

Abstract
Treatment of advanced cancers has benefited from new agents that supplement or bypass conventional therapies. However, even effective therapies fail as cancer cells deploy a wide range of resistance strategies. We propose that evolutionary dynamics ultimately determine survival and proliferation of resistant cells. Therefore, evolutionary strategies should be used with conventional therapies to delay or prevent resistance. Using an agent-based framework to model spatial competition among sensitive and resistant populations, we applied anti-proliferative drug treatments to varying ratios of sensitive and resistant cells. We compared a continuous maximum tolerated dose schedule with an adaptive schedule aimed at tumor control via competition between sensitive and resistant cells. Continuous treatment cured mostly sensitive tumors, but with any resistant cells, recurrence was inevitable. We identified two adaptive strategies that control heterogeneous tumors: dose modulation controls most tumors with less drug, while a more vacation-oriented schedule can control more invasive tumors. These findings offer potential modifications to treatment regimens that may improve outcomes and reduce resistance and recurrence.
Keywords: tumor progression, invasion, and metastasis; drug resistance; adaptive therapy; tumor heterogeneity; agent-based model
Quick Guide to Model and Major Assumptions
Off-lattice agent-based model
We used an off-lattice agent-based model to investigate the spatial evolutionary dynamics of different treatment schedules on heterogeneous tumors. Space is the limiting factor, such that at carrying capacity, a cell will enter quiescence due to contact inhibition. Space is deemed available if any contiguous set of integer angles are empty and sufficient to allow a cell to fit without overlap (described previously in (1)). When a cell is quiescent, we assume that it cannot proliferate and is not affected by the drug. We also assume that there is no cell death due to regular turnover, and resources are abundant when cell densities are below carrying capacity. Model flowchart and setup are shown in Fig. S1A-B.
We initially seeded 100 cells in 2 configurations: randomly scattered throughout a 1.5mm radius circular domain, representing an in vitro cell culture distribution, and tightly clustered in the center, representing a dense tumor mass. We started with 100 cells to keep the initial population small but also large enough to capture a sufficiently diverse pre-existing distribution of phenotypes. When these were grown with 2 distinct sensitive and resistant phenotypes, we used the average cell cycle times for the sensitive and resistant cells found at the beginning of the in vitro experiment (MCF7: 40h, MCF7Dox: 60h). With the heterogeneous tumors, the initial distribution of phenotypes varied and was reported. The simulation was stopped when the number of cells reached 15,000, which is around 1.5 mm in diameter when grown as a dense mass.
Treatment
For continuous therapy, we applied the maximum tolerated dose (MTD) the entire duration, but only if a cell is capable of dividing does it become sensitive to drug toxicity. For adaptive therapy, each dose was instantaneously effective, ignoring any short-term pharmacokinetics. Regardless of the treatment schedule, a cell’s response to drug exposure was defined as a probability of death, Pdeath, which depends on its sensitivity, s, and the dose, D:
| (1) |
Here, D is a value from 0-1 where D=1 at the MTD. Due to the resistance cost, we assigned the cells with the shortest cycle times, Tmin, 100% sensitivity, and in a linear fashion, the longest cycle times, Tmax, as completely resistant, i.e. s(T)=1-(T-Tmin)/(Tmax-Tmin). This provides a simple linear trade-off between a cell’s fitness in the absence and presence of drug (2–6). We used a Hill function to describe the dose effect (7,8), setting the Hill coefficient and half-maximal activity to n=1.5 and K0.5=0.25. These values are not specific, but generically assume a response function that gives nearly 100% probability of death at the highest drug concentrations, dropping off slowly at mid ranges and then quicker as it gets to lower concentrations. The dose varies from its previous value D0 based on the relationship of the current number of cells N to the previous number of cells N0 according to the following equation:
| (2) |
where α and β are fixed values that affect how much the dose changes due to population size changes. In Eq. 2, if the number of cells is below half of the original, a treatment vacation occurs. If there is a fractional increase in number of cells greater than β, there will be a fractional increase in the dose by α, and if there is a fractional decrease in population size greater than β there is a fractional decrease in dose by α. Otherwise, the dose remains the same. The dose always began at the MTD, never exceeds the MTD, and each new dose was determined every 3 days in accordance with the experiments reported in (9). If a cell was killed by the drug, there was a 15-30h delay, randomly chosen, before it was removed from the domain, to account for the time it takes for apoptosis to occur (10,11).
