

Abstract
Proponents for electronic cigarettes (E-cigs) claim that they are a safe alternative to tobacco-based cigarettes; however, little is known about the long-term effects of exposure to E-cig vapor on vascular function. The purpose of this study was to determine the cardiovascular consequences of chronic E-cig exposure. Female mice (C57BL/6 background strain) were randomly assigned to chronic daily exposure to E-cig vapor, standard (3R4F reference) cigarette smoke, or filtered air (n = 15/group). Respective whole body exposures consisted of four 1-h-exposure time blocks, separated by 30-min intervals of fresh air breaks, resulting in intermittent daily exposure for a total of 4 h/day, 5 days/wk for 8 mo. Noninvasive ultrasonography was used to assess cardiac function and aortic arterial stiffness (AS), measured as pulse wave velocity, at three times points (before, during, and after chronic exposure). Upon completion of the 8-mo exposure, ex vivo wire tension myography and force transduction were used to measure changes in thoracic aortic tension in response to vasoactive-inducing compounds. AS increased 2.5- and 2.8-fold in E-cig- and 3R4F-exposed mice, respectively, compared with air-exposed control mice (P < 0.05). The maximal aortic relaxation to methacholine was 24% and 33% lower in E-cig- and 3R4F-exposed mice, respectively, than in controls (P < 0.05). No differences were noted in sodium nitroprusside dilation between the groups. 3R4F exposure altered cardiac function by reducing fractional shortening and ejection fraction after 8 mo (P < 0.05). A similar, although not statistically significant, tendency was also observed with E-cig exposure (P < 0.10). Histological and respiratory function data support emphysema-associated changes in 3R4F-exposed, but not E-cig-exposed, mice. Chronic exposure to E-cig vapor accelerates AS, significantly impairs aortic endothelial function, and may lead to impaired cardiac function. The clinical implication from this study is that chronic use of E-cigs, even at relatively low exposure levels, induces cardiovascular dysfunction.
NEW & NOTEWORTHY Electronic cigarettes (E-cigs) are marketed as safe, but there has been insufficient long-term exposure to humans to justify these claims. This is the first study to report the long-term in vivo vascular consequences of 8 mo of exposure to E-cig vapor in mice (equivalent to ~25 yr of exposure in humans). We report that E-cig exposure increases arterial stiffness and impairs normal vascular reactivity responses, similar to other risk factors, including cigarette smoking, which contribute to the development of cardiovascular disease.
Keywords: aortic stiffness, smoking, transthoracic echocardiography, vaping, vascular reactivity
INTRODUCTION
Smoking is the most prevalent source of preventable mortality in modern history and accounts for one of every five deaths in the United States each year (2, 11). Electronic cigarettes (E-cigs), which are also known as electronic nicotine delivery devices, are advertised as a “safe” alternative to conventional tobacco cigarettes (45, 55). Proponents for E-cigs suggest that these devices should be considered a harm-reduction device to assist with smoking cessation (33, 46, 56), in part due to tobacco industry-sponsored animal studies that have concluded that E-cigs have no adverse effects on pulmonary structure and function (34, 54). However, meta-analysis and systemic reviews collectively state that there is limited robust evidence of the impact of the E-cigs on tobacco smoking cessation (18) and that there is even evidence to suggest that E-cigs may negatively impact smoking cessation (32). Accordingly, the value of E-cigs for smoking cessation remains controversial, particularly as it relates to nicotine dependence/addiction. Importantly, there is also considerable concern about the overall health consequences related to short- and long-term E-cig use.
Counter to the notion that E-cigs are safe is the recognition that E-cig vapor contains chemicals, such as nicotine, formaldehyde, acetaldehyde, acrolein, and acetone, as well as other compounds, that are known to have deleterious health effects in humans (4, 30, 35, 60). Indeed, in vitro studies have found that E-cigs are cytotoxic to epithelial cells (59, 74), increase oxidative stress in the lung (59), and likely suppress host defenses and promote the virulence of colonizing bacteria (28, 73). Animal studies are also finding evidence of oxidative stress in the lung following short-term daily exposure (i.e., 3–14 days) to E-cigs (41, 62). Studies of the acute effects of E-cig vapor in humans show increases in airway resistance (65) and diastolic blood pressure (17), greater sympathetic activity (50), higher oxidative stress (10, 50), acute increases in aortic arterial stiffness (AS) (68), and impaired flow-mediated dilation (FMD, a measure of arterial health and function) (10). The changes in AS and FMD are consistent with the development of premature or accelerated cardiovascular disease (CVD). While there is growing evidence of the long-term pulmonary toxicity related to E-cigs [see recent review (16)], to our knowledge there are no interventional studies that have reported on the long-term effects of E-cig exposure on cardiac and vascular function. Although observational studies (up to 24 mo) have reported few cardiovascular events in E-cig users (9, 19, 44), it is too early to know the consequences of decades of E-cig use in terms of human cardiovascular health. This issue is particularly important, as the most recent US Surgeon General report states that E-cigs have replaced all other forms of smoking or tobacco products to become the leading product used by 12- to 17-yr-old youths and that use of E-cigs among individuals in this age group increased 900% between 2011 and 2015 (1). While animal smoking models can be controversial (38), they have proven useful and broadly reflect the functional cardiopulmonary and vascular outcomes observed in humans (22, 71, 72). Given the rapid increase in popularity of E-cigs among young adults, and particularly in youth, it is critical to investigate and understand the long-term health consequences associated with habitual E-cig use before these devices are deemed safe. Therefore, the purpose of our study was to evaluate the cardiovascular effects following 8 mo of chronic E-cig exposure in young mice as they advance to middle age to gain insight into the long-term consequences and potential progression of CVD that humans might face after decades of E-cig use.
We hypothesized that if E-cigs are safe, we would not observe cardiovascular dysfunction in mice chronically exposed to E-cig vapor for 8 mo. Based on the life span of the mouse (~2 yr), an 8-mo exposure paradigm represents ~33% of the animal’s life, which in human terms would equate to chronic exposure for a period of ∼25 yr (assuming an average life expectancy of 78 yr). Since E-cigs were only first introduced in the United States in 2006–2007, the earliest possible time frame to study this level of exposure in humans would theoretically be ~2032 (assuming sufficient numbers of reliable early adopters from 2007 could be recruited and studied). Rather, it is more likely that it will be many decades (perhaps closer to 2050 or beyond) before a large enough study population of humans can be recruited to robustly determine the long-term impact of daily E-cig use on cardiovascular health.
METHODS
Study design.
For this environmentally controlled animal study, 10-wk-old female C57BL/6J mice (n = 45) were purchased (stock no. 000664, Jackson Laboratory, Bar Harbor, ME) and randomly assigned (n = 15/group) to chronic exposure to 1) E-cig vapor (cappuccino-flavored, 18 mg/ml nicotine), 2) 3R4F reference cigarette smoke, or 3) filtered air. Mice were allowed 1 wk to adapt to the new vivarium before baseline testing (see below) and randomization to one of the three treatment groups. The starting age at exposure (~13–14 wk old) followed by 8 mo of exposure (ending at ~12 mo of age) represents exposure beginning at adolescence and continuing into adulthood. In human terms, this equates to an individual starting to smoke at ~11 yr of age and continuing to smoke until 35 yr of age, or similar to middle school age through early adulthood (assuming a total life span of 2 yr for mice and 78 yr for humans).
Mice were group-housed (4–5 animals per cage with the same exposure group) in a temperature-controlled (22 ± 4°C, relative humidity 39 ± 6%) pathogen-free vivarium room and maintained on a 12:12-h light-dark cycle. Standard chow (Teklad diet; 18% fat, 24% protein, and 58% carbohydrates) and tap water were provided ad libitum. All procedures were approved by the West Virginia University Institutional Animal Care and Use Committee.
Exposure.
Mice were exposed as single respective treatment groups (i.e., E-cig, 3R4F cigarette, or filtered air) at the same time (n = 15/group) in separate identical 15.1-liter whole body exposure chambers. E-cig vapor and tobacco smoke were gradually introduced during the first 8 wk, after which the mice were consistently exposed to four 1-h-exposure time blocks, with each exposure separated by 30-min intervals of fresh air breaks, resulting in an intermittent exposure pattern for a total exposure of 4 h/day (occurring over a 6-h window each day). The animals were subjected to this daily regimen for 5 days/wk for a total of 8 mo. Urine analysis for cotinine (cotinine ELISA, Calbiotech, El Cajon, CA) suggested that our daily exposure paradigm tended to produce higher nicotine exposure in 3R4F- than E-cig-exposed groups [47.4 ± 16.3 vs. 24.3 ± 0.6 (SD) ng/ml, P = 0.07]. Urine cotinine levels were undetectable in the air-exposed (control) group.
