

Abstract
ANG II-salt hypertension selectively increases splanchnic sympathetic nerve activity (sSNA), but the extent to which this reflects increased respiratory versus cardiac rhythmic bursting is unknown. Here, integrated sSNA was elevated in ANG II-infused rats fed a high-salt (2% NaCl) diet (ANG II-HSD) compared with vehicle-infused rats fed a normal-salt (0.4% NaCl) diet (Veh-NSD; P < 0.01). Increased sSNA was not accompanied by increased inspiratory or expiratory bursting, consistent with no group difference in central inspiratory drive. Consistent with preserved inhibitory baroreflex entrainment of elevated sSNA in ANG II-HSD rats, the time integral (P < 0.05) and amplitude (P < 0.01) of cardiac rhythmic sSNA were increased. Consistent with activity of hypothalamic paraventricular nucleus (PVN) neurons supporting basal SNA in ANG II-salt hypertension, inhibition of PVN with the GABA-A receptor agonist muscimol reduced mean arterial pressure (MAP) and integrated sSNA only in the ANG II-HSD group (P < 0.001). PVN inhibition had no effect on respiratory rhythmic sSNA bursting in either group but reduced cardiac rhythmic sSNA in ANG II-HSD rats only (P < 0.01). The latter likely reflected reduced inhibitory baroreflex entrainment subsequent to the fall of MAP. Of note is that MAP as well as integrated and rhythmic burst patterns of sSNA were similar in vehicle-infused rats whether they were fed a normal or high-salt diet. Findings indicate that PVN neurons support elevated sSNA in ANG II-HSD rats by driving a tonic component of activity without altering respiratory or cardiac rhythmic bursting. Because sSNA was unchanged in Veh-HSD rats, activation of PVN-driven tonic sSNA appears to require central actions of ANG II.
NEW & NOTEWORTHY ANG II-salt hypertension is strongly neurogenic and depends on hypothalamic paraventricular nucleus (PVN)-driven splanchnic sympathetic nerve activity (sSNA). Here, respiratory and cardiac bursts of sSNA were preserved in ANG II-salt rats and unaltered by PVN inhibition, suggesting that PVN neurons drive a tonic component of sSNA rather than modulating dominant patterns of burst discharge.
Keywords: arterial baroreceptor reflex, high blood pressure, phrenic nerve activity, respiratory sympathetic coupling
INTRODUCTION
Systemic infusion of the peptide hormone ANG II leads to sustained hypertension (HTN), the magnitude of which increases in direct proportion to the amount of NaCl consumed in the diet. Studies in rats have indicated that this ANG II-dependent, salt-sensitive model of HTN (ANG II-salt HTN) depends on a regionally specific and persistent increase of splanchnic sympathetic nerve activity (sSNA) (29, 57), which is consistent with evidence that rats with ANG II-salt HTN exhibit 1) an exaggerated fall of mean arterial pressure (MAP) during ganglionic blockade (27, 30, 31), 2) increased splanchnic norepinephrine (NE) spillover (28), and 3) an attenuated rise of MAP after surgical interruption of sSNA by celiac ganglionectomy (29).
Of special importance for the present study is that splanchnic nerve action potentials, like those of most sympathetic nerves, occur mainly in bursts synchronized with the respiratory and cardiac cycles (3, 15, 16, 39). Respiratory rhythmic bursts of sympathetic nerve activity (SNA) result from excitatory and inhibitory synaptic inputs mainly to neurons of the rostral ventrolateral medulla (RVLM) from brain stem and hypothalamic elements of the extended respiratory network (19, 34, 35). Cardiac rhythmic SNA oscillations dominantly reflect periodic entrainment of RVLM vasomotor neurons by pulse synchronous inhibitory synaptic input to RVLM neurons from arterial baroreceptors (15, 49). Blunted arterial baroreflex inhibition of SNA in HTN has been documented in numerous animal models (8, 9, 11, 20) as well as in humans (42, 51). To the extent that baroreflex function is attenuated in rats with ANG II-salt HTN, we hypothesized that the cardiac rhythmic oscillation amplitude of sSNA would be blunted relative to normotensive controls.
There is now considerable evidence that enhanced respiratory rhythmic SNA bursting contributes to other models of neurogenic HTN. Enhanced respiratory rhythmic bursting contributes to HTN in spontaneously hypertensive rats (14, 32, 47) and rats exposed to chronic intermittent hypoxia (37, 58, 59). A similar pattern has been reported in hypertensive humans (32). We previously reported that the activity of neurons in the hypothalamic paraventricular nucleus (PVN) is required to maintain sSNA in ANG II-salt HTN (7). Here, we sought to determine the impact of PVN inhibition on respiratory and cardiac rhythmic bursting of sSNA in ANG II-salt HTN. To accomplish this, averages of respiratory and cardiac rhythmic sSNA were constructed using the onset of bursts of phrenic nerve activity (PNA) and the occurrence of ECG R-waves as (time 0) trigger events. We determined that inhibition of PVN neurons in the ANG II-HSD group significantly reduced ongoing integrated sSNA and MAP without significantly reducing either respiratory or cardiac rhythmic sSNA bursting. Our findings indicate that PVN neuronal activity in the ANG II-HSD group supported a tonic component of sSNA that was largely absent from vehicle (Veh)-treated rats regardless of whether they consumed a normal-salt diet (NSD) or a high-salt diet (HSD).
METHODS
Animals.
Adult male Sprague-Dawley rats (250–400 g, Charles River Laboratories, Wilmington, MA) were housed in a temperature-controlled room (22–23°C) with a 14:10-h light-dark cycle (lights on at 0700 hours). Of three experimental groups studied, one group was fed a HSD (2% NaCl, Research Diets, New Brunswick, NJ) and made hypertensive by systemic infusion of ANG II (Sigma, St. Louis, MO). Of the two remaining groups, both groups were normotensive and received systemic infusion of Veh (normal saline) with one group consuming a NSD (0.4% NaCl; Veh-NSD) and the other consuming the HSD (2% NaCl; Veh-HSD). Experimental and surgical procedures complied with National Institutes of Health and ARRIVE guidelines and were approved by the University of Texas Health at San Antonio Institutional Animal Care and Use Committee.
Treatment groups.
After 1 wk of consuming the NSD or HSD, rats were anesthetized with isoflurane (3% in O2) and surgically instrumented with telemetry transmitters (model PA-C40, Data Sciences, St. Paul, MN) for conscious, unrestrained recording of arterial blood pressure (ABP) and heart rate (HR), as previously described (7, 44, 45). After 5–7 days of recovery, baseline values of MAP and HR were recorded for 5 days. Osmotic minipumps (2ML2, ALZET) were then implanted subcutaneously. In the ANG II-HSD group, pumps delivered ANG II at a rate of 150 ng·kg−1·min−1 for 14 consecutive days (7). Control rats received minipumps filled with normal saline Veh. Rats in all groups had continuous ad libitum access to tap water.
Acute experiments.
Rats of each treatment group (Veh-NSD, ANG II-HSD, and Veh-HSD) were anesthetized by an intraperitoneal injection of α-chloralose (80 mg/kg) and urethane (800 mg/kg, Sigma-Aldrich), and catheters (PE-50 tubing) were implanted in the left femoral artery and both femoral veins for recording ABP and administering drugs, respectively. The trachea was cannulated, and rats were mechanically ventilated with oxygen-enriched room air. Rats were then paralyzed by infusion of gallamine triethiodide (25 mg·kg−1·h−1 iv), and end-tidal CO2 was maintained at ~5.5% by adjusting ventilation rate (85–95 breaths/min) and/or tidal volume (2–3 ml). Vagus nerves were transected to prevent respiratory network entrainment to the ventilator by rhythmic activation of pulmonary stretch receptors (38). Aortic depressor nerves were left intact. HR was obtained from a lead I ECG. Rats were placed in a stereotaxic frame, and body temperature was maintained at 37 ± 1°C with a ventrally located water-circulating pad. Supplements of anesthetic (10% of the initial dose) were given when needed, as assessed by limb withdrawal to noxious foot pinch before paralysis and by pressor responses to foot pinch thereafter.
Phrenic and sympathetic nerve recording.
PNA and sSNA were recorded as previously described (14, 21, 32). Briefly, the left phrenic nerve was isolated near the brachial plexus and transected. The left greater splanchnic nerve was isolated proximal to the adrenal gland. The central cut end of each nerve bundle was placed on a bipolar electrode and insulated from body fluid by coating with a silicon-based gel. Amplified signals were filtered (30–1,000 Hz), full-wave rectified, series resistor-capacitor (RC) integrated (time constant = 10 ms for PNA and 5 s for sSNA), and digitized at 1.5 kHz.
