed.
Fig. 9.

Mean arterial pressure (MAP) and heart rate (HR) responses to acute air jet stress in PTP1Bflox/flox (n = 8) and PTP1Bflox/flox/POMC-Cre (n = 8) mice fed a HFD. A: MAP (mmHg) at baseline and during acute stress. B: change in MAP from baseline in PTP1Bflox/flox (males, n = 3; females, n = 5) and PTP1Bflox/flox/POMC-Cre (males, n = 3; females, n = 5) during air jet stress. C: HR (beats/min) at baseline and during acute stress. D: change in HR from baseline in PTP1Bflox/flox (males, n = 3; females, n = 5) and PTP1Bflox/flox/POMC-Cre (males, n = 3; females, n = 5) during air jet stress. Data are expressed as means ± SE. *P < 0.05, paired Student’s t-test comparing MAP and HR values during air jet stress with prestress values; #P < 0.05, unpaired Student’s t-test comparing male vs. female changes in MAP and HR during air jet stress in PTP1Bflox/flox or PTP1Bflox/flox/POMC-Cre mice.
DISCUSSION
An important goal of this study was to test the hypothesis that POMC neuronal-specific PTP1B deficiency protects against the adverse metabolic effects of dietary-induced obesity, including glucose intolerance and liver steatosis, while exacerbating increases in BP and HR. We also tested whether there were sex differences in the cardiometabolic effects of POMC neuronal-specific deletion of PTP1B mice fed a HFD.
Our most important findings are that POMC neuron PTP1B-deficient (PTP1Bflox/flox/POMC-Cre) mice fed a chronic HFD had attenuated weight gain and decreased whole body fat accumulation without measurable decreases in daily food intake compared with control mice fed a HFD. We also found important sex differences in the effect of POMC neuron PTP1B deficiency on glucose tolerance and liver lipid accumulation in mice fed a HFD. Male PTP1Bflox/flox/POMC-Cre mice fed a HFD exhibited marked improvements in glucose tolerance and reduced liver lipid accumulation compared with male control mice fed a HFD. In contrast, POMC neuron PTP1B deficiency did not protect female mice from the detrimental effects of a HFD on glucose tolerance or lipid liver accumulation. There were no significant sex differences in any other metabolic parameter analyzed in these studies.
Another important, albeit surprising, finding of our study was that PTP1B deficiency in POMC neurons did not significantly enhance BP and HR responses to a HFD or to acute stress in male or female mice compared with control mice fed a HFD. These findings suggest that blockade of the actions of PTP1B in POMC neurons may offer beneficial metabolic effects in dietary-induced obesity, especially in male mice, without significantly raising BP.
Diet-induced obesity is associated with resistance to many of the metabolic effects of leptin, including its ability to suppress appetite, enhance glucose tolerance, and protect against lipid accumulation in various tissues such as the liver (25). However, the effect of leptin to enhance sympathetic nervous system (SNS) activity and, therefore, to increase BP and HR appears to be preserved, resulting in “selective” leptin resistance in obese subjects (24). Multiple mechanisms have been proposed to explain leptin resistance (16, 24, 26), but the factors contributing to selectivity of the effects of leptin on SNS activity, BP, food intake, glucose regulation, and liver lipid accumulation in obesity remain unclear.
Because many of the cardiometabolic responses to LR activation are initiated in the CNS, considerable effort has been focused on factors that may induce leptin resistance in the brain. As a negative regulator of LR signaling, PTP1B has been considered as a potential contributor to diet-induced leptin resistance as well as a modulator of the metabolic responses to other hormones such as insulin (9, 21). Global and CNS-specific PTP1B deficiency in mice fed a normal or HFD have been reported to reduce adiposity, increase energy expenditure, increase insulin sensitivity, and improve glucose tolerance (3, 20). However, VMH-specific PTP1B deficiency appears to increase rather than decrease weight gain and adiposity in female mice (8). These observations suggest that PTP1B may have different modes of action on body weight regulation and fat metabolism, depending on the neuronal population in which PTP1B is expressed, although sex differences could also be a potential contributing factor (8). To our knowledge, there have been no previous studies that have explored potential sex differences in the role of neuronal-specific PTP1B in regulating body weight, adiposity, glucose tolerance, and liver lipids.