Treatment was stopped when either the tumor was cured (i.e. the number of current cells N=0), recurs (i.e. N=4/3N0=20,000 cells), or the tumor reached an age of 2 years post-treatment. We assumed tumor control if the final number of cells was below 10,000, which is half the value that determines recurrence and accounts for fluctuations during adaptive treatments.
Cell migration
To examine cell migration, we assumed all cells move in a persistent random walk as follows. Each cell was given a random angle of movement and a persistence time (randomly chosen from a normal distribution with 80±40 minutes) during which moved in a straight line at 5 μm/h. After this time, it turned at a random angle and started again with a new persistence time. All cells migrated in this way as long as they were not in the quiescent state and did not contact another cell. Upon contact, the cells turned by a random angle and started again with a new persistence time.
Phenotypic drift
We examined phenotypic drift by allowing daughter cells to inherit a slightly different proliferation rate upon division. At each division, there was a 10% probability that the cell’s proliferation rate could change by randomly choosing 1 of 3 options: increase its cycle time by 1 h, decrease its cycle time by 1 h, or stay the same (while keeping the cell cycle bounded within the range of 10-50h). The other 90% of the time the proliferation rate stayed the same. Daughter cells had the possibility of inheriting different proliferation rates.
Introduction
Despite major advances in cancer therapies, most metastatic cancers remain fatal because tumor cells have a remarkable capacity to evolve drug resistance, both through genetic and non-genetic mechanisms (12). Most investigations of cancer treatment resistance have focused on identifying and targeting the molecular mechanisms that confer resistance. However, defeat of one resistance strategy often results in the deployment of another (13).
An alternative approach focuses on the population-level dynamics governed by Darwinian evolutionary principles that define the fitness of each cell within the local environmental context. For example, cancer cells often employ multidrug resistance pumps, in which the synthesis, maintenance, and operation require considerable investment of resources (up to 50% of the cell’s total energy budget) (14). In the harsh tumor microenvironment this investment in survival will likely require diversion of resources that would ordinarily be devoted to invasion or proliferation. Thus, while tumor cells may possess the molecular mechanisms necessary for therapy resistance, proliferation of resistant cells is governed by complex interactions that include the cost/benefit ratio of the resistance mechanism(s) and competition with other tumor subpopulations.
A common maxim in cancer treatment is to “hit hard and fast” through maximum dose dense strategies that administer the highest possible drug dose in the shortest possible time period. The maximum tolerated dose (MTD) principle has been the standard of care for cancer treatment for several decades and is the basis for clinical evaluation for most Phase I cancer drug trials. It has not, however, resulted in consistent cures in patients with most disseminated cancers (15). An evolutionary flaw in this strategy is the assumption that resistant populations are not present prior to therapy. It is now clear that cancer cells can be insensitive even to treatments that they have never “seen” before. Therefore, MTD therapy designed to kill as many cancer cells as possible, although intuitively appealing, may be evolutionarily unwise. This is because of a well-recognized Darwinian dynamic from ecology termed “competitive release”, which is observed, for example, when high doses of pesticide are applied for pest eradication (16); competitive release allows rapid emergence of resistant populations because of the combination of intense selection pressure and elimination of all potential competitors (17). When well timed, lower drug doses are also less toxic and can normalize vasculature, which may improve drug delivery and immune response (18–21).
Despite the growing recognition that heterogeneity and evolution play a significant role in driving treatment failure, explicit inclusion of Darwinian principles in clinical trial design is rare (22–25). However, both clinical and pre-clinical studies have shown promising results. Enriquez-Navas et al. used an evolution-guided treatment strategy to control breast cancer tumors in mice (9). They found that progression free survival can be prolonged when paclitaxel treatment schedules incorporate dose modulations and treatment holidays such that less drug is given to a responding tumor and more to a rebounding tumor (Fig. 1A). An ongoing clinical trial at Moffitt Cancer Center (NCT02415621) tests these evolutionary principles in patients with metastatic castration resistant prostate cancer to try to prevent the evolution of resistance to abiraterone therapy (26,27). In this trial, abiraterone is discontinued when the blood Prostate Specific Antigen (PSA) concentration falls below 50% of the initial value and does not resume until the PSA returns to the pre-treatment level (Fig. 1B). It is important to note that: 1) each patient serves as their own control to calibrate the PSA as a relative value, and 2) the adaptive schedules effectively personalize the treatment to patient response so that while one patient has only 2 courses of treatment in a year, another gets 3 in 10 months. Yet both remain under control.
