

Abstract
Purpose
Schlafen 11 (SLFN11), a putative DNA/RNA helicase is a dominant genomic determinant of response to DNA damaging agents and is frequently not expressed in cancer cells. Whether histone deacetylase (HDAC) inhibitors can be used to release SLFN11 and sensitize SLFN11-inactivated cancers to DNA-targeted agents is tested here.
Experimental Design
SLFN11 expression was examined in The Cancer Genome Atlas (TCGA), in cancer cell line databases and in patients treated with romidepsin. Isogenic cells overexpressing or genetically inactivated for SLFN11 were used to investigate the effect of HDAC inhibitors on SLFN11 expression and sensitivity to DNA damaging agents.
Results
SLFN11 expression is suppressed in a broad fraction of common cancers and cancer cell lines. In cancer cells not expressing SLFN11, transfection of SLFN11 sensitized the cells to camptothecin, topotecan, hydroxyurea and cisplatin but not to paclitaxel. SLFN11 mRNA and protein levels were strongly induced by class I (romidepsin, entinostat), but not class II (roclinostat) HDAC inhibitors in a broad panel of cancer cells. SLFN11 expression was also enhanced in peripheral blood mononuclear cells of patients with circulating cutaneous T-cell lymphoma treated with romidepsin. Consistent with the epigenetic regulation of SLFN11, camptothecin and class I HDAC inhibitors were synergistic in many of the cell lines tested.
Conclusion
This study reports the prevalent epigenetic regulation of SLFN11 and the dominant stimulatory effect of HDAC inhibitors on SLFN11 expression. Our results provide a rationale for combining class I HDAC inhibitors and DNA damaging agents to overcome epigenetic inactivation of SLFN11-mediated resistance to DNA-targeted agents.
Keywords: SLFN11, TCGA, epigenetic, histone deacetylase inhibitors, precision medicine, CCLE, NCI-60, GDSC
Introduction
Despite the promise of kinase inhibitors and immunotherapy, DNA damaging agents remain widely used for a broad range of solid tumors and hematologic malignancies. DNA damaging agents have a wide range of anti-tumor activity but are associated with toxicities related to their effects on normal tissue. Use of predictive biomarkers to maximize treatment efficacy and to minimize the risk of deleterious toxicities has long been recognized as an important goal in the clinical application of these drugs. However, at the present time, there are no reliable biomarkers of activity of DNA-targeted agents that have proven useful for patient stratification.
Recent studies have identified Schlafen 11 (SLFN11), a nuclear protein belonging to the Schlafen family of mammalian proteins, as a causal and dominant genomic determinant of response to topoisomerase (TOP) 1 inhibitors (camptothecin, topotecan and irinotecan), TOP2 inhibitors (etoposide, doxorubicin, mitoxantrone), alkylating agents (cisplatin and carboplatin, nitrogen mustards, ifosfamide), DNA synthesis inhibitors (gemcitabine, cytarabine, hydroxyurea) and poly-(ADP)-ribose polymerase (PARP) inhibitors (olaparib, rucaparib, niraparib and talazoparib) (1–6). In addition to the role of SLFN11 in sensitizing cancer cells to DNA damaging agents, Schlafen proteins play important roles in regulating biological functions including cellular proliferation, induction of immune response and suppression of viral replication (7,8). In patient tumors and cancer cell lines, SLFN11 demonstrates a broad range of expression in a wide range of cancer types (Fig. 1 and Supplementary Fig. S1), which suggests its use as a predictive biomarker of response to DNA damaging agents. Indeed, SLFN11 has been identified as a predictor of response to chemotherapy in ovarian cancer, small cell and non-small cell lung cancer, colorectal cancer and Ewing sarcoma (5,6,9–13). For example, retrospective analyses show that high SLFN11 expression is positively correlated with tumor-free survival in Ewing sarcoma and colorectal and ovarian cancer patients (1,5,12).
Fig. 1.
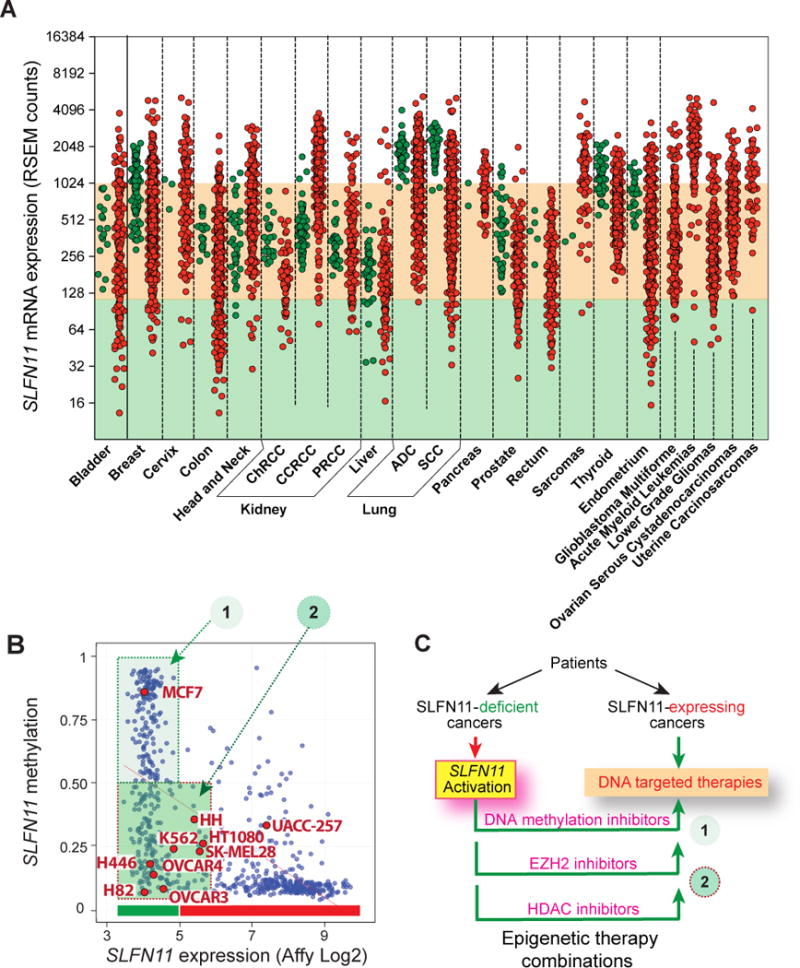
SLFN11 is both repressed and overexpressed across the various cancer types of the TCGA database and is epigenetically regulated in the 1,000 cancer cell line databases. (A) When available, for each tissue type, normal samples are shown in green and cancer samples in red. SLFN11 transcript expression was determined by RNASeq and is represented as the log2 of the RSEM-quantified transcript abundance level. The salmon color shaded area corresponds to average normal transcript levels. Abbreviations: ChRCC: Chromophobe renal cell carcinoma; CCRCC: clear cell renal cell carcinoma; PRCC: papillary renal cell carcinoma; ADC: adenocarcinoma; SCC: squamous cell carcinoma. (B) SLFN11 expression in the cancer cell lines of the CCLE collection (http://www.broadinstitute.org/ccle/) matched with promoter methylation determined from the data from the GDSC database (http://www.cancerrxgene.org/) for the individual common cell lines across the two databases (blue dots) obtained with CellMiner (http://discover.nci.nih.gov/cellminercdb/). Cell lines in groups 1 do not express SLFN11 and have promoter methylation. Cell lines in group 2 do not express SLFN11 presumably due to chromatin epigenetic drivers such as histone deacetylation. Cell lines used in this study are marked in red (see also Supplementary Fig. S1). (C) Scheme for the rationale of the study. Recent studies have shown that SLFN11 can be derepressed by inhibiting DNA methylation and by EZH2 inhibitors (9,13). The present study explores the relevance of epigenetic acetylation by using HDAC inhibitors.