Inhibition of the PVN.
Neuronal activity in the PVN was inhibited bilaterally by local injection of the long-acting GABA-A receptor agonist muscimol (100 pmol in 50 nl/side, Sigma), as previously described (7, 45). Briefly, rats were mounted in a stereotaxic device with the skull leveled between the bregma and lambda, and a craniotomy was performed to access the PVN. Using a pneumatic pump (World Precision Instruments, Sarasota, FL) connected to a single-barreled glass micropipette (tip: ~50-μm outer diameter), muscimol was injected bilaterally into the PVN at the following coordinates with respect to the bregma: posterior: 1.8 to 2.1 mm, lateral: ±0.2 to 0.3 mm, and ventral: 7.5 mm from the dura. Each injection was completed over a period of 20–30 s, first on one side of PVN and then on the other. Injections were separated by 2–3 min, and sites were marked by including 0.2% rhodamine beads in the injected solution.
Histology.
At the end of the experiments, rats received an overdose of α-chloralose-urethane anesthetic. Brains were removed, postfixed in 4% paraformaldehyde for 24–48 h, cryoprotected in 30% sucrose in 0.1 M PBS, and sectioned at 50 μm with a freezing microtome. Injection sites were identified by the distribution of fluorescent beads, as previously described (19, 29). Images from similar rostral-caudal levels of the PVN from all subjects within each treatment group were overlaid, such that the outermost distribution of beads in the overlaid image revealed the full range of injection sites within each group.
Data analysis.
Values of MAP, sSNA, and PNA were determined from 5-min segments of stable data recorded just before and 30 min after PVN injections. Values of sSNA were expressed in microvolts after subtracting the voltage due to noise, which was determined as the average voltage during a 3-min data segment recorded 5 min after administration of the ganglionic blocker hexamethonium (30 mg/kg iv). MAP was calculated as diastolic pressure + [(systolic pressure – diastolic pressure) × 3−1].
To quantify respiratory rhythmic bursting of sSNA, averages were constructed using the onset of 150 consecutive phrenic nerve bursts as (time 0) trigger events before and 30 min after PVN injections of muscimol. Each PNA-triggered sSNA average consisted of a 0.3-s pretrigger and 1.6-s posttrigger period, with the latter being equal to the average duration of the respiratory cycle, which was taken as the average PNA burst interval. Respiratory rhythmic sSNA oscillation amplitudes were defined as the difference between the mean voltage of the triggered average and the voltage value of each posttrigger peak or trough (see the inset in Fig. 2A, left). Amplitudes were expressed in microvolts. Area under the curve (AUC) of each peak or trough was calculated by multiplying its mean amplitude by its duration. AUC was expressed in units of microvolts·seconds.
Fig. 2.

Effect of graded salt intake and systemic infusion of ANG II on respiratory rhythmic splanchnic sympathetic nerve activity (sSNA). A: representative phrenic nerve activity (PNA) burst-triggered averages of sSNA from 1) a vehicle (Veh)-infused rat that consumed a normal-salt (0.4% NaCl) diet (Veh-NSD; left), 2) a Veh-infused rat that consumed a high-salt (2% NaCl) diet (Veh-HSD; middle) and 3) an ANG II-infused rat that consumed a high-salt (2% NaCl) diet (ANG II-HSD; right). The timing of neural inspiration is indicated by the PNA burst-triggered average of integrated PNA shown in each graph (gray dashed lines). The inset in A, left, shows analyzed parameters of respiration-modulated sSNA. A prominent inspiratory peak (IP) of sSNA occurred nearly synchronous with the PNA burst in each example. The IP was followed by a variably sized expiratory trough (ET) and expiratory peak (EP). B: summary data (n = 6/group) revealed that the mean voltage of sSNA was significantly greater in ANG II-HSD rats (black bar) compared with Veh-NSD (open bar) or Veh-HSD (gray bar) rats. In contrast, the amplitude (top right) and area under the curve (AUC; bottom left) of the IP, ET, and EP were similar across groups. Rhythmicity index values (bottom right) indicate that respiratory modulation of sSNA across the entire respiratory cycle (Total) was similar across groups. The IP was the major contributor to total respiration-modulated sSNA in each group, but the contribution of IP, ET, or EP to total respiratory modulation was not different between groups. Triggered sSNA averages were constructed from 150 consecutive PNA bursts. Summary data are means ± SE. *P < 0.05 vs. Veh-NSD and Veh-HSD groups.
Changes in central respiratory drive in response to PVN muscimol were quantified from PNA burst-triggered averages of integrated PNA. The same 150 triggers used for constructing PNA burst-triggered averages of sSNA were used. Neural inspiration and expiration were defined as the duration of the PNA burst and interburst interval, respectively. PNA burst amplitudes were quantified as the difference between peak inspiratory phase and end-expiratory phase voltage. PNA burst amplitude was expressed in microvolts. The average AUC of PNA bursts was also calculated, and expressed in units of microvolt·seconds. PNA burst frequency was derived from the mean frequency of phrenic burst trigger events and expressed as bursts per minute.
To quantify cardiac rhythmic bursting of sSNA, triggered averages were constructed from ~1600 ECG R waves concurrently recorded with the sSNA used for the construction of PNA burst-triggered averages. Each R wave-triggered average consisted of a 0.3-s posttrigger period (>2 cardiac cycles). The amplitude of cardiac rhythmic sSNA, an index of the strength of inhibitory baroreflex entrainment, was calculated as the voltage difference between the peak and trough of the oscillation (Fig. 2A, left). Amplitudes were expressed in microvolts. The average AUC was calculated across one cardiac cycle period and expressed in microvolts·seconds.
Calculating respiratory and cardiac rhythmicity index values.
The overall level of respiratory or cardiac rhythmicity of sSNA was quantified from PNA burst- and R wave-triggered averages by calculating a rhythmicity index, as previously described (23). Briefly, respiratory rhythmicity was calculated from sSNA signals from which the direct current offset was zeroed before full-wave rectification and RC integrations. First, the AUC of the inspiratory peak (IP), expiratory (postinspiratory) trough (ET), and expiratory peak (EP) were each determined and expressed as a ratio to the total AUC. The total AUC was determined as the product of the mean voltage and duration of the post-trigger period (~1.6 s). Cardiac rhythmicity was calculated from R wave-triggered averages of the same sSNA data. The AUC of signal deflections above and below the mean voltage were summed and expressed as a ratio of the total AUC (mean voltage × cardiac cycle duration). A respiratory or cardiac rhythmicity index value of 100% indicates that all recorded sSNA was rhythmic with respect to the trigger event (PNA burst or ECG R wave). Likewise, an index value of 0% indicates that no component of recorded sSNA was rhythmic with respect to the trigger.
Statistics.
Baseline sSNA, MAP, PNA (amplitude, AUC, and frequency), end-tidal CO2, hematocrit, osmotic pressure, and plasma protein concentration were each compared across Veh-NSD, Veh-HSD, and ANG II-HSD groups with one-way ANOVA. The same analysis was used to compare the amplitude and AUC of R wave- and PNA burst-triggered sSNA across groups. Across-group comparison of effects of PVN-injected muscimol on sSNA, MAP, HR, PNA bursts (amplitude, AUC, and frequency), neural inspiratory and expiratory duration, and rhythmic sSNA burst amplitudes were analyzed by two-way ANOVA. When significant F values were obtained, Bonferroni post hoc tests were used to assign significance to pairwise comparisons. Statistical tests were performed using Prism software (version 7.0, GraphPad). In all cases, a critical value of P < 0.05 was considered statistically significant. Data in the text and figures are expressed as means ± SE.
RESULTS
Baseline hemodynamics and SNA.
As previously reported (7, 52), MAP measured before vehicle or ANG II infusion was not different in conscious rats regardless of whether they consumed a NSD (n = 5) or HSD (n = 7 or 8; Fig. 1). By day 14 of the infusion protocol, MAP of vehicle-infused rats remained normotensive regardless of whether they were on a NSD or HSD. Compared with baseline and daily values in Veh-infused rats, MAP increased significantly by day 2 of ANG II infusion in rats that consumed a HSD and remained elevated throughout the remainder of the 14-day infusion protocol (P < 0.05 to P < 0.001). Table 1 shows that the elevated MAP in the ANG II-HSD group was not fully maintained under anesthesia, although resting sSNA was elevated compared with Veh-infused groups (P < 0.05 to P < 0.01). Compared with the Veh-NSD group, HR was elevated under anesthesia in the ANG II-HSD group (P < 0.05) but was not different between Veh-infused rats that consumed the HSD versus the NSD. Resting tachycardia was present only when ANG II-HSD and Veh-NSD groups were compared and only while under anesthesia. The latter likely reflects greater baroreceptor unloading due to the greater fall of MAP induced by anesthesia in the ANG II-HSD group.
