

Abstract
Systemic inflammation in breast cancer correlates with poor prognosis but the molecular underpinnings of this connection are not well understood. In this study, we explored the relationship between HER2 overexpression, inflammation and expansion of the mammary stem/progenitor and cancer stem-like cell (CSC) population in breast cancer. HER2-positive epithelial cells initiated and sustained an inflammatory milieu needed to promote tumorigenesis. HER2 induced a feed-forward activation loop of IL-1α and IL-6 that stimulated NF-κB and STAT3 pathways for generation and maintenance of breast CSC. In mice, Il1a genetic deficiency delayed MMTV-Her2-induced tumorigenesis and reduced inflammatory cytokine expression as well as CSC in primary tumors. In clinical specimens of human breast tumor tissues, tissue microarray analysis revealed a strong positive correlation between IL-1α/IL-6 expression and CSC-positive phenotype. Pharmacologic blockade of IL-1α signaling reduced the CSC population and improved chemotherapeutic efficacy. Our findings suggest new therapeutic or prevention strategies for HER2-positive breast cancers.
Keywords: HER2, Inflammation, Cancer stem cells, Interleukin 1α, Interleukin 6
Introduction
Considerable clinical evidence have established a strong association between inflammation states and cancer development (1). Furthermore, levels of chronic inflammation are correlated with the risk of breast cancer recurrence after primary therapy (2). Inflammatory cytokines have been shown to link oncogenic signaling to generation and maintenance of cancer stem cells (CSCs) (3,4). Human epidermal growth factor receptor 2 (HER2) is overexpressed in about 25% of all breast cancers and is associated with an aggressive behavior, high rate of relapse, and poor prognosis (5). MMTV-Her2 transgenic animal developed tumors with inflammatory patterns based on gene expression profiling that corresponded to the proinflammatory gene expression patterns found in human tumors (6,7). Several in vitro studies have demonstrated that HER2 overexpression in breast cancer cells increased IL-6 production (8). Consistently, Korkaya et al. concluded that activation of an IL-6-NF-κB inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the CSC population (9). It has been reported that HER2 downstream PI3-kinase/AKT pathway is activated in the putative CSC subpopulations of several HER2 positive cell lines (10). It seems to be logical to propose that HER2 activates the IL-6-NF-κB signaling loop via its canonical downstream PI3/AKT pathway in breast CSCs. In contrast, there is evidence suggesting that HER2 activates NF-κB independent of the PI3/AKT pathway in breast cancer cells (11). In animal models, the canonical NF-κB pathway governs HER2-induced tumorigenesis and CSC expansion. However, the precise mechanisms to link HER2-induced inflammation and tumorigenesis in vivo have not been established.
In this study, we found that HER2 overexpression induced IL-1α secretion to trigger cancer-cell driven inflammatory circuits, which is required for sequential activation of NF-κB-IL6-STAT3 axis for generation and maintenance of CSCs both in vitro and in vivo. Given that the feed-forward inflammation loop can be sustained by small percentages of HER2-positive breast cancer and even surrounding normal epithelial cells, our study may provide an alternative explanation for the efficacy of HER2-blocking agents in HER2-negative breast cancer cells classified by the arbitrary cutoff (12,13).
Materials and Methods
The detailed procedures of plasmid construction, cell culture, antibody and immunoblot, flow cytometry, microarray analysis, qRT-PCR, ChIP-qPCR, shRNA-mediated knockdown, luciferase reporter assay, microarray, tumorsphere formation assay, CRISPR/Cas9-mediated genomic editing, immunochemical staining, animal injections as well as key reagents are described in Supplemental Experimental Procedures.
Cell lines
MCF10A, MCF7 and HCC1954 were purchased from ATCC in 2013. MCF10A-vector and HER2 cell lines were provided by Dr. Shizhen Emily Wang (University of California at San Diego). MDA-MB361 cells were obtained from Dr. Saraswati Sukuma, Johns Hopkins University, in 2008. Cells were grown under standard conditions. Cell lines were tested for Mycoplasma via PCR using the Universal Mycoplasma Detection Kit (ATCC).
Animals
All animal experiments were approved by the institutional animal care and use committee at the University of South Carolina. Il1a−/− mice (14) were backcrossed to wild type FVB/N mice for at least 6 generations and then crossed to N-Tg(MMTV-Neu) 202Mul/J mice (JAX lab) to generate MMTV-Her2/Il1a+/− mice. The MMTV-Her2/Il1a+/− mice were intercrossed to generate MMTV-Her2/Il1a+/+, MMTV-Her2/Il1a+/− and MMTV-Her2/Il1a−/− mice.
Tissue microarray
TMAs used de-identified tumor samples and were considered exempt by the Institutional Review Boards of the University of South Carolina. The tissue array samples from breast cancer patients were provided by the Chonnam National University Hwasun Hospital National Biobank of Korea, which is supported by the Ministry of Health, Welfare and Family Affairs. Characterization of clinicopathological features of patients was based on the Cancer Staging System from the American Joint Committee (Table S1). The core tissue biopsies exhibiting carcinoma with a diameter of 2 mm were punched from individual donor paraffin-embedded tissue blocks and precisely arrayed into a new recipient block. Four-micron sections of TMA blocks were cut and used for IHC analysis.
Statistics
All results were confirmed in at least three independent experiments, and all quantitative data are presented as mean ± SD or SEM as indicated. Student’s t test or one-way ANOVA test was employed for analyzing quantitative variables. Drug synergy effects were determined using CompuSyn software. The association between IHC staining and the clinicopathologic parameters of the breast cancer patients was evaluated by the Fisher extract or Chi-square test. Survival curves were evaluated using Kaplan-Meier method and the differences between those survival curves were tested by log-rank test.
Results
HER2 overexpression triggers inflammatory circuits to enhance stem-like properties in pre-neoplastic tissues and breast epithelial cells
In addition to direct oncogenic effect, a chronically inflamed microenvironment possessed many other mechanisms to promote progression of a preneoplastic lesion. Cancer prone transgenic mice serve as models for autochthonous tumor formation. 100 % of MMTV-Her2 heterozygous transgenic mice develop palpable tumors starting at around 6 months old and with a median incidence of around 8 months (15). We hypothesized that HER2-induced inflammation promotes tumorigenesis by enhancing stem-like properties. Consistent with our previously published data (16,17), overexpression of Her2 resulted in expansion of epithelial progenitor cell population (defined as CD49fmedCD24hi cells) in the pre-neoplastic tissues (Fig. 1A). Previous gene profiling data have indicated that tumorigenesis in Her2-transgenic mice are associated with activation of pro-inflammatory pathways (6). To verify this observation, we examined cytokine and chemokine expression in pre-neoplastic and age-matched normal mammary glands, and found that the expression of many cytokines/chemokines were significantly upregulated in mammary gland tissues of MMTV-Her2 transgenic mice compared to normal mice (Fig. 1B). Because the mammary gland is not commonly susceptible to chronic inflammation, we examined the expression of these cytokines in isolated mammary epithelial cells. Similarly, the expression of several cytokines including IL-1α and IL-6 were upregulated (Fig. 1C), suggesting that HER2 overexpressing mammary epithelial cells may be responsible for initiation and maintenance of a pro-inflammatory environment during tumorigenesis.
Figure 1. HER2 overexpression induces inflammatory circuits to enhance stem-like properties.
