

Abstract
Immune checkpoint inhibitors have unique toxicities and response kinetics compared to cytotoxics and gene-targeted anti-cancer agents. We investigated the impact of innovative/accelerated immunotherapy drug development/approval models on the accuracy of safety and efficacy assessments by searching the US Food and Drug Administration (FDA) website. Initial phase I trials for each agent were reviewed and safety and efficacy data compared to that found in later trials leading to regulatory approvals of the same agents. As of June 2017, the FDA approved six checkpoint inhibitors for a variety of cancer types. All checkpoint inhibitors received a priority review status and access to at least two additional FDA special access programs, more often breakthrough designation and accelerated approval. Median clinical development time (investigational new drug application to approval) was 60.77 months (avelumab had the shortest timeline (52.33 months)). Response rates during early phase I trials (median =16%) are higher than for phase I trials of other agents (with the exception of gene-targeted agents tested with a biomarker). Doses approved were usually not identical to doses recommended on phase I trials. Approximately 50% of types of immune-related and 43% of types of clinically relevant toxicities from later trials were identified in early phase trials. Even so, treatment-related mortality remains exceedingly low in later studies (0.33% of patients). In conclusion, efficacy and safety of immune checkpoint inhibitors appear to be reasonably predicted from the dose-finding portion of phase I trials, indicating that the fast-track development of these agents is safe and justified.
INTRODUCTION
Therapeutic manipulation of the immune system has been attempted in oncology for many years. Numerous trials tested cytokines, vaccines and other immunostimulating agents in patients with cancer. Overall, this wave of development led to Food and Drug Administration (FDA) approval of a few, first-generation agents, including interferon and interleukin-2 for kidney cancer and melanoma(1–3) and sipuleucel-T for prostate cancer.(4)
More recently, an enhanced understanding of the mechanisms underlying immune responses against cancer cells led to the description of negative immunologic regulators (checkpoints) preventing effective immune eradication of tumors. As a result, monoclonal antibodies blocking immune checkpoints started clinical development.(5) Two of the main targets of these agents are cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and the programmed cell death protein pathway (PD1/PD-L1). Responses to these antibodies have been impressive, especially since some patients with advanced malignancies achieve long-term remissions. This new surge of immunotherapeutic agents is characterized by relatively rapid FDA approvals in diverse solid malignancies.
Many challenges unique to immunotherapy are, however, emerging, and important unanswered questions need to be explored.(6) The pertinent issues include an evaluation of how the traditional drug development model performs, as well as assessment of the regulatory timeline for these agents. Of interest, early marketing of these checkpoint inhibitors has occurred, including approval of pembrolizumab for melanoma, after a phase I trial.(7) The recent approval of pembrolizumab based on a tumor biomarker test regardless of tissue origin (approval for microsatellite unstable solid tumors) has also challenged historical approval models in oncology.(8) Therefore, drug development paradigms in the era of immunotherapy are evolving.
In order to better understand the impact of emerging drug development models, we performed a systematic review of FDA-approved immune checkpoint inhibitors, exploring their development timeline, and the correlations between toxicities, dosing and efficacy from early phase I trials, with similar information from later trials leading to approvals.
METHODS
Search Strategy
Immune checkpoint inhibitors newly approved for anticancer treatment prior to June 1st 2017 were identified on the FDA website.(9) Agents approved for the treatment of solid and hematologic malignancies were selected for further analysis. Original and updated package inserts for each agent were reviewed along with review documents available at the FDA website. Development milestones, drug indications, dose scheduling, and clinical trials leading to each immunotherapeutic agent approval were evaluated.
Selection of trials
An extensive search was concomitantly done through MEDLINE to identify phase I trials for each checkpoint inhibitors selected from the FDA database. Studies were obtained from publications in oncology journals. Alternatively, if data was not published yet, abstracts presented during oncology conferences were selected.
Phase I trials of single agent or different approved combinations and schedules were selected for evaluation, excluding phase Ib studies. Data was extracted preferentially from dose escalation and dose expansion, for dose finding purposes. It has become common in modern phase I trials to have different amendments to include expansion cohorts beyond dose finding, aiming to better define efficacy. For the purpose of our analysis, we excluded the information from the latter cohorts in order to evaluate the performance of a traditional dose-finding design of a phase I trial. In order to match the results of phase I trials with those from registration trial we selected one phase I trial representative of the initial development of each checkpoint inhibitor. The criteria for selection were: the phase I trial enrolled non-pediatric cancer patients and explored either monotherapy (as FDA-approved) or the same combination and schedule as described in the FDA package insert; they started before the registration trial; when more than one trial met these criteria we selected the one that started first after Investigational New Drug (IND) approval.
When referring to “later trials”, we considered the pivotal trial used for the first approval of a drug, in each tumor type. If a larger trial was published after drug approval (such as a phase III trial as part of an accelerated approval requirement), this trial was preferentially used for our analysis. This approach was utilized in order to have a more precise comparison between phase I and phase III trials. Both phase I and later trials were compared for each checkpoint inhibitor in regard to dosing, safety and efficacy. All correlations were summarized using descriptive statistics.
Data extraction and definitions
Toxicities were graded according to the criteria adopted in each trial. Considering the different terms used to describe adverse events, similar toxicities were categorized under the same group (types of toxicities), as long as they were not exclusionary (Supplemental Tables S1 and S2). All deaths reported by investigators as “possibly”, “probably”, or “definitely” related to treatment were considered toxicity-related deaths.
We defined Clinically Significant Toxicities in later trials as: treatment-related toxicities leading to death, treatment delays and discontinuations, as well as toxicities among the three most frequent grade 3/4 laboratory and non-laboratory toxicities, with an overall incidence of at least 1%.