Figure 1.
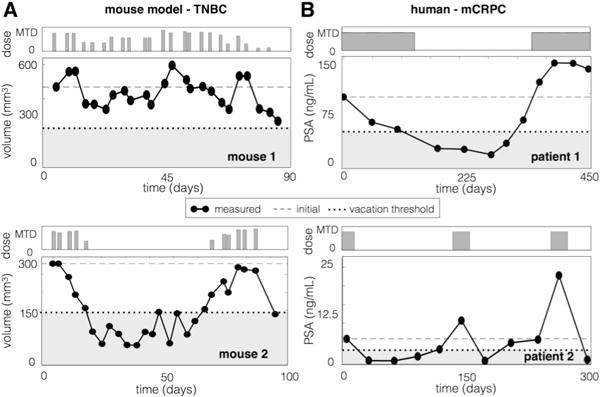
Adaptive therapy in the laboratory and the clinic. A) Mice implanted with triple negative human breast cancer were treated with adaptive paclitaxel treatment. If the volume dips below 150 mm3, a treatment vacation occurs, if there is a 20% tumor volume decrease (or increase), there is a 50% dose decrease (or increase), and otherwise the dose remains the same (see (10) for details). B) Patients with metastatic castrate resistant prostate cancer are treated with abiraterone such that treatment is stopped if PSA falls below 50% of the original and resumes when the PSA exceeds the original value.
Two important questions emerge from these results: (i) For which cancers is continuous (MTD) treatment the best strategy, and when is adaptive better? (ii) For adaptive treatments, when should a patient receive treatment holidays instead of dose modulation? While selection for resistance through application of continuous cytotoxic therapy seems inevitable, proliferation of those cells may be controlled using evolutionary principles. Importantly, multiple experimental models have shown that drug-resistant cancer cells proliferate slower than sensitive cells in the absence of drug (2–6). This is because resistant tumor cells, like most drug-resistant bacteria, incur a fitness cost due to the energy costs involved (28). Here, we demonstrated that fitness costs can be observed in vitro and computationally investigated these questions under the hypothesis that these costs can be exploited to delay or prevent proliferation of resistant cells in the tumor.
Materials & Methods
In-vitro co-culture experiment
The MCF7 human breast cancer cells were acquired from the Physical Sciences in Oncology Network (PSON) cell library. Prior to beginning the study and after completion, the cells were authenticated by the Moffitt Cancer Center Molecular Genomics Core using short tandem repeat DNA typing according to ATCC’s guidelines in Reid et al. (29) and tested for mycoplasma using MycoAlert™ Mycoplasma Detection Kit. At all times, the cells tested negative for mycoplasma and were determined to be greater than 90% identical to MCF7 human breast adenocarcinoma cells (ATCC HTB-22). Cells were used in the described experiments within 5 passages from thawing. MCF7 doxorubicin (Dox) resistance was selected and maintained by collecting the survivors of high-dose Dox treatments (1μM). Parental MCF7 and MCF7 Dox were tagged with green fluorescent and red fluorescent protein, respectively. 5×105 cells were plated in complete growth media at 0:1, 1:1, and 0:1 ratios. Cells were harvested every 3-4 days and counted. 5×105 of the cells were re-plated each time and the remaining cells were analyzed by flow cytometry for viability, GFP, and RFP expression. Fraction of population in mixed cultures was measured as percent positive of all viable cells.
Results
Fitness differences and space limitations affect competition and selection
The cost of resistance and the selection force imposed by competition and space limitations can be demonstrated through a combination of in vitro and in silico models. For the in vitro experiment, the MCF7Dox cell line was used, which is highly resistant to many chemotherapy agents due to upregulation of the membrane efflux pump P-glycoprotein or Multiple Drug Resistance (MDR1) proteins. When plated in the absence of chemotherapy (doxorubicin) in the media, the MCF7Dox cell line was observed to grow much slower than the parental MCF7 line (initial doubling times were MCF7:40h and MCF7Dox:60h). When co-cultured, the sensitive MCF7 cells rapidly outcompeted the resistant MCF7Dox line after only a few generations (Fig. 2 top left). This clearly illustrates the cost to resistance. We then investigated these evolutionary dynamics through our computational model system using the same initial cell cycle times for the sensitive (40h) and resistant cells (60h) from the in vitro experiment using both a scattered distribution analogous to the in vitro setting and a more clustered distribution analogous to a more in vivo setting e.g. a solid tumor. With the scattered distribution, we observed selection for the faster growing sensitive cells, but there was ample space for most cells to grow unimpeded due to the low density (Fig. 2 middle left). In contrast, when cells were clustered closely together, they were less free to move and proliferate (Fig. 2 bottom left). Proliferation was thus confined to a very narrow advancing edge, so the population grew much slower than when each separated colony had its own “edge”. This added additional selection pressure, causing sensitive cells to quickly take over the expanding front, trapping the resistant cells within.