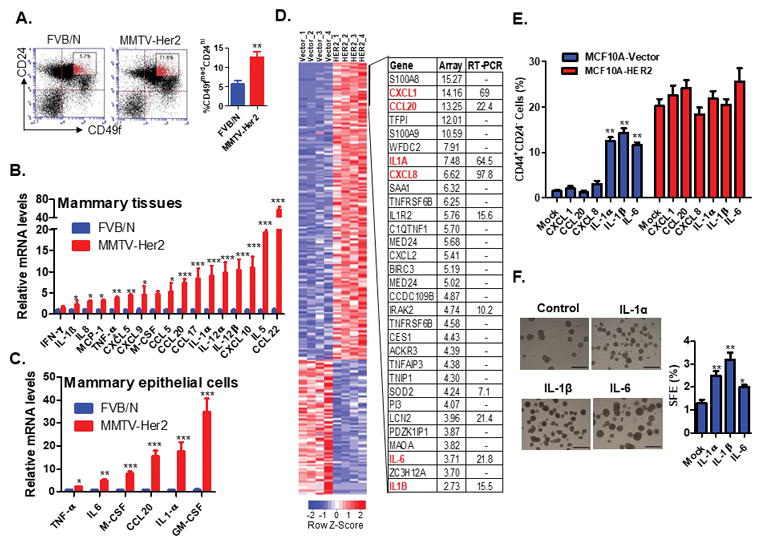
A. FACS analysis was performed to examine the luminal progenitor cell population (CD49fmedCD24hi cells) in mammary glands of 6-month old FVB/N and MMTV-Her2 transgenic mice (n=3). **, p < 0.01 (Unpaired Student’s t test). B–C. RT-PCR analysis of inflammatory cytokine and chemokine expression in mammary gland tissues (B) and purified epithelial cells (C) of 6-month old FVB/N and MMTV-Her2 transgenic mice (n=3). Data are reported as mean fold changes after intrasample normalization to the levels of GAPDH for n = 3 animals per group. *p < 0.05, **p < 0.01, ***, p< 0.001 (unpaired Student’s t test). D. Microarray analysis of gene expression profiles of MCF10A-vector and MCF10A-HER2 cells. Quadruplicate samples were used for array analysis and RT-PCR validation analysis was performed for selected genes. E. FACS analysis of the CD44+CD24− populations in MCF10A-vector and MCF10A-HER2 cells treated with different cytokines at the concentration of 10ng/ml for six days. Data are presented as mean ± SD. **p < 0.01 (Unpaired Student’s t test). F. MCF10A cells were treated with cytokines for six days followed with tumorsphere formation assay. Independent stimulation experiments were repeated three times. *p < 0.05, **p < 0.01 (Unpaired Student’s t test). See also Figure S1.
To confirm that HER2 induces inflammatory circuits in breast epithelial cells, we performed microarray analysis to obtain the gene expression profiles of HER2-overexpressing normal epithelial MCF10A and control cells. Ontology analysis of microarray data revealed that a high proportion of these significantly affected genes were inflammatory genes (37 of 616 probes, p<0.0001) (Fig. 1D and S1A). We further validated the expression levels of the selected inflammatory genes including IL1A, IL1B, CXCL1, CCL20, IL6, and CXCL8 using real-time RT-PCR analysis (Fig. 1D). Given that IL-6 has been shown to induce breast CSCs (3,9,18,19), we examined the effect of these cytokines on induction of a stem-like (CD44+CD24−) population in MCF10A cells. We treated MCF10A-vector and MCF10A-HER2 cells with these cytokines for 6 days and measured the percentage of CD44+CD24− cells. We found that treatment with IL-1α, IL-1β and IL-6, but not with CXCL1, CCL20, or CXCL-8, caused expansion of the CD44+CD24− population in MCF10A-vector cells (Fig. 1E). However, none of these cytokines or chemokines further increased the percentage of CD44+CD24− cells in MCF10A-HER2 cells, presumably because MCF10A-HER2 cells already express high levels of both IL-1α and IL-6. Indeed, gene set enrichment analysis (GSEA) indicated that IL-1α production and IL-6 downstream signaling pathways were significantly enriched in MCF10A-HER2 cells compared to control MCF10A cells (Fig. S1A–S1E). Additionally, we utilized mammosphere formation assay, a surrogate reporter of stem cell activity (20), and confirmed that MCF10A-HER2 cells treated with IL-1α, IL-1β or IL-6 promoted tumor sphere formation (Fig. 1F). Taken together, these data imply that HER2-induced pro-inflammatory circuits, including activation of IL-1α, IL-1β and IL-6 pathways may promote tumorigenesis by increasing stem-like properties of pre-neoplastic and transforming breast epithelial cells.
Secreted IL-1α and IL-6 in HER2-positive epithelial and cancer cells are responsible for constitutive activation of NF-kB and STAT3, respectively
Given that IL-1α, IL-1β and IL-6 were reported to activate multiple downstream signaling pathways including MAPK, PI3K/AKT, NF-κB, and STAT3 in some cultured cells (21), we investigated whether these pathways are activated by IL-1α, IL-1β or IL-6 in breast cancer cells. Interestingly, none of these cytokines significantly activated AKT, relative to their basal level expression; however, IL-1α and IL-1β modestly induced ERK phosphorylation, peaking at 15 min post treatment (Fig. 2A and Fig. S2). Both IL-1α and IL-1β strongly induced the activation of NF-κB pathways within 5 min, but IL-1α-mediated NF-κB activation was more sustainable (Fig. 2A). Although the IL-6-NF-κB signaling loop has been proposed to be critical for maintenance of breast CSCs (3,9), we did not observe a significant activation of NF-κB signaling by IL-6 at any time point. However, IL-6 stimulated a potent, rapid, and transient phosphorylation of STAT3 that reached a maximum level by 15 min after treatment (Fig. 2A). In comparison, STAT3 phosphorylation stimulated by IL-1α and IL-1β was delayed for 60 min and relatively less robust. Collectively, these findings suggest that HER2 overexpression upregulates IL-1 and IL-6 expression, which seem to mainly stimulate NF-κB and STAT3, respectively, in MCF10A cells.
Figure 2. Secreted IL-1α and IL-6 are responsible for constitutive activation of NF-κB and STAT3 in HER2-positive breast cancer cells.
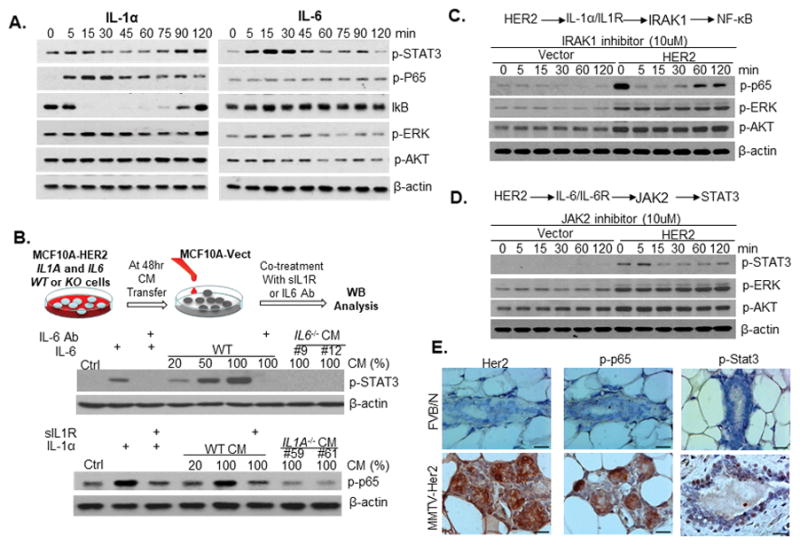
A. Western blot analysis of HER2 signaling kinetics in MCF10A cells after 10ng/ml of IL-1α treatment. B. Serum-free conditioned medium (CM) was harvested from parental and two clones of IL1A−/− or IL6−/− MCF-HER2 cell lines at 48hrs. MCF10A cells were treated with different amount of CM or cytokines for 15 min and then harvested for western blot analysis. IL-6 Ab, IL-6 neutralizing antibody; sIL1R, soluble IL-1 receptor antibody. One representative experiment out of two similar results is shown. C–D. MCF10A-vector and MCF10A-HER2 cells were treated with IRAK1 inhibitor (C) or JAK2 inhibitor (D) at the final concentration of 10μM. E. Representative IHC staining images of total HER2, phosphorylated p65 and Stat3 levels in mammary gland cells of 6-week old FVB/N and MMTV-Her2 transgenic mice. Similar results were observed in tissue sections of three animals in each group. Scale bar, 20μM. See also Figures S2–S7.