Fig. 1.
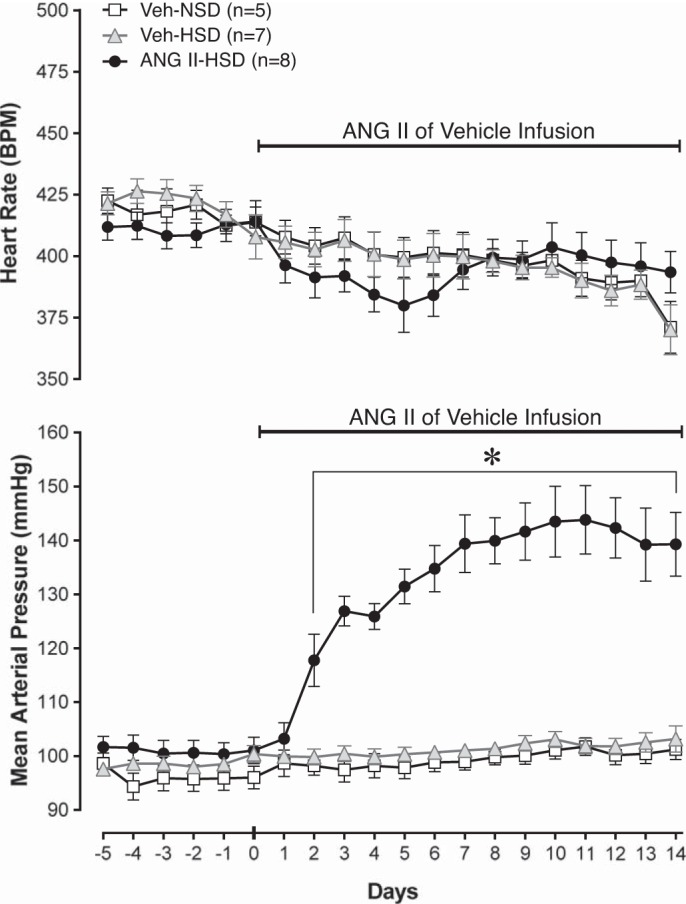
Effects of graded salt intake and systemic ANG II on heart rate (HR) and mean arterial pressure (MAP). Summary data of HR and MAP over 5 days of baseline and 14 days of subcutaneous infusion of ANG II or vehicle (Veh; normal saline) are shown. Baseline MAP and HR were similar across groups that consumed a normal salt (0.4% NaCl) diet (NSD) and a high-salt (2% NaCl) diet (HSD). Within ~2 days of ANG II infusion (150 ng·kg−1·min−1 sc), MAP increased significantly and remained stably elevated thereafter. ANG II infusion reduced HR similarly in all groups, but the effects were not statistically significant. *P < 0.05 vs. baseline and vs. Veh-NSD and Veh-HSD groups.
Table 1.
Baseline hemodynamics and splanchnic sympathetic nerve activity
| Group | n | Mean Arterial Pressure, mmHg | Heart Rate, beats/min | Splanchnic Sympathetic Nerve Activity, µV |
|---|---|---|---|---|
| Veh-NSD | 6 | 100 ± 4 | 415 ± 14 | 4.3 ± 0.8 |
| Veh-HSD | 6 | 99 ± 5 | 450 ± 14 | 6.9 ± 1.8 |
| ANG II-HSD | 6 | 106 ± 5 | 486 ± 14* | 11.6 ± 1.0*† |
Values are expressed as means ± SE; n, number of rats. Veh, vehicle; NSD, normal-salt diet; HSD, high-salt diet.
P < 0.01 vs. NSD;
P < 0.05 vs. HSD.
Effects on respiration and respiratory rhythmic sSNA.
Table 2 shows that values of end-expiratory CO2, duration of neural inspiration and expiration as well as PNA burst frequency, amplitude, and AUC were all similar at baseline across treatment groups (n = 6/group). Thus, central respiratory drive was unaffected by consumption of a HSD alone or by prior induction of HTN in the ANG II-HSD group. Representative PNA-burst triggered averages of sSNA recorded from a Veh-NSD rat (Fig. 2A, left), Veh-HSD rat (Fig. 2A, middle), and ANG II-HSD rat (Fig. 2A, right) are shown; data were analyzed according to Fig. 2A, inset. Note that the strength of neural inspiration in each rat is shown as the simultaneously recorded PNA burst-triggered average of integrated PNA (dashed traces in Fig. 2A). As we have previously reported (21–23), each sSNA average featured a prominent IP followed by a variably sized ET and EP. Figure 2B, top left, shows that the mean voltage of sSNA in the ANG II-HSD group was elevated compared with Veh-infused rats regardless of which NaCl diet they consumed. Figure 2B shows that in contrast to the greater mean sSNA in ANG II-HSD rats (Fig. 2B, top left; see also Table 1), neither the amplitude (Fig. 2B, top right) nor AUC (Fig. 2B, bottom left) of the IP, ET, or EP of sSNA differed across treatment groups. The latter indicates that treatments did not affect the duration of sSNA bursts during the IP (Veh-NSD: 0.34 ± 0.02 s, Veh-HSD: 0.30 ± 0.03 s, and ANG II-HSD: 0.30 ± 0.02 s), ET (Veh-NSD: 0.43 ± 0.07 s, Veh-HSD: 0.21 ± 0.03 s, and ANG II-HSD: 0.23 ± 0.06 s), or EP (Veh-NSD: 0.48 ± 0.05 s, Veh-HSD: 0.44 ± 0.02 s, and ANG II-HSD: 0.40 ± 0.05 s). Collectively, the above findings indicate that elevated mean sSNA segregated with ANG II treatment and not with the level of dietary salt per se. Note that the lack of group differences in the duration of respiratory phase-specific sSNA bursts is consistent with the lack of treatment effects on the frequency of PNA bursts as well as the amplitude and duration of the respiratory cycle phases (Table 2).
Table 2.
Baseline values of expired CO2 and PNA
| Treatment | n | End-Expiratory CO2, % | Inspiratory Duration, s | Expiratory Duration, s | PNA Burst Amplitude, µV | PNA Burst AUC, µV·s | PNA Burst Frequency, bursts/min |
|---|---|---|---|---|---|---|---|
| Veh-NSD | 6 | 5.5 ± 0.1 | 0.40 ± 0.06 | 1.16 ± 0.08 | 13 ± 2 | 3.3 ± 0.5 | 34 ± 1 |
| Veh-HSD | 6 | 5.5 ± 0.1 | 0.38 ± 0.03 | 1.00 ± 0.05 | 14 ± 2 | 3.9 ± 0.6 | 39 ± 1 |
| ANG II-HSD | 6 | 5.4 ± 0.1 | 0.36 ± 0.02 | 1.08 ± 0.08 | 15 ± 2 | 3.9 ± 0.5 | 37 ± 1 |
Values are expressed as means ± SE; n, number of rats. Veh, vehicle; NSD, normal-salt diet; HSD, high-salt diet; PNA, phrenic nerve activity; AUC, area under the curve.
A rhythmicity index (22, 23), which quantifies the extent to which PNA-triggered sSNA signals deviate from mean voltage, was used for across-group comparisons of overall respiratory rhythmicity of sSNA. Figure 2B, bottom right, shows that the rhythmicity of sSNA across the entire respiratory cycle in Veh-NSD rats (29 ± 6%) was not different in rats that consumed a HSD alone (23 ± 3%) or by the combination of a HSD with systemic ANG II (29 ± 3%). Rhythmicity index values for the IP, ET, and EP phases of the respiratory cycle were also similar across treatment groups. Collectively, data indicate that the greater average value of integrated sSNA observed in ANG II-HSD rats was not due to enhancement of respiratory rhythmic bursting.
Effects of ANG II-HSD on cardiac rhythmic sSNA.