While the expression of SLFN11 is linked to sensitivity to DNA-targeted agents, lack of SLFN11 expression observed in approximately half of the 60 cancer cell lines in the US National Cancer Institute (NCI) Human Tumor Cell Line Screen (NCI-60) (Supplementary Fig. S1) is related to resistance to these agents (1,9,14,15). DNA methylation analysis of the NCI-60 cell line panel identified CpG promoter island hypermethylation of SLFN11 as a biomarker of resistance to platinum drugs as well as a broad range of DNA-targeted anticancer therapies (9,15). A significant association was observed between SLFN11 CpG promoter island hypermethylation and diminished SLFN11 mRNA levels in cancer cells. The epigenetic silencing of SLFN11 and the fact that promoter hypermethylation only accounts for a fraction of cancer cells that lack SLFN11 expression (Fig. 1B and Supplementary Fig. S1A), and are therefore likely chemotherapy resistant, raises the question of whether epigenetic modifying drugs can derepress SLFN11 and sensitize SLFN11-inactivated cancer cells to chemotherapy. Consistent with this possibility, two recent studies showed that the DNA demethylating drug, 5-aza-2′-deoxycytidine (decitabine) and the EZH2 inhibitor, EPZ011989 induced SLFN11 expression both at the transcript and protein levels (9,13). Moreover, treatment with the EZH2 inhibitor was shown to enhance the cytotoxicity and antitumor activity of topotecan in small cell lung cancer (SCLC) models (13).
In this study, we investigated the effect of histone deacetylase (HDAC) inhibitors on SLFN11 expression and sensitivity to DNA-damaging agents across a panel of 10 cancer cell lines from various tissues of origin. Nine of them did not express SLFN11 and did not exhibit CpG promoter methylation (2 SCLC cell lines: H446 and H82; 2 ovarian cell lines: OVCAR3 and OVCAR4; 2 melanoma cell lines: SK-MEL28 and UACC-257; one fibrosarcoma cell line: HT1080; two leukemia cell lines: K562 and HH). We also tested one cell line with high promoter methylation (breast cancer MCF7) (Fig. 1B and Supplementary Fig. S1A). Our rationale (see Fig. 1C) was to determine whether epigenetic activation by clinically used HDAC inhibitors could derepress SLFN11 and sensitize cells to the prototypical TOP1 inhibitor camptothecin (CPT).
Materials and Methods
Cell lines, culture and drugs
The K562, HCT116, OVCAR-3, SK-MEL-28, OVCAR-4, UACC-257, MCF7 and CCRF-CEM cell lines were obtained from the Division of Cancer Treatment and Diagnosis (DCTD), Developmental Therapeutics Program (DTP, NCI). HT1080 cells were kindly provided by Dr. Lee Helman (NCI/NIH). H82 (HTB-175), H446 (HTB-171) and HH (CRL-2105) cells were obtained from ATCC®. All cells were grown in RPMI medium with 10% fetal bovine serum (FBS) (Gibco-BRL) at 37°C in 5% CO2. Camptothecin, topotecan, cisplatin, hydroxyurea, romidepsin, entinostat and rocilinostat were obtained from the DCTD. Actinomycin D (A9415) was obtained from Sigma-Aldrich.
Generation of SLFN11-expressing cells
SLFN11 cDNA was amplified using the forward primer (5′- ATCGGATCC GCGGCCAACATGGAGGCAAATCAGTGC-3′) and the reverse primer with the sequence for the Flag tag (5′-ATTGTCGACGCGGCCCTACTTATCGT CGTCAT CCTTGTAATCATGGCCACCCCACGGAA-3′) and cloned into the pCDH-EF1-MCS-(PGK-copGFP) lentiviral expression vector (System Biosciences) using the In-Fusion HD cloning kit (Clontech). The lentiviral SLFN11-expressing vector and the pPACKH1 lentivector packaging plasmids were cotransfected into 293TN cells (System Biosciences) and the viral particles were collected to infect K562 cells with TransduxTM (System Biosciences). The SLFN11-expressing cells with GFP signal were sorted using a Fluorescence Activated Cell Sorter (FACS).
Cell viability assays
Cells were treated or untreated with different concentrations of HDAC inhibitors for 16 hours prior to adding camptothecin. After the pretreatment, cells were collected, washed and seeded in 96-well white plates (#6005680 Perkin Elmer Life Sciences) in 100 μl of medium per well with the following cell number; 30,000 cells for K562, 3,000 cells for HT1080 and HCT116, 5,000 cells for H446, H82 and HH cells. Cells were continuously exposed to the indicated drug concentrations for 48-72 hours in triplicate. Cellular viability was determined using ATPlite 1-step kits (PerkinElmer). The ATP level in untreated cells was defined as 100%. Viability of treated cells was defined as: (ATP in treated cells)/(ATP in untreated cells)x 100.