Figure 2.
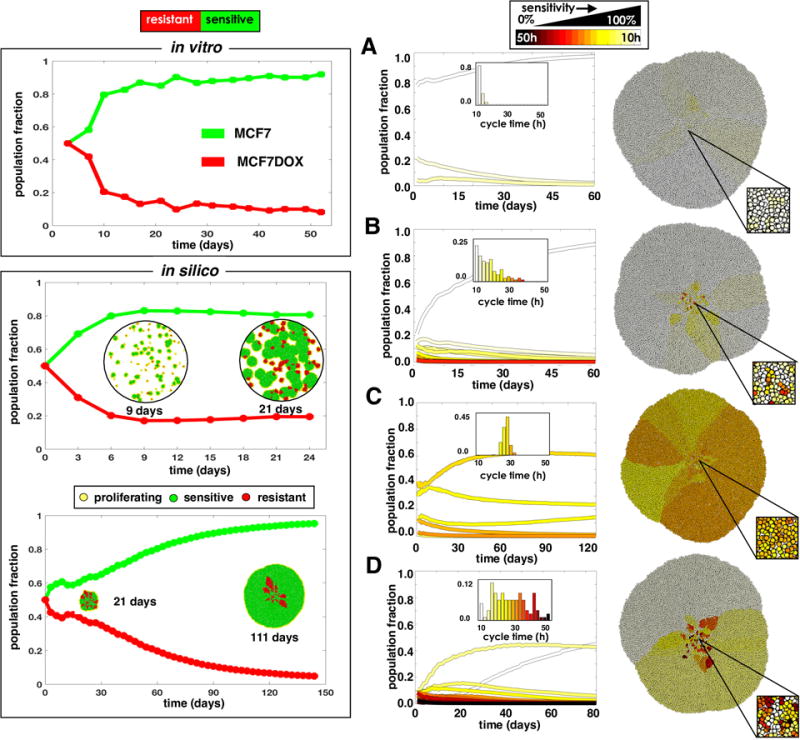
Competition between sensitive and resistant cells using in vitro and in silico models. Binary sensitive and resistant populations (left) and populations with a spectrum of sensitivity (right) are considered. Top left: Cells sensitive (MCF7) and resistant (MCF7Dox) to doxorubicin are co-cultured in vitro and replated every 3-4 days. Lower left: An in silico co-culture of sensitive (40h cell cycle) and resistant (60h cell cycle) cells seeded randomly throughout the domain or clustered in the center. The spatial distributions of cells are shown at several time points. Right: In silico simulations were initialized with different phenotypic distributions, characterized by a mean sensitivity s and standard deviation σs: A) s=100%, σs=5%; B) s=100%, σs=25%; C) s=60%, σs=5%; and D) s=60%, σs =25%. The initial distributions are shown as histograms in the plot insert; the final spatial layout is shown to the right.
Sensitivity to drugs is often viewed as binary, where a cell that is exposed to drug simply dies if sensitive or survives if resistant. However, in reality tumors have a more nuanced mix of phenotypes. We investigated how various mixtures of sensitive and resistant cells compete to form a solid tumor mass, starting with cell cycle times randomly drawn from a normal distribution characterized by a mean, s, and a standard deviation, σs. Figure 2A-D shows the final spatial configuration of some tumors grown from different initial populations. In each case, the more resistant cells got trapped in the interior of the tumor by the more sensitive cells, which took over the invading edge. Coexistence between different phenotypes was seen, but given enough time the more proliferative phenotypes should take over the invading edge. Similar changes in phenotype distributions are seen over a variety initial conditions (Fig. S2). Overall, the trend shows that for a heterogeneous distribution of phenotypes, the faster proliferators will dominate the population, but the timing depends on the relative fitness differences.