ECG R wave-triggered sSNA averages from Veh-NSD (Fig. 3A, left), Veh-HSD (Fig. 3A, middle), and ANG II-HSD (Fig. 3A, right) rats each featured prominent cardiac rhythmic oscillations. Data were analyzed according to Fig. 3A, left. Mean sSNA values shown in Fig. 3B, top left, again revealed greater activity in ANG II-HSD rats than in Veh-NSD or Veh-HSD rats (P < 0.05; see also Fig. 2B, top left). Cardiac rhythmic sSNA oscillation amplitude (Fig. 2B, top right) and AUC (Fig. 2B, bottom left) were similar across Veh-infused rats regardless of whether they consumed a NSD or HSD. As with mean sSNA, cardiac rhythmic oscillation amplitude and AUC were significantly greater in the ANG II-HSD group (amplitude: 8.1 ± 1.1 µV and AUC: 0.33 ± 0.04 µV·s) compared with the Veh-NSD group (ampliture: 2.8 ± 0.8 µV, P < 0.05, and AUC: 0.13 ± 0.03 µV·s, P < 0.05) and Veh-HSD group (amplitude: 4.6 ± 1.0 µV, P < 0.05, and AUC: 0.12 ± 0.04 µV·s, P < 0.05). Note that the duration of the cardiac oscillation period was significantly greater in the Veh-NSD group (0.15 ± 0.01 s) than in either the Veh-HSD (0.13 ± 0.01 s, P < 0.05) or ANG II-HSD (0.12 ± 0.02 s, P < 0.01) groups, consistent with the lower HR in Veh-NSD rats (see Table 1). Consistent with the greater cardiac rhythmic oscillation amplitude in the ANG II-HSD group, the rhythmicity index values for R wave-triggered sSNA averages (Fig. 3B, bottom right) indicate greater cardiac rhythmicity in ANG II-HSD (18.8 ± 0.7%) compared with Veh-NSD (11.9 ± 1.7%, P < 0.05) or Veh-HSD (12.0 ± 1.7%, P < 0.05) rats. The cardiac rhythmicity of sSNA was similar in Veh-NSD and Veh-HSD rats, indicating that HSD alone did not alter baroreflex entrainment of sSNA. Collectively, the above data indicate that combined treatment with ANG II and a HSD is required to increase the mean level of sSNA and its cardiac rhythmicity.
Fig. 3.

Effects of graded salt intake and systemic infusion of ANG II on cardiac rhythmic splanchnic sympathetic nerve activity (sSNA). A: representative ECG R wave-triggered averages of sSNA from 1) a vehicle (Veh)-infused rat that consumed a normal salt (0.4% NaCl) diet (Veh-NSD; left), 2) a Veh-infused rat that consumed a high-salt (2% NaCl) diet (Veh-HSD; middle), and 3) an ANG II infused rat that consumed a high-salt (2% NaCl) diet (ANG II-HSD; right). ECG R wave triggers occurred at time 0, and the time domain of each sSNA average shows >2 cardiac cycles. The inset in A, left, shows analyzed parameters of arterial pulse-modulated sSNA. Data are from the same sSNA segments as in Fig. 2A. Note that the cycle duration of sSNA oscillations was similar in each group, reflecting similar levels of heart rate. Oscillation amplitude was increased and shifted to a higher mean voltage in the ANG II-HSD rat compared with either Veh-treated rat regardless of the level of dietary salt consumed. B: summary data (n = 6/group) show that the mean level of sSNA (top left) as well as the amplitude (top right) and area under the curve (AUC; bottom left) of its cardiac rhythmic oscillation were significantly greater in the ANG II-HSD group (black bar) compared with Veh-NSD (open bar) or Veh-HSD (gray bar) groups. Mean sSNA voltage and cardiac oscillations were similar across Veh-infused rats regardless of the level of salt intake. Cardiac rhythmicity index values (bottom right) indicate that the overall degree of cardiac modulation of sSNA was greater in ANG II-HSD rats compared with control rats, consistent with greater cardiac oscillation amplitude and AUC. Triggered averages of sSNA in A were constructed using ~1,600 consecutive R waves. Summary data are expressed as means ± SE. *P < 0.05 vs. the Veh-NSD group; †P < 0.05 vs. the Veh-HSD group.
PVN support of MAP, sSNA, and PNA.
The contribution of PVN neuronal activity to rhythmic bursting components of sSNA was investigated by inhibiting discharge by local injection of the long-acting GABA-A receptor agonist muscimol (100 pmol in 50 nl/side). As shown in Fig. 4A, baseline sSNA was greatest in the ANG II-HSD rat (see also Table 1), which also showed the greatest fall of sSNA and MAP in response to PVN-injected muscimol. As shown in Fig. 4B, integrated sSNA was elevated in the ANG II-HSD group, and only in this group did PVN-injected muscimol reduce MAP and sSNA. Of note, the reduction of integrated PNA by muscimol in the ANG II-HSD rat shown in Fig. 4A, right, reflected offsetting changes in PNA burst amplitude and duration, as PVN inhibition did not alter PNA burst frequency (Fig. 4A, right) or AUC (Fig. 4A, left).
Fig. 4.

Effects of inhibition of paraventricular nucleus (PVN) neuronal activity on splanchnic sympathetic nerve activity (sSNA), blood pressure, and phrenic nerve activity (PNA). A: nanoinjection of the long-acting GABA-A receptor agonist muscimol bilaterally into the PVN (100 pmol in 50 nl/side) of vehicle (Veh)-infused rats that consumed either a normal-salt diet (NSD; left) or high-salt diet (HSD; middle) had little effect on the level of ongoing sSNA, arterial blood pressure (ABP), heart rate (HR), or PNA amplitude. In contrast, in an ANG II-infused rat that consumed a HSD (right), PVN-injected muscimol reduced sSNA, ABP, and integrated PNA without significantly altering HR. Arrows indicate the timing of muscimol injections. B: summary data from anesthetized rats (n = 6/group) indicate that baseline mean arterial pressure (MAP; open bars) was similar across treatment groups (top left). PVN inhibition with muscimol (solid bars) significantly reduced MAP only in ANG II-HSD rats (P < 0.01). PVN-injected muscimol also reduced mean sSNA (P < 0.01), but again only in the ANG II-HSD group. PVN-injected muscimol did not affect the area under the curve of PNA bursts (bottom left) or the frequency of their occurrence (bottom right) in any group. Values are expressed as means ± SE. *P < 0.01 vs. baseline; †P < 0.01 vs. Veh-NSD and Veh-HSD groups.
PVN support of respiratory rhythmic sSNA.
Figure 5A shows representative PNA burst-triggered averages of integrated sSNA before and after PVN inhibition with muscimol. Note that whereas PVN inhibition reduced mean sSNA in an ANG II-HSD rat (Fig. 5A, right), no effect was observed in either Veh-NSD or Veh-HSD rats (Fig. 5A, left and middle) on mean sSNA or on the amplitude of the IP, ET, or EP in these animals. Summary data (n = 6/group) shown in Fig. 5B indicate that the average sSNA voltage from PNA burst-triggered averages (Fig. 5B, left) was significantly greater in the ANG II-HSD (10.1 ± 1.2 µV) group compared with Veh-treated groups (NSD: 4.3 ± 0.8 µV and HSD: 6.9 ± 1.8 µV, P < 0.05). PVN-injected muscimol significantly reduced mean sSNA in the ANG II-HSD group (−3.6 ± 0.4 µV, P < 0.001) but failed to do so in either Veh-treated group (NSD: +0.4 ± 0.4 µV and HSD: +0.2 ± 0.2 µV). The average reduction of mean sSNA by muscimol in the ANG II-HSD group was significantly greater than in Veh-NSD (P < 0.05) and Veh-HSD (P < 0.01) groups. Examination of respiratory phase-specific bursting of sSNA (Fig. 5B, middle) revealed that the AUC of the IP was greater in ANG II-HSD rats compared with vehicle-infused rats (NSD: P < 0.05 and HSD: P < 0.01), but PVN-injected muscimol was without effect on the IP, ET, or EP in any group. Analysis further revealed that PVN-dependent sSNA (Fig. 5B, right) was greater in ANG II-HSD rats than Veh-infused rats that consumed a NSD (P < 0.05) or HSD (P < 0.05). The PVN-dependent contribution to sSNA during each respiratory phase was also significantly greater in the ANG II-HSD group compared with Veh-NSD (P < 0.05) or Veh-HSD (P < 0.05) groups. Collectively, data indicate that PVN neuronal activity in ANG II-HSD rats maintains elevated sSNA by supporting a component of activity that does not have respiratory rhythmicity.
Fig. 5.