Immune-related toxicities were extracted according to each trial assignment, including either high or low-grade immune-related toxicity, regardless of incidence. Safety profile was extracted from all later trials for different tumor types of immunotherapies selected for analysis.
Drug Development Information
Review documents for the newly approved checkpoint inhibitors were obtained for analysis through the FDA website(9). We evaluated data from IND submission, first and subsequent new drug application (NDA) or biologic license application (BLA) submission and approval, and access to FDA expedited programs. Definitions and further explanation about FDA programs data are depicted on Supplemental Table S3. We considered US clinical phase as the time between first IND submission and NDA/BLA submission. Approval phase was defined as the time of first NDA/BLA submission to approval. Total clinical development time was considered as the sum of both clinical and approval phases. Information about European Medicines Agency (EMA) approvals was obtained through the EMA website (http://www.ema.europa.eu/ema/).
RESULTS
Checkpoint inhibitors and approval history
Ipilimumab, a CTLA-4 inhibitor, was the first checkpoint inhibitor approved by the FDA (NDA approval date = March 2011). Since then, five additional checkpoint inhibitors were approved for the treatment of advanced cancer (first approval of nivolumab in 09/2014, pembrolizumab in 12/2014, atezolizumab in 05/2016, avelumab in 03/2017 and durvalumab in 05/2017). Ipilimumab together with nivolumab is the only combined treatment approved. For the first three checkpoint inhibitors the first registration was initially granted for metastatic melanoma, while for the more recent drugs for urothelial carcinoma (atezolizumab and durvalumab) and Merkel cell carcinoma (avelumab). Subsequent approvals for other tumor types were obtained for nivolumab, pembrolizumab, atezolizumab and avelumab (Table 1 and Supplemental Table S4). Currently, urothelial cancer is the tumor type with most checkpoint inhibitors approved (five in total), followed by melanomas and lung cancer (3 drugs for each).
Table 1.
Phase I trial as well as approval stage characteristics of immune checkpoint inhibitors
| Characteristic | Ipilimumab* (36) | Nivolumab (37) | Pembrolizumab (24) | Atezolizumab (25) | Avelumab (38) | Durvalumab (39) | Ipilimumab +Nivolumab (20) |
|---|---|---|---|---|---|---|---|
| Phase I stage | |||||||
| Trial Location | USA | USA | USA | USA, Europe | USA, Europe, Asia | USA, Europe, Asia | USA |
| Number of centers | 2 | 11 | 2 | 20 | 10 | 4 | 4 |
| Tumor types | 1 (non-metastatic melanoma) | 8 solid tumors | All solid tumors | All solid tumors | All solid tumors | All solid tumors | 1 (metastatic melanoma) |
| Trial design (40) | Pre-assigned dose levels | Accelerated titration design transitioned to a 3+3 design | 3+3 design | 3+3 design | 3+3 design | 3+3 design | 3+3 design |
| N patients | 19 | 207 | 30 | 171 | 27 | 27 | 86 |
| N dose levels | 3 | 4 | 3 | 5 | 4 | 6 | 7 |
| Response Criteria | N/A | Recist 1.1(10) | Recist 1.1(10) | Recist 1.1(10) | Recist 1.1(10) | Immune-related | Modified WHO(41) |
| N DLTs | 3 | 0 | 0 | 0 | 1 | 0 | 6 |
| MTD reached | Yes | No | No | No | No | No | Yes |
| Dose recommendation (parameter) | Yes (toxicities) | No clear dose recommended | Yes (lowest dose with efficacy) | Yes (PK) | Yes (PK) | Yes (PK/PD/safety data) | Yes (toxicities) |
| Approval Stage | |||||||
| First FDA approval | Melanoma | Melanoma | Melanoma | Urothelial carcinoma | Merkel cell carcinoma(42) | Urothelial carcinoma | Melanoma |
| Design of first pivotal trial leading to approval | Phase III | Phase III | Phase Ib | Phase II | Phase II | Phase II | Phase III |
| Subsequent approvals | No | 5 (NSCLC, Kidney cancer, Hodgkin Lymphoma, HNSCC, Urothelial) | 5 (NSCLC, HNSCC, Hodgkin Lymphoma, Urothelial, MSI-H cancers) | 1 (NSCLC) | 1 (Urothelial) | No | No |
| Response Criteria Pivotal Trials | Modified WHO(41) | Recist 1.1(10) and International Working Group (Hodgkin)(43) | Recist 1.1(10) and revised response criteria for Lymphomas(43) | Recist 1.1(10) | Recist 1.1(10) | Recist 1.1(10) | Recist 1.1(10) |
| Selection biomarker | No | No | Yes (PDL1 expression is required for NSCLC indication) and MSI-H tested cancers | No | No | No | No |
This phase I trial included patients with completely resected melanoma. Hence, response evaluation was not performed
Abbreviations: DLTs: dose limiting toxicities; HNSCCC: head and neck small cell carcinoma; MSI-H: microsatellite instability-high; MTD: maximum tolerated dose; N/A: not applicable; NSCLC: non small cell lung cancer; PD: pharmacodynamics; PK: pharmacokinetics; WHO: world health organization
Evidence for first approval was obtained from a phase III trial only for ipilimumab, nivolumab and the combination of both agents, while atezolizumab, avelumab and durvalumab authorization relied on phase II data, and pembrolizumab registration was based on a phase Ib trial. Among the 21 later trials included in our analysis, 18 (86%) used RECIST 1.1(10) as the response criteria for efficacy analysis (the other criteria adopted are described on Table 1). Additionally, 14 of these 21 (67%) trials defined response rate (RR) or progression-free survival (PFS) as the primary or co-primary endpoint. Pembrolizumab is a unique case, since it is the only drug that had approvals based on a biomarker-based rational. The metastatic non-small cell lung cancer (NSCLC) indication required a biomarker-based companion diagnosis (FDA-approved test for PD-L1 expression) for patient selection. More innovative was the recent approval for microsatellite instability high cancers (MSI-H) of pembrolizumab, the first tissue/site agnostic approval in oncology. All the remaining tumor type approvals for checkpoint inhibitors were for unselected, non-biomarker based cancer population.