In accordance with our above results, previous studies have detected constitutively activated NF-κB and STAT3 in breast tumors, especially HER2-positive tumors (22,23). We hypothesized that overexpression of HER2 in breast cancer cells may activate NF-κB and STAT3 pathways through secreted IL-1α, IL-1β and IL-6. To test this hypothesis, we performed ELISA analysis to examine the expression levels of IL-1α, IL-1β and IL-6 in the culture medium of HER2-overexpresing MCF10A cells and control cells. As expected, IL-1α and IL-6 were upregulated in MCF10A-HER2 cells compared to control cells, while the expression of IL-1β was barely detectable (Fig.S3A–C and S4A–B). Since IL-1β is solely active as a secreted product, it may not play a critical role in our system. To further elucidate the signaling pathways, we generated IL1A and IL6 knockout MCF10A-HER2 cells using CRISP/Cas9 system (Fig. S3A and S4A–C). We then tested whether secreted IL-1α and IL-6 were responsible for activation of NF-κB and STAT3, respectively, by measuring the signaling response of MCF10A cells cultured in conditioned medium from wild type, IL1A−/−, or IL6−/− MCF10A-HER2 cells. We found that conditioned medium from parental MCF10A-HER2 cells strongly induced activation of STAT3, and that addition of IL-6 neutralizing antibody to the conditioned medium greatly diminished this effect (Fig. 2B). In contrast, conditioned medium from parental MCF10A-HER2 cells induced NF-κB activation, which was largely blocked by addition of soluble IL-1 receptor (Fig. 2B). Consistently, conditioned medium from two clones of IL1A−/− or IL6−/− MCF10A-HER2 cells were unable to activate NF-κB or STAT3 pathways, respectively (Fig. 2B).
To verify that IL-1α and IL-6 signaling are required for maintenance of constitutive activation of NF-κB and STAT3 pathways in HER2-positive breast cancer cells, we blocked IL-1α and IL-6 signaling with IRAK1 and JAK2 inhibitors (24) (25). As expected, treatment with IRAK1 and JAK2 inhibitors resulted in a rapid decrease of p65 and STAT3 phosphorylation levels in MCF10A-HER2 (Fig. 2C and 2D) and several endogenous HER2-overexpressing breast cancer cell lines (Fig. S5A–B). In contrast, treatment with Lapatinib, a dual inhibitor of EGFR and HER2, ablated HER2-mediated phosphorylation of ERK and AKT within 15 minutes, but had no effect on the phosphorylation status of p65 and STAT3 over two hours of treatment (Fig. S6A). Additionally, inhibition of NF-κB and STAT3 signaling did not occur until 8–12 hours of lapatinib treatment (Fig. S6B). In a complementary experiment, we treated HER2-positive SKBR3 breast cancer cells with HER2-agonist (26), and found that activation of HER2 signaling induced phosphorylation of AKT and ERK1/2 within 10 minutes, but failed to activate NF-κB and STAT3 pathways even after 90 minutes of treatment (Fig. S6C). Collectively, these findings suggest that NF-κB and STAT3 pathways are not directly activated by either MAPK or PI3K/AKT kinases, which are immediately downstream of HER2. Instead, we propose that HER2 overexpression mediates the upregulation and secretion of IL-1α and IL-6 to initiate and maintain the constitutive activation of NF-κB and STAT3, respectively, in breast cancer cells. Consistent with our in vitro data, immunohistochemical (IHC) staining and western blot assay confirmed increased phosphorylation levels of p65 and STAT3 in mammary epithelial cells of MMTV-Her2 transgenic mice (Fig. 2E and S7).
NF-κB and STAT3 signaling pathways are critical for HER2-induced expansion of CSC-like cells
To determine the contribution of different HER2-downstream signaling pathways to expand the CSCs population, a variety of chemical inhibitors were used to specifically block HER2 (Lapatnib), MAPK (PD0325901), PI3K/AKT (LY290042), SRC (PP2), NF-κB (JSH23), and STAT3 (Stattic) pathways. All of these inhibitors effectively blocked the corresponding pathways (Fig. S8A). Treatment of MCF10A-HER2 and HER2-positive HCC1954 breast cancer cells with STAT3 or NF-κB inhibitor resulted in a significant reduction in the percentage of CD44+CD24− cells (Fig. 3A). Treatment with MAPK or PI3K inhibitors moderately decreased the percentage of CD44+CD24− cells. The effects of drug treatment on tumor sphere formation efficiency also reflected the changes in the proportion of CD44+CD24− cell populations (Fig. 3B). To exclude possible off-target effects associated with the reduction of CSC-like cells by STAT3 and NF-κB inhibitors, we utilized siRNA to specifically knockdown the expression of p65 and STAT3 (Fig. 3C). Consistently, specific knockdown of p65 and STAT3 in HCC1954 cells resulted in a reduction of CD44+CD24− cells (Fig. 3C). Interestingly, no direct correlations existed between the inhibition of cell growth and reduction of CSCs after drug treatment, although all of the drug treatments had negative effects on cell growth to some extent (Fig. S8B). Together, these data suggest that a modest level of MAPK and PI3K/AKT activation may be necessary for cell proliferation and survival, whereas NF-κB and STAT3 signaling pathways are critical for HER2-mediated expansion of CSCs.
Figure 3. Sequential activation of NF-κB and STAT3 is required for HER2-mediated expansion of cancer stem-like cells.