Effects of inhibition of paraventricular nucleus (PVN) neuronal activity on phrenic nerve activity (PNA) burst-triggered averages of splanchnic sympathetic nerve activity (sSNA). A: in a vehicle (Veh)-treated rat fed a normal-salt diet (Veh-NSD; left) and a Veh-treated rat fed a high-salt diet (Veh-HSD; middle), nanoinjection of muscimol into the PVN caused little change in the mean level of sSNA or its inspiratory peak (IP), expiratory trough (ET), or expiratory peak (EP). In contrast, in an ANG II-HSD rat (right), mean sSNA voltage was higher at baseline compared with Veh-treated controls, and PVN inhibition caused a short-latency, prolonged reduction of mean voltage without significantly changing the amplitude of the IP, ET, or EP. Note that the burst-triggered average of integrated PNA in each graph (gray dashed line) indicates that treatments had no effect on the strength of neural inspiration. B: group data (n = 6/group) showing that mean sSNA voltage at baseline (open bars) was significantly greater in the ANG II-HSD group compared with Veh-NSD or Veh HSD groups (P < 0.05). PVN-injected muscimol reduced mean voltage only in ANG II-HSD rats (P < 0.01). PVN-injected muscimol did not affect the area under the curve (AUC) of the IP, ET, or EP (middle) in any group. PVN-dependent mean sSNA (right) was significantly greater in ANG II-HSD rats (P < 0.05) compared with Veh-treated control rats whether they consumed a NSD or HSD. The contribution of PVN neuronal activity to the IP, ET, and EP phases of respiration-modulated sSNA were not different and similar to the PVN contribution to the mean voltage of sSNA. †P < 0.05 vs. baseline in Veh-NSD and Veh-HSD groups; ‡P < 0.01 vs. baseline in the ANG II-HSD group; +P < 0.05 vs. postmuscimol values in Veh-NSD and Veh-HSD groups; *P < 0.01 vs. the corresponding parameters in Veh-NSD and Veh-HSD groups. NS, not significant. Values are expressed as means ± SE.
PVN support of cardiac rhythmic sSNA.
To determine the contribution of cardiac rhythmic oscillation in the fall of integrated sSNA in response to PVN muscimol, R wave-triggered averages of sSNA were constructed at baseline and during the maximal effect of PVN-injected muscimol. Figure 6A shows representative responses of R wave-triggered averages before (black line) and after PVN-injected muscimol (gray line) in a Veh-NSD rat (Fig. 6A, left), Veh-HSD rat (Fig. 6A, middle), and ANG II-HSD rat (Fig. 6A, right). Cardiac oscillation amplitudes were similar between Veh-treated rats regardless of which NaCl diet was consumed, but the mean voltage of the HSD-treated rat was slightly greater. Both cardiac oscillation amplitude and mean activity were greater in ANG II-HSD rats compared with Veh-treated rats. The oscillation period was less in the ANG II-HSD rat compared with the Veh-NSD or Veh-HSD rat.
Fig. 6.

Effects of inhibition of paraventricular nucleus (PVN) neuronal activity on R wave-triggered averages of splanchnic sympathetic nerve activity (sSNA). A: representative responses to PVN injections of muscimol showed little effect on the mean voltage of sSNA or the amplitude of its cardiac rhythmic bursting in a vehicle (Veh)-treated rat fed a normal-salt diet (Veh-NSD; left) or a Veh-treated rat fed a high-salt diet (Veh-HSD; middle). In contrast, in an ANG II-HSD rat, PVN-injected muscimol caused the mean level of sSNA and the amplitude of its cardiac rhythmic oscillation to fall. B: summary data (n = 6/group) showing that the mean voltage (left) and area under the curve (AUC) of the cardiac oscillation (middle) of sSNA were greater in ANG II-HSD rats compared with Veh-NSD or Veh-HSD rats (P < 0.01). PVN-injected muscimol significantly (P < 0.01) reduced the mean voltage and AUC of cardiac rhythmic sSNA only in ANG II-HSD rats. Further analysis revealed, as expected, that sSNA driven by PVN neuronal activity, i.e., PVN-dependent sSNA (right), was significantly (P < 0.01) greater in ANG II-HSD rats than controls. †P < 0.01 vs. the Veh-NSD group; +P < 0.01 vs. the Veh-HSD group; *P < 0.01 vs. baseline. Values are expressed as means ± SE.
The summary data in Fig. 6B (n = 6/group) show baseline mean voltage of R wave-triggered averages of sSNA was significantly greater in ANG II-HSD (11.8 ± 1.3 µV) rats compared with Veh-NSD (4.9 ± 0.5 µV, P < 0.01) but not Veh-HSD (6.8 ± 0.7 µV, P > 0.05) rats. Baseline mean voltage tended to be greater in rats that consumed the HSD compared with the NSD (P = 0.07) but did not reach statistical significance. PVN-injected muscimol significantly reduced mean voltage only in ANG II-HSD rats (−2.3 ± 0.7 µV, P < 0.05), not in Veh-infused rats that consumed either the NSD (−0.4 ± 0.3 µV) or HSD (+0.1 ± 0.2 µV). Reductions of mean voltage were significantly greater in ANG II-HSD rats compared with Veh-NSD (P < 0.05) or Veh-HSD (P < 0.05) rats. Baseline cardiac oscillation AUC (Fig. 6B, middle) was significantly greater in ANG II-HSD rats (0.34 ± 0.04 µV⋅s) compared with Veh-infused rats that consumed a NSD (0.13 ± 0.03 µV·s, P < 0.01) or HSD (0.12 ± 0.04 µV·s, P < 0.05). PVN-injected muscimol significantly reduced cardiac oscillation AUC in ANG II-HSD (−0.10 ± 0.01 µV·s, P < 0.01) rats but not Veh-NSD (−0.01 ± 0.01 µV·s) or Veh-HSD (−0.02 ± 0.01 µV·s) rats. Reductions in the cardiac oscillation of sSNA were significantly greater in the ANG II-HSD group compared with either Veh-treated group (NSD: P < 0.01, HSD: P < 0.05). The greater reduction of the cardiac rhythmic oscillation of sSNA by muscimol in ANG II-HSD rats is likely a secondary effect of reduced arterial baroreceptor input given the greater fall of MAP that occurred in this group. Collectively, data indicate that PVN neuronal activity in ANG II-HSD rats maintains elevated sSNA by supporting a component of activity that is not intrinsically cardiac rhythmic.
Histology.
Figure 7A shows representative micrographs, and summary depictions of the location of PVN injection sites are shown in Fig. 7B. Injection sites are indexed by the distribution of fluorescence intensity of coinjected rhodamine-containing latex nanospheres in Veh-NSD (Fig. 7B, left), Veh-HSD (Fig. 7B, middle), and ANG II-HSD rats (Fig. 7B, right). As in our previous studies using PVN nanoinjections, injections were consistently located within the dorsal and lateral parvocellular regions throughout the rostral-caudal extent of the PVN and encompassed most of the central magnocellular region as well.
Fig. 7.

Histological verification of paraventricular nucleus microinjection sites. A: representative photomicrographs of histological sections through the PVN showing the location and distribution of fluorescent (rhodamine) nanospheres coinjected with muscimol. B: schematic drawings of microinjection sites targeting the PVN in rats (n = 6/group) infused with vehicle (Veh) and consumed a normal-salt diet (NSD; left) or high-salt diet (HSD; middle) or infused with ANG II and consumed a HSD (right). Gray regions indicate the overlapping distributions of PVN-injected nanospheres within each group. Nanosphere distribution was similar across groups. Stereotaxic coordinates on the right are with respect to bregma.
DISCUSSION
The present study provides several novel findings with regard to the mechanisms of persistently elevated sSNA in rats fed a HSD together with chronic infusion of ANG II. Firstly, even under anesthesia, ANG II-HSD rats had increased sSNA compared with rats that only consumed a NSD or HSD. Secondly, elevated sSNA in ANG II-HSD rats was dominantly due to increased tonic nerve activity with comparatively minor changes in respiratory phase-specific bursting. Thirdly, cardiac rhythmic inhibitory entrainment of sSNA was enhanced in ANG II-HSD rats consistent with preserved arterial baroreflex phasic inhibition of exaggerated tonic discharge. Finally, inhibition of neurons in the PVN effectively normalized enhanced tonic sSNA in ANG II-HSD rats and reduced the ongoing level of MAP, responses that were not observed in Veh-infused rats that consumed a NSD or HSD. When considered with previous studies, the present findings indicate that neural mechanisms in the PVN (46, 55) support elevated MAP in ANG II-salt HTN through the generation of a tonic component of splanchnic SNA without significantly altering respiratory or cardiac rhythmic burst discharge.
The overall increase of sSNA observed in this study is consistent with previous reports of significantly increased whole body NE spillover in the ANG II-salt hypertensive rat (28). According to a study in humans (6), nearly 40% of whole body NE spillover normally derives from nonhepatic splanchnic organs, which suggests that increased sSNA is likely to have significant impact on systemic hemodynamics and blood pressure. The splanchnic vascular bed and its sympathetic innervation are important for the development of ANG II-salt HTN, as indicated by evidence that increased sSNA (30) is strongly associated with increased mean circulatory filling pressure (MCFP) (27, 29). Evidence indicates that splanchnic veins account for the majority of active capacitance for the circulation, and, hence, the degree of venoconstriction dominantly determines MCFP, which is a principle driver of venous return and thus cardiac output and, depending on the total peripheral resistance, the level of MAP (18, 43). In this regard, it is noteworthy that the elevated MCFP in ANG II-salt hypertensive rats (36) was reported not to be accompanied by increased blood volume, indicating increased tonus of capacitance vessels (27). Consistent with the latter contributing to the development of ANG II-salt HTN, bilateral celiac ganglionectomy normalized MCFP and attenuated the rise of MAP (27, 29, 30).