Our results indicate that PTP1B deficiency specifically in POMC neurons only modestly attenuated weight gain in male and female mice fed a high-fat diet and that this was accounted for predominantly by reductions in fat mass. There was a slight but significant effect of POMC neuron PTP1B deficiency to reduce cumulative food intake but not on the acute effect of leptin injections to reduce food intake. Careful analysis of weekly food intake over the duration of the study revealed that on occasion PTP1Bflox/flox/POMC-Cre mice ate slightly less than PTP1Bflox/flox controls, which accounts for only a small part of the attenuated weight gain in mice with PTP1B deficiency in POMC neurons. Consistent with these data, multiple studies have reported that reductions in body weight of PTP1Bflox/flox/POMC-Cre mice fed a HFD may be due mainly to increased energy expenditure and reductions in feed efficiency rather than major reductions in food intake (3, 6, 10). Another study also noted the importance of sex differences in POMC neuronal regulation of body weight, energy expenditure, and obesity (7). Metabolic phenotyping data collected on the animals examined in this study suggest that PTP1Bflox/flox/POMC-Cre may have increased motor activity compared with controls (data not shown), supporting the results of Banno et al. (3).
In another study using male mice fed a normal diet, De Jonghe et al. (10) showed that deficiency of PTP1B in POMC neurons enhanced the effects of hindbrain (4th ventricle) administration of leptin to reduce food intake and body weight compared with control mice. Whether hindbrain-mediated appetite suppression occurs with physiological levels of leptin or whether this effect remains intact after chronic exposure to a HFD and obesity was not tested in these studies. Our current study demonstrated that the appetite-suppressing effects of physiological levels of systemically administered leptin, which better mimics the normal route of leptin access from the blood to the brain, were not enhanced by POMC neuronal-specific PTP1B deficiency in male or female mice fed a HFD.
Although male and female mice with POMC neuron PTP1B deficiency had reductions in body weight compared with controls, we found a sex difference in liver lipid accumulation. PTP1B deficiency in POMC neurons reduced liver lipids by 40% in male mice, as assessed by three different methods: Oil Red O staining, Echo-MRI, and biochemical measurement of triacylglycerol (TAG) in the liver. This large reduction in liver lipids was not apparent in female mice with PTP1B deficiency in POMC neurons compared with controls. Thus, PTP1B deficiency in POMC neurons appears to have an important sex-specific protective effect against liver steatosis in dietary-induced obesity. This finding suggests that PTP1B may play a major role in contributing to the development of fatty liver in obesity in males but may be of lesser importance in females. However, pair-feeding studies would be needed to completely rule out a potential effect of the small reduction in body weight and overall adiposity as a potential cause of reduced liver lipids.
Another important finding of the present study is that there were important sex differences in the effect of POMC neuronal PTP1B deficiency on glucose regulation. A chronic HFD often causes impaired glucose regulation associated with insulin resistance in the liver as well as in other tissues, such as skeletal muscle and fat (22, 30). In our study, male but not female PTP1Bflox/flox/POMC-Cre mice fed a chronic HFD had substantial improvements in glucose tolerance compared with control mice fed a HFD. This finding suggests an important role for POMC neuron PTP1B in development of HFD-induced glucose intolerance in males but not in females. These results complement those presented by Shi et al. (28), who demonstrated that LR deletion in POMC neurons resulted in glucose intolerance and insulin insensitivity in males, but not in females, compared with controls.
Fatty liver is a well-recognized cause of insulin resistance and impaired glucose regulation. Sex differences in glucose tolerance caused by POMC neuronal-specific PTP1B deficiency may, therefore, be related in part to differences in liver lipids since only males with PTP1B deficiency in POMC neurons were protected against liver steatosis. However, the mechanisms responsible for these sex differences in glucose regulation caused by POMC neuronal PTP1B deficiency are still unclear and warrant further investigation. Also, it is important to note that control female mice fed a HFD had considerably better glucose tolerance than males fed a HFD, warranting further investigation of these sex differences.