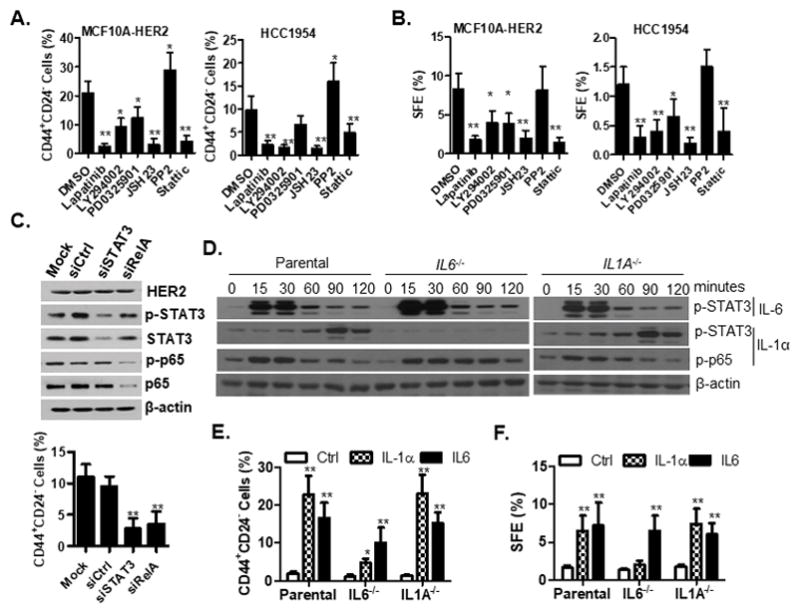
A–B. Cells were treated with lapatinib (2μM), LY294002 (10μM), PD0325901 (100nM), JSH (20μM), PP2 (10μM), Stattic (1μM) and DMSO for 6 days, followed by FACS analysis of the CD44+CD24- population (A) and tumorsphere formation assay (B). C. Knockdown of STAT3 and p65 reduced the proportion of CD44+CD24- cells. STAT3 and P65 siRNA-mediated knockdown efficiency was evaluated by western blot analysis. siCtrl, control siRNA; siSTAT3, STAT3 siRNA; siRelA, RelA siRNA. Knockdown of STAT3 and p65 reduced the proportion of CD44+CD24- cells. D–F. Parental and IL1A−/− or IL6−/− MCF10A cells were treated with exogenous IL-1α or IL-6 cytokines followed by signaling and functional analysis. Western blot analysis of phosphorylated p65 and STAT3 (D), FACS analysis of the CD44+CD24− populations (E) and tumorsphere formation assay of cells (F) were performed after cytokine treatment for six days. SFE, sphere formation efficiency. For A, B, C, E and F, all experiments were repeated three times and data are shown as mean ± SD. *p < 0.05, **p < 0.01 (Unpaired Student’s t test). See also Figures S3 and S8.
HER2 induces sequential activation of IL-1α and IL-6 signaling for maintenance of CSCs
Since we demonstrated that i) inhibition of either NF-κB or STAT3 pathway dramatically reduced the proportion of CSC-like populations in HER2-overexpressing cells (Fig. 3C); ii) IL-1α treatment delayed STAT3 activation (Fig. 2A); and iii) IL-6 treatment accelerated STAT3 activation (Fig. 2A); we sought to explain our results by HER2 inducing a sequential activation of IL-1α and IL-6 signaling. To test this sequential activation model, we treated wild type, IL1A−/−, and IL6−/− MCF10A cells with either IL-1α or IL-6 cytokine to monitor the signaling response for two hours (Fig. S3A–S3C and 3E). As expected, exogenous IL-1α and IL-6 were sufficient to activate NF-κB and STAT3 in both IL1A−/− and IL6−/− cells (Fig. 3D). However, knockout of IL6 completely abolished IL-1α-induced delayed activation of STAT3, indicating that delayed activation of STAT3 by IL-1α is mediated via induction of IL-6 expression (Fig. 3D). To investigate the relative contribution of IL-1α and IL-6 signaling in regulation of CSCs, we measured the CD44+CD24− population and sphere formation abilities after cytokine treatment of parental, IL1A−/− and IL6−/− MCF10A cells for 6 days. As shown in Fig. 3F, IL-1α or IL-6 cytokine treatment of wild type cells induced a 10-fold increase in the CD44+CD24− population. Both IL-1α and IL-6 cytokine treatment of IL1A−/− cells were sufficient to induce the expansion of CD44+CD24− population to the same level as wild type cells (Fig. 3E). However, in IL6−/− cells, IL-1α treatment only slightly increased the proportion of CD44+CD24− cells while IL-6 treatment largely restored its function (Fig. 3E). A very similar pattern was observed in the sphere formation assay (Fig. 3F). These data reinforces our hypothesis that IL-1α acts upstream of IL-6 in breast cells.
To explore the molecular mechanisms of HER2-mediated sequential upregulation of IL-1α and IL-6 expression, we treated MCF10A-HER2 cells with a variety of chemicals to specifically block putative HER2-downstream pathways and measured IL1A and IL6 mRNA levels (Fig. 4A). ERK1/2 and NF-κB inhibition reduced IL-1α expression by 80% and 50%, respectively. In contrast, NF-κB inhibitor treatment silenced IL-6 expression, while ERK1/2 inhibition reduced its expression by about 70% (Fig. 4A). PI3K kinase inhibitor slightly increased the expression levels of IL-1α and IL-6, possibly due to the inhibitory effects of PI3K/AKT activity on MAPK kinase pathway (27,28). To identify the specific transcriptional factors that directly regulate IL1A and IL6 expression, we performed transient transfection assays to examine the effects of several candidate transcriptional factors on the promoter activity of IL1A and IL6 in HER2-overexpressing and control MCF10A cells (Fig. 4B). IL1A promoter activities were increased by 5–6 fold by PU.1 or NF-κB in control cells and further enhanced by another 4–5 folds in HER2-overexpressing cells (Fig. 4C), implying that HER2-signaling may post-transcriptionally regulate the activities of PU.1 and NF-κB. ChIP analysis confirmed that both PU.1 and NF-κB could bind to the IL1A promoter (Fig. 4D). Given that transcriptional activation ability of PU.1 may be activated by ERK1/2 kinase (29), we treated HER2-positive cells with MAPK inhibitor. Indeed, MAPK kinase inhibitor treatment completely blocked the binding of PU.1 to IL1A promoter (Fig. 4D). Among the transcriptional factors we studied, only that of NF-κB could bind to the IL6 promoter (Fig. 4E), which was partially blocked by MAPK inhibitors (Fig. 4F and 4G). Collectively, these data support a model of HER2-induced sequential activation of IL-1α and IL-6 signaling pathways, as further evidenced by: i) HER2 upregulating IL-1α expression via MAPK-mediated activation of PU.1 transcription factor; ii) secreted IL-1α binding to its receptor and activating NF-κB, which subsequently binds to and activates IL1A and IL-6 promoters via a feedback mechanism; iii) Secreted IL-6 binding to its receptor and activating the downstream STAT3 transcriptional factor (Fig. 4H).
Figure 4. HER2-mediated sequential activation of IL1A and IL6 transcription.
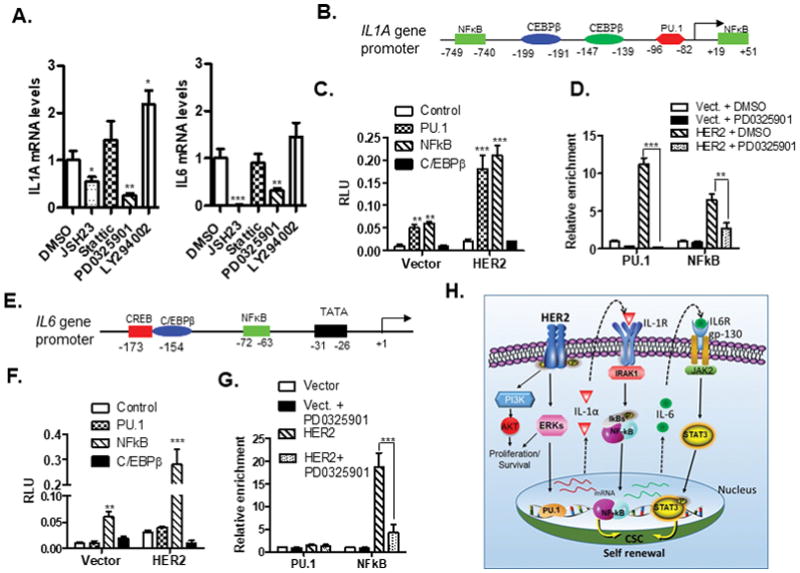
A. Real-time RT-PCR analysis of IL1A and IL6 mRNA levels in MCF10A-HER2 cells treated with several HER2-downstream signaling pathway inhibitors for 24 hours. B and E. Schematic depiction of the potential transcriptional binding sites on the IL1A (B) and IL6 (E) promoter. C and F. MCF10A-vector and MCF10A-HER2 cells were co-transfected with IL1A (C) or IL6 (F) promoter reporter and several transcriptional factors-expression plasmids. At 48hrs post-transfection, luciferase assays were performed. RLU, relative luciferase unit; Vector, MCF10A-vector; HER2, MCF10A-HER2 cells. D and G. ChIP analysis of the relative binding abilities of PU.1 and NF-κB to IL1A (D) or IL6 (G) promoter in MCF10A-HER2 cells treated with DMSO and PD0325901 for 24 hours. The IP experiments were repeated and all of qRT-PCR reactions were performed in triplicate. H. A proposed model for HER2-mediated sequential upregulation of IL1A and IL6 expression in the context of HER2-downstream pathways and functions. For A, C, D, and F, the experiment were repeated three times and results were shown as mean ± SD. *p < 0.05, **p < 0.01. ***p < 0.001 (unpaired Student’s t test).