The E-cig device, a third-generation, tank-style device purchased online (eGrip OLED, Joyetech, www.joyetech.com), was controlled using a custom-made electronic cradle (i.e., artificial hand and thumb) that allowed precise and reliable control of the frequency and duration of E-cig button activation without modifications to the E-cig device. The E-cig voltage was set to 4.9 V, and the device was activated every 99 s for a 5-s duration, resulting in ~38–39 puffs each hour.
The 3R4F reference cigarettes were purchased from the University of Kentucky Center for Tobacco Reference Products, stored at 4°C for the duration of the study, and set in room air 1 wk before use. One cigarette was loaded and lighted on the ventilator inlet every 10 min, resulting in smoke generated from six cigarettes each hour (for a total of 24 cigarettes each day over the 4-h exposure).
Vapor/smoke was generated and delivered to the respective chambers with independent, but identical, rodent ventilators (Harvard Apparatus, Natick, MA) using a 55-ml tidal puff volume. Control mice received filtered air (Carbon Cap 150, Whatman) from a central compressed air line. Each chamber had a bias flow of ~3 l/min, and all exposures occurred simultaneously each day.
In vivo measurements.
A VisualSonics Vevo 2100 high-frequency, high-resolution micro-ultrasound system (with color-Doppler mode) was used to perform transthoracic echocardiography to assess cardiac function (64) and in vivo Doppler ultrasonography was used to assess AS (52) at three separate times points: 1) before the exposure started, 2) halfway through the exposure (i.e., at ~4 mo), and 3) after 8 mo of exposure. Assessment of AS comprised imaging of the common carotid artery from its insertion on the aorta to the bifurcation of the common carotid artery at the internal and external branches to measure pulse wave velocity (PWV). The theory and details of PWV measurements have been previously described (21, 52). Briefly, we used a 15-MHz linear-array transducer and color-flow Doppler probe (VisualSonics Vevo 2100) to scan the aorta to the bifurcation of the common carotid artery into its internal and external branches. Color-flow Doppler was employed to help locate the arteries and guide placement of the sample gate for obtaining pulse waveforms. The probe was directed parallel to blood flow. ECG and Doppler signals were then recorded simultaneously at a sweep speed of 200 mm/s for several cardiac cycles, and the data were stored for subsequent offline analysis. The distance (D, measured in mm) between the points of probe applanation over the aorta and the carotid bifurcation was measured using an on-screen digital caliper. The time intervals between the R wave of the ECG and the foot of the Doppler carotid and aortic waveforms were averaged over three cardiac cycles, and the pulse-transit time from the carotid to the aorta was calculated by subtracting the mean R wave-to-carotid foot time interval from the mean R wave-to-aortic foot time interval. PWV was then calculated as follows: PWV = D/T, where D is the distance (in mm) and T = R wave-to-aortic foot interval − R wave-to-carotid foot interval (in ms). During both assessments (i.e., cardiac function and AS), mice are lightly anesthetized with inhaled isoflurane, and body temperature was monitored and maintained at 37°C.
Noninvasive whole body in vivo plethysmography was used to measure respiratory parameters in awake, unanesthetized mice under basal (resting) conditions (26, 31) prior to and just before completion of the 8-mo exposure. Upon completion of the 8-mo exposure, animals were deeply anesthetized (intraperitoneal ketamine-xylazine), tracheally intubated, and ventilated after administration of a paralytic agent (pancuronium bromide, 0.8 mg/kg) to assess airway reactivity to an aerosolized methacholine challenge using a ventilator (flexiVent, Scireq, Montreal, QC, Canada). Thereafter, major body organs were removed, and the thoracic aorta was surgically dissected, sectioned into rings, and mounted onto an ex vivo wire tension myograph system.
Ex vivo measurements.
Force transduction was used to measure changes in aortic tension in response to vasoactive compounds (i.e., phenylephrine, methacholine, and sodium nitroprusside). The thoracic aorta was removed, rinsed in physiological salt solution, cleared of surrounding tissue, and cut into 3-mm ring segments. Each ring was mounted in a myobath chamber between a fixed point and a force transducer (World Precision Instruments) and stretched to 0.5-g tension for 45 min for equilibration; then the final experimental baseline tension was adjusted to 0.25 g. The organ baths contained Krebs-Henseleit buffer at 37°C and were aerated with 95% O2-5% CO2. Ring viability and maximal constriction were tested with 50 mM KCl, which was washed out, and baseline tension was reestablished. Subsequently, endothelial function was assessed by preconstriction with phenylephrine (10−7 M) followed by increasing concentrations (10−9–10−5 M) of methacholine; data are represented as percent return to baseline from preconstriction. A washout time of ≥10 min was allowed between pharmacological agents (and verified by return to baseline tension). Aortic rings were then exposed to increasing concentrations (10−9–10−5 M) of phenylephrine, and data are represented as percentage of KCl maximal constriction. After a final washout period and return to baseline tension, endothelium-independent relaxation was tested with phenylephrine (10−7 M) preconstriction followed by sodium nitroprusside (10−9–10−5 M).
Histological assessments.
Prior to fixation, both lungs were carefully excised from the chest cavity. One lung was clamped, tied off, removed, and flash-frozen. The remaining lung was fixed with 1 ml of fixative (4% paraformaldehyde), which was kept in the lung with use of a closed-off stopcock attached to the tracheal catheter. The entire lung was submerged in a 4% paraformaldehyde bath for 48–72 h and then processed at the West Virginia University Pathology Core Facility using standardized automated tissue-processing techniques. A pathologist with experience in evaluating mouse lung tissue and blinded to the treatment groups used the following scoring criteria to quantify the presence and severity of abnormalities in fixed lung tissue: 0, none; 1, minimal; 2, mild; 3, moderate; and 4, marked.
Statistical analysis.
Initially, there were 15 mice in each group and no significant group differences in prestudy measures. Despite the appearance of health, it was expected that, over the course of the 8-mo study, some mice would die prematurely due to unexpected or unknown conditions. A total of eight mice died at varying time points during the study due to physical trauma/accidents (n = 4) or unexplained causes (n = 4). Postmortem examination of one mouse revealed nonsuppurative meningoencephalitis with mild hydrocephalus, but necropsies of the remaining three animals with unexplained death did not provide further insight into cause of death. Deaths occurred across all groups, resulting in a final number of mice (with data from all study time points) used for statistical analysis in each group as follows: 13 air, 11 E-cig, and 13 3R4F. Although there were a greater number of deaths in the E-cig group than the other two groups, no obvious pathological cause of death was identified at necropsy in E-cig mice.
Repeated-measures ANOVA was used to assess group and time differences and group × time interactions when multiple measures were obtained from the same animal. ANOVA was utilized for cross-sectional group comparison for single time-point data (e.g., aortic reactivity measured at the final time point). If significant main effects were observed, post hoc Student’s t-test was used to identify individual group differences. Kruskal-Wallis (nonparametric) analysis was used for discontinuous variables (i.e., histological score rankings).
All data were analyzed without imputing missing data, when this concurred. Continuous variables are presented as means ± SE, unless otherwise noted. All analyses were conducted using the StatView software package (version 5.0.01, SAS Institute, Cary, NC). Significance was set with α ≤ 0.05.
RESULTS
Body mass was not different between the groups before exposure (Table 1). 3R4F-exposed mice gained significantly less body mass (P < 0.05) and exhibited a tendency toward lower body mass after the 8-mo exposure than E-cig- and air-exposed mice. Body mass and weight gain between E-cig and air-exposed mice were not different (Table 1).
Table 1.