The role of SNA to other vascular beds was not investigated in the present study, but previously published evidence has indicated that denervation of the kidneys has no effect on the development of ANG II-salt HTN (45), and hindlimb NE spillover is unchanged in this model (56). These functional data are consistent with direct nerve recordings, which indicate that renal SNA falls and lumbar SNA is unchanged in ANG II-HSD rats (56). Given that this pattern of regional SNA occurs with elevated MAP in ANG II-HSD rats, the possibility arises that increased baroreflex inhibition at the level of the brain stem could occlude transmission of renal and lumbar sympathoexcitatory signals driven in a feedforward fashion by higher-order central nervous system neurons known to be activated by the combination of systemic ANG II and a HSD. However, determining whether this occurs will require that those central nervous system neurons that specifically govern lumbar and renal SNA are at least partially separable from those that control splanchnic SNA, and studies would have to identify and segregate these separate neuronal populations and quantify the relative strength of their baroreflex inhibition. This is presently beyond the reach of available technology and, hence, beyond the scope of the present study.
Results further indicate that the contribution of respiratory rhythmic bursting to total sSNA was not different between ANG II-HSD and Veh-treated control groups regardless of their level of salt intake. This is consistent with data indicating that central inspiratory drive was similar between groups and with data showing that end-expiratory CO2 along with PNA burst amplitude, AUC, and frequency were similar between groups (Table 2). It is worth noting, however, that salt loading by itself has been reported to increase SNA and blood pressure responses to chemical excitation (1, 2, 24) and inhibition of the RVLM (1). On the basis of this, it might be expected that excitatory and inhibitory respiratory network input to bulbospinal sympathoexcitatory neurons in the RVLM would be more effective in rats that consumed a HSD whether treated with systemic ANG II or Veh. Our failure to observe this could be explained by the fact that all rats in the present study had continuous access to drinking water compared with saline, as in the aforementioned studies. Consistent with the present findings, it is also possible that a specific subset of RVLM sympathoexcitatory neurons receives respiratory network input and that these respiratory modulated neurons are unaffected by elevated dietary salt. Of special interest in the latter regard is that a subgroup of sympathoexcitatory RVLM neurons has been identified that shows no evidence of respiratory modulation (36), raising the possibility that these neurons could serve as a substrate for the increased tonic SNA in the ANG II-HSD group.
Treatment either with ANG II or consumption of a HSD has been reported to attenuate baroreflex control of SNA (5, 10, 12, 33). However, the cardiac rhythmic oscillation of sSNA in the present study, which largely reflects arterial pulse rhythmic synaptic inhibition of the RVLM, was exaggerated, not attenuated, in ANG II-HSD rats. Our data (Fig. 3A) indicate that the pulse rhythmic nadirs of cardiac oscillations of sSNA were elevated, suggesting that inhibition of sSNA with each arterial pressure pulse was incomplete, which is consistent with the notion of an attenuated arterial baroreflex. The fact that mean sSNA voltage and the amplitude of the cardiac rhythmic sSNA oscillation were concurrently increased indicates that peak cardiac rhythmic bursting increased more than did sSNA voltage at the nadir within each cardiac cycle, which indicates that sSNA was significantly elevated while arterial baroreceptors were relatively unloaded during diastolic phases of the cardiac cycle. Of special importance is that HSD alone did not increase cardiac rhythmic oscillation amplitude or increase the minimum voltage recorded with each cardiac cycle. The latter findings indicate that consumption of a 2% NaCl HSD alone is not sufficient to effectively attenuate arterial baroreflex control of sSNA.
From the present study, it appears that the bulk of exaggerated SNA in anesthetized ANG II-HSD rats reflects activation of a tonic component of sSNA. Xing and Pilowsky (56) reported a similar response among rats exposed to acute intermittent hypoxia (AIH). These authors reported that AIH-induced long-term facilitation of sSNA was independent of central respiratory network activity. They further reported that respiratory rhythmic bursting of sSNA was unaffected by AIH, whereas mean sSNA was increased. These observations are remarkably consistent with findings of the present study and with our previous report in water-deprived rats (23). Thus, it appears that activation of the sympathetic network during a subchronic physiological challenge (water deprivation), chronic disease model (ANG II-salt HTN), or acute repetitive activation of arterial chemoreceptors by hypoxia selectively increases a tonic component of sSNA.
As noted above, PVN neuronal activity is well known to support ongoing SNA and MAP during a variety of acute and chronic homeostatic challenges (4, 7, 40, 45, 48, 53, 54). A second major goal of this study was to determine the role of PVN neuronal activity in supporting specific rhythmic components of sSNA. As expected, inhibition of PVN reduced integrated SNA and MAP in ANG II-HSD rats (Fig. 4 and see Ref. 7) but not Veh-treated rats that consumed a NSD or HSD. Reductions of integrated sSNA were primarily due to reductions of tonic sSNA, as respiratory rhythmic (Fig. 5) and cardiac rhythmic (Fig. 6) bursting was largely unaltered in the ANG II-HSD group. It should be stressed that the above conclusion with respect to cardiac rhythmic sSNA seems to run contrary to the fact that our data indicate that PVN-injected muscimol significantly reduced the cardiac rhythmic oscillation of sSNA. However, the latter is consistent with the muscimol causing a significant fall in MAP with an ensuing reduction of arterial baroreceptor inhibitory input. Therefore, the reduced amplitude of cardiac rhythmic sSNA appears to reflect a secondary effect of reflex disinhibition.
The concept that heightened PVN neuronal activity is capable of generating increased tonic SNA is not new. Indeed, studies using power spectral analysis have previously demonstrated that even acute PVN activation through blockade of GABA-A receptors increased SNA without affecting respiratory or cardiac frequency components (25, 26). Similarly, a previous report (13) from our laboratory identified a subgroup of spinally projecting neurons with SNA-related tonic firing. Findings raise the possibility that increased nonrespiratory and noncardiac rhythmic bursting of SNA contributes to sympathetic vasomotor tone. The latter, however, has yet to be demonstrated. No study, for example, has yet to demonstrate that tonic SNA results in vasoconstriction or a reduction of regional/organ blood flow. Moreover, specific cellular mechanisms driving discharge of PVN neurons that support tonic SNA are unknown, and, until elucidated, it will not be possible to selectively interfere with PVN-driven tonic SNA to definitively demonstrate the resulting hemodynamic consequences.
A caveat to interpreting the present findings is that rats were anesthetized, which could have reduced central respiratory and sympathetic drive. We chose a cocktail of urethane and α-chloralose for anesthesia to minimize this possibility (17, 41), but two observations suggest that anesthesia could have influenced experimental outcomes. First, anesthesia reduced hypertensive MAP of conscious ANG II-HSD rats to a normotensive level, consistent with reducing sympathetic control of blood pressure. Second, respiratory rate (phrenic burst frequency) under anesthesia was ~35–40 bursts/min, which is significantly less than when rats are conscious (50). However, our rats were also artificially ventilated with 100% O2, which also reduces central respiratory drive (52), making it difficult to evaluate the extent to which reduced respiratory drive reflected effects of anesthesia per se. To the extent that anesthesia differentially reduced sympathetic and respiratory drive in the ANG II-HSD group, it is possible that respiratory-sympathetic coupling may have also been differentially diminished, preventing us from detecting a contribution of PVN neuronal activity to any enhancement of respiratory-sympathetic coupling that might have been present in conscious ANG II-HSD rats (52).
In summary, treatment of rats with systemic ANG II and a HSD leads to exaggerated sSNA that is not explained by enhanced respiratory or cardiac rhythmic bursting. The exaggerated tonic sSNA in ANG II-HSD rats was strongly dependent on PVN neuronal activity. Over the past several years, studies investigating mechanisms that drive exaggerated SNA during neurogenic HTN have focused on the possibility of enhanced coupling between the respiratory and sympathetic networks leading to enhanced respiratory rhythmic burst discharge (5, 6, 17, 23, 50, 53). The present findings suggest that a component of SNA driven by the hypothalamic PVN that has neither respiratory nor cardiac rhythmicity may play a previously unrecognized role in arterial HTN. Further studies are needed to determine the role that tonic sSNA plays during the development of neurogenic HTN and to identify neuronal populations and underlying mechanisms that generate the tonic component of SNA.