Quantitative real-time PCR
The RNAs of drug-treated or -untreated cells were extracted using RNeasy Mini RNA kit (Qiagen) and the complementary DNA was generated by SuperScript™ II Reverse Transcriptase kit (Invitrogen) according to the manufacturer’s instructions. The primers specific for SLFN11, FLI-1 and GAPDH and materials for quantitative real-time PCR were described in our previous study(5). The melting curve was measured to show the amplification specificity. The expression levels of GAPDH was considered as the reference gene. The relative expression of SLFN11 and FLI-1 to reference gene GAPDH was determined using the 2(−ΔΔCt) method.
To measure the half-life of mRNA for SLFN11, GAPDH, PUMA and Cyclin E, CCRF-CEM cells were treated or untreated with actinomycin D (1 μM) for 6 hours, and total RNA was extracted. The expression level of 18S ribosomal RNA was considered as the reference gene. Primers for each gene was designed by PrimerBank (https://pga.mgh.harvard.edu/primerbank/).
Western blotting
Cells were treated with the indicated concentrations of HDAC inhibitors for 16 hours and and lysed in 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.1% SDS supplemented with protease inhibitor cocktail (Roche). The lysates were separated in 8-15% of SDS-PAGE and transferred to polyvinylidene fluoride membranes (Millipore). The antibodies specific to SLFN11 (sc-374339 or sc-515071, Santa Cruz Biotechnology), acetyl histone H3 (ab4441, Abcam), FLI1 (sc-356, Santa Cruz Biotechnology), actin (MAB1501, Chemicon International), or GAPDH (2118S, Cell Signaling Technology) were applied to the membranes 4°C overnight followed by horseradish peroxidase-conjugated secondary antibodies (GE Healthcare Life Sciences). The signals were developed by ECL Western Blotting Substrate (Thermo Scientific). Actin was used as the loading control.
Microarray and RNA-Seq data
Peripheral blood mononuclear cells from patients with circulating tumor cells were obtained before infusion (pre), and at 4 hours after start of the infusion of the first cycle of treatment (16) The normalized gene expression from microarray analysis (16) were downloaded from the Gene Expression Omnibus website (GSE45405, http://www.ncbi.nlm.nih.gov/geo) (17). The RNA-Seq data for SLFN11 gene expression in the datasets of various cancers and normal tissues were retrieved from TCGA portal (http://cancergenome.nih.gov).
Generation of SLFN11-deleted cells
To disrupt the SLFN11 gene, we designed a guide RNA targeting just downstream of the start codon in the 4th exon using the CRISPR design tool (http://crispr.mit.edu) (18). A human codon-optimized SpCas9 and chimeric guide RNA expression plasmid (pX330: pX330-U6-chimeric_BB-CBh-hSpCas9) was purchased from Addgene. Each guide RNA was inserted into the pX330 plasmid (pX330-SLFN11). The gene-targeting constructs harboring homology arms and a puromycin-resistance gene were prepared. Briefly, ~1 kb genomic sequences just upstream and downstream of the Cas9 cleavage sites were amplified by PCR methods from genomic DNA. The PCR products of the upstream site (left homology arm) and downstream site (right homology arm) were subcloned into pCR2.1-TOPO vector (Invitrogen) at the TA cloning site and ApaI/XhoI restriction endonuclease site, respectively in the desired direction. Puromycin resistance gene was finally subcloned between the homology arms at the NotI restriction endonuclease site. The targeting construct and pX330-SLFN11 were co-transfected into K562 cells by electroporation. After transfection, cells were released into drug-free medium for 48 hours followed by puromycin selection until single colonies were formed. Single clones were expanded, and gene-deletion was confirmed by western blotting. PCR primers and guide RNA sequences will be provided on request22.
Statistical and bioinformatics analyses
Level 3 RSEM-quantified RNASeq gene expression data (platform code IlluminaHiSeq_RNASeqV2) was downloaded from the TCGA data portal for tumor and normal samples of the presented tissue types. IC50 (inhibition concentration 50%) was calculated as the treated cell counts relative to untreated control cells by Nonlinear regression using GraphPad Prism5. The differences between two experimental groups was determined by the two-tailed independent-samples t-test for the results from quantitative real-time PCR and microarray analysis. Synergism was assessed using the CompuSyn Software. Combination index between 0.7 and 0.3, combination index between 0.3 and 0.1, and combination index less than 0.1 indicate synergy, strong synergy, and very strong synergy, respectively.
Results
SLFN11 expression is suppressed in many human cancers and cancer cell lines
SLFN11 expression has previously been shown to vary broadly across cancer cell lines with some lines expressing no or very low SLFN11 whereas others such as sarcomas and leukemias express very high levels of SLFN11 transcripts (1,2,5). Analyzing the TCGA database, we found that SLFN11 exhibits a broad dynamic range of expression within and across the collection of available tissues (Fig. 1A). Notably some normal tissues such as lung and thyroid, and to a lesser extent some breast and endometrium samples exhibit high SLFN11 transcripts. Also, acute myeloid leukemias, sarcomas and clear cell renal cell carcinomas (CCRCC) show high SLFN11 expression in the majority of patients. By contrast, a large fraction of the bladder, colon, liver, prostate and endometrial carcinomas show tumor samples with notably low SLFN11 expression (Fig. 1; shaded green area below the shaded salmon color area corresponding to the range of expression in normal tissues).
SLFN11 expression was also widely distributed across the cell lines of the cancer cell line databases from the Broad Institute (CCLE; http://www.broadinstitute.org/ccle/), the Welcome Trust Sanger Institute-Massachusetts General Hospital Cancer Center (GDSC; http://www.cancerrxgene.org/), and the NCI-60 (http://discover.nci.nih.gov/cellminercdb). Figure 1B and Supplementary Figure S1A show that approximately 45-50% of the cell lines repress SLFN11 (green-shaded areas), and that among them a significant fraction could not be linked to promoter hypermethylation (dark green-shading). Whole exome sequencing (19) and copy number analyses (20) of the NCI-60 (http://discover.nci.nih.gov/) showed that the SLFN11-non-expressing cells had no detectable copy loss or deleterious mutation. This led us to test the role of epigenetic acetylation (Fig. 1C).