Mouse mass and echocardiography data
| Groups | ||||
| Air | E-cig | 3R4F | P Value (ANOVA) | |
| n | 8 | 9 | 12 | |
| Body mass, g | ||||
| Pre | 19.6 ± 0.2 | 19.4 ± 0.3 | 19.8 ± 0.3 | 0.59 |
| Post | 28.9 ± 0.7 | 29.8 ± 1.0‡ | 27.1 ± 0.7 | 0.08 |
| Δ | 9.3 ± 0.5 | 10.2 ± 0.9† | 7.4 ± 0.6¶ | 0.02 |
| Heart mass, mg | 115.1 ± 3.2 | 118.5 ± 2.4 | 109.4 ± 2.5 | 0.09 |
| Heart-to-body mass ratio | 4.0 ± 0.1 | 4.2 ± 0.2 | 4.2 ± 0.1 | 0.65 |
| LV mass,a mg | 109 ± 10 | 126 ± 5†¶ | 102 ± 3 | 0.02 |
| LV corrected,a mg | 87 ± 8 | 101 ± 4†¶ | 83 ± 2 | 0.03 |
| Lung, mg | 84.8 ± 4.1 | 97.3 ± 8.7 | 91.2 ± 5.6 | 0.36 |
| Spleen, mg | 84.5 ± 3.7 | 89.3 ± 3.9† | 72.6 ± 2.7* | <0.01 |
| Echocardiography | ||||
| n | 11 | 12 | 13 | |
| Heart rate, beats/min | 461 ± 16 | 472 ± 18 | 477 ± 17 | 0.84 |
| Stroke volume, µl | 31.0 ± 3.0 | 26.9 ± 2.4 | 25.4 ± 2.3 | 0.33 |
| Cardiac output, ml/min | 12.5 ± 1.1 | 12.0 ± 1.4 | 12.8 ± 1.5 | 0.91 |
| Stroke volume-to-body mass ratio, µl/g | 1.10 ± 0.14 | 0.87 ± 0.08 | 0.94 ± 0.09 | 0.34 |
| Cardiac output-to-body mass ratio, ml/min/g | 0.44 ± 0.03 | 0.41 ± 0.05 | 0.47 ± 0.06 | 0.70 |
| FS, % | 24.9 ± 3.2 | 21.4 ± 2.5‡ | 15.2 ± 2.0* | 0.04 |
| EF, % | 72 ± 3 | 63 ± 3¶ | 56 ± 4* | 0.01 |
| Area, mm2 | ||||
| Systole | 8.40 ± 0.79 | 10.00 ± 1.03 | 11.67 ± 0.93 | 0.07 |
| Diastole | 18.61 ± 0.91 | 18.30 ± 1.14 | 19.13 ± 0.81 | 0.83 |
| Volume, µl | ||||
| Systole | 13.5 ± 1.3 | 19.5 ± 2.1 | 20.4 ± 2.6 | 0.07 |
| Diastole | 44.0 ± 3.5 | 43.9 ± 4.3 | 45.9 ± 2.8 | 0.91 |
Values are means ± SE. E-cig, electronic cigarette; EF, ejection fraction; FS, fractional shortening; LV, left ventricle; 3R4F, reference cigarette. Boldface indicates statistical significance.
Values are from echocardiographic analysis.
P < 0.05 vs. air,
P < 0.05 vs. 3R4F,
P < 0.10 vs. air, and
P < 0.10 vs. 3R4F.
Vascular responses.
Assessment of AS revealed no significant change in PWV between the first (prestudy) and halfway (after 4.5 mo of exposure) assessment time points (Fig. 1A) but a nearly threefold greater increase in E-cig- and 3R4F-exposed than air-exposed mice after 8 mo of exposure (P < 0.05, by repeated-measures ANOVA; Fig. 1).
Fig. 1.

B-mode Doppler ultrasound in vivo data from the carotid artery of mice under anesthesia (inhaled isoflurane) before, during (at ~4.5 mo), and after 8 mo of chronic exposure to electronic cigarette (E-cig) vapor and reference tobacco (3R4F) cigarette smoke. A: significant increase in arterial stiffness [measured as pulse wave velocity (PWV)] for E-cig and 3R4F groups following 8-mo exposure. B: significantly greater change in PWV (translating to greater arterial stiffness) after 8 mo in E-cig- and 3R4F-exposed than control (air-exposed) mice. Slight, nonsignificant, rise in PWV in control mice following 8 mo is consistent with the normal aging effect. n = 5–8 mice/group. *P < 0.05 vs. air.
Ex vivo assessment of the aortic vessel constrictor response to phenylephrine (an α1-adrenergic receptor agonist) was greater in E-cig- and 3R4F- than air-exposed mice (P < 0.05; Fig. 2A). The vasodilatory response to methacholine (i.e., a muscarinic receptor agonist) was reduced in E-cig- and 3R4F-exposed mice compared with air-exposed mice (P < 0.05), suggesting endothelium-dependent impaired dilation (Fig. 2B). However, the nitric oxide (NO) donor (sodium nitroprusside), representing a non-endothelial-derived source of NO, dilated aortic rings equally in all groups (Fig. 2C). At the highest respective dosage, the adrenergic constriction response was 52 ± 4% greater (P < 0.05), while the muscarinic vasodilatory effect was 24 ± 2% lower (P < 0.05), in E-cig- than air-exposed (control) mice, demonstrating significant vascular endothelial dysfunction in E-cig-exposed compared with control mice. Like E-cig-exposed mice, 3R4F-exposed mice also exhibited a higher constriction (63 ± 3%) and a lower relaxation (33 ± 2%) response than control mice (P < 0.05).
Fig. 2.
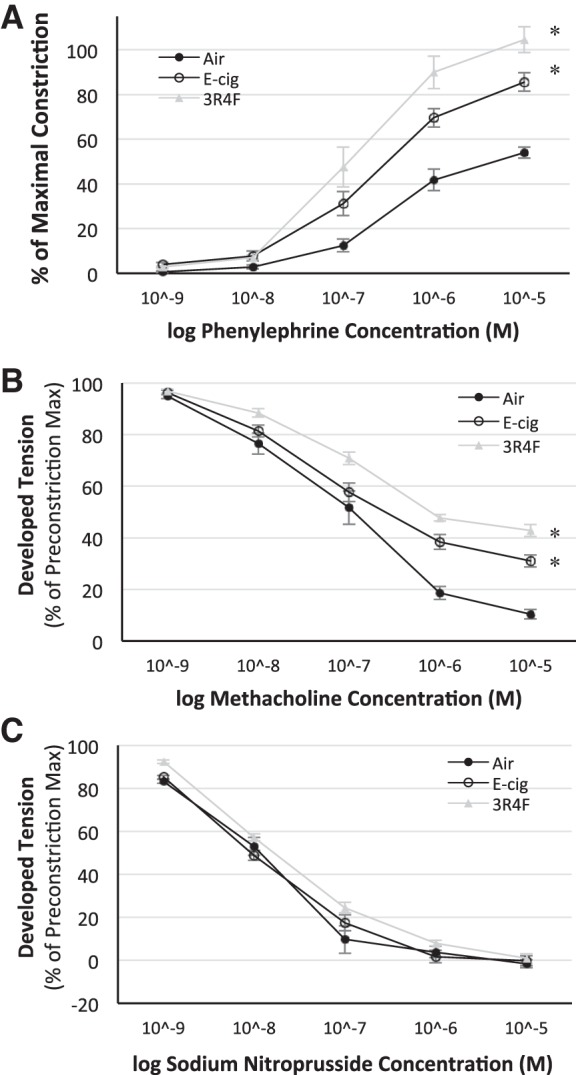
Ex vivo dose-response curves for phenylephrine (A), methacholine (B), and sodium nitroprusside (C) obtained from thoracic aorta ring segments following 8 mo of exposure to E-cig vapor, reference tobacco (3R4F) cigarette smoke, and filtered air. α-Adrenergic vasoconstrictor response was greater (A), while the endothelium-mediated vasodilatory response was impaired (B), following 8 mo of exposure to E-cig vapor and 3R4F cigarette smoke. Non-endothelium-dependent response (C) to sodium nitroprusside was not altered or different between groups, demonstrating that exposure to E-cig vapor and 3R4F smoke resulted in direct harm to endothelial cell-mediated mechanisms. n = 5 mice/group. *P < 0.05 vs. air.
Cardiac responses.
After 8 mo of exposure, whole heart mass was not statistically different but tended to be higher in E-cig- than air- and 3R4F-exposed mice. In support of this finding, the estimation of left ventricular (LV) mass from echocardiographic dimensional measurements revealed greater LV mass for E-cig-exposed than 3R4F-exposed mice (P < 0.05) and a tendency similar to air-exposed mice (P < 0.10; Table 1). Cardiac function from echocardiography revealed no effects on heart rate, stroke volume, and cardiac output but significantly lower fractional shortening (FS%) and ejection fraction (EF%) in 3R4F-exposed mice (P < 0.05), with a similar tendency only for EF% (P < 0.10) in E-cig-exposed mice, compared with air-exposed mice (Table 1).
Pulmonary responses.
Respiratory function assessed by flexiVent measurements (assessing airway reactivity to methacholine challenge) revealed slightly greater respiratory compliance in 3R4F- than E-cig- or air-exposed groups, which is consistent with a developing emphysema phenotype (Fig. 3). Consistent with this finding, we observed 1) a trend (P = 0.08) for peak expiratory flow to be lower in 3R4F- than E-cig- and air-exposed mice, as assessed via whole body plethysmography (Table 2), and 2) histology of the fixed lung tissue demonstrating a higher emphysematous score (air space enlargement) and more pigmented macrophages in the lungs of 3R4F-exposed mice (P < 0.05) but no effect in E-cig-exposed compared with air-exposed mice (Table 3).