Knockout of IL1A inhibits HER2-induced tumorigenesis via reducing CSCs and dampening inflammatory microenvironment in vivo
To investigate the role of the sequential activation of IL-1α and IL-6 signaling on maintenance of breast CSCs, IL1A−/−, IL6−/−, and HER2 knockdown (HER2 KD as a positive control) HCC1954 cell lines were generated using CRISP9 system (Fig. S9A). The loss of cytokine production in the supernatants from these knockout (KO) cells was confirmed by ELISA analysis (Fig. S9B). The effect of IL1A and IL6 deficiency on CD44+CD24− population was assessed by FACS. As shown in Figure 5A, the percentage of CD44+CD24− population was greatly diminished in IL1A −/− cells than in wild type HCC1954. Similarly, IL6 deficiency resulted in a drastic reduction in the percentage of CD44+CD24− population (Fig. 5A). Further, the role for IL-1α and IL-6 on maintenance of breast CSCs was verified by sphere formation assay (Fig. 5B). To further evaluate the effects of IL1A and IL6 deficiency on the tumorigenicity of HCC1954 cells in vivo, we performed a series dilution transplantation assay. KO of either IL1A or IL6 drastically reduced tumor initiation efficiency (Supplementary Table S1), which was consistent with tumors from IL1A−/− and IL6−/− cells growing much slower than those from parental line HCC1954 (Fig. 5C). Taken together, both IL-1α and IL-6 are required for maintenance of CSC population in vitro and the tumorigenicity of HCC1954 cells in vivo.
Figure 5. IL-1α loss reduces CSCs, turns off the inflammatory signaling circuits and inhibits HER2-induced tumorigenicity in vivo.
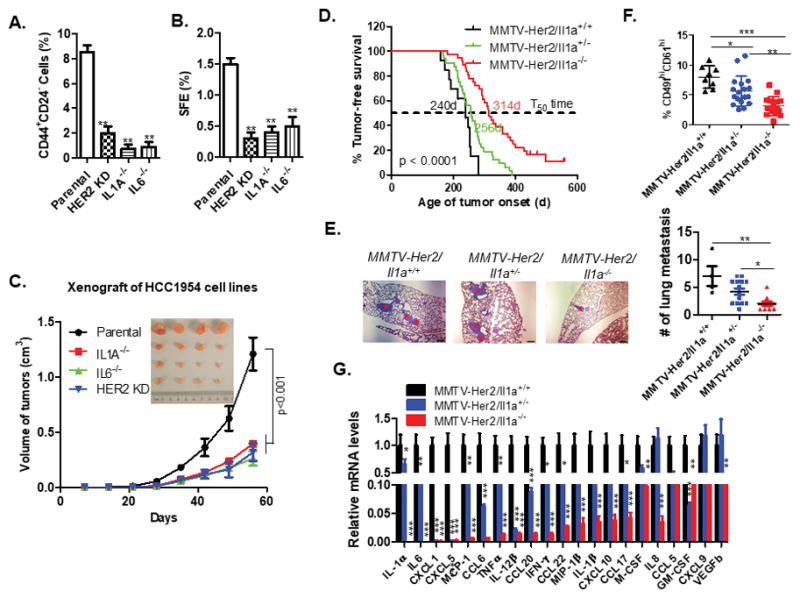
A–B. FACS analysis of the CD44+CD24− populations (A) and tumorsphere formation assay (B) were performed using parental, IL1A−/−, IL6−/− and HER2KD HC1954 cells. HER2KD, HER2 stably knocked down HCC1954 cells. The experiment were repeated three times and Data represents means ± SD from three separate experiments. **, p < 0.01 (unpaired Student’s t test). C. Tumor growth in mice injected with 1 × 105 cells (n = 8). Tumor growth was monitored every 7 days; tumor size and weight were recorded. Data are represented as mean ± SEM. Tumor sizes of each group at the end point were compared with the parental control mice group by ANOVA test, p < 0.001. D. Tumor incidence in MMTV-Her2/Il1a+/+ (n =13), MMTV-Her2/Il1a+/− (n = 32) and MMTV-Her2/Il1a−/− (n = 31) mice. The p value was calculated using the long-rank test. E. Lung metastasis were assessed in animals sacrificed at 60 days post tumor onset. The number of metastatic foci in the three group of animals with lung metastasis were compared using One-way ANOVA followed with Tukey’s multiple comparison test of each pair. *p < 0.05, **p < 0.01. F. FACS analysis of the putative CSC cells (CD49fhiCD61hi population) in the first tumor of each animal. One-way ANOVA test was performed, *p < 0.05, **p < 0.01; ***, p < 0.001. G. Inflammatory chemokine and cytokines expression profiling of primary tumor samples from animals. Data are reported as mean fold changes ± SEM after intrasample normalization to the levels of GAPDH for n = 3 animals per group. *p < 0.05, **p < 0.01, ***, p < 0.001 (unpaired student’s t test). See also Figures S9 and S10.
To further evaluate the role of IL-1α in HER2-induced tumorigenesis at a physiologically relevant condition, we crossed Il1a−/− mice with MMTV-Her2 transgenic mice. Knockout of Il1a significantly delayed Her2-induced tumor onset and partially inhibited tumor metastasis (Fig. 5D and 5E). FACS analysis revealed a significant reduction in the proportion of CSCs (CD49fhiCD61hi cells) in both primary tumors from MMTV-Her2/Il1a+/− and MMTV-Her2/Il1a−/− mice compared to control mice (Fig. 5F) (17). In addition, loss of Il1a expression caused a systematic reduction of a panel of inflammatory cytokines and chemokines (Fig. 5G), with Il6 being one of the most significantly downregulated cytokine genes in Il1a-knockout mice, thus supporting the sequential activation mechanism as proposed. Consistently, NF-κB and Stat3 signaling pathways was partially blocked in the primary tumor samples from MMTV-Her2/Il1a+/− mice and almost completely blocked in MMTV-Her2/Il1a−/− mice (Fig. S10). Together, these data suggest that Il1a promotes Her2-induced tumor onset and progression via maintenance of an inflammatory microenvironment and population of CSCs
IL-1α and IL-6 expression are positively associated with the CSC-positive phenotype in primary breast tumors and serve as prognostic biomarkers
To investigate the clinical relevance of our findings, we examined the expression status of HER2, IL-1α, and IL-6 by IHC staining analysis of 136 human breast cancer tissues on a tissue array (Supplementary Fig. S11A–L and Table S2). As shown in Fig. 6A, IL-1α and IL-6 proteins were highly expressed in 55.2% and 44.1% of breast cancer samples, respectively, and exhibited a strong positive correlation with each other (p<0.0001), corroborating their aforementioned linear relationship. Interestingly, HER2 expression status positively correlated with IL-1α (p = 0.001) (Fig. 6A and Table S3), which is also conclusive with our hypothesis that HER2 directly regulates IL-1α expression in breast cancer cells. Considering the role of IL-1α and IL-6 in expansion of CSCs in vitro and in animal models, we also analyzed CD44 and CD24 expression to identify the CSC phenotype (CD44+CD24−) in the breast cancer tissues (30,31). CSC-positive phenotype (CD44+CD24−) was detected in 50.7% (69/136) of breast cancer samples (Fig. 6B). The CSC prevalence was significantly associated with the positive expression status of IL-1α (p=0.0002) and IL-6 (p<0.0001) alone or both (Fig. 6B and Table S4). More importantly, both IL-1α and IL6 high expression at protein level predicted distant metastasis-free survival of breast cancer patients (Fig. 6C–D and Fig. S12 A–B) and therefore had the potential to be new prognostic biomarkers in breast cancer.