Enhancing SLFN11 expression in cancer cells increases sensitivity to DNA-damaging agents
To investigate whether forced SLFN11 expression sensitizes cancer cells to DNA-damaging agents, SLFN11 was exogenously expressed in leukemia K562 cells, which express a very low level of SLFN11 (Fig. 2A and Figs. 1B and S1A) (1). As shown in Figure 2B, overexpression of SLFN11 strongly sensitized K562 cells to DNA-damaging agents including camptothecin (IC50 = 272.5 nM vs. 11.5 nM in vector control cells and SLFN11-expressing cells, respectively), topotecan (IC50 = 192.6 nM vs. 23.9 nM), hydroxyurea (IC50 = 6.4 μM vs. 0.4 μM) and cisplatin (IC50 = 15.6 μM vs. 2.2 μM), but not to the anti-mitotic agent paclitaxel (IC50 = 27.5 nM vs. 18.1 nM). A similar phenomenon was observed in the colorectal cancer cell line HCT116 with SLFN11 overexpression (Supplementary Fig. 2). These results demonstrate that enhancing SLFN11 expression in cancer cells increases sensitivity to DNA-damaging agents.
Fig. 2.
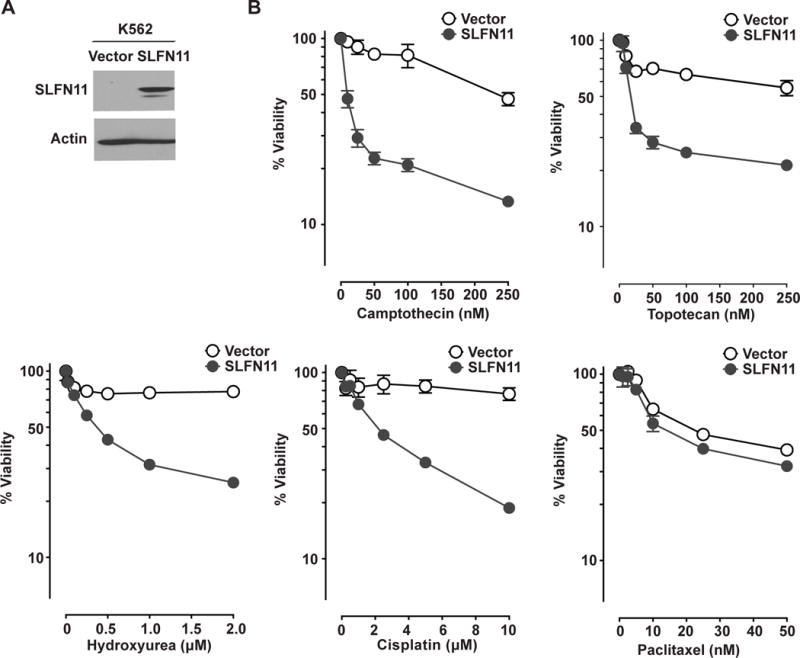
Forcing SLFN11 expression sensitizes K562 cells to DNA-damaging drugs. (A) Protein levels of SLFN11 in K562/Vector and K562/SLFN11 cells determined by Western blotting using antibodies against SLFN11 (98 kDa). Actin (42 kDa) was used as loading control. (B) K562/Vector (empty circles) and K562/SLFN11 (solid circles) cells were treated with camptothecin, topotecan, hydroxyurea, cisplatin or paclitaxel for 2 days before assessing cell viability. Representative results in triplicate from three independent experiments are shown as mean ± SD.
Class I HDAC inhibitors induce SLFN11 expression both in vitro and in vivo
HDAC inhibitors have been shown to regulate gene expression. To date, the US Food and Drug Administration has approved four HDAC inhibitors- vorinostat, romidepsin, belinostat and panobinostat- for cutaneous/peripheral T-cell lymphoma and multiple myeloma. HDAC inhibitors as monotherapy and in combination with other agents are in different stages of clinical development for the treatment of hematological malignancies and solid tumors (21–23). To examine the effect of HDAC inhibitors on SLFN11 expression, K562 cells were treated with two class I HDAC inhibitors, romidepsin and entinostat, and one class IIb HDAC inhibitor rocilinostat. Figure 3A shows that SLFN11 mRNA and protein levels were strongly enhanced by romidepsin and entinostat, but not with rocilinostat. We also observed the induction of SLFN11 expression both at the RNA and protein levels in the fibrosarcoma cell line HT1080 by the class I HDAC inhibitors, romidepsin and entinostat but not by the class IIb HDAC inhibitor rocilinostat (Fig. 3B).
Fig. 3.
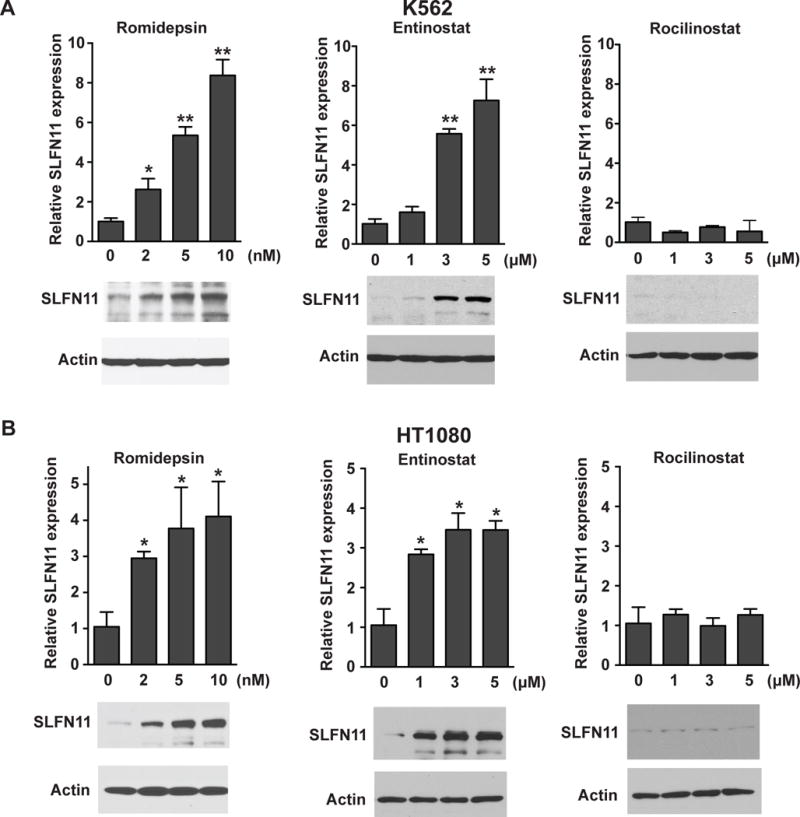
Class I HDAC inhibitors induce SLFN11 expression. K562 (A) and HT1080 (B) cells were treated with romidepsin, entinostat or rocilinostat for 16 h. Upper panels: SLFN11 mRNA levels were measured by quantitative real-time PCR. The y-axis represents relative SLFN11 expression normalized to untreated cells (0). Representative results in triplicate from three independent experiments are shown as mean ± SD. *, p < 0.05; **, p < 0.001 by t test. Lower panels: SLFN11 protein levels were determined by western blotting using antibodies against SLFN11 (98 kDa). Actin (42 kDa) was used as loading control.