Fig. 3.
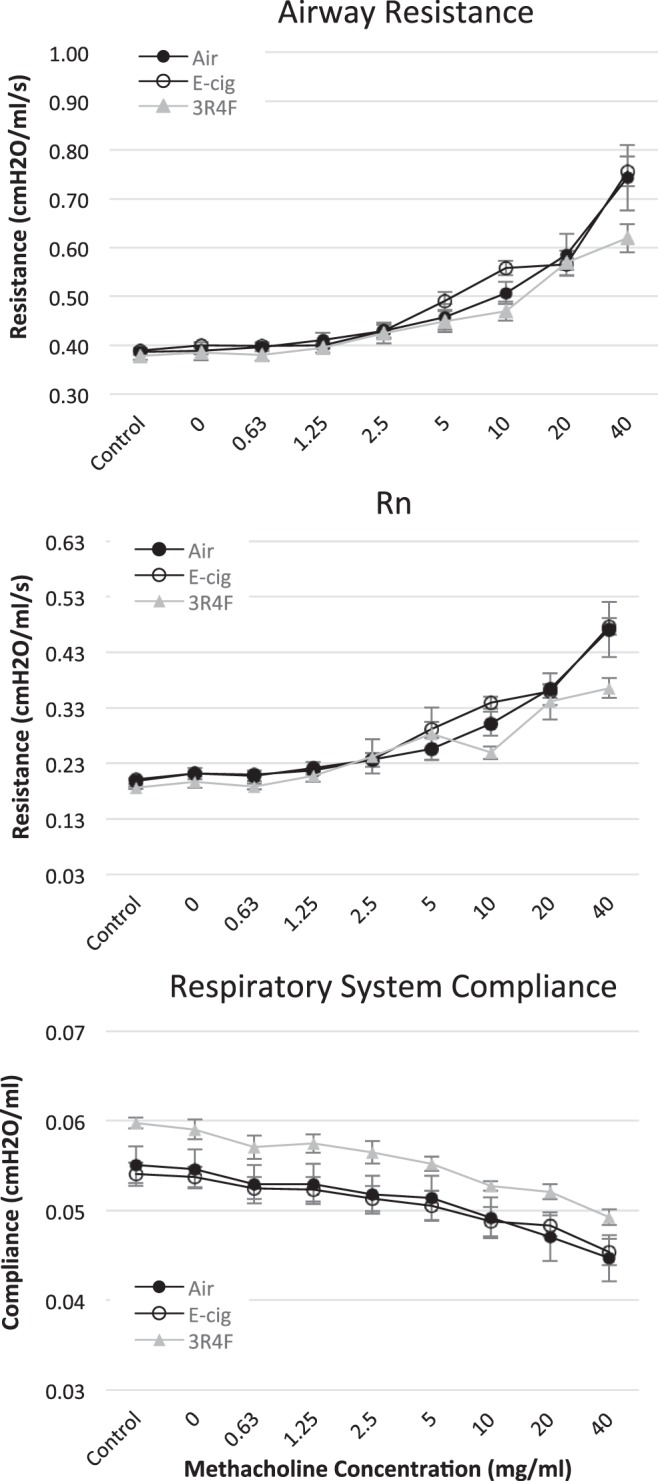
Airway resistance, lung compliance (Rn), and respiratory system compliance in response to inhaled methacholine dose challenge in anesthetized (intraperitoneal ketamine-xylazine) and paralyzed mice on a flexiVent ventilator following 8 mo of exposure to E-cig vapor, reference tobacco (3R4F) smoke, and/or filtered air. n = 8–10 mice/group.
Table 2.
Respiratory function measured in awake, resting, mice at ~8 mo of chronic exposure
| Groups | P Value (ANOVA) | |||
| Air (n = 13) | E-cig (n = 11) | 3R4F (n = 13) | ||
| Frequency, breaths/min | 384 ± 10 | 408 ± 10 | 384 ± 9 | 0.15 |
| Tidal volume, ml | 0.72 ± 0.08 | 0.70 ± 0.04 | 0.59 ± 0.05 | 0.26 |
| Minute ventilation, ml/min | 271 ± 28 | 281 ± 12 | 223 ± 18 | 0.12 |
| Peak inspiratory flow | 16.8 ± 1.4 | 18.1 ± 0.8 | 15.0 ± 0.8 | 0.14 |
| Peak expiratory flow | 17.4 ± 0.6 | 18.6 ± 0.9 | 15.5 ± 1.2 | 0.08 |
Values are means ± SE.
Table 3.
Histological scoring of fixed lung tissue
| Groups | ||||
| Air (n = 12) | E-cig (n = 11) | 3R4F (n = 13) | P Value (Kruskal-Wallis) | |
| Inflammation | ||||
| Acute | 0.0 ± 0.0 | 0.09 ± 0.09 | 0.15 ± 0.10 | 0.805 |
| Chronic | 1.33 ± 0.26 | 1.09 ± 0.16 | 1.62 ± 0.27 | 0.462 |
| Emphysematous changes (air space enlargement) | 0.0 ± 0.0 | 0.18 ± 0.12 | 1.00 ± 0.28*§ | 0.019 |
| Alveolar macrophages | 1.3 ± 0.3 | 1.0 ± 0.0 | 1.6 ± 0.2 | 0.132 |
| Pigmented macrophages | 0.0 ± 0.0 | 0.0 ± 0.0 | 1.69 ± 0.26*§ | 0.0002 |
| Smooth muscle hyperplasia (main stem bronchus) | 0.08 ± 0.08 | 0.27 ± 0.14 | 0.62 ± 0.24 | 0.251 |
| Fibrosis | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.08 ± 0.08 | 0.931 |
Values are means ± SE. Scoring criteria are as follows: 0 = none, 1 = minimal, 2 = mild, 3 = moderate, and 4 = marked. Boldface indicates statistical significance.
P < 0.05 vs. air and
P < 0.05 vs. E-cig.
DISCUSSION
The principal finding of this study is that habitual E-cig use leads to vascular dysfunction, such as a significant increase in AS, reduced vascular relaxation to vasodilators, and enhanced responses to vascular constrictor agents. These findings are associated with the development of CVD (14) and qualitatively relate to other well-known CVD risk factors, including smoking traditional cigarettes.
Vascular and cardiac responses.
The key observation from this study is that even low levels (see below) of exposure to E-cig vapor increased AS and impaired ex vivo vascular responses. The clinical relevance of our findings can be demonstrated by relating the degree of the arterial dysfunction we observed in the present study to other well-known CVD risk factors (Fig. 4). 1) Previous reports indicate that smoking increases central AS from 0.6 to 1.1 m/s (6, 40, 57, 66). In context, a 1-m/s increase in central PWV corresponds to an age-, sex-, and risk factor-adjusted risk increase of 15% in cardiovascular and all-cause mortality (67). Thus the 1.14- and 1.28-m/s increase in AS (i.e., ΔPWV shown in Fig. 1B) we observed in the present study would reflect an ∼17–19% increased risk of all-cause mortality with chronic use of E-cigs and conventional cigarettes in humans. 2) When comparing our results with other studies assessing aortic reactivity in rodents with either overt CVD (e.g., hypertension and atherosclerosis) or known CVD risk factors (e.g., stress, hyperlipidemia, and diabetes), we found that E-cig exposure for 8 mo created a risk for CVD similar to several other well-known risk factors, including smoking (Fig. 4).
Fig. 4.

Magnitude of aortic vascular dysfunction from E-cig vapor compared with other rodent studies (using the same or similar ex vivo methodology) evaluating aortic responses to various knowm cardiovascular disease (CVD) risk factors and/or overt CVD (e.g., atherosclerosis and hypertension). All data represent maximal methacholine dose reported (e.g., 10−5 M) compared with control conditions within each respective study, where controls were set to equal 100 and change in the treatment/disease condition is calculated. UCMS, unpredictable chronic mild stress; IP, intraperotineal; SHR, spontaneously hypertensive rats; HSD, high-salt diet. **Data from the present study.