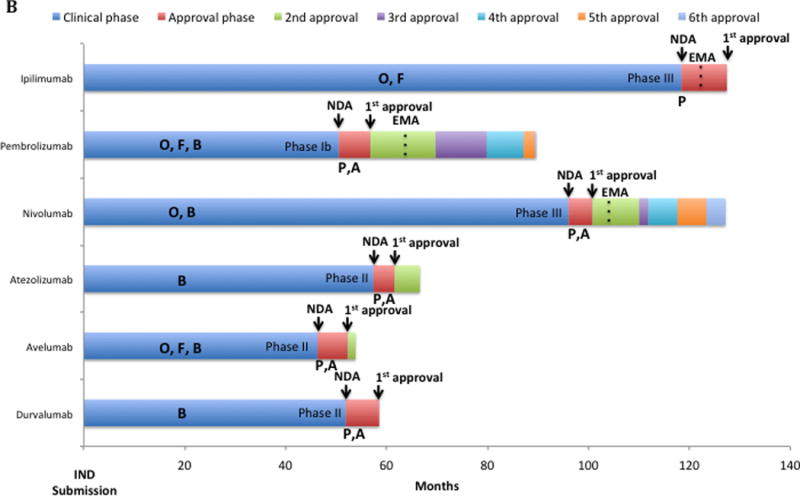
Total time for development of approved checkpoint inhibitors was a median of 60.77 months from the time of IND submission to the time of NDA approval (54.65 months for the clinical phase and 6.12 months for the approval phase). This timeline compared favorably to that of other anticancer agents approved by the FDA between 09/1999 and 07/2014 (median clinical and approval times of 75.4 and 6 months, respectively) (Figure 1A). Nonetheless, the specific timelines differed between checkpoint inhibitors as depicted in Figure 1B. All the drugs received a priority review status and access to at least two additional FDA special access programs. The five PD1/PD-L1 inhibitors received the more recent Breakthrough Designation and were first approved under Accelerated Approval status. Consequently, ipilimumab had the longest total development (127.4 months); avelumab, the shortest, since it was approved only after 52.33 months from IND submission (Figures 1B). None of the PD-L1 inhibitors (atezolizumab, avelumab and durvalumab) obtained European Medical Agency (EMA) approval yet, in contrast to the CTLA-4 and PD-1 inhibitors. The time gap between first FDA and EMA approval was longer for nivolumab and pembrolizumab (5.9 and 10.4 months, respectively) compared to ipilimumab (3.6 months).
Figure 1.
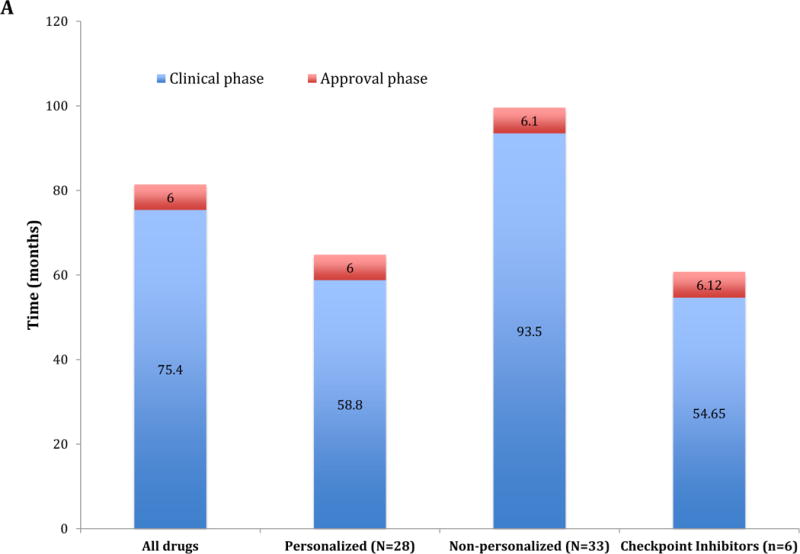
A: Comparison of the drug development timeline (Clinical and Approval phase) between 61 anticancer drugs approved by the FDA between 09/1999 and 07/2014 and immune checkpoint inhibitors approved until 06/2017. The 61 drugs were also stratified under personalized (n = 28 drugs) and non-personalized (n = 33 drugs) according to the development strategy. “Personalized” indicates biomarker-based approval.
B: Drug development Timeline of Immune Checkpoints inhibitors approved by the FDA. Each bar represents in months the time for each step of clinical development. Letters inside clinical phase and below approval phase represent access to each FDA-expedited program. At the end of blue bars the type of registration trial submitted for first approval is described (e.g., Phase I, Phase II or Phase III trial). Dashed lines inside bars are the moment in time of EMA approval (if received). Time from first FDA to first EMA approval: ipilimumab, 3.6 months; pembrolizumab, 10.4 months; nivolumab, 5.9 months. Clinical phase is time from IND submission to the FDA (necessary before first-in-human trial initiates) to the submission for new drug application (NDA). Approval phase is the time between the NDA submission to the FDA and the approval by the FDA of the drug for marketing.