The increase in the SLFN11 mRNA level was observed as early as 3 hours post-treatment, peaked at 24 hours and returned to low levels 24 hours after romidepsin removal (Supplementary Fig. S3A and C). This transient upregulation is at least in part due to the relatively rapid turnover of SLFN11 transcripts. Using actinomycin D to block RNA synthesis and comparing SLFN11 with transcripts known to have short half-life (CCNE and PUMA) and long half-life (GADPH), we determined the half-life of SLFN11 mRNA to be relatively short (approximately 4 hours; Supplementary Fig. S3D) in CCRF-CEM cells that express high SLFN11 (1,24).
Our recent publication showed FLI1, which is a member of ETS transcriptional factor family, to be a cellular regulator of SLFN11 expression in cancer cells from various tissues of origin (5). However, we did not observe the induction of FLI1 expression by romidepsin in K562 cells (Supplementary Fig. S3B) under conditions where SLFN11 was strongly induced (see Fig. 3A, left). Therefore, the derepression of SLFN11 by the HDAC inhibitors is not due to FLI1 activation.
We also examined whether romidepsin could activate SLFN11 expression in vivo by assessing gene expression microarray data of PBMCs obtained before and after romidepsin from patients with cutaneous T-cell lymphoma (CTCL) and circulating malignant T-cells (25). Four out of 7 cases exhibited more than 3-fold induction of SLFN11 expression in response to romidepsin therapy. The induction of SLFN11 expression tended to be higher at 4 than 24 hours after romidepsin (Fig. 4), which is likely related to the rapid elimination of romidepsin (half-life around 3 hours) and to the short half-life of SLFN11 transcripts (Supplementary Fig. S3D). No obvious romidepsin-induced changes in SLFN11 expression were observed in two cases (Fig. 4). These results indicate that class I HDAC inhibitors activate SLFN11 expression both in cancer cell line models and patients.
Fig. 4.
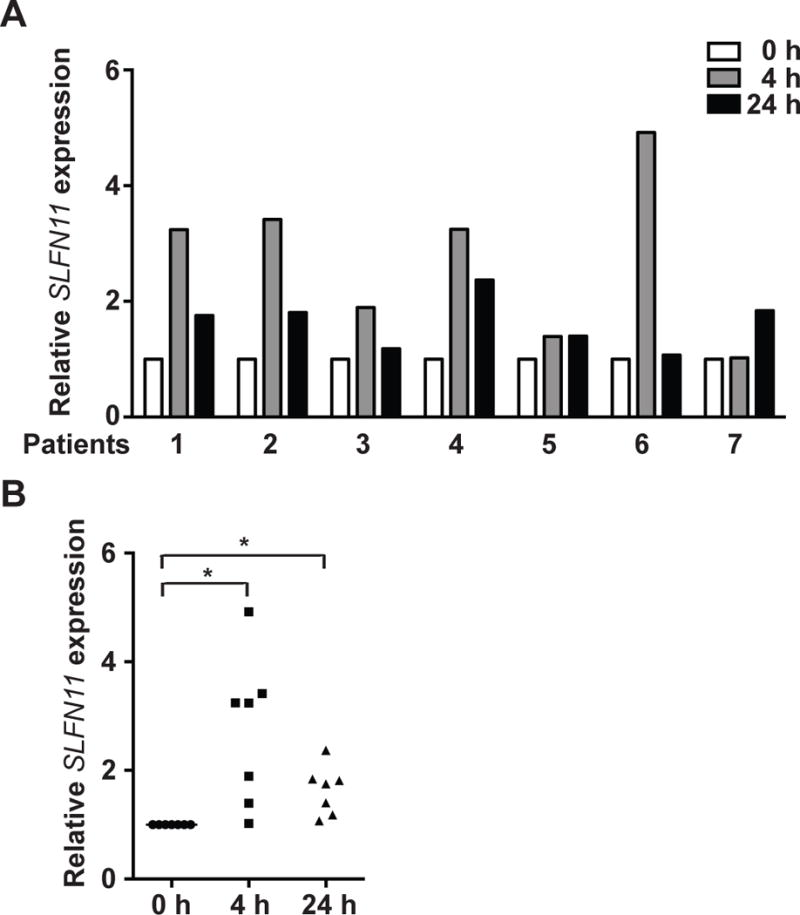
SLFN11 expression is induced in response to romidepsin in patient samples. Microarray analysis of total RNA isolated from PBMCs of seven patients with significant CTCL blood involvement. Samples were obtained pre-romidepsin infusion and at 4 h and 24 h after the initiation of romidepsin therapy (at romidepsin infusion end). The y-axis represents relative SLFN11 expression normalized to the samples before romidepsin infusion (open bars).
Class I HDAC inhibitors are synergistic with camptothecin
Because SLFN11 expression determines the sensitivity of cancer cells to different classes of DNA damaging agents (see Fig. 2) (1,6,24), we examined whether class I HDAC inhibitors, which activate SLFN11 expression show synergy with camptothecin, a specific TOP1 inhibitor (26) representative of the DNA-damaging drugs whose activity is under the control of SLFN11 expression level (1,2). K562 and HT1080 cells were pretreated with HDAC inhibitors for 16 hours, washed out, and then incubated with various doses of camptothecin. Pretreatment with romidepsin or entinostat, but not rocilinostat, sensitized K562 and HT1080 cells to camptothecin in a dose-dependent manner (Fig. 5A and B). Combination index values were calculated to evaluate synergy using the CompuSyn software (bottom panels of Fig. S5A and B). Romidepsin (2-10 nM) and entinostat (1-5 μM) showed high synergistic values with camptothecin in K562 and HT1080 cells (except 1 μM entinostat in K562 cells). As expected, based on the fact that SLFN11 does not determine response to tubulin inhibitors (see Fig. 2) (1,9,15), the combination of class I HDAC inhibitors with paclitaxel did not show synergy under similar conditions (Fig. 5C).