Because a measurable deficit in AS was only seen in our mice after 4.5 mo of exposure, it might be tempting to think that the same relative duration in humans (i.e., ~15 yr) will be the time frame needed to achieve vascular dysfunction in humans. This notion should be viewed with caution, since it is likely that our mice experienced much lower levels of E-cig vapor than are likely to occur in humans. For chronic smoke chamber studies, daily total particulate matter (TPM) in a range of 100–250 mg/m3 has typically been used to induce chronic obstructive pulmonary disease (COPD) in small animals (20, 39, 69). Our average daily chamber TPM from the E-cigs was much lower [i.e., 59 ± 14 (SD) mg/m3]. This means that our exposure was less than half of the average concentration range typically used to elicit COPD symptoms with cigarette smoke in animal studies (20, 39, 69). Yet the fact that vascular dysfunction was observed, despite the lower exposure level, suggests that the threshold to induce vascular injury may be “very low.” This finding is consistent with evidence showing a marked increase in cardiovascular risk even at “low levels” of cigarette exposure (7, 37). A recent study in humans also reports an acute effect of E-cig exposure (to just 9 puffs) of impaired FMD, a noninvasive measure of endothelium-mediated vascular function linked to NO bioavailability that is frequently used to assess vascular function in humans (10). Given that endothelial dysfunction, even if temporary, can be seen early in atherogenesis (14), we believe that these data collectively suggest that the threshold for damage from E-cigs is likely low with respect to the vascular system, similar to that observed with traditional cigarettes (7, 37).
A potential explanation for increased risk from low-level exposures may be that E-cigs produce elevated levels of particulate matter (PM) in the ultrafine (<100 nm) and PM2.5 (<2.5 μm) range (47, 48). While some studies indicate similar distribution of the particle size from E-cigs and tobacco cigarettes in the submicrometer range (~125–160 nm) (29, 70, 76), there is also evidence that E-cigs deliver more ultrafine (<1-µm) particles (23, 42, 43). Ultrafine and submicrometer particles are more easily brought into and out of the lung and penetrate more deeply than larger (micrometer) particles (42). Nanoparticles also easily traverse the alveolar-capillary interface and gain direct access to vascular endothelial cells and the bloodstream, which could explain the robust and rapid systemic vascular effects that have recently been observed in response to acute E-cig use (10).
One aspect that cannot be addressed by our study is determination of the component(s) in E-cig vapor responsible for mediating these vascular effects. For example, nicotine is known to induce significant effects on the cardiovascular system. In humans and rodents, nicotine increases blood pressure and has been linked to arterial remodeling (75). The arterial response to phenylephrine-induced contraction is greater in nicotine-treated than control rats, and nicotine-treated animals showed impaired endothelium-dependent relaxation to acetylcholine compared with control rats (75), demonstrating that nicotine alone is capable of inducing vascular dysfunction. However, the role of nicotine in CVD risk is controversial, since CVD risk is low (or lower) in individuals who use nicotine medications or smokeless tobacco products compared with active smokers (5). However, very few long-term exposure studies have been conducted with inhaled or aerosolized nicotine, and this route involves less contact with other cells and nicotine metabolism before contact with the vascular endothelium. So, while nicotine replacement therapies do not appear to increase CVD risk, the long-term effects of inhaling nicotine (in the absence of combustion of tobacco) are still poorly understood.
Since nicotine is capable of acutely increasing vascular wall stiffness (due to its effects on the central nervous system), temporal increases in AS that do not reflect vessel remodeling can be observed immediately following acute exposures. However, the changes in AS and ex vivo aortic ring tension observed in our study are not likely related to the acute exposure effects, since all our assessments were made ≥24 h after smoke/vapor exposure. Moreover, the halfway time-point assessment for AS (i.e., PWV; Fig. 1) does not show significant change compared with baseline; therefore, we do not believe that our data are the result of lingering acute effects. Also, the level of urine cotinine (a stable by-product of nicotine used as a biomarker for nicotine due to its short half-life) in our E-cig-exposed mice was almost half that in 3R4F-exposed mice, yet the degree of vascular dysfunction was very similar between the groups. This could suggest that some component of the E-cig liquid (other than nicotine) may have a greater influence on vascular impairment. Further studies are required to elucidate these effects.
Our chronic exposure resulted in small, but statistically significant, decreases in FS% and EF% in 3R4F-exposed mice, with a similar (although not significant) trend for E-cig-exposed mice (P < 0.10; Table 1). Based on these data, this level of E-cig exposure did not result in overt cardiac dysfunction. However, LV mass was greater in the E-cig- than air- or 3R4F-exposed mice. Although this finding can signify cardiac remodeling, its significance is unclear, as reduced cardiac performance was observed only in 3R4F-exposed mice (in which LV mass was not different from controls). It may be tempting to speculate that E-cigs have little impact on cardiac function, but we cannot rule out the possibility that the subtle changes we observed could progress to pathological outcomes with more intense and/or longer exposure. Further research is needed to elucidate such effects, as well as other responses (i.e., development of hypertension and histological assessment of aortic remodeling) that was not determined from our present study.
Respiratory responses.
We observed changes in lung histology (Table 3) and respiratory system compliance (Fig. 4) in 3R4F-exposed, but not E-cig- or air-exposed, mice. The changes in 3R4F-exposed mice (i.e., higher compliance, more pigmented macrophages, and higher emphysema score) are consistent with the well-known effects of cigarette smoke. While our pulmonary findings are consistent with findings from at least one other study that used the C57BL/6 murine model (54), studies using other mouse strains (e.g., A/J or BALB/c) demonstrate that E-cig vapor does induce histological changes and impairment in airway and lung mechanical properties similar to a COPD phenotype (24, 36). Thus it is possible that the lack of pulmonary effects from E-cigs in our study may be due to 1) the selected inbred mouse strain we used and/or 2) the relatively mild TPM exposure in our paradigm (see above). Given growing evidence from cellular and in vivo animal studies, as well as acute studies in humans, showing the toxicity of E-cig vapor in airway cells and respiratory function [see review (16)], we would caution against the interpretation (based on our data) that E-cigs are safe for the lung.
Clinical relevance to humans and study limitations.
Some might argue that intermittent E-cig use for a total of 4 h/day seems too high or unrealistic for the average E-cig user. However, when examining data anonymously volunteered from >180,000 Evolv DNA-series E-cig devices via the ECigStats data collection program (www.ecigstats.org, accessed April 6, 2017) that records user usage characteristics, we found that the average number of puffs per day reported across all devices is 172 ± 131 (SD). This is actually higher than the 152–156 puffs/day used in this study (38–39 puffs per hour × 4 h). However, the ECigStats data also report that devices/users have an average puff duration time of 2.31 ± 2.11 (SD) s. The nearly identical mean and SD of the puff duration (2.31 and 2.11, respectively) indicates a wide variability in individual usage characteristics, with nearly one-third of the average users adopting long (up to 4.4-s) puff durations. Our E-cig usage characteristics (i.e., 5-s, 55-ml puff with the device set at 4.9 V, 14.1 W) are actually similar to those of several recent scientifically controlled studies examining E-cig puff topography, showing that the average “experienced” E-cig user adopts longer (e.g., 4–8 s) puff durations (61, 63). Moreover, an “average-experienced” E-cig user (with a 4-s puff duration) is reported to generate 29.4–152.7 mg of TPM, depending on puff velocity and voltage (i.e., 3.3 vs. 5.2 V, respectively), but an “extremely experienced” E-cig user (with an 8-s puff duration) can generate 68.8–333.2 mg of TPM (63). So, when comparing the total daily TPM achieved by experienced E-cig users with our chamber exposure, we find that the mice (with a daily TPM average of 59 mg) received the lower end of TPM compared with most experienced E-cig users. Thus, despite the similarities in E-cig topology between our animal exposure and human usage, it is possible that our exposure paradigm underestimates the effect that will be experienced by a human E-cig user. Our use of a chamber exposure paradigm may also have a dampening effect on our outcomes, because rodents are obligate nasal breathers, and the nose can effectively filter many airborne particles compared with direct inhalation via the mouth (as humans would experience). Together with the caveat that our animals experienced a lower level of TPM exposure than the average E-cig user, this could also potentially explain why we saw minimal pulmonary abnormalities in our study. Nevertheless, the mice developed significant vascular dysfunction, suggesting that the degree of vascular dysfunction may not be substantially different between E-cigs and conventional cigarettes. The fact that we observed minimal cardiac and pulmonary changes could mean that these organs have a greater threshold and/or resilience to functional impairments and that endothelial and vascular dysfunction is simply the first step and harbinger in the etiology of CVD (15, 25, 49).
An additional clinical consideration is that we studied only female mice. The chamber exposure method we used necessitated the use of female mice to reduce fighting and injury when mice (otherwise housed in separate cages) were temporarily grouped communally for several hours each day during the exposures. We do not know if male mice would have exhibited the same level of vascular dysfunction. Consistent with human studies (53), rodent studies have also found that females are less sensitive than males to the pharmacological effects of nicotine (27, 58), in part because of protective effects of female hormones (8). Thus one could speculate that female mice may have a dampened response and that E-cig exposure in males may result in greater CVD risk and worse outcomes (3, 8). Future studies must include both sexes in these evaluations.