Figure 6. Expression of IL-1α and IL-6 is associated with CSC-positive phenotype and other prognostic factors.
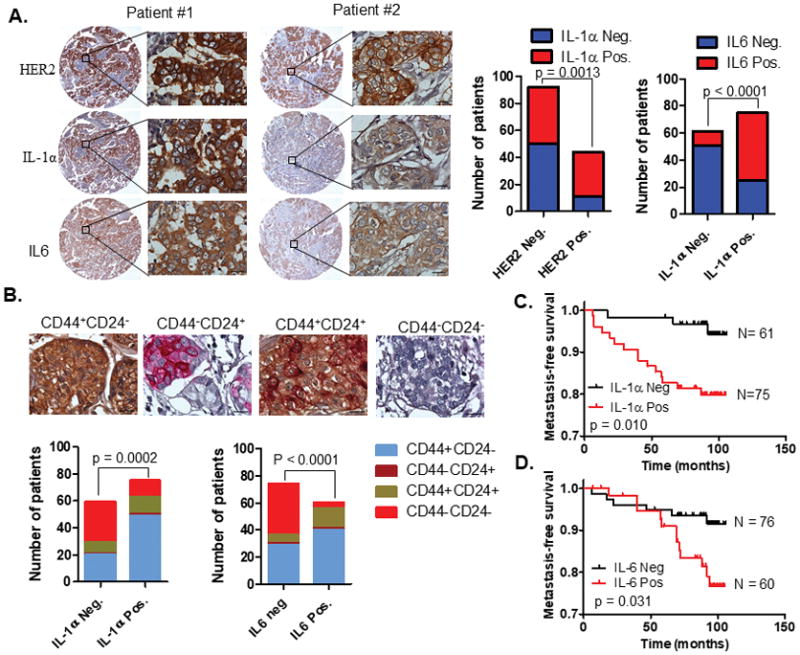
A. Representative IHC detection of IL-1α, IL-6, and HER2 expression in primary breast tumors. Tissue microarray (TMA) containing 136 breast cancer samples were used for immunohistochemical analysis. Association of IL-1α positive expression status with HER2 and IL-6 expression was assessed by Chi-square analysis. Scale bar, 20μM. B. Representative IHC images of CD44 and CD24 double-staining breast tumors. Association of IL-1α or IL-6-positve expression with CD44/CD24 phenotypes was assessed by Chi-square analysis. Scale bar, 20μM. C–D. Kaplan-Meier plots of distant metastasis-free survival of patients, stratified by expression of IL-1α (C) and IL-6 (D). The p values were obtained using log-rank (Mantel-Cox) test. See also Figure S11 and S12, Table S2–4.
Pharmacological blockage of IL-1α signaling reduces breast CSCs and increases the efficacy of chemotherapy in vivo
CSCs have been proposed to be responsible for chemotherapeutic resistance and tumor recurrence (32,33). Given the sequential activation of IL-1α and IL-6 in HER2-positive breast cancer cells, we reasoned that blockade of IL-1α signaling may sensitize tumor cells to chemotherapies. We evaluated the sensitivity of IL1A −/− HCC1954 cells to the treatment of two classic chemotherapeutic drugs: cisplatin (CPL) and paclitaxel (PTX). Knockout of IL1A in HCC1954 cell line significantly decreased the IC50 values of both CPL and PTX compared to parental cells and HER2 KD cells (Fig. 7A). Considering the potential translational relevance of these findings to clinical benefits, we also tested whether chemical inhibition of IL-1α signaling could improve chemotherapy efficacy. IRAK1 inhibitor alone at a concentration of 10uM had no significant effect on cell growth. However, this level of IRAK1 inhibitor synergized with either CPL or PTX significantly induced cell death compared to either inhibitor alone (Fig. 7B and Fig. S13A–B). Using an orthotropic xenograft mouse model, we observed significant tumor growth inhibition by IRAK1 inhibitor or PTX treatment alone compared to controls (Fig. 7C and 7D); but detected a further 60% reduction in tumor size upon the combination of these treatments compared to PTX alone (p<0.001) (Fig. 7D). In addition, all of the long-term follow-up tumors in the combined treatment group exhibited either consistent reduction in tumor size or slow growth rate after the last treatment, in contrast to rapid tumor growth observed in 3 out of 5 mice in the PTX group (Fig. S14A–D).
Figure 7. Blockade of IL-1α signaling sensitizes breast cancer cells to chemotherapy.
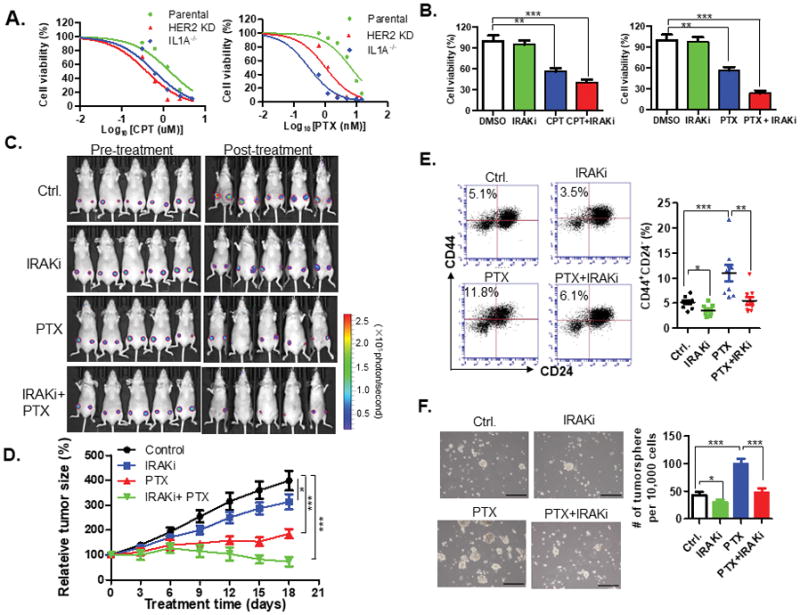
A. MTT assay of parental, IL1A−/− or HER2KD HCC1954 cells treated with indicated concentration of cisplatin or paclitaxel for 7 days. IC50 values (mean ± SD) were calculated from three independent experiments. B. MTT assay of HCC1954 cells treated with IRAK inhibitor (10 μM), cisplatin (2.5 μM), paclitaxel (10 nM) alone or in combination for 7 days. Samples were prepared in triplicate. Data are presented as the mean ±SD and compared with DMSO control in each group. **, p < 0.01; ***, p < 0.001 (one-way ANOVA test). C. Luciferase-labeled HCC1954 cells were injected into the number 4 mammary fat pads of NOD-SCID mice. Mice were treated with i.p. injection of IRAK inhibitor (4mg/Kg), paclitaxel (10 mg/Kg), combination of IRAK1 inhibitor and paclitaxel, or solvent control, respectively. Bioluminescence imaging before (0 day) or after (18 days) treatment is shown. D. Tumor growth was monitored every three days and normalized to tumor volume at treatment start point. Data are represented as a mean ± SEM from 5 mice (2 injection sites/mouse). Tumor sizes of each group at the end point (5 weeks) were compared with the parental control mice group by ANOVA test, *p < 0.05, **p < 0.01, ***p < 0.001. E. FACS analysis of the CD44+CD24− populations in the xenograft tumors as described above. F. Tumorsphere formation assay of xenograft tumor cells. Scale bar, 200μM. For E and F, two-tailed t tests were performed to obtain the p values. *p < 0.05, **p < 0.01; ***p < 0.001. CPT, cisplatin; PTX, paclitaxel; IRAKi, IRAK1 inhibitor. See also Figure S13 and S14.