Our previous studies demonstrated that POMC neurons mediate most of the chronic effects of leptin to raise BP (11, 13, 18, 19). For example, LR deficiency specifically on POMC neurons completely abolished the rise in BP that occurred in control mice during 7 days of leptin infusion (13). Because PTP1B is a negative regulator of LR activation, we hypothesized that selective deficiency of PTP1B in POMC neurons would increase BP in obese mice fed a HFD. In contrast to our hypothesis, we did not observe any major differences in MAP or HR in mice with POMC-specific PTP1B deficiency compared with control mice. This was true for male as well as for female mice. Furthermore, PTP1B deficiency in POMC neurons did not enhance the HR and BP responses to an acute air jet stress in male or female mice.
These surprising results are difficult to explain if one assumes that PTP1B inactivation only enhances LR signaling since leptin-mediated activation of POMC neurons has been clearly demonstrated to stimulate SNS activity and raise BP (11, 13, 15, 18). A possible explanation for these findings is that PTP1B signaling in POMC neurons may modulate the effects of additional factors that either inactivate POMC neurons or attenuate the effects of leptin. Another possibility is that PTP1B does not substantially reduce activity of the downstream pathways associated with chronic SNS and BP effects of leptin in POMC neurons. Consistent with this possibility are the results of Bruder-Nascimento et al. (6), who reported that PTP1B deficiency in POMC neurons did not exacerbate the BP responses to chronic leptin infusion in male mice fed a normal chow diet. Although their results are not strictly comparable with our findings since we investigated the impact of PTP1B deficiency in obese mice fed a HFD, it is clear that PTP1B deficiency in POMC neurons does not raise BP in male or female mice on a normal or HFD. A potential limitation of these findings, however, is the possibility that reduced body weight associated with PTP1B deficiency in POMC neurons may partially offset a rise in BP despite enhanced leptin signaling, although we did not find any differences in day or night BP or BP responses to stress. Further experiments utilizing weight-matched mice may be useful in determining whether POMC-specific PTP1B deficiency may have effects on BP independent of changes in body weight.
Belin de Chantemèle et al. (4) previously reported that whole body deficiency of PTP1B in male mice increases MAP and amplifies the BP response to leptin infusion mainly by increasing sympathetic tone. However, PTP1B deficiency did not enhance the effects of a behavioral stress (cage switching) on BP. Additional studies have shown in rats that blockade of PTP1B within the NTS may be important for maintaining normal baroreflex sensitivity (1). Taken together, these results suggest that global deficiency of PTP1B may have multiple adverse cardiovascular effects, including increased BP and inhibition of baroreflex sensitivity, that do not appear to be mediated via POMC neurons. However, the mechanisms involved and importance of peripheral and CNS effects of PTP1B in chronic BP regulation await further investigation.
Perspectives and Significance
These data indicate that PTP1B deficiency in POMC neurons attenuates weight gain, adiposity, and liver lipid accumulation and improves glucose tolerance without significantly altering BP or HR in male mice fed a HFD. PTP1B deficiency in POMC neurons also attenuated weight gain and adiposity in female mice fed a HFD but did not protect against liver lipid accumulation or glucose intolerance. Therefore, our findings suggest that PTP1B in POMC neurons may exacerbate the adverse metabolic effects of obesity induced by a chronic HFD in male mice to a greater extent than females, although the mechanisms for these sex differences remain unknown. Taken together, these data indicate important sex differences in the regulation of glucose and liver lipid accumulation by PTP1B in POMC neurons.
Although our observations indicate that deficiency of PTP1B in POMC neurons does not increase BP, global PTP1B deficiency appears to cause hypertension via mechanisms that remain to be elucidated. The potential adverse cardiovascular effects of PTP1B blockade may limit development of this therapeutic approach for obesity and associated metabolic abnormalities such as liver steatosis, insulin resistance, and diabetes mellitus. Further studies are needed to determine whether novel therapeutic strategies can be developed to avoid deleterious cardiovascular effects while retaining beneficial metabolic actions of PTP1B blockade.