A broad concern is the relevance of E-cig-related cardiovascular responses in mice compared with humans, especially given the potential difference we have noted with respect to mouse strains. Indeed, it is too early to know if vaping effects in rodents will faithfully recapitulate those ultimately seen in humans. Given the wide variations and options possible with E-cig vapor exposures (i.e., varying quality of devices, different device settings, and varying concentration of constituents in the E-cig liquid and/or formulation of the base solution), it is likely that varying and, possibly, divergent outcomes may be observed, depending on the exposure paradigm. However, broadly speaking, we would emphasize that decades of evidence from cigarette smoking demonstrate good fidelity with pulmonary and cardiovascular outcomes observed between rodents and humans (22, 71, 72). We would also suggest, even given the limited evidence that currently exists, from the preponderance of data obtained from interventional studies (including this study), that it seems counterintuitive to believe or expect that long-term use of E-cigs will likely prove to be safe in terms of overall human health.
Conclusions.
To our knowledge, these are the first interventional data to show the long-term cardiovascular health consequences of chronic E-cig use. While our exposure paradigm resulted in no significant changes in the pulmonary outcomes we measured in E-cig-exposed mice and only a small change in cardiac function, we caution that the potential interpretation that E-cigs are safe is likely short-sighted, especially given the relatively low level of exposure with our exposure paradigm. Rather, despite the relatively low daily exposure level, we found significant increases in AS and adrenergic-mediated vasoconstriction and impaired endothelium-dependent vasodilation. These data indicate significant vascular dysfunction induced by E-cig vapor exposure. Based on existing evidence, the level of vascular dysfunction is similar to that observed for other known risk factors leading to CVD, including smoking tobacco cigarettes.
The clinical implication is that chronic use of E-cigs impairs vascular function. Future animal studies will be able to determine the potential time-course effects of varying exposure/usage levels and the contribution of specific components of the E-cig liquid/vapor (e.g., carbonyl compounds, nicotine, and flavorings) to the etiology toward arterial dysfunction. These data should be viewed as a harbinger of the potential effects on humans, such that E-cig use should not be considered safe and perhaps even questionable as a harm-reduction device, given the low potential threshold for inducing vascular injury. Diligent clinical monitoring of vascular health should be encouraged in adolescent and adult E-cig users.
GRANTS
This work was supported by a West Virginia University/Marshall University Health Cooperative Initiative Award (I. M. Olfert and P. Dasgupta); National Institute of General Medical Sciences Grants P20 GM-103434 (West Virginia IDeA Network for Biomedical Research Excellence), U54 GM-104942 (West Virginia Clinical Translational Science Institute), and P20 GM-109098 (P. D. Chantler), and American Heart Association Grant 16PRE30820000 (E. DeVallance).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.M.O., Z.-X.W., P.K., B.H.E., R.W.B., and P.D. conceived and designed research; I.M.O., E.D., H.H., K.W.B., S.C., C.R.P., D.P.S., M.J.B., Z.-X.W., W.K.M., B.H.E., B.S.D., and P.D.C. analyzed data; I.M.O., E.D., K.W.B., D.P.S., M.J.B., Z.-X.W., W.K.M., B.S.D., R.W.B., and P.D.C. interpreted results of experiments; I.M.O. and E.D. prepared figures; I.M.O. drafted manuscript; I.M.O. and P.D.C. edited and revised manuscript; I.M.O., E.D., H.H., K.W.B., S.C., C.R.P., D.P.S., M.J.B., Z.-X.W., P.K., W.K.M., B.H.E., B.S.D., R.W.B., P.D., and P.D.C. approved final version of manuscript; E.D., H.H., K.W.B., S.C., C.R.P., M.J.B., and B.H.E. performed experiments.
ACKNOWLEDGMENTS
We are grateful to Dr. Eiman Aboaziza, Mireia Fabrega, Morgan Pelley, Richard Nolan, and Gregory Ede for help in conducting the daily exposure. We also thank Dr. Sarah McLaughlin for help with echocardiographic data collection and analysis and Lennie Samsell (Pediatrics Research) for advice regarding flexiVent measurements. We thank Dr. Clay Marsh for advice and critique of the manuscript.
REFERENCES
- 1.Anonymous E-Cigarettes Use Among Youth and Young Adults: A Report of the Surgeon General. Atlanta, GA: US Dept. of Health and Human Services, 2016. [Google Scholar]
- 2.Anonymous The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. Atlanta, GA: US Dept. of Health and Human Services, 2014. [Google Scholar]
- 3.Barrett-Connor E, Wingard DL. Sex differential in ischemic heart disease mortality in diabetics: a prospective population-based study. Am J Epidemiol 118: 489–496, 1983. doi: 10.1093/oxfordjournals.aje.a113654. [DOI] [PubMed] [Google Scholar]
- 6.Bekki K, Uchiyama S, Ohta K, Inaba Y, Nakagome H, Kunugita N. Carbonyl compounds generated from electronic cigarettes. Int J Environ Res Public Health 11: 11192–11200, 2014. doi: 10.3390/ijerph111111192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benowitz NL, Burbank AD. Cardiovascular toxicity of nicotine: implications for electronic cigarette use. Trends Cardiovasc Med 26: 515–523, 2016. doi: 10.1016/j.tcm.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binder S, Navratil K, Halek J. Chronic smoking and its effect on arterial stiffness. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub 152: 299–302, 2008. doi: 10.5507/bp.2008.047. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn H. Environmental tobacco smoke exposure was associated with an increased risk of ischemic heart disease. Evid Based Cardiovasc Med 2: 43–44, 1998. doi: 10.1016/S1361-2611(98)80084-5. [DOI] [PubMed] [Google Scholar]
- 8.Bolego C, Poli A, Paoletti R. Smoking and gender. Cardiovasc Res 53: 568–576, 2002. doi: 10.1016/S0008-6363(01)00520-X. [DOI] [PubMed] [Google Scholar]
- 9.Caponnetto P, Campagna D, Cibella F, Morjaria JB, Caruso M, Russo C, Polosa R. EffiCiency and Safety of an eLectronic cigAreTte (ECLAT) as tobacco cigarettes substitute: a prospective 12-month randomized control design study. PLoS One 8: e66317, 2013. doi: 10.1371/journal.pone.0066317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carnevale R, Sciarretta S, Violi F, Nocella C, Loffredo L, Perri L, Peruzzi M, Marullo AG, De Falco E, Chimenti I, Valenti V, Biondi-Zoccai G, Frati G. Acute impact of tobacco vs. electronic cigarette smoking on oxidative stress and vascular function. Chest 150: 606–612, 2016. doi: 10.1016/j.chest.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention QuickStats: number of deaths from 10 leading causes—National Vital Statistics System, United States, 2010. Morb Mortal Wkly Rep 62:155, 2013. [Google Scholar]
- 14.Cheng KS, Baker CR, Hamilton G, Hoeks AP, Seifalian AM. Arterial elastic properties and cardiovascular risk/event. Eur J Vasc Endovasc Surg 24: 383–397, 2002. doi: 10.1053/ejvs.2002.1756. [DOI] [PubMed] [Google Scholar]
- 15.Chow B, Rabkin SW. The relationship between arterial stiffness and heart failure with preserved ejection fraction: a systemic meta-analysis. Heart Fail Rev 20: 291–303, 2015. doi: 10.1007/s10741-015-9471-1. [DOI] [PubMed] [Google Scholar]
- 16.Chun LF, Moazed F, Calfee CS, Matthay MA, Gotts JE. Pulmonary toxicity of e-cigarettes. Am J Physiol Lung Cell Mol Physiol 313: L193–L206, 2017. doi: 10.1152/ajplung.00071.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cooke WH, Pokhrel A, Dowling C, Fogt DL, Rickards CA. Acute inhalation of vaporized nicotine increases arterial pressure in young non-smokers: a pilot study. Clin Auton Res 25: 267–270, 2015. doi: 10.1007/s10286-015-0304-z. [DOI] [PubMed] [Google Scholar]