Fig. 5.
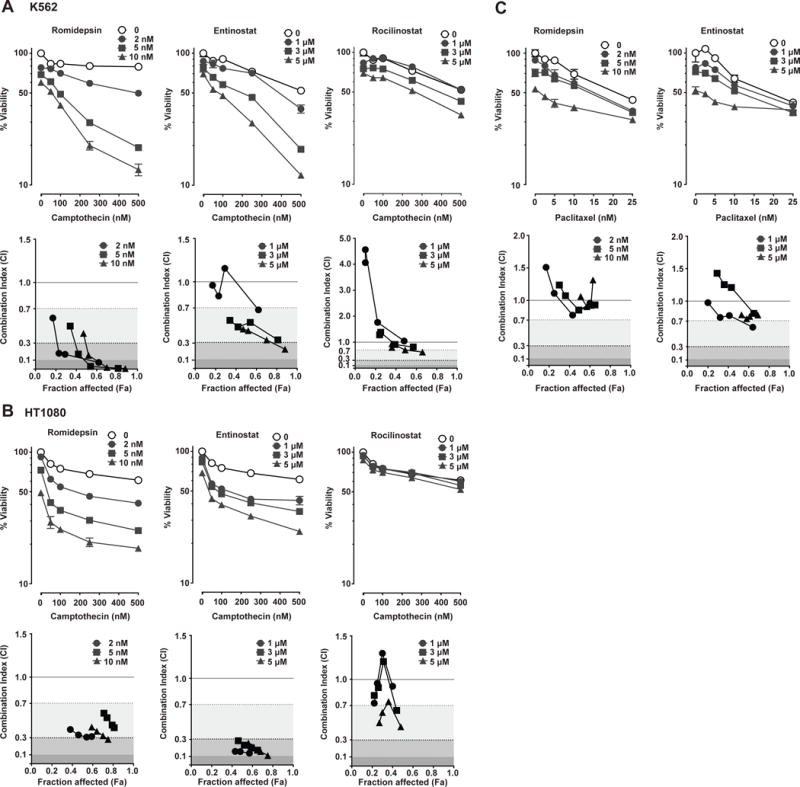
Class I HDAC inhibitors show synergistic effects with camptothecin but not with paclitaxel. K562 (A) and HT1080 (B) cells were pretreated with romidepsin (left), entinostat (middle) or rocilinostat (right) for 16 h, washed and then treated with the indicated concentrations of camptothecin for another 2 days before assessing cell viability (upper panel). Representative results in triplicate from three independent experiments are shown as mean ± SD. Lower panels: graphs of combination index versus Fa (fraction affected) for data points of romidepsin (left), entinostat (middle) or rocilinostat (right) in combination with camptothecin. (C) K562 cells were pretreated with romidepsin (left) or entinostat (right) for 16 h, washed and then treated with camptothecin for another 2 days before assessing cell viability (upper panels). Representative results in triplicate from three independent experiments are shown as mean ± SD. Lower panels: graphs of combination index versus Fa (fraction affected) for data points of romidepsin (left) or entinostat (right) in combination with camptothecin. Shading represents the levels of synergism. Combination index between 0.7 and 0.3, combination index between 0.3 and 0.1, and combination index less than 0.1 indicate synergy, strong synergy, and very strong synergy, respectively.
We extended these experiments to three additional cancer cell lines: two small cell lung cancer (SCLC) cell lines, H82 and H446 that were used recently to demonstrate the epigenetic regulation of SLFN11 by EZH2 (13), and the HH cutaneous T cell lymphoma cell line (Supplementary Fig. S4). The results show that entinostat derepressed SLFN11 in all three cell lines and produced synergy with camptothecin in H82 and to a lesser extent in HH cells. Entinostat produced only additional cytotoxicity in H446 cells in spite of SLFN11 induction. Together these results demonstrate the synergy of class I HDAC inhibitors with camptothecin, which is consistent with the epigenetic derepression of SLFN11 by HDAC inhibitors.
Induction of SLFN11 expression is a dominant factor for the synergy of class I HDAC inhibitors with camptothecin
To investigate the causal contribution of SLFN11 to the synergistic effect of class I HDAC inhibitors with DNA-damaging agents, we generated SLFN11-knockout (SLFN11-KO) K562 cells using CRISPR/Cas9. As expected, entinostat induced acetylation of histone H3 but did not increase SLFN11 expression in SLFN11-KO cells (Fig. 6A). We further examined the synergistic effect of entinostat and camptothecin in SLFN11-WT and –KO K562 cells. Combination of entinostat and camptothecin showed much weaker synergy in SLFN11-KO cells than in SLFN11-WT cells (Fig 6B), indicating that enforced SLFN11 expression by class I HDAC inhibitors dominantly drives the synergy of class I HDAC inhibitors with camptothecin.
Fig. 6.