Abbreviations: A: accelerated approval; B: breakthrough designation; EMA: European Medical Agency; F: fast-track: IND: investigational new drug; NDA: new drug application; O: orphan drug status; P: priority review
Definitions for these types of special FDA designations are given in Supplemental Table 3.
Correlation of response between early and later trials
In order to define how the tumor RR compared in phase I versus later registration trials, we assessed RR for the same tumor type from phase I and later trials (Figure 2). Some approvals were excluded from this analysis, as the phase I trials selected herein did not include the tumor types later approved based on registration trials. Median RR in phase I trials was 16%; phase I RRs in tumor types that were later granted FDA approval ranged from 0 to 57%. It is important to emphasize that RR was analyzed in our study only for the dose-defining portion of the phase I trials. In five of the eight (63%) comparisons, the RR of the later trial was higher compared to the phase I trial. The absolute difference in the RR in these five comparisons ranged from 9% to 18%. RR of pembrolizumab in melanoma and atezolizumab in urothelial and lung cancer during phase I was higher compared to later trials.
Figure 2.
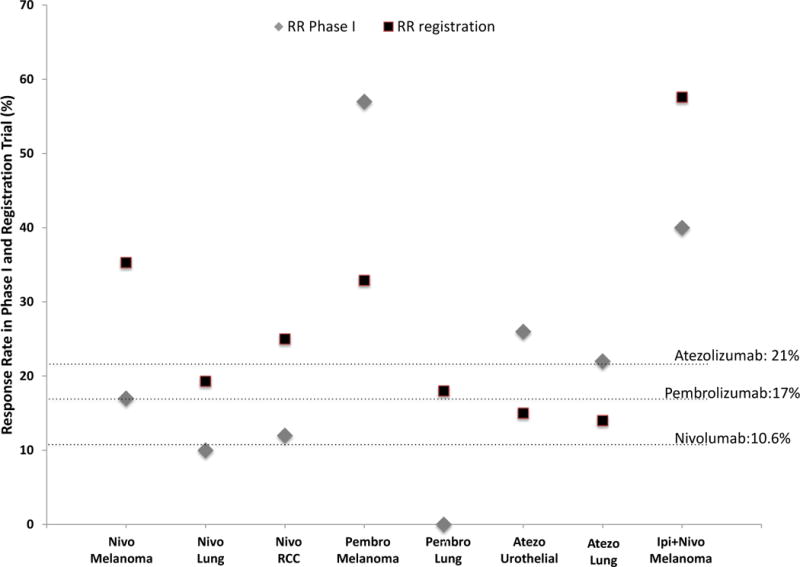
Correlation between the response rate (%) in a particular tumor included during the phase I trial (gray rhombus) and the later registration trial (black square). Horizontal axis depicts the immune checkpoint inhibitor in each approved indication. Response rate information for phase I was only included if a metastatic tumor of the same histology from the approval was tested. Dashed lines represent the overall response rate in the phase I trial (including all tumor types treated).
The figure shows that the response rates in later trials were generally higher than in the phase I trials; however, in the cases of pembrolizumab in melanoma and atezolizumab in urothelial and lung cancer, response rates in phase I trials were higher than in later trials. Some approvals were excluded as the phase I did not included the tumor types (Nivolumab for Hodgkin lymphoma and HNSCC; Pembrolizumab for Hodgkin Lymphoma, HNSCC, urothelial cancer and MSI-H tumors; Avelumab for Merkel Cell Carcinoma and urothelial cancer and Durvalumab for urothelial cancer).
Abbreviations: atezo = atezolizumab; ipi = ipilumumab; nivo = nivolumab; pembro = pembrolizumab; MSI-H: microsatellite instability high cancers; RCC = renal cell cancer: RR = response rate
Correlation of toxicities seen in Phase I trials of checkpoint inhibitors with those seen in later trials
We included seven phase I trials representing early phase of development from each checkpoint inhibitor and the combination treatment of ipilumumab and nivolumab (Table 1). The phase I trials were located exclusively in the USA in four instances, while for the development of three agents (atezolizumab, avelumab and durvalumab) the phase I trial also included sites in Europe and Asia. Number of patients included in these trials varied from 19 to 207. For the majority of them, dose escalation schema was a traditional 3+3, aiming to define the recommended phase II dose (RP2D) based on toxicities. Interestingly, dose-limiting toxicities (DLTs) were seen in the phase I trials testing ipilimumab together with nivolumab as well as ipilimumab and avelumab as a single agent, but not in the other phase I trials. As a result, only in two phase I trials a maximum tolerated dose (MTD) was found (both with ipilimumab). The phase I trials with PD1/PD-L1 inhibitors deployed as monotherapy used other parameters for RP2D definition (including pharmacodynamics and dose-efficacy curves). For nivolumab, the optimal dosing was not clearly defined after the phase I trial and the drug was excluded of dose comparison analysis. Later trials with checkpoint inhibitors adopted a dose that varied from 50% to 400% of RP2D. In four out of 13 (31%) matched comparisons (atezolizumab, durvalumab and ipilimumab together with nivolumab) the later trials dosage was exactly the same (100% of the RP2D) as that recommended based on phase I.
We identified a total of 65 types of clinically significant toxicities in the later trials with checkpoint inhibitors, of which 28 (43%) were at least cited in respective phase I trials (Table 2). The avelumab phase I trial described 25% of types of clinically relevant toxicities documented in later trials; it was one of the smallest phase I trial (n=27 patients). The number of types of toxicities considered to be immune related in later trials was 57. Of these, 29 (50.9%) were described during phase I trials. In our group of matched comparisons, the total number of patients included in a phase I did not correlate with an improved description of clinically significant toxicities during phase I trials (Figure 3). However, it appeared that a better description of types of immune-related toxicities in phase I trials was associated with more patients included in the phase I trial. Finally, a more robust correlation between the ability of the phase I trials describing types of clinically significant and immune-related toxicity was seen according to the ratio of the number of patients included in a phase I versus the later trial.