- 18.El Dib R, Suzumura EA, Akl EA, Gomaa H, Agarwal A, Chang Y, Prasad M, Ashoorion V, Heels-Ansdell D, Maziak W, Guyatt G. Electronic nicotine delivery systems and/or electronic non-nicotine delivery systems for tobacco smoking cessation or reduction: a systematic review and meta-analysis. BMJ Open 7: e012680, 2017. doi: 10.1136/bmjopen-2016-012680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farsalinos KE, Romagna G, Tsiapras D, Kyrzopoulos S, Voudris V. Characteristics, perceived side effects and benefits of electronic cigarette use: a worldwide survey of more than 19,000 consumers. Int J Environ Res Public Health 11: 4356–4373, 2014. doi: 10.3390/ijerph110404356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Finch GL, Lundgren DL, Barr EB, Chen BT, Griffith WC, Hobbs CH, Hoover MD, Nikula KJ, Mauderly JL. Chronic cigarette smoke exposure increases the pulmonary retention and radiation dose of 239Pu inhaled as 239PuO2 by F344 rats. Health Phys 75: 597–609, 1998. doi: 10.1097/00004032-199812000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Fleenor BS, Eng JS, Sindler AL, Pham BT, Kloor JD, Seals DR. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell 13: 576–578, 2014. doi: 10.1111/acel.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fricker M, Deane A, Hansbro PM. Animal models of chronic obstructive pulmonary disease. Expert Opin Drug Discov 9: 629–645, 2014. doi: 10.1517/17460441.2014.909805. [DOI] [PubMed] [Google Scholar]
- 23.Fuoco FC, Buonanno G, Stabile L, Vigo P. Influential parameters on particle concentration and size distribution in the mainstream of e-cigarettes. Environ Pollut 184: 523–529, 2014. doi: 10.1016/j.envpol.2013.10.010. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Arcos I, Geraghty P, Baumlin N, Campos M, Dabo AJ, Jundi B, Cummins N, Eden E, Grosche A, Salathe M, Foronjy R. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 71: 1119–1129, 2016. doi: 10.1136/thoraxjnl-2015-208039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giamouzis G, Schelbert EB, Butler J. Growing evidence linking microvascular dysfunction with heart failure with preserved ejection fraction. J Am Heart Assoc doi: 10.1161/JAHA.116.003259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goineau S, Rompion S, Guillaume P, Picard S. Ventilatory function assessment in safety pharmacology: optimization of rodent studies using normocapnic or hypercapnic conditions. Toxicol Appl Pharmacol 247: 191–197, 2010. doi: 10.1016/j.taap.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 27.Hatchell PC, Collins AC. The influence of genotype and sex on behavioral sensitivity to nicotine in mice. Psychopharmacology (Berl) 71: 45–49, 1980. doi: 10.1007/BF00433251. [DOI] [PubMed] [Google Scholar]
- 28.Hwang JH, Lyes M, Sladewski K, Enany S, McEachern E, Mathew DP, Das S, Moshensky A, Bapat S, Pride DT, Ongkeko WM, Crotty Alexander LE. Electronic cigarette inhalation alters innate immunity and airway cytokines while increasing the virulence of colonizing bacteria. J Mol Med (Berl) 94: 667–679, 2016. doi: 10.1007/s00109-016-1378-3. [DOI] [PubMed] [Google Scholar]
- 29.Ingebrethsen BJ, Cole SK, Alderman SL. Electronic cigarette aerosol particle size distribution measurements. Inhal Toxicol 24: 976–984, 2012. doi: 10.3109/08958378.2012.744781. [DOI] [PubMed] [Google Scholar]
- 30.Jensen RP, Luo W, Pankow JF, Strongin RM, Peyton DH. Hidden formaldehyde in e-cigarette aerosols. N Engl J Med 372: 392–394, 2015. doi: 10.1056/NEJMc1413069. [DOI] [PubMed] [Google Scholar]
- 31.Johnston RA, Schwartzman IN, Flynt L, Shore SA. Role of interleukin-6 in murine airway responses to ozone. Am J Physiol Lung Cell Mol Physiol 288: L390–L397, 2005. doi: 10.1152/ajplung.00007.2004. [DOI] [PubMed] [Google Scholar]
- 32.Kalkhoran S, Glantz SA. E-cigarettes and smoking cessation in real-world and clinical settings: a systematic review and meta-analysis. Lancet Respir Med 4: 116–128, 2016. doi: 10.1016/S2213-2600(15)00521-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khoudigian S, Devji T, Lytvyn L, Campbell K, Hopkins R, O’Reilly D. The efficacy and short-term effects of electronic cigarettes as a method for smoking cessation: a systematic review and a meta-analysis. Int J Public Health 61: 257–267, 2016. doi: 10.1007/s00038-016-0786-z. [DOI] [PubMed] [Google Scholar]
- 34.Kogel U, Schlage WK, Martin F, Xiang Y, Ansari S, Leroy P, Vanscheeuwijck P, Gebel S, Buettner A, Wyss C, Esposito M, Hoeng J, Peitsch MC. A 28-day rat inhalation study with an integrated molecular toxicology endpoint demonstrates reduced exposure effects for a prototypic modified risk tobacco product compared with conventional cigarettes. Food Chem Toxicol 68: 204–217, 2014. doi: 10.1016/j.fct.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 35.Kosmider L, Sobczak A, Fik M, Knysak J, Zaciera M, Kurek J, Goniewicz ML. Carbonyl compounds in electronic cigarette vapors: effects of nicotine solvent and battery output voltage. Nicotine Tob Res 16: 1319–1326, 2014. doi: 10.1093/ntr/ntu078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larcombe AN, Janka MA, Mullins BJ, Berry LJ, Bredin A, Franklin PJ. The effects of electronic cigarette aerosol exposure on inflammation and lung function in mice. Am J Physiol Lung Cell Mol Physiol 313: L67–L79, 2017. doi: 10.1152/ajplung.00203.2016. [DOI] [PubMed] [Google Scholar]
- 37.Law MR, Wald NJ. Environmental tobacco smoke and ischemic heart disease. Prog Cardiovasc Dis 46: 31–38, 2003. doi: 10.1016/S0033-0620(03)00078-1. [DOI] [PubMed] [Google Scholar]
- 38.Leberl M, Kratzer A, Taraseviciene-Stewart L. Tobacco smoke induced COPD/emphysema in the animal model—are we all on the same page? Front Physiol 4: 91, 2013. doi: 10.3389/fphys.2013.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee KM, Renne RA, Harbo SJ, Clark ML, Johnson RE, Gideon KM. 3-week inhalation exposure to cigarette smoke and/or lipopolysaccharide in AKR/J mice. Inhal Toxicol 19: 23–35, 2007. doi: 10.1080/08958370600985784. [DOI] [PubMed] [Google Scholar]
- 40.Lemogoum D, Van Bortel L, Leeman M, Degaute JP, van de Borne P. Ethnic differences in arterial stiffness and wave reflections after cigarette smoking. J Hypertens 24: 683–689, 2006. doi: 10.1097/01.hjh.0000217850.87960.16. [DOI] [PubMed] [Google Scholar]
- 41.Lerner CA, Rutagarama P, Ahmad T, Sundar IK, Elder A, Rahman I. Electronic cigarette aerosols and copper nanoparticles induce mitochondrial stress and promote DNA fragmentation in lung fibroblasts. Biochem Biophys Res Commun 477: 620–625, 2016. doi: 10.1016/j.bbrc.2016.06.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manigrasso M, Buonanno G, Fuoco FC, Stabile L, Avino P. Aerosol deposition doses in the human respiratory tree of electronic cigarette smokers. Environ Pollut 196: 257–267, 2015. doi: 10.1016/j.envpol.2014.10.013. [DOI] [PubMed] [Google Scholar]
- 43.Manigrasso M, Buonanno G, Fuoco FC, Stabile L, Avino P. Electronic cigarettes: age-specific generation-resolved pulmonary doses. Environ Sci Pollut Res Int 24: 13068–13079, 2017. doi: 10.1007/s11356-017-8914-8. [DOI] [PubMed] [Google Scholar]
- 44.Manzoli L, Flacco ME, Ferrante M, La Vecchia C, Siliquini R, Ricciardi W, Marzuillo C, Villari P, Fiore M; ISLESE Working Group . Cohort study of electronic cigarette use: effectiveness and safety at 24 months. Tob Control 26: 284–292, 2017. doi: 10.1136/tobaccocontrol-2015-052822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McNeil A, Brose L, Calder R, Hitchamn S. E-Cigarettes: An Evidence Update. A Report Commissioned by Public Health England. London, UK: Public Health England, 2015. [Google Scholar]
- 46.McRobbie H, Bullen C, Hartmann-Boyce J, Hajek P. Electronic cigarettes for smoking cessation and reduction. Cochrane Database Syst Rev CD010216, 2014. [DOI] [PubMed] [Google Scholar]
- 47.Melstrom P, Koszowski B, Thanner MH, Hoh E, King B, Bunnell R, McAfee T. Measuring PM2.5, ultrafine particles, nicotine air and wipe samples following the use of electronic cigarettes. Nicotine Tob Res 19: 1055–1061, 2017. doi: 10.1093/ntr/ntx058. [DOI] [PubMed] [Google Scholar]