GRANTS
This work was supported by grants from the National Heart, Lung, and Blood Institute (P01-HL-51971) and the National Institute of General Medical Sciences (P20-GM-104357 and U54-GM-115428) of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.A., J.M.d.C., Z.W., and J.E.H. conceived and designed research; N.A., R.J.D., J.M.d.C., and L.E.M. performed experiments; N.A., R.J.D., L.E.M., and H.A.D. analyzed data; N.A., J.M.d.C., Z.W., H.A.D., and J.E.H. interpreted results of experiments; N.A., R.J.D., L.E.M., and H.A.D. prepared figures; N.A., R.J.D., J.M.d.C., Z.W., L.E.M., and J.E.H. drafted manuscript; N.A., R.J.D., J.M.d.C., Z.W., L.E.M., H.A.D., and J.E.H. edited and revised manuscript; N.A., R.J.D., J.M.d.C., Z.W., L.E.M., H.A.D., and J.E.H. approved final version of manuscript.
REFERENCES
- 1.Arnold AC, Nautiyal M, Diz DI. Protein phosphatase 1b in the solitary tract nucleus is necessary for normal baroreflex function. J Cardiovasc Pharmacol 59: 472–478, 2012. doi: 10.1097/FJC.0b013e31824ba490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC Jr, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42: 983–991, 2004. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J Clin Invest 120: 720–734, 2010. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belin de Chantemèle EJ, Muta K, Mintz J, Tremblay ML, Marrero MB, Fulton DJ, Stepp DW. Protein tyrosine phosphatase 1B, a major regulator of leptin-mediated control of cardiovascular function. Circulation 120: 753–763, 2009. doi: 10.1161/CIRCULATIONAHA.109.853077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12: 917–924, 2006. [Erratum in Nat Med 16: 237, 2010. 10.1038/nm0210-237a.] doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 6.Bruder-Nascimento T, Butler BR, Herren DJ, Brands MW, Bence KK, Belin de Chantemèle EJ. Deletion of protein tyrosine phosphatase 1b in proopiomelanocortin neurons reduces neurogenic control of blood pressure and protects mice from leptin- and sympatho-mediated hypertension. Pharmacol Res 102: 235–244, 2015. doi: 10.1016/j.phrs.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 7.Burke LK, Doslikova B, D’Agostino G, Greenwald-Yarnell M, Georgescu T, Chianese R, Martinez de Morentin PB, Ogunnowo-Bada E, Cansell C, Valencia-Torres L, Garfield AS, Apergis-Schoute J, Lam DD, Speakman JR, Rubinstein M, Low MJ, Rochford JJ, Myers MG, Evans ML, Heisler LK. Sex difference in physical activity, energy expenditure and obesity driven by a subpopulation of hypothalamic POMC neurons. Mol Metab 5: 245–252, 2016. doi: 10.1016/j.molmet.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiappini F, Catalano KJ, Lee J, Peroni OD, Lynch J, Dhaneshwar AS, Wellenstein K, Sontheimer A, Neel BG, Kahn BB. Ventromedial hypothalamus-specific Ptpn1 deletion exacerbates diet-induced obesity in female mice. J Clin Invest 124: 3781–3792, 2014. doi: 10.1172/JCI68585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dadke S, Kusari J, Chernoff J. Down-regulation of insulin signaling by protein-tyrosine phosphatase 1B is mediated by an N-terminal binding region. J Biol Chem 275: 23642–23647, 2000. doi: 10.1074/jbc.M001063200. [DOI] [PubMed] [Google Scholar]
- 10.De Jonghe BC, Hayes MR, Zimmer DJ, Kanoski SE, Grill HJ, Bence KK. Food intake reductions and increases in energetic responses by hindbrain leptin and melanotan II are enhanced in mice with POMC-specific PTP1B deficiency. Am J Physiol Endocrinol Metab 303: E644–E651, 2012. doi: 10.1152/ajpendo.00009.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension 57: 918–926, 2011. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.do Carmo JM, da Silva AA, Dubinion J, Sessums PO, Ebaady SH, Wang Z, Hall JE. Control of metabolic and cardiovascular function by the leptin-brain melanocortin pathway. IUBMB Life 65: 692–698, 2013. doi: 10.1002/iub.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.do Carmo JM, da Silva AA, Ebaady SE, Sessums PO, Abraham RS, Elmquist JK, Lowell BB, Hall JE. Shp2 signaling in POMC neurons is important for leptin’s actions on blood pressure, energy balance, and glucose regulation. Am J Physiol Regul Integr Comp Physiol 307: R1438–R1447, 2014. doi: 10.1152/ajpregu.00131.