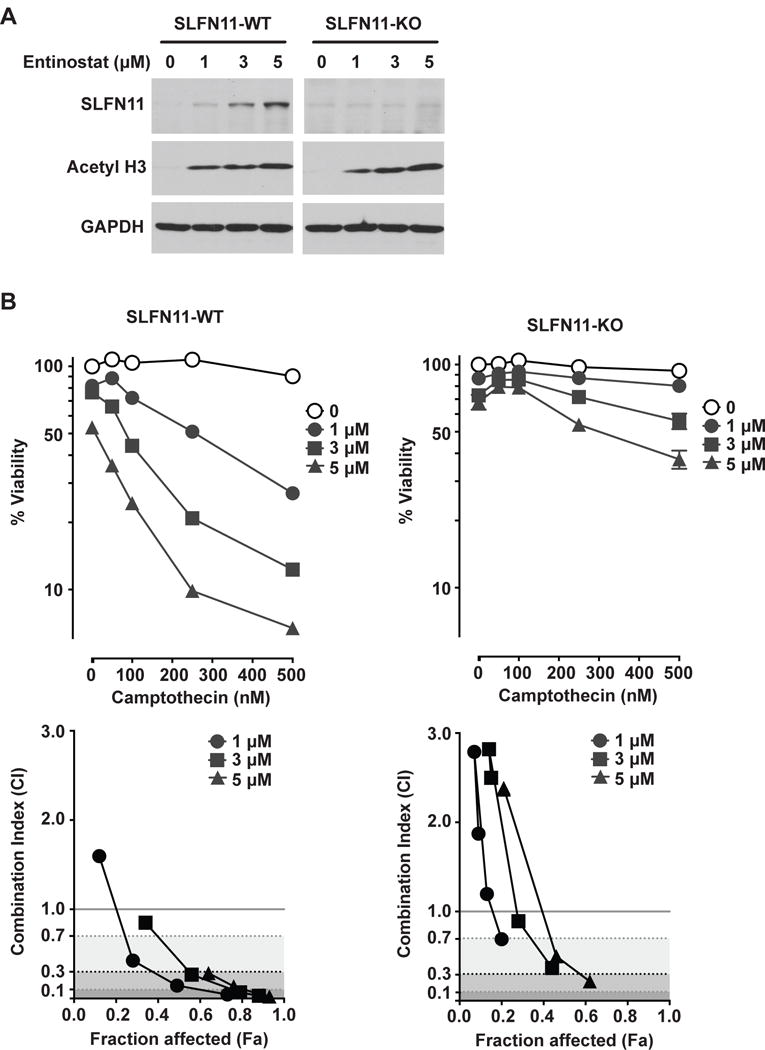
SLFN11 induction is crucial for the synergy of class I HDAC inhibitors with camptothecin. (A) Protein levels of SLFN11 and acetyl histone H3 (K9) in K562 WT and K562/SLFN11-knockout cells determined by Western blotting using antibodies against SLFN11 (98 kDa) and acetyl histone H3 (17 kDa). Actin (42 kDa) was used as loading control. (B) K562 WT (left) and K562/SLFN11-knockout (right) cells were pretreated with entinostat for 16 h, washed and then treated with the indicated concentrations of camptothecin for another 2 days before assessing cell viability (upper panels). Representative results in triplicate from three independent experiments are shown as mean ± SD. Lower panels: graphs of combination index versus Fa (fraction affected) for data points of entinostat in combination with camptothecin.
Discussion
Availability of robust biomarkers is an unmet need to optimize the efficacy of DNA-damaging agents and maximize their impact on cancer treatment. Yet, despite their broad utility in multiple cancers, clinically useful predictive biomarkers of response and resistance to DNA-damaging agents are lacking (14).
We initially discovered expression of SLFN11 as a genetic determinant of response to a broad range of DNA damaging agents (1), a finding that has since been confirmed in multiple independent studies (2–5,9,13,24). Recently, we demonstrated that epigenetic inactivation of SLFN11 expression by promoter hypermethylation confers resistance to DNA damaging therapies (9,15). Based on these considerations, in this study we investigated the effect of epigenetic modifiers on SLFN11 expression and sensitivity of cancer cells to DNA damaging agents. Our experiments show that class I HDAC inhibitors induce SLFN11 expression in epigenetically SLFN11-inactivated cells both in vitro and in vivo. We further demonstrate that pre-treatment with class I HDAC inhibitors enhance the cell killing by DNA damaging agents via a SLFN11-dependent mechanism. Our study reports the effect of HDAC inhibitors on SLFN11 expression and provides a proof of concept for novel combination therapy based on genomic determination of SLFN11 status of tumors. This concept is further supported by recent findings by Gardner et al. (13) that SLFN11 expression is suppressed in resistant tumors after exposure to chemotherapy and that EZH2 inhibitor activates SLFN11 expression to re-sensitize SCLC cells and PDX murine models to topotecan. In Figure 1C, we summarize the rationale and outline the strategy for combining class I HDAC inhibitors and DNA-targeted therapies to overcome the resistance of cells lacking both SLFN11 expression and SLFN11promoter methylation. Notably, a recent publication by Zeng et al. (27) suggested the rationale for combining HDAC and BRD4 inhibitors. Further studies are warranted to determine whether the combination of HDAC inhibitors with other epigenetic therapies (EZH2 and/or BRD4 inhibitors) will improve the activity of DNA-targeted therapies for tumors that do not express SLFN11 and do not show promoter hypermethylation.
Synergistic effects of HDAC inhibitors and DNA-damaging agents with respect to growth inhibition and apoptosis are well-established in preclinical studies (28). This synergistic effect is thought to be due to HDAC inhibition-mediated increase in chromatin accessibility which allows access of DNA-damaging agents to their targets. Alternately, HDAC inhibition may result in down-regulation of the DNA damage response. Genes involved in non-homologous end-joining (NHEJ) and homologous recombination (HR) including Ku70, Ku86, DNA-PKc, RAD51, BRCA1 and BRCA2 are downregulated by HDAC inhibition (29,30). Our study for the first time implicates HDAC inhibition-mediated direct activation of SLFN11 in sensitization of cancer cells to DNA-targeted therapies. That this effect is specific for class I HDAC inhibitors may be a function of the distinct cellular and subcellular expression patterns, signaling pathways and substrates associated with class I and class II HDACs.
It is critical to generate and validate reliable techniques to assess SLFN11 expression in patient tumors to translate these findings to a clinical setting. SLFN11 promoter methylation has been found to explain SLFN11 gene silencing in cancer cell lines (9). In earlier studies, we have found significant association between SLFN11 CpG island methylation and diminished SLFN11 RNA levels as well as SLFN11 RNA levels and protein expression (9,15,24). However, based on the cancer cell line data (see Figs. 1B and S1A), promoter methylation is clearly insufficient to determine lack of SLFN11 expression. Quantitative assessment of SLFN11 transcripts and SLFN11 protein expression by immunohistochemistry should provide clinically translatable assays to measure SLFN11 transcripts and protein expression in formalin-fixed paraffin-embedded tumor samples.
The precise mechanism by which SLFN11 sensitizes cancer cells to DNA-targeted agents is not fully established. Studies to date have provided seemingly contradictory data in this regard. Mu et al found defective checkpoint maintenance and HR as the likely cause of SLFN11-dependent cell death (31). In contrast, our recent study showed that SLFN11-induced cell death was mediated by HR-independent irreversible and prolonged S-phase arrest (24). In our model, SLFN11 inhibits replication and forces cell cycle arrest at mid S-phase in response to treatment with DNA-damaging agents. Ongoing efforts are focused on elucidating the molecular mechanisms of SLFN11-dependent replication arrest and cancer cell death.