- 48.Mikheev VB, Brinkman MC, Granville CA, Gordon SM, Clark PI. Real-time measurement of electronic cigarette aerosol size distribution and metals content analysis. Nicotine Tob Res 18: 1895–1902, 2016. doi: 10.1093/ntr/ntw128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mitchell GF. Arterial stiffness and hypertension: chicken or egg? Hypertension 64: 210–214, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moheimani RS, Bhetraratana M, Yin F, Peters KM, Gornbein J, Araujo JA, Middlekauff HR. Increased cardiac sympathetic activity and oxidative stress in habitual electronic cigarette users: implications for cardiovascular risk. JAMA Cardiol 2: 278–284, 2017. doi: 10.1001/jamacardio.2016.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morgan EE, Casabianca AB, Khouri SJ, Kalinoski ALN. In vivo assessment of arterial stiffness in the isoflurane anesthetized spontaneously hypertensive rat. Cardiovasc Ultrasound 12: 37, 2014. doi: 10.1186/1476-7120-12-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perkins KA, Donny E, Caggiula AR. Sex differences in nicotine effects and self-administration: review of human and animal evidence. Nicotine Tob Res 1: 301–315, 1999. doi: 10.1080/14622299050011431. [DOI] [PubMed] [Google Scholar]
- 54.Phillips B, Veljkovic E, Peck MJ, Buettner A, Elamin A, Guedj E, Vuillaume G, Ivanov NV, Martin F, Boué S, Schlage WK, Schneider T, Titz B, Talikka M, Vanscheeuwijck P, Hoeng J, Peitsch MC. A 7-month cigarette smoke inhalation study in C57BL/6 mice demonstrates reduced lung inflammation and emphysema following smoking cessation or aerosol exposure from a prototypic modified risk tobacco product. Food Chem Toxicol 80: 328–345, 2015. doi: 10.1016/j.fct.2015.03.009. [DOI] [PubMed] [Google Scholar]
- 55.Polosa R. Electronic cigarette use and harm reversal: emerging evidence in the lung. BMC Med 13: 54, 2015. doi: 10.1186/s12916-015-0298-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rahman MA, Hann N, Wilson A, Mnatzaganian G, Worrall-Carter L. E-cigarettes and smoking cessation: evidence from a systematic review and meta-analysis. PLoS One 10: e0122544, 2015. doi: 10.1371/journal.pone.0122544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rehill N, Beck CR, Yeo KR, Yeo WW. The effect of chronic tobacco smoking on arterial stiffness. Br J Clin Pharmacol 61: 767–773, 2006. doi: 10.1111/j.1365-2125.2006.02630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schechter MD, Rosecrans JA. C.N.S. effect of nicotine as the discriminative stimulus for the rat in a T-maze. Life Sci I 10: 821–832, 1971. doi: 10.1016/0024-3205(71)90037-3. [DOI] [PubMed] [Google Scholar]
- 59.Schober W, Szendrei K, Matzen W, Osiander-Fuchs H, Heitmann D, Schettgen T, Jörres RA, Fromme H. Use of electronic cigarettes (e-cigarettes) impairs indoor air quality and increases FeNO levels of e-cigarette consumers. Int J Hyg Environ Health 217: 628–637, 2014. doi: 10.1016/j.ijheh.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Sleiman M, Logue JM, Montesinos VN, Russell ML, Litter MI, Gundel LA, Destaillats H. Emissions from electronic cigarettes: key parameters affecting the release of harmful chemicals. Environ Sci Technol 50: 9644–9651, 2016. doi: 10.1021/acs.est.6b01741. [DOI] [PubMed] [Google Scholar]
- 61.Spindle TR, Breland AB, Karaoghlanian NV, Shihadeh AL, Eissenberg T. Preliminary results of an examination of electronic cigarette user puff topography: the effect of a mouthpiece-based topography measurement device on plasma nicotine and subjective effects. Nicotine Tob Res 17: 142–149, 2015. doi: 10.1093/ntr/ntu186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sussan TE, Gajghate S, Thimmulappa RK, Ma J, Kim JH, Sudini K, Consolini N, Cormier SA, Lomnicki S, Hasan F, Pekosz A, Biswal S. Exposure to electronic cigarettes impairs pulmonary anti-bacterial and anti-viral defenses in a mouse model. PLoS One 10: e0116861, 2015. doi: 10.1371/journal.pone.0116861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Talih S, Balhas Z, Eissenberg T, Salman R, Karaoghlanian N, El Hellani A, Baalbaki R, Saliba N, Shihadeh A. Effects of user puff topography, device voltage, and liquid nicotine concentration on electronic cigarette nicotine yield: measurements and model predictions. Nicotine Tob Res 17: 150–157, 2015. doi: 10.1093/ntr/ntu174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tanaka N, Dalton N, Mao L, Rockman HA, Peterson KL, Gottshall KR, Hunter JJ, Chien KR, Ross J Jr. Transthoracic echocardiography in models of cardiac disease in the mouse. Circulation 94: 1109–1117, 1996. doi: 10.1161/01.CIR.94.5.1109. [DOI] [PubMed] [Google Scholar]
- 65.Vardavas CI, Anagnostopoulos N, Kougias M, Evangelopoulou V, Connolly GN, Behrakis PK. Short-term pulmonary effects of using an electronic cigarette: impact on respiratory flow resistance, impedance, and exhaled nitric oxide. Chest 141: 1400–1406, 2012. doi: 10.1378/chest.11-2443. [DOI] [PubMed] [Google Scholar]
- 66.Vlachopoulos C, Alexopoulos N, Panagiotakos D, O’Rourke MF, Stefanadis C. Cigar smoking has an acute detrimental effect on arterial stiffness. Am J Hypertens 17: 299–303, 2004. doi: 10.1016/j.amjhyper.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 67.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol 55: 1318–1327, 2010. doi: 10.1016/j.jacc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 68.Vlachopoulos C, Ioakeimidis N, Abdelrasoul M, Terentes-Printzios D, Georgakopoulos C, Pietri P, Stefanadis C, Tousoulis D. Electronic cigarette smoking increases aortic stiffness and blood pressure in young smokers. J Am Coll Cardiol 67: 2802–2803, 2016. doi: 10.1016/j.jacc.2016.03.569. [DOI] [PubMed] [Google Scholar]
- 69.Weissmann N, Lobo B, Pichl A, Parajuli N, Seimetz M, Puig-Pey R, Ferrer E, Peinado VI, Domínguez-Fandos D, Fysikopoulos A, Stasch JP, Ghofrani HA, Coll-Bonfill N, Frey R, Schermuly RT, García-Lucio J, Blanco I, Bednorz M, Tura-Ceide O, Tadele E, Brandes RP, Grimminger J, Klepetko W, Jaksch P, Rodriguez-Roisin R, Seeger W, Grimminger F, Barberà JA. Stimulation of soluble guanylate cyclase prevents cigarette smoke-induced pulmonary hypertension and emphysema. Am J Respir Crit Care Med 189: 1359–1373, 2014. doi: 10.1164/rccm.201311-2037OC. [DOI] [PubMed] [Google Scholar]
- 70.Williams M, Villarreal A, Bozhilov K, Lin S, Talbot P. Metal and silicate particles including nanoparticles are present in electronic cigarette cartomizer fluid and aerosol. PLoS One 8: e57987, 2013. doi: 10.1371/journal.pone.0057987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wright JL, Churg A. Animal models of cigarette smoke-induced COPD. Chest 122 Suppl: 301S–306S, 2002. doi: 10.1378/chest.122.6_suppl.301S. [DOI] [PubMed] [Google Scholar]
- 72.Wright JL, Cosio M, Churg A. Animal models of chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol 295: L1–L15, 2008. doi: 10.1152/ajplung.90200.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu Q, Jiang D, Minor M, Chu HW. Electronic cigarette liquid increases inflammation and virus infection in primary human airway epithelial cells. PLoS One 9: e108342, 2014. doi: 10.1371/journal.pone.0108342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu V, Rahimy M, Korrapati A, Xuan Y, Zou AE, Krishnan AR, Tsui T, Aguilera JA, Advani S, Crotty Alexander LE, Brumund KT, Wang-Rodriguez J, Ongkeko WM. Electronic cigarettes induce DNA strand breaks and cell death independently of nicotine in cell lines. Oral Oncol 52: 58–65, 2016. doi: 10.1016/j.oraloncology.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zainalabidin S, Budin SB, Ramalingam A, Lim YC. Aortic remodelling in chronic nicotine-administered rat. Korean J Physiol Pharmacol 18: 411–418, 2014. doi: 10.4196/kjpp.2014.18.5.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang Y, Sumner W, Chen DR. In vitro particle size distributions in electronic and conventional cigarette aerosols suggest comparable deposition patterns. Nicotine Tob Res 15: 501–508, 2013. doi: 10.1093/ntr/nts165. [DOI] [PubMed] [Google Scholar]