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.do Carmo JM, da Silva AA, Sessums PO, Ebaady SH, Pace BR, Rushing JS, Davis MT, Hall JE. Role of Shp2 in forebrain neurons in regulating metabolic and cardiovascular functions and responses to leptin. Int J Obes 38: 775–783, 2014. doi: 10.1038/ijo.2013.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dubinion JH, do Carmo JM, Adi A, Hamza S, da Silva AA, Hall JE. Role of proopiomelanocortin neuron Stat3 in regulating arterial pressure and mediating the chronic effects of leptin. Hypertension 61: 1066–1074, 2013. doi: 10.1161/HYPERTENSIONAHA.111.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105: 1827–1832, 2000. doi: 10.1172/JCI9842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 68: 545–560, 1992. doi: 10.1016/0092-8674(92)90190-N. [DOI] [PubMed] [Google Scholar]
- 18.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, Smith G, Stec DE. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem 285: 17271–17276, 2010. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res 116: 991–1006, 2015. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 20: 5479–5489, 2000. doi: 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koren S, Fantus IG. Inhibition of the protein tyrosine phosphatase PTP1B: potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract Res Clin Endocrinol Metab 21: 621–640, 2007. doi: 10.1016/j.beem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Kraegen EW, Clark PW, Jenkins AB, Daley EA, Chisholm DJ, Storlien LH. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 40: 1397–1403, 1991. doi: 10.2337/diab.40.11.1397. [DOI] [PubMed] [Google Scholar]
- 23.Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, Sowers J. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension. J Clin Hypertens (Greenwich) 15: 14–33, 2013. doi: 10.1111/jch.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mark AL. Selective leptin resistance revisited. Am J Physiol Regul Integr Comp Physiol 305: R566–R581, 2013. doi: 10.1152/ajpregu.00180.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Myers MG Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, Tschöp MH, Yanovski JA. Challenges and opportunities of defining clinical leptin resistance. Cell Metab 15: 150–156, 2012. doi: 10.1016/j.cmet.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Myers MG Jr, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab 21: 643–651, 2010. doi: 10.1016/j.tem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Panzhinskiy E, Ren J, Nair S. Protein tyrosine phosphatase 1B and insulin resistance: role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS One 8: e77228, 2013. doi: 10.1371/journal.pone.0077228. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Shi H, Sorrell JE, Clegg DJ, Woods SC, Seeley RJ. The roles of leptin receptors on POMC neurons in the regulation of sex-specific energy homeostasis. Physiol Behav 100: 165–172, 2010. doi: 10.1016/j.physbeh.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song GJ, Jung M, Kim J-H, Park H, Rahman MH, Zhang S, Zhang Z-Y, Park DH, Kook H, Lee I-K, Suk K. A novel role for protein tyrosine phosphatase 1B as a positive regulator of neuroinflammation. J Neuroinflammation 13: 86, 2016. doi: 10.1186/s12974-016-0545-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamauchi T, Kamon J, Waki H, Terauchi Y, Kubota N, Hara K, Mori Y, Ide T, Murakami K, Tsuboyama-Kasaoka N, Ezaki O, Akanuma Y, Gavrilova O, Vinson C, Reitman ML, Kagechika H, Shudo K, Yoda M, Nakano Y, Tobe K, Nagai R, Kimura S, Tomita M, Froguel P, Kadowaki T. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 7: 941–946, 2001. doi: 10.1038/90984. [DOI] [PubMed] [Google Scholar]
- 31.Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem 283: 14230–14241, 2008. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]