Table 2.
Information about doses and important toxicities present in later trials and correlation with early phase I trials
| Ipilimumab | Nivolumab | Pembrolizumab | Atezolizumab | Avelumab | Durvalumab | Ipilimumab +Nivolumab | |
|---|---|---|---|---|---|---|---|
| Dose used in later trial (% of RP2D from phase I) | 3mg/kg q3wks (400%) | 3 mg/kg q2wks (no RP2D) | Different doses (66% to 333%) | 1200 mg q3wks (100%) | 10 mg/kg q2wks (50%) | 10 mg/kg q2wks (100%) | Nivolumab 1mg/kg Ipilumab 3mg/kg Q3 wks × 4 doses (100%) |
| Number of patients in phase I trial* | 19 | 207 | 30 | 171 | 27 | 27 | 86 |
| Number of patients in later trials** | 137 | 1904 | 1334 | 1160 | 337 | 191 | 314 |
| Number of types of clinically significant toxicities in later trials# | 5 | 18 | 21 | 6 | 6 | 4 | 5 |
|
| |||||||
| Number of types of toxicities described in Phase I trial (% of types of toxicities described in later trials) | 2 (40) | 7 (39) | 6 (29) | 3 (50) | 4 (67) | 1 (25) | 5 (100) |
|
| |||||||
| Number of types of immune-related toxicities in later trials# | 7 | 9 | 15 | 4 | 8 | 7 | 7 |
|
| |||||||
| Number of types of immune-related toxicities described in Phase I trials (% of types of immune-related toxicities described in later trials) | 3 (42.9) | 6 (66.7) | 4 (26.6) | 3 (75) | 3 (37.5) | 3 (43) | 7 (100) |
| Treatment related mortality in phase I trials (% of patients in the trials) All trials = 0.18% |
0 | 0 | 3.33 | 0 | 0 | 0 | 0 |
| Treatment-related mortality later trials (% of patients in the trials) All Trials = 0.33% |
2.9 | 0.32 | 0.37 | 0 | 0.3 | 1.05 | 0 |
Patients included during dose escalation and dose expansion cohorts for dose-finding purposes only
Data obtained from all later trials for the different FDA-approved indications
Clinically significance toxicities were defined as: treatment-related toxicities leading to death, treatment delays and discontinuations, and toxicities among the three most frequent grade 3/4 laboratory and non-laboratory toxicities, with an overall incidence of at least 1%(33); in the case of immune-related toxicities, all types of toxicities were included regardless of grade
Abbreviations: RP2D: recommended phase II dose
Figure 3.
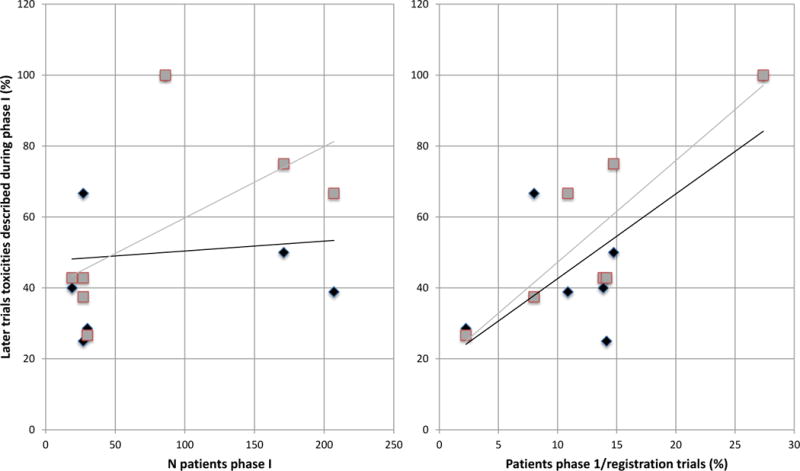
Correlation between toxicities in phase I and in later trials.
Y axis is the types of toxicities described in phase I trials as a percent of the types of toxicities described in the registration trials.
Left panel: correlation between the number of patients included during the phase I trial and the description of all clinically significant toxicities (black diamond), as well as immune-related toxicities (gray square) from later trials. This panel shows that there is an increase in the ability to identify the immune-related toxicities as the number of patients in phase I increases. The number of patients in phase I did not, however, correlate with the ability to identify all types of clinically relevant toxicities.
Right panel: correlation between the ratio of patients included during phase I over patients included in registration trials and the description of clinically significant (black diamond) and immune-related toxicities (gray square) from later trials.
This panel shows that the ratio of the number of patients in phase I over the number of patients in the registration trial(s) correlated with the ability to identify either immune-related or all clinically relevant toxicities.
Treatment-related mortality in phase I trials with checkpoint inhibitors was low (0.18%), and accurately predicted a low treatment-related mortality rate in later trials (0.33%).
DISCUSSION
Checkpoint inhibitors represent a new wave of successful immunotherapies in oncology. Indeed, based on their striking results, cancer immunotherapy was heralded as the science breakthrough of 2013.(11) Only a few years later, we have six checkpoint inhibitors approved by the FDA. In this comprehensive assessment of FDA-approved immune checkpoint inhibitors, we aimed to evaluate how the drug development paradigm performed for these agents.