To confirm whether blockade of IL-1α signaling reduces CSCs in vivo, we first analyzed the proportion of CD44+/CD24− population in tumors harvested three days after the last treatment. We found that PTX significantly increased CD44+/CD24− CSCs by about two-fold compared to controls (Fig. 7E). We also observed a significant reduction in the CSCs population by IRAK inhibitor alone, and moreover, IRAK1 inhibitor in combination with PTX reduced the PTX-induced CSC population to control level (Fig. 7E). The PTX-induced CSC increase correlated with the increased sphere formation efficiency of xenograft tumors compared to the controls (Fig. 7F). The combined treatment significantly inhibited the PTX-induced sphere formation efficiency of the xenograft tumors to levels comparable to control. IRAK1 inhibitor alone significantly, although not as evident, impaired the sphere forming ability (Fig. 7F). Immunochemical staining analysis of phosphor-p65 positive cells in the treated tumors confirmed that IRAK1 inhibitor effectively blocked IL-1α signaling (Fig. S14A and S14B). Together, these results strongly support the hypothesis that pharmacological blockade of IL-1α signaling in breast cancer cells can reduce CSCs in vivo and improve chemotherapy efficacy.
Discussion
Inflammation is now recognized to be important in the pathogenesis of many types of malignancies (1,34). Specifically, HER2-positive breast cancers exhibit higher expressions of tumor infiltrated lymphocytes than ER-positive/HER2-negative breast cancers (35). Accordingly, HER2-targeted treatments can induce lymphoctic infiltrates (36), suggesting that HER2 signaling may be a mechanism to modulate immune responses. Although the role of inflammation in HER2-induced tumorigenesis remains controversial, our studies support that HER2 overexpression induces a tumor cell-derived cytokine production to trigger and maintain a local inflammatory environment. Mechanistically, HER2 overexpression activated expression and secretion of IL-1α which functions as a pro-inflammatory signal to sequentially trigger expression of other cytokines including IL-6. Knockout of Il1a alone dramatically dampened the inflammatory microenvironment and delayed Her2-induced tumor onset and progression. These findings support a new hypothesis that IL-1α functions as one of the newly defined class of ‘Sressorin’ cytokines (37), which can be actively released in response to numerous physiological or cellular stress, e.g. oncogene overexpression, and contribute to initiation and maintenance of chronic inflammation. In contrast to the conventional view that chronic sterile inflammation is generally triggered by passively released damage or danger associated molecular patterns (DAMPs) from dead cells (38), our studies reveal a new mechanism for HER2-induced IL-1α expression and secretion to maintain chronic inflammation for promotion of cancer initiation and progression.
This present work also sheds light on the molecular mechanisms of HER2-induced expansion of breast CSCs. Korkaya et al. proposed that HER2 overexpression triggered the NF-κB-IL-6 loop to induce expansion of CSCs. (9). However, we found that IL-1α, not IL-6, can swiftly activate NF-κB. These results led us to hypothesize that IL-1α is the missing link between HER2 and IL-6 pathways. In support of our hypothesis, we found that IL-1α induced a delayed activation of STAT3 in control cells, but not in IL6−/− MCF10A cells. Our data support the model that HER2 sequentially activates the IL-1α-NF-κB-IL-6 signaling axis in breast cancer cells. As a canonical HER2 downstream signaling molecule, AKT can activate NF-κB via degrading IkB in immune cells (39). We initially assumed that AKT plays an indispensable role in the activation of NF-κB pathway in HER2-positive breast cancer cells. Contrary to our expectation, we found that manipulation of HER2-downtream MAPK and AKT signaling with lapatinib or HER2 agonist had no obvious effect on p65 phosphorylation. These observations are consistent with another previous work indicating that AKT inhibition has no effect on inactivation of NF-κB in breast cancer cell lines (11). Instead, our findings revealed that IL-1α strongly activates NF-κB in breast cancer cells. Knockout of IL-1α or chemical blockade of its signaling in HER2-overexpressing breast cancer cells resulted in the dramatic reduction of p65 phosphorylation and NF-κB transcriptional activity. To our knowledge, this current study is the first to demonstrate that IL-1α mediates HER2-induced activation of the NF-κB pathway and the sequential activation of IL-1α and IL-6, connecting the function of NF-κB and STAT3 in regulation of breast CSCs. Consistently, there is a strong positive correlation of IL-1α expression with HER2 status and CSCs-positive phenotype in primary breast cancer tissues. However, one of the caveats of our study is that the CD44+CD24− phenotypes used as the CSC-identification biomarkers may not necessarily reflect the characteristics of CSCs and unfavorable clinical outcomes in patients.
Our studies exemplifies the need for further investigation of the central role of IL-1α in regulating breast CSCs. This may open up a new avenue for treatment, considering that chemotherapy alone increases the CSC fraction in breast cancer tumors, leading to drug resistance and tumor recurrence (40). Our in vitro data indicated that either knockout of the IL1A gene or blockade of downstream signaling with IRAK1 inhibitor can significantly improve the chemotherapy efficacy. Perhaps the most intriguing aspect of this work is that the IRAK1 inhibitor in combination with paclitaxel resulted in complete remission in 20% of mice and continued to shrink tumor sizes after the last treatment. FACS analysis of post-treatment tumor samples indicated that paclitaxel alone increases the CSC fraction, whereas IRAK1 inhibitor alone or in combination has opposite effects. Our data supports the hypothesis that HER2-elicited IL-1α signaling can promote drug resistance via expansion of CSCs. Of note, HER2 genetic heterogeneity is frequently detected among HER2 equivocal and negative breast cancer tissues (41). Korkaya et al. (2008) has previously proposed that even ‘HER2-negative’ tumors contain a small percentage of HER2 expressing CSCs that may account for HER2-negative tumor patients’ sensitivity to trastuzumab treatment (42). Our study suggested that HER2-positive tumor or surrounding normal epithelia cells could secrete a variety of cytokines to generate a pro-inflammatory environment for maintenance and/or expansion of CSCs. These observations provide a new biological explanation of the unexpected findings from two separate prospective randomized trials that the benefits of adjuvant trastuzumab in women with HER2-negative cancers is roughly the same as those with HER2-positve cancers (12,13). Therefore, targeting our proposed tumor cell-derived pro-inflammatory IL-1α-IL-6 circuit triggered by HER2 may represent an effective therapeutic avenue for breast cancer prevention and treatment.