In conclusion, our study reveals the epigenetic modulation of SLFN11 expression and its relevance to sensitivity to DNA-damaging agents. These promising results warrant future clinical investigation. Developing assays for assessing SLFN11 expression status as a predictive marker for tumor response to DNA-damaging agents and clarifying the molecular mechanisms underlying SLFN11 biology are important future considerations.
Supplementary Material
Translational Relevance.
SLFN11 is a predictive biomarker of sensitivity and a determinant of response to a wide range of DNA-targeted therapies. Epigenetic inactivation of SLFN11 confers resistance to DNA-damaging agents and is a frequent occurrence. We report here that class I HDAC inhibitors derepress SLFN11 expression and are synergistic with DNA damaging drugs. These findings provide a SLFN11-dependent epigenetic mechanism in the synergy between DNA-targeted agents and HDAC inhibitors, and present a rational therapeutic combination to overcome drug resistance.
Acknowledgments
Financial support: This work was supported by the Center for Cancer Research, Intramural NCI program (Z01 BC006150; Z01 BC011793)
JBT received research funding from Syndax Pharmaceuticals.
Footnotes
Conflict of interest: The other authors declare no conflicts of interest
References
- 1.Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(37):15030–5. doi: 10.1073/pnas.1205943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polley E, Kunkel M, Evans D, Silvers T, Delosh R, Laudeman J, et al. Small Cell Lung Cancer Screen of Oncology Drugs, Investigational Agents, and Gene and microRNA Expression. Journal of the National Cancer Institute. 2016;108(10) doi: 10.1093/jnci/djw122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nature chemical biology. 2016;12(2):109–16. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang SW, Bilke S, Cao L, Murai J, Sousa FG, Yamade M, et al. SLFN11 is a Transcriptional Target of EWS-FLI1 and a Determinant of Drug Response in Ewing Sarcoma. Clinical Cancer Research. 2015;21(18):4184–93. doi: 10.1158/1078-0432.CCR-14-2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, et al. PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin Cancer Res. 2017;23(2):523–35. doi: 10.1158/1078-0432.CCR-16-1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 2012;491(7422):125–8. doi: 10.1038/nature11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mavrommatis E, Fish EN, Platanias LC. The Schlafen Family of Proteins and Their Regulation by Interferons. J Interf Cytok Res. 2013;33(4):206–10. doi: 10.1089/jir.2012.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, et al. Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget. 2016;7(3):3084–97. doi: 10.18632/oncotarget.6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, et al. Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anti-cancer drugs. 2014;25(10):1175–81. doi: 10.1097/CAD.0000000000000151. [DOI] [PubMed] [Google Scholar]
- 11.Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, et al. Activity of MM-398, Nanoliposomal Irinotecan (nal-IRI), in Ewing’s Family Tumor Xenografts Is Associated with High Exposure of Tumor to Drug and High SLFN11 Expression. Clinical Cancer Research. 2015;21(5):1139–50. doi: 10.1158/1078-0432.CCR-14-1882. [DOI] [PubMed] [Google Scholar]
- 12.Deng Y, Cai Y, Huang Y, Yang Z, Bai Y, Liu Y, et al. High SLFN11 expression predicts better survival for patients with KRAS exon 2 wild type colorectal cancer after treated with adjuvant oxaliplatin-based treatment. BMC cancer. 2015;15:833. doi: 10.1186/s12885-015-1840-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell. 2017;31(2):286–99. doi: 10.1016/j.ccell.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reinhold WC, Thomas A, Pommier Y. DNA-Targeted Precision Medicine; Have We Been Caught Sleeping? Trends in Cancer. 2017 doi: 10.1016/j.trecan.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinhold WC, Varma S, Sunshine M, Rajapakse V, Luna A, Kohn KW, et al. The NCI-60 Methylome and Its Integration into CellMiner. Cancer Research. 2017;77:601–12. doi: 10.1158/0008-5472.CAN-16-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakraborty AR, Robey RW, Luchenko VL, Zhan Z, Piekarz RL, Gillet JP, et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: rationale to combine romidepsin with an MEK inhibitor. Blood. 2013;121(20):4115–25. doi: 10.1182/blood-2012-08-449140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–90. doi: 10.1093/nar/gkn764. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abaan OD, Polley EC, Davis SR, Zhu YJ, Bilke S, Walker RL, et al. The Exomes of the NCI-60 Panel: A Genomic Resource for Cancer Biology and Systems Pharmacology. Cancer Research. 2013;73(14):4372–82. doi: 10.1158/0008-5472.CAN-12-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Varma S, Pommier Y, Sunshine M, Weinstein JN, Reinhold WC. High resolution copy number variation data in the NCI-60 cancer cell lines from whole genome microarrays accessible through CellMiner. PloS one. 2014;9(3):e92047. doi: 10.1371/journal.pone.0092047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. The Journal of clinical investigation. 2014;124(1):30–9. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oronsky BT, Oronsky AL, Lybeck M, Oronsky NC, Scicinski JJ, Carter C, et al. Episensitization: Defying Time’s Arrow. Front Oncol. 2015;5:134. doi: 10.3389/fonc.2015.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13(9):673–91. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 24.Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, et al. Resistance to PARP inhibitors by SLFN11 inactivation can be overcome by ATR inhibition. Oncotarget. 2016;7(47):76534–50. doi: 10.18632/oncotarget.12266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bates SE, Eisch R, Ling A, Rosing D, Turner M, Pittaluga S, et al. Romidepsin in peripheral and cutaneous T-cell lymphoma: mechanistic implications from clinical and correlative data. Brit J Haematol. 2015;170(1):96–109. doi: 10.1111/bjh.13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol. 2010;17(5):421–33. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng H, Qu J, Jin N, Xu J, Lin C, Chen Y, et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell. 2016;30(3):459–73. doi: 10.1016/j.ccell.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 28.Eot-Houllier G, Fulcrand G, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase inhibitors and genomic instability. Cancer letters. 2009;274(2):169–76. doi: 10.1016/j.canlet.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Adimoolam S, Sirisawad M, Chen J, Thiemann P, Ford JM, Buggy JJ. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(49):19482–7. doi: 10.1073/pnas.0707828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M. Attenuated DNA damage repair by trichostatin a through BRCA1 suppression. Radiat Res. 2007;168(1):115–24. doi: 10.1667/Rr0811.1. [DOI] [PubMed] [Google Scholar]
- 31.Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO reports. 2016;17(1):94–109. doi: 10.15252/embr.201540964. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.