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grants HL-102310 and HL-088052 and by American Heart Association Grant-in-Aid GRNT25710176 (to G. M. Toney).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W.W.H. and G.M.T. conceived and designed research; W.W.H., M.B.B., and M.A.A. performed experiments; W.W.H., M.B.B., M.A.A., and G.M.T. analyzed data; W.W.H. and G.M.T. interpreted results of experiments; W.W.H. and G.M.T. prepared figures; W.W.H. and G.M.T. drafted manuscript; W.W.H., M.B.B., M.A.A., and G.M.T. approved final version of manuscript; G.M.T. edited and revised manuscript.
ACKNOWLEDGMENTS
The authors gratefully acknowledge Alfredo S. Calderon and Myrna Herrera-Rosales for excellent technical assistance.
REFERENCES
- 1.Adams JM, Madden CJ, Sved AF, Stocker SD. Increased dietary salt enhances sympathoexcitatory and sympathoinhibitory responses from the rostral ventrolateral medulla. Hypertension 50: 354–359, 2007. doi: 10.1161/HYPERTENSIONAHA.107.091843. [DOI] [PubMed] [Google Scholar]
- 2.Adams JM, McCarthy JJ, Stocker SD. Excess dietary salt alters angiotensinergic regulation of neurons in the rostral ventrolateral medulla. Hypertension 52: 932–937, 2008. doi: 10.1161/HYPERTENSIONAHA.108.118935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adrian ED, Bronk DW, Phillips G. Discharges in mammalian sympathetic nerves. J Physiol 74: 115–133, 1932. doi: 10.1113/jphysiol.1932.sp002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen AM. Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension 39: 275–280, 2002. doi: 10.1161/hy0202.104272. [DOI] [PubMed] [Google Scholar]
- 5.Andresen MC. High-salt diet elevates baroreceptor pressure thresholds in normal and Dahl rats. Circ Res 64: 695–702, 1989. doi: 10.1161/01.RES.64.4.695. [DOI] [PubMed] [Google Scholar]
- 6.Aneman A, Eisenhofer G, Olbe L, Dalenbäck J, Nitescu P, Fändriks L, Friberg P. Sympathetic discharge to mesenteric organs and the liver. Evidence for substantial mesenteric organ norepinephrine spillover. J Clin Invest 97: 1640–1646, 1996. doi: 10.1172/JCI118590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bardgett ME, Holbein WW, Herrera-Rosales M, Toney GM. Ang II-Salt Hypertension Depends on neuronal activity in the hypothalamic paraventricular nucleus but not on local actions of tumor necrosis factor-α. Hypertension 63: 527–534, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrett CJ, Ramchandra R, Guild SJ, Lala A, Budgett DM, Malpas SC. What sets the long-term level of renal sympathetic nerve activity: a role for angiotensin II and baroreflexes? Circ Res 92: 1330–1336, 2003. doi: 10.1161/01.RES.0000078346.60663.A0. [DOI] [PubMed] [Google Scholar]
- 9.Bishop VS, Ryuzaki M, Cai Y, Nishida Y, Cox BF. Angiotensin II-dependent hypertension and the arterial baroreflex. Clin Exp Hypertens 17: 29–38, 1995. doi: 10.3109/10641969509087052. [DOI] [PubMed] [Google Scholar]
- 10.Boscan P, Allen AM, Paton JF. Baroreflex inhibition of cardiac sympathetic outflow is attenuated by angiotensin II in the nucleus of the solitary tract. Neuroscience 103: 153–160, 2001. doi: 10.1016/S0306-4522(00)00559-5. [DOI] [PubMed] [Google Scholar]
- 11.Burke SL, Lukoshkova EV, Head GA. Characteristics of renal sympathetic nerve single units in rabbits with angiotensin-induced hypertension. Exp Physiol 101: 50–66, 2016. doi: 10.1113/EP085472. [DOI] [PubMed] [Google Scholar]
- 12.Casto R, Phillips MI. Angiotensin II attenuates baroreflexes at nucleus tractus solitarius of rats. Am J Physiol Regul Integr Comp Physiol 250: R193–R198, 1986. doi: 10.1152/ajpregu.1986.250.2.R193. [DOI] [PubMed] [Google Scholar]
- 13.Chen QH, Toney GM. Identification and characterization of two functionally distinct groups of spinal cord-projecting paraventricular nucleus neurons with sympathetic-related activity. Neuroscience 118: 797–807, 2003. doi: 10.1016/S0306-4522(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 14.Czyzyk-Krzeska MF, Trzebski A. Respiratory-related discharge pattern of sympathetic nerve activity in the spontaneously hypertensive rat. J Physiol 426: 355–368, 1990. doi: 10.1113/jphysiol.1990.sp018142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev 74: 323–364, 1994. doi: 10.1152/physrev.1994.74.2.323. [DOI] [PubMed] [Google Scholar]
- 16.Darnall RA, Guyenet P. Respiratory modulation of pre- and postganglionic lumbar vasomotor sympathetic neurons in the rat. Neurosci Lett 119: 148–152, 1990. doi: 10.1016/0304-3940(90)90820-Y. [DOI] [PubMed] [Google Scholar]
- 17.Dorward PK, Burke SL, Jänig W, Cassell J. Reflex responses to baroreceptor, chemoreceptor and nociceptor inputs in single renal sympathetic neurones in the rabbit and the effects of anaesthesia on them. J Auton Nerv Syst 18: 39–54, 1987. doi: 10.1016/0165-1838(87)90133-0. [DOI] [PubMed] [Google Scholar]
- 18.Greenway CV. Role of splanchnic venous system in overall cardiovascular homeostasis. Fed Proc 42: 1678–1684, 1983. [PubMed] [Google Scholar]
- 19.Guyenet PG, Darnall RA, Riley TA. Rostral ventrolateral medulla and sympathorespiratory integration in rats. Am J Physiol Regul Integr Comp Physiol 259: R1063–R1074, 1990. doi: 10.1152/ajpregu.1990.259.5.R1063. [DOI] [PubMed] [Google Scholar]
- 20.Hayashi J, Takeda K, Kawasaki S, Nakamura Y, Oguro M, Nakata T, Tanabe S, Lee LC, Sasaki S, Nakagawa M. Central attenuation of baroreflex by angiotensin II in normotensive and spontaneously hypertensive rats. Am J Hypertens 1: 15S–22S, 1988. doi: 10.1093/ajh/1.3.15S. [DOI] [PubMed] [Google Scholar]
- 21.Holbein WW, Bardgett ME, Toney GM. Blood pressure is maintained during dehydration by hypothalamic paraventricular nucleus-driven tonic sympathetic nerve activity. J Physiol 592: 3783–3799, 2014. doi: 10.1113/jphysiol.2014.276261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holbein WW, Toney GM. Activation of the hypothalamic paraventricular nucleus by forebrain hypertonicity selectively increases tonic vasomotor sympathetic nerve activity. Am J Physiol Regul Integr Comp Physiol 308: R351–R359, 2015. doi: 10.1152/ajpregu.00460.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holbein WW, Toney GM. Sympathetic network drive during water deprivation does not increase respiratory or cardiac rhythmic sympathetic nerve activity. J Appl Physiol (1985) 114: 1689–1696, 2013. doi: 10.1152/japplphysiol.00078.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito S, Gordon FJ, Sved AF. Dietary salt intake alters cardiovascular responses evoked from the rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol 276: R1600–R1607, 1999. doi: 10.1152/ajpregu.1999.276.6.R1600. [DOI] [PubMed] [Google Scholar]
- 25.Kenney MJ, Weiss ML, Haywood JR. The paraventricular nucleus: an important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol Scand 177: 7–15, 2003. doi: 10.1046/j.1365-201X.2003.01042.x. [DOI] [PubMed] [Google Scholar]
- 26.Kenney MJ, Weiss ML, Mendes T, Wang Y, Fels RJ. Role of paraventricular nucleus in regulation of sympathetic nerve frequency components. Am J Physiol Heart Circ Physiol 284: H1710–H1720, 2003. doi: 10.1152/ajpheart.00673.2002. [DOI] [PubMed] [Google Scholar]
- 27.King AJ, Fink GD. Chronic low-dose angiotensin II infusion increases venomotor tone by neurogenic mechanisms. Hypertension 48: 927–933, 2006. doi: 10.1161/01.HYP.0000243799.84573.f8. [DOI] [PubMed] [Google Scholar]
- 28.King AJ, Novotny M, Swain GM, Fink GD. Whole body norepinephrine kinetics in ANG II-salt hypertension in the rat. Am J Physiol Regul Integr Comp Physiol 294: R1262–R1267, 2008. doi: 10.1152/ajpregu.00819.2007. [DOI] [PubMed] [Google Scholar]