Supplementary Material
Acknowledgments
We acknowledge Dr. for kindly providing the pAH 9614 plasmid, Dr. Feng Zhang for the CRISPRv2 lentiviral vector, Dr. Robert Weinberg for the pVSV-G plasmid, Dr. Didier Trono for the psPAX2 plasmid, Dr. Emily Wang for the MCF10A-vector and MCF10A-HER2 cells and Dr. Rachel Schiff for MCF7/neo and MCF7/HER2-18 cell lines. This work was supported by the NIH grant (4R01 CA178386-04) and the USC ASPIRE-1 grant to H. Chen, 5P30 GM103336-05 to the Biorepository Core Facility of the USC Center for Colon Cancer Research to D. Reisman, the USC ASPIRE Fellowship to S. Liu.
Footnotes
Competing financial interests: none
References
- 1.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 2.Pierce BL, Ballard-Barbash R, Bernstein L, Baumgartner RN, Neuhouser ML, Wener MH, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol. 2009;27:3437–44. doi: 10.1200/JCO.2008.18.9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693–706. doi: 10.1016/j.cell.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proceedings of the National Academy of Sciences of the United States of America. 108:1397–402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slamon DJ, Clark GM. Amplification of c-erbB-2 and aggressive human breast tumors? Science. 1988;240:1795–8. doi: 10.1126/science.3289120. [DOI] [PubMed] [Google Scholar]
- 6.Calogero RA, Cordero F, Forni G, Cavallo F. Inflammation and breast cancer. Inflammatory component of mammary carcinogenesis in ErbB2 transgenic mice. Breast Cancer Res. 2007;9:211. doi: 10.1186/bcr1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome biology. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hartman ZC, Yang XY, Glass O, Lei G, Osada T, Dave SS, et al. HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011;71:4380–91. doi: 10.1158/0008-5472.CAN-11-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47:570–84. doi: 10.1016/j.molcel.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korkaya H, Paulson A, Iovino F, Wicha MS. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene. 2008;27:6120–30. doi: 10.1038/onc.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merkhofer EC, Cogswell P, Baldwin AS. Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha. Oncogene. 2010;29:1238–48. doi: 10.1038/onc.2009.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paik S, Kim C, Wolmark N. HER2 status and benefit from adjuvant trastuzumab in breast cancer. N Engl J Med. 2008;358:1409–11. doi: 10.1056/NEJMc0801440. [DOI] [PubMed] [Google Scholar]
- 13.Perez EA, Reinholz MM, Hillman DW, Tenner KS, Schroeder MJ, Davidson NE, et al. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J Clin Oncol. 2010;28:4307–15. doi: 10.1200/JCO.2009.26.2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horai R, Asano M, Sudo K, Kanuka H, Suzuki M, Nishihara M, et al. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J Exp Med. 1998;187:1463–75. doi: 10.1084/jem.187.9.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:10578–82. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lo PK, Chen H. Cancer stem cells and cells-of-origin in MMTV-Her2/neu induced mammary tumorigenesis. Oncogene. 2012 doi: 10.1038/onc.2012.456. in press. [DOI] [PubMed] [Google Scholar]
- 17.Lo PK, Kanojia D, Liu X, Singh UP, Berger FG, Wang Q, et al. CD49f and CD61 identify Her2/neu-induced mammary tumor-initiating cells that are potentially derived from luminal progenitors and maintained by the integrin-TGFbeta signaling. Oncogene. 2012;31:2614–26. doi: 10.1038/onc.2011.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iliopoulos D, Hirsch HA, Wang G, Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:1397–402. doi: 10.1073/pnas.1018898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res. 2015;13:651–8. doi: 10.1158/1541-7786.MCR-14-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 21.Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Science signaling. 2010;3:cm1. doi: 10.1126/scisignal.3105cm1. [DOI] [PubMed] [Google Scholar]
- 22.Biswas DK, Shi Q, Baily S, Strickland I, Ghosh S, Pardee AB, et al. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc Natl Acad Sci U S A. 2004;101:10137–42. doi: 10.1073/pnas.0403621101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berishaj M, Gao SP, Ahmed S, Leslie K, Al-Ahmadie H, Gerald WL, et al. Stat3 is tyrosine-phosphorylated through the interleukin-6/glycoprotein 130/Janus kinase pathway in breast cancer. Breast Cancer Res. 2007;9:R32. doi: 10.1186/bcr1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akira S, Takeda K. Toll-like receptor signalling. Nature reviews Immunology. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 25.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. The Biochemical journal. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang TH, Morrison SL. A trimeric anti-HER2/neu ScFv and tumor necrosis factor-alpha fusion protein induces HER2/neu signaling and facilitates repair of injured epithelia. J Pharmacol Exp Ther. 2006;316:983–91. doi: 10.1124/jpet.105.095513. [DOI] [PubMed] [Google Scholar]
- 27.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B) Science. 1999;286:1741–4. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 28.Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–41. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 29.Hamdorf M, Berger A, Schule S, Reinhardt J, Flory E. PKCdelta-induced PU. 1 phosphorylation promotes hematopoietic stem cell differentiation to dendritic cells. Stem cells (Dayton, Ohio) 2011;29:297–306. doi: 10.1002/stem.564. [DOI] [PubMed] [Google Scholar]
- 30.Makki J, Myint O, Wynn AA, Samsudin AT, John DV. Expression distribution of cancer stem cells, epithelial to mesenchymal transition, and telomerase activity in breast cancer and their association with clinicopathologic characteristics. Clin Med Insights Pathol. 2015;8:1–16. doi: 10.4137/CPath.S19615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Honeth G, Bendahl PO, Ringner M, Saal LH, Gruvberger-Saal SK, Lovgren K, et al. The CD44+/CD24- phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008;10:R53. doi: 10.1186/bcr2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 33.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–5. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 34.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31:860–7. doi: 10.1200/JCO.2011.41.0902. [DOI] [PubMed] [Google Scholar]
- 36.Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Science translational medicine. 2015;7:315ra188. doi: 10.1126/scitranslmed.aac4925. [DOI] [PubMed] [Google Scholar]
- 37.Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. Alarmins: Feel the Stress. J Immunol. 2017;198:1395–402. doi: 10.4049/jimmunol.1601342. [DOI] [PubMed] [Google Scholar]
- 38.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nature reviews Immunology. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Current biology : CB. 1999;9:601–4. doi: 10.1016/s0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 40.Yu F, Yao H, Zhu P, Zhang X, Pan Q, Gong C, et al. let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell. 2007;131:1109–23. doi: 10.1016/j.cell.2007.10.054. [DOI] [PubMed] [Google Scholar]
- 41.Kurozumi S, Padilla M, Kurosumi M, Matsumoto H, Inoue K, Horiguchi J, et al. HER2 intratumoral heterogeneity analyses by concurrent HER2 gene and protein assessment for the prognosis of HER2 negative invasive breast cancer patients. Breast Cancer Res Treat. 2016;158:99–111. doi: 10.1007/s10549-016-3856-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ithimakin S, Day KC, Malik F, Zen Q, Dawsey SJ, Bersano-Begey TF, et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013;73:1635–46. doi: 10.1158/0008-5472.CAN-12-3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.