Among our findings, total clinical development of checkpoint inhibitors took a median of 60.77 months, which compared favorably to other anticancer agents approved by the FDA (Figure 1). The checkpoint inhibitors timeline is more similar to the faster approval for targeted agents approved under a biomarker-based rationale, a finding that could represent a contemporary shift by the FDA. Indeed, after ipilimumab approval, there was a trend towards shortening the development approval process (Figure 2). It is noteworthy that these agents, especially PD1/PD-L1 inhibitors, are also benefitting from access to FDA programs for expedited development. Of note, all five PD-1/PD-L1 inhibitors received breakthrough designation and accelerated approval designations, and pembrolizumab was approved for melanoma after a phase IB study.(7)
Recent publications demonstrate that a biomarker-based strategy was an independent factor predicting faster development of anticancer agents.(12–14) Interestingly, pembrolizumab was the only immune checkpoint inhibitor approved with a biomarker for patient selection, including the NSCLC (requiring PD-L1 expression) and the most recent tissue agnostic microsatellite instability tumor indication.(8) Despite the absence of a widespread clinical use of biomarkers for checkpoint inhibitors,(15), this later approval represents a regulatory shift paradigm in oncology, especially since the approval used a genomic marker for regulatory authorization across all solid tumors.
Data from early trials are also serving as the basis for regulatory initial approval of these agents. It is important to note that this observation is more related to a changing paradigm adopted by the FDA in recent years, rather than a special privilege for immunotherapies. As examples, approvals of crizotinib and ceritinib used very early trial data for approval, including phase I data alone for ceritinib.(16, 17) Consequently, if other regulatory agencies do not adopt a similar pathway, the time gap between FDA approvals compared to other worldwide agencies for checkpoint inhibitor approvals might increase. Herein, we described a longer gap between FDA and EMA approvals of PD-L1 inhibitors nivolumab and pembrolizumab (5.9 and 10.4 months, respectively), compared to ipilimumab (3.6 months).
Registration adoption of checkpoint inhibitors based only after early phase trials could raise concerns regarding safety and performance in later trials. Nevertheless, it is important that there is not a large safety issue that is discovered in later clinical trials. Regarding dosing and schedule, our analysis suggests that phase I studies of checkpoint inhibitors define doses that are usually different than those later adopted. Doses accepted in later trials were 400% (ipilumumab), 66% to 333% (pembrolizumab), 100% for atezolizumab, durvalumab and for combined iplilumumab and 50% (avelumab) of the recommended dose in phase I (Table 2). Therefore, phase I testing did not clearly establish a dose definition for checkpoint inhibitors. There is also uncertainty regarding final optimal dose, as illustrated by the variation of approved doses within package inserts of ipilimumab(18) and pembrolizumab,(19) even after FDA approval. Many of the phase I trials of checkpoint inhibitors were designed using traditional pre-assigned dose levels design (3+3 dose escalation) and defining toxicities (DLTs) as the main outcome for dose definition. Nonetheless, DLTs were often not found for these agents, especially concerning PD1/PD-L1 inhibitors.(6) The determination of a MTD might be more important for immunotherapy combinations, which are usually associated with greater toxicity.(20) The concept of a DLT window (usually about four weeks) in phase I trials must also adapt to immunotherapy, as immune-related toxicities may occur only after weeks or months of administration.(21) Although the current model is so far not compromising safety, a longer period of toxicity assessment could lead to a more precise definition of the toxicity profile among checkpoint inhibitors. Finally, it is not unexpected that new challenges might arise after approval, regardless of the type of study leading to approval. An example of such a challenge is the recent recognition of accelerated progression (hyper-progression) in a subset of patients treated with anti-PD1/PD-L1 agents.(22, 23) However, there is no evidence that this phenomenon, which occurs in less that 10 percent of patients, would be more identifiable with a different development/approval pathway, nor does its recognition obviate the substantial benefit derived by significant subgroups of patients from checkpoint blockade.
For checkpoint inhibitors, RP2D recommendations are often based on maximum administered doses.(6) As part of phase I trials, pharmacokinetic studies and understanding immune target engagement may help to more precisely define a dose, with less inter-individual variation.(24, 25) This information, as well as efficacy data, could be used to designate a ”minimal effective dose.” Otherwise, post-phase I and even post-approval testing can explore different doses and provide updates to approval documents, as has occurred with nivolumab.(26–28)
An interesting aspect of checkpoint inhibitor development is that anti-tumor activity, including durable complete remissions, were observed in phase I trials. Overall RRs, however, remained low—about 16%, which is still higher than those in historical phase I studies of genomically targeted agents or chemotherapy performed without a biomarker (approximately 5%).(14, 29) RRs in phase I trials of checkpoint inhibitors often, but not always, mirrored the activity observed in tumor types tested in later trials (Figure 2). Profound clinical activity was described for some patients, both in early and later trials, including complete and long -lasting responses. Nevertheless, a significant number of patients do not yet derive benefit from the current approved checkpoint inhibitors. Primary and acquired resistance to checkpoint inhibitors are major challenges for the future development of immunotherapies. Resistance may be due to modulation of antigen-presenting proteins, as well as genetic abnormalities in tumor and lack of T-cell infiltrate; genomic deletions in beta2 microgloblobulin and JAK2 genes may be operative. (30, 31) Hyperprogression (accelerated progression) after checkpoint blockade may also occur and has been associated with MDM2 amplification and EGFR alterations.(22, 23) Although only one checkpoint combination is currently FDA-approved (combining anti-PD-1 and anti-CTLA-4), emerging combinations of checkpoint inhibitors with a variety of other agents might be one strategy to overcome treatment resistance.(32)
During the dose definition portion of phase I trials of checkpoint inhibitors, 43% of types of clinically relevant toxicities seen in later trials were described. Previously, we have shown that phase I studies from the pre-immunotherapy era predicted about 70% of types of toxicities identified in later studies.(33) The lower proportion of toxicities uncovered in phase I immunotherapy trials as compared to previous trials of other drugs could be due to many factors, including, but not limited to, the comparative side effect profile of immunotherapy versus other agents and the relatively few phase I trials of approved checkpoint inhibitors. Immune related toxicities are characteristic of checkpoint inhibitors, and, although generally mild, they can be life-threatening.(34) Overall, we found that 50.9% of the types of immune-related toxicities detected in later trials were already evident in the phase I studies. The occurrence of delayed toxicities with immune checkpoint inhibitors might also account for the fewer descriptions of clinically relevant and immune-related toxicities in phase I trials. In addition to higher numbers of patients included, treatment duration and toxicity assessment window can be longer on later trials. Encouragingly, treatment-related mortality remained low in early trials as well as in later studies of checkpoint inhibitors. A recent paper reassured the similarity of immune toxicity profile between phase I and late trials, with a higher concordance according to the increased sample size of the former.(35)
The findings reported here are limited to the few numbers of checkpoint inhibitors approved so far by the FDA. Many new immune checkpoint modulators, including agonists and antagonists, are currently in development and might take advantages of the first conclusions regarding the drug development paradigm discussed here. Future systematic reviews might be needed according to the approval of new classes of immune modulatory drugs.