- 29.King AJ, Osborn JW, Fink GD. Splanchnic circulation is a critical neural target in angiotensin II salt hypertension in rats. Hypertension 50: 547–556, 2007. doi: 10.1161/HYPERTENSIONAHA.107.090696. [DOI] [PubMed] [Google Scholar]
- 30.Kuroki MT, Guzman PA, Fink GD, Osborn JW. Time-dependent changes in autonomic control of splanchnic vascular resistance and heart rate in ANG II-salt hypertension. Am J Physiol Heart Circ Physiol 302: H763–H769, 2012. doi: 10.1152/ajpheart.00930.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McBryde FD, Guild SJ, Barrett CJ, Osborn JW, Malpas SC. Angiotensin II-based hypertension and the sympathetic nervous system: the role of dose and increased dietary salt in rabbits. Exp Physiol 92: 831–840, 2007. doi: 10.1113/expphysiol.2007.037473. [DOI] [PubMed] [Google Scholar]
- 32.Menuet C, Le S, Dempsey B, Connelly AA, Kamar JL, Jancovski N, Bassi JK, Walters K, Simms AE, Hammond A, Fong AY, Goodchild AK, McMullan S, Allen AM. Excessive Respiratory Modulation of Blood Pressure Triggers Hypertension. Cell Metab 25: 739–748, 2017. doi: 10.1016/j.cmet.2017.01.019. [DOI] [PubMed] [Google Scholar]
- 33.Miyajima E, Buñag RD. Exacerbation of central baroreflex impairment in Dahl rats by high-salt diets. Am J Physiol Heart Circ Physiol 252: H402–H409, 1987. [DOI] [PubMed] [Google Scholar]
- 34.Miyawaki T, Goodchild AK, Pilowsky PM. Evidence for a tonic GABA-ergic inhibition of excitatory respiratory-related afferents to presympathetic neurons in the rostral ventrolateral medulla. Brain Res 924: 56–62, 2002. doi: 10.1016/S0006-8993(01)03025-6. [DOI] [PubMed] [Google Scholar]
- 35.Miyawaki T, Minson J, Arnolda L, Chalmers J, Llewellyn-Smith I, Pilowsky P. Role of excitatory amino acid receptors in cardiorespiratory coupling in ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol 271: R1221–R1230, 1996. doi: 10.1152/ajpregu.1996.271.5.R1221. [DOI] [PubMed] [Google Scholar]
- 36.Miyawaki T, Pilowsky P, Sun QJ, Minson J, Suzuki S, Arnolda L, Llewellyn-Smith I, Chalmers J. Central inspiration increases barosensitivity of neurons in rat rostral ventrolateral medulla. Am J Physiol Regul Integr Comp Physiol 268: R909–R918, 1995. doi: 10.1152/ajpregu.1995.268.4.R909. [DOI] [PubMed] [Google Scholar]
- 37.Moraes DJ, Zoccal DB, Machado BH. Medullary respiratory network drives sympathetic overactivity and hypertension in rats submitted to chronic intermittent hypoxia. Hypertension 60: 1374–1380, 2012. doi: 10.1161/HYPERTENSIONAHA.111.189332. [DOI] [PubMed] [Google Scholar]
- 38.Moreira TS, Takakura AC, Colombari E, West GH, Guyenet PG. Inhibitory input from slowly adapting lung stretch receptors to retrotrapezoid nucleus chemoreceptors. J Physiol 580: 285–300, 2007. doi: 10.1113/jphysiol.2006.125336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Numao Y, Koshiya N, Gilbey MP, Spyer KM. Central respiratory drive-related activity in sympathetic nerves of the rat: the regional differences. Neurosci Lett 81: 279–284, 1987. doi: 10.1016/0304-3940(87)90396-X. [DOI] [PubMed] [Google Scholar]
- 40.Patel KP. Role of paraventricular nucleus in mediating sympathetic outflow in heart failure. Heart Fail Rev 5: 73–86, 2000. doi: 10.1023/A:1009850224802. [DOI] [PubMed] [Google Scholar]
- 41.Pórszász J, Gibiszer KP. Effect of anaesthetics on the sympathetic reflex. Acta Physiol Acad Sci Hung 46: 141–150, 1975. [PubMed] [Google Scholar]
- 42.Rea RF, Hamdan M. Baroreflex control of muscle sympathetic nerve activity in borderline hypertension. Circulation 82: 856–862, 1990. doi: 10.1161/01.CIR.82.3.856. [DOI] [PubMed] [Google Scholar]
- 43.Rothe CF. Reflex control of veins and vascular capacitance. Physiol Rev 63: 1281–1342, 1983. doi: 10.1152/physrev.1983.63.4.1281. [DOI] [PubMed] [Google Scholar]
- 44.Sharpe AL, Andrade MA, Herrera-Rosales M, Britton SL, Koch LG, Toney GM. Rats selectively bred for differences in aerobic capacity have similar hypertensive responses to chronic intermittent hypoxia. Am J Physiol Heart Circ Physiol 305: H403–H409, 2013. doi: 10.1152/ajpheart.00317.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharpe AL, Calderon AS, Andrade MA, Cunningham JT, Mifflin SW, Toney GM. Chronic intermittent hypoxia increases sympathetic control of blood pressure: role of neuronal activity in the hypothalamic paraventricular nucleus. Am J Physiol Heart Circ Physiol 305: H1772–H1780, 2013. doi: 10.1152/ajpheart.00592.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen XZ, Li Y, Li L, Shah KH, Bernstein KE, Lyden P, Shi P. Microglia participate in neurogenic regulation of hypertension. Hypertension 66: 309–316, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol 587: 597–610, 2009. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stocker SD, Simmons JR, Stornetta RL, Toney GM, Guyenet PG. Water deprivation activates a glutamatergic projection from the hypothalamic paraventricular nucleus to the rostral ventrolateral medulla. J Comp Neurol 494: 673–685, 2006. doi: 10.1002/cne.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun MK, Guyenet PG. GABA-mediated baroreceptor inhibition of reticulospinal neurons. Am J Physiol Regul Integr Comp Physiol 249: R672–R680, 1985. 10.1152/ajpregu.1985.249.6.R672. [DOI] [PubMed] [Google Scholar]
- 50.Takakura AC, Moreira TS, De Paula PM, Menani JV, Colombari E. Control of breathing and blood pressure by parafacial neurons in conscious rats. Exp Physiol 98: 304–315, 2013. doi: 10.1113/expphysiol.2012.065128. [DOI] [PubMed] [Google Scholar]
- 51.Takeshita A, Tanaka S, Kuroiwa A, Nakamura M. Reduced baroreceptor sensitivity in borderline hypertension. Circulation 51: 738–742, 1975. doi: 10.1161/01.CIR.51.4.738. [DOI] [PubMed] [Google Scholar]
- 52.Toney GM, Pedrino GR, Fink GD, Osborn JW. Does enhanced respiratory-sympathetic coupling contribute to peripheral neural mechanisms of angiotensin II-salt hypertension? Exp Physiol 95: 587–594, 2010. doi: 10.1113/expphysiol.2009.047399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 57: 435–441, 2011. doi: 10.1161/HYPERTENSIONAHA.110.160671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei SG, Yu Y, Weiss RM, Felder RB. Endoplasmic reticulum stress increases brain MAPK signaling, inflammation and renin-angiotensin system activity and sympathetic nerve activity in heart failure. Am J Physiol Heart Circ Physiol 311: H871–H880, 2016. doi: 10.1152/ajpheart.00362.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winklewski PJ, Radkowski M, Demkow U. Neuroinflammatory mechanisms of hypertension: potential therapeutic implications. Curr Opin Nephrol Hypertens 25: 410–416, 2016. doi: 10.1097/MNH.0000000000000250. [DOI] [PubMed] [Google Scholar]
- 56.Xing T, Pilowsky PM. Acute intermittent hypoxia in rat in vivo elicits a robust increase in tonic sympathetic nerve activity that is independent of respiratory drive. J Physiol 588: 3075–3088, 2010. doi: 10.1113/jphysiol.2010.190454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshimoto M, Miki K, Fink GD, King A, Osborn JW. Chronic angiotensin II infusion causes differential responses in regional sympathetic nerve activity in rats. Hypertension 55: 644–651, 2010. doi: 10.1161/HYPERTENSIONAHA.109.145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zoccal DB, Machado BH. Sympathetic overactivity coupled with active expiration in rats submitted to chronic intermittent hypoxia. Respir Physiol Neurobiol 174: 98–101, 2010. doi: 10.1016/j.resp.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 59.Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF, Machado BH. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol 586: 3253–3265, 2008. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]