In conclusion, approval of checkpoint inhibitors in a variety of tumor types is rapidly changing the landscape of cancer treatment. Their development is being characterized by the increased importance of early trials. Indeed, many of these phase I trials are being expanded to include diverse patient cohorts, leading to expedited regulatory approval. Our analysis suggests that the current clinical trial paradigms work reasonably well for predicting safety and efficacy of immunotherapy in later studies, though dosing based on phase I trials appears to be variable compared to final dosing. Although the dose-finding portion of phase I studies did not report on a significant percentage of the types of toxicities that are detected with broader use of these agents, this does not appear to have affected drug-related mortality in later trials. Indeed, drug-related mortality in later trials remains exceedingly low, demonstrating that rapid approvals of appropriate immunotherapy agents are justifiable.
Supplementary Material
Acknowledgments
Grant Support:
Funded in part by National Cancer Institute grant P30 CA016672 and the Joan and Irwin Jacobs Fund philanthropic fund (all funds received by Razelle Kurzrock).
Author’s Disclosures
Denis L. Jardim received honoraria from Bristol-Myers Squibb, Merck-Sharpe Dohme and Roche.
Débora de Melo Gagliato received honoraria from Merck-Sharpe Dohme and Roche.
Razelle Kurzrock receives research funds from Sequenom, Guardant, Foundation Medicine, Genentech, Pfizer, and Merck Serono, consultant fees from XBiotech and Actuate Therapeutics, and has an ownership interest in Curematch, Inc.
References
- 1.Summers J, Cohen MH, Keegan P, Pazdur R. FDA drug approval summary: bevacizumab plus interferon for advanced renal cell carcinoma. Oncologist. 2010;15:104–11. doi: 10.1634/theoncologist.2009-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coventry BJ, Ashdown ML. The 20th anniversary of interleukin-2 therapy: bimodal role explaining longstanding random induction of complete clinical responses. Cancer Manag Res. 2012;4:215–21. doi: 10.2147/CMAR.S33979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickler MN, Coit DG, Meyers ML. Adjuvant therapy of malignant melanoma. Surg Oncol Clin N Am. 1997;6:793–812. [PubMed] [Google Scholar]
- 4.Gardner TA, Elzey BD, Hahn NM. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum Vaccin Immunother. 2012;8:534–9. doi: 10.4161/hv.19795. [DOI] [PubMed] [Google Scholar]
- 5.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33:1974–82. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Postel-Vinay S, Aspeslagh S, Lanoy E, Robert C, Soria JC, Marabelle A. Challenges of phase 1 clinical trials evaluating immune checkpoint-targeted antibodies. Ann Oncol. 2016;27:214–24. doi: 10.1093/annonc/mdv550. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384:1109–17. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 8.First Tissue-Agnostic Drug Approval Issued. Cancer Discov. 2017 doi: 10.1158/2159-8290.CD-NB2017-078. [DOI] [PubMed] [Google Scholar]
- 9.Administration UFaD. FDA approved drug products. 2016 [cited 2016 08/2016]. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/
- 10.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–47. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–3. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 12.Jardim DL, Schwaederle M, Hong DS, Kurzrock R. An appraisal of drug development timelines in the Era of precision oncology. Oncotarget. 2016;7:53037–46. doi: 10.18632/oncotarget.10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwaederle M, Zhao M, Lee JJ, Eggermont AM, Schilsky RL, Mendelsohn J, et al. Impact of Precision Medicine in Diverse Cancers: A Meta-Analysis of Phase II Clinical Trials. J Clin Oncol. 2015;33:3817–25. doi: 10.1200/JCO.2015.61.5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwaederle M, Zhao M, Lee JJ, Lazar V, Leyland-Jones B, Schilsky RL, et al. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysis. JAMA Oncol. 2016;2:1452–9. doi: 10.1001/jamaoncol.2016.2129. [DOI] [PubMed] [Google Scholar]
- 15.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhillon S, Clark M. Ceritinib: first global approval. Drugs. 2014;74:1285–91. doi: 10.1007/s40265-014-0251-3. [DOI] [PubMed] [Google Scholar]
- 17.Gandhi L, Janne PA. Crizotinib for ALK-rearranged non-small cell lung cancer: a new targeted therapy for a new target. Clin Cancer Res. 2012;18:3737–42. doi: 10.1158/1078-0432.CCR-11-2393. [DOI] [PubMed] [Google Scholar]
- 18.FDA. Ipilimumab Package Insert. 2015 Available from: http://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125377s073lbl.pdf.
- 19.FDA. Pembrolizumab Package Insert. 2016 [Google Scholar]
- 20.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eigentler TK, Hassel JC, Berking C, Aberle J, Bachmann O, Grunwald V, et al. Diagnosis, monitoring and management of immune-related adverse drug reactions of anti-PD-1 antibody therapy. Cancer Treat Rev. 2016;45:7–18. doi: 10.1016/j.ctrv.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Kato S, Goodman A, Walavalkar V, Barkauskas DA, Sharabi A, Kurzrock R. Hyperprogressors after Immunotherapy: Analysis of Genomic Alterations Associated with Accelerated Growth Rate. Clin Cancer Res. 2017;23:4242–50. doi: 10.1158/1078-0432.CCR-16-3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin Cancer Res. 2017;23:1920–8. doi: 10.1158/1078-0432.CCR-16-1741. [DOI] [PubMed] [Google Scholar]
- 24.Patnaik A, Kang SP, Rasco D, Papadopoulos KP, Elassaiss-Schaap J, Beeram M, et al. Phase I Study of Pembrolizumab (MK-3475; Anti-PD-1 Monoclonal Antibody) in Patients with Advanced Solid Tumors. Clin Cancer Res. 2015;21:4286–93. doi: 10.1158/1078-0432.CCR-14-2607. [DOI] [PubMed] [Google Scholar]
- 25.Herbst RS, Gordon MS, Fine GD, Sosman JA, Soria J-C, Hamid O, et al. A study of MPDL3280A, an engineered PD-L1 antibody in patients with locally advanced or metastatic tumors. ASCO Meeting Abstracts. 2013;31:3000. [Google Scholar]
- 26.Motzer RJ, Rini BI, McDermott DF, Redman BG, Kuzel TM, Harrison MR, et al. Nivolumab for Metastatic Renal Cell Carcinoma: Results of a Randomized Phase II Trial. J Clin Oncol. 2015;33:1430–7. doi: 10.1200/JCO.2014.59.0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T, et al. Safety and Antitumor Activity of Anti-PD-1 Antibody, Nivolumab, in Patients With Platinum-Resistant Ovarian Cancer. J Clin Oncol. 2015;33:4015–22. doi: 10.1200/JCO.2015.62.3397. [DOI] [PubMed] [Google Scholar]
- 28.FDA. Modification of The Dosage Regimen for Nivolumab. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm520871.htm2016 [cited 2016 11/04]
- 29.Horstmann E, McCabe MS, Grochow L, Yamamoto S, Rubinstein L, Budd T, et al. Risks and benefits of phase 1 oncology trials, 1991 through 2002. N Engl J Med. 2005;352:895–904. doi: 10.1056/NEJMsa042220. [DOI] [PubMed] [Google Scholar]
- 30.Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer. 2016;16:121–6. doi: 10.1038/nrc.2016.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375:819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris SJ, Brown J, Lopez J, Yap TA. Immuno-oncology combinations: raising the tail of the survival curve. Cancer Biol Med. 2016;13:171–93. doi: 10.20892/j.issn.2095-3941.2016.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jardim DL, Hess KR, Lorusso P, Kurzrock R, Hong DS. Predictive value of phase I trials for safety in later trials and final approved dose: analysis of 61 approved cancer drugs. Clin Cancer Res. 2014;20:281–8. doi: 10.1158/1078-0432.CCR-13-2103. [DOI] [PubMed] [Google Scholar]
- 34.Bourke JM, O’Sullivan M, Khattak MA. Management of adverse events related to new cancer immunotherapy (immune checkpoint inhibitors) Med J Aust. 2016;205:418–24. doi: 10.5694/mja16.00586. [DOI] [PubMed] [Google Scholar]
- 35.Costa R, Costa RB, Talamantes SM, Helenoswki I, Carneiro BA, Chae YK, et al. Analyses of selected safety endpoints in phase 1 and late-phase clinical trials of anti-PD-1 and PD-L1 inhibitors: prediction of immune-related toxicities. Oncotarget. 2017 doi: 10.18632/oncotarget.18847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanderson K, Scotland R, Lee P, Liu D, Groshen S, Snively J, et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J Clin Oncol. 2005;23:741–50. doi: 10.1200/JCO.2005.01.128. [DOI] [PubMed] [Google Scholar]
- 37.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–65. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heery CR, Coyne GHOS, Madan RA, Schlom J, Heydebreck Av, Cuillerot J-M, et al. Phase I open-label, multiple ascending dose trial of MSB0010718C, an anti-PD-L1 monoclonal antibody, in advanced solid malignancies. Journal of Clinical Oncology. 2014;32:3064. [Google Scholar]
- 39.Lutzky J, Antonia SJ, Blake-Haskins A, Li X, Robbins PB, Shalabi AM, et al. A phase 1 study of MEDI4736, an anti–PD-L1 antibody, in patients with advanced solid tumors. Journal of Clinical Oncology. 2014;32:3001. [Google Scholar]
- 40.Le Tourneau C, Lee JJ, Siu LL. Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst. 2009;101:708–20. doi: 10.1093/jnci/djp079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.James K, Eisenhauer E, Christian M, Terenziani M, Vena D, Muldal A, et al. Measuring response in solid tumors: unidimensional versus bidimensional measurement. J Natl Cancer Inst. 1999;91:523–8. doi: 10.1093/jnci/91.6.523. [DOI] [PubMed] [Google Scholar]
- 42.Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016;17:1374–85. doi: 10.1016/S1470-2045(16)30364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheson BD, Pfistner B, Juweid ME, Gascoyne RD, Specht L, Horning SJ, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25:579–86. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.