Continuous treatments can cure some tumors; adaptive treatments can control most tumors
We compared a conventional continuous MTD treatment strategy (CT) with an “adaptive therapy” (AT), wherein we adapted the next treatment based upon the tumor’s previous response to treatment in terms of change in population size. We used an adaptive scheme that changes the dose by 25% (α=0.25) if the population size changes by 5% (β=0.05), further we apply a treatment vacation if the population is below half the original, otherwise the dose stays the same (see Eq. 2). Treatment was applied to the tumors from Fig. 2A-D until either the tumor was cured, recurred, or the tumor reached an age of 2 years post-treatment.
In general, we found that there was not one therapy strategy that works best for all tumors, but the response to the treatment depended on the tumor composition (Fig. 3). For the most sensitive and homogeneous tumor (Fig. 3A), CT killed the tumor completely while AT kept the tumor under control by using an average dose of 34% of the MTD. While the total dose for AT was ~2 times that of CT, it was distributed over a time period ~6 times as long. The tumor consisting primarily of sensitive cells, but with some treatment-resistant phenotypes (Fig. 3B), initially responded very well to CT, but this was followed by recurrence of a completely resistant population after ~1 year. The same tumor under the AT could be controlled with an average of only 35% of the MTD with a total dose that was 70% of that given using CT. The more resistant tumor (Fig. 3C) should yield a slight net negative growth rate with the MTD applied. However, after a modest initial response to CT treatment, resistant cells took over enough to yield a positive net growth rate, which led to tumor progression. Using the AT strategy, the change in the population was too slow to induce dose modulation, however, when the population halved at around 1 year, a treatment vacation was applied. At this point, most of the sensitive cells had already been eliminated and the tumor was spatially diffuse. Consequently, a large portion of actively proliferating cells quickly expanded, and the tumor recurred 1 month prior than when using the CT strategy. For the more resistant and heterogeneous tumor (Fig. 3D), both treatments led to eventual recurrence, but the total number of cells was kept stable for a while using AT. Notably, AT gave ~2 times the dose but over a time period that was ~2.4 times longer. Recurrence was delayed using an average of 79% of the MTD. Phenotypic dynamics during CT and AT schedules for more initial conditions are compared in Fig. S3.
Figure 3.
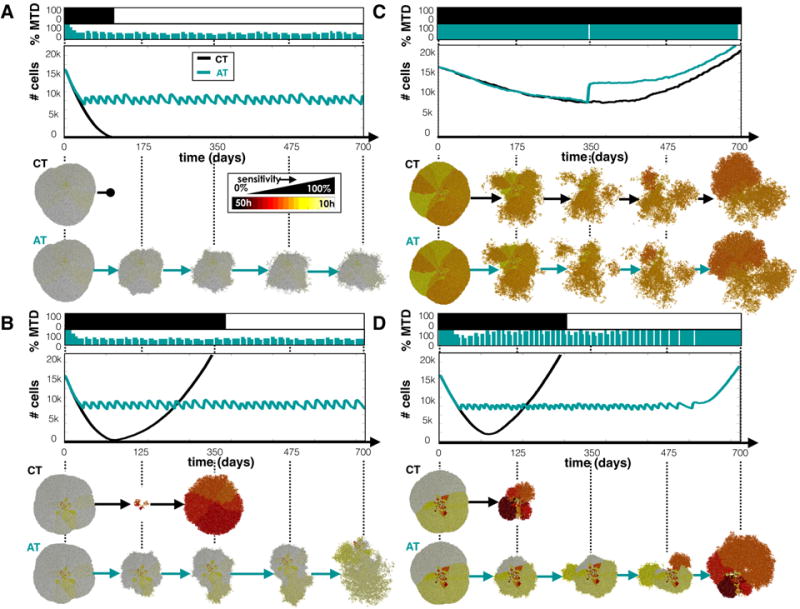
CT and AT schedules applied to tumors with a variety of phenotypes. Tumor phenotypes were initialized by normal distributions with mean sensitivity s and standard deviation σs: A) s=100% and σs=5%, B) s=100% and σs=25%, C) s=60% and σs=5%, and D) s=60% and σs=25%. Top panels show the dose schedules for each treatment strategy, middle panels, the population dynamics, and lower panels, the spatial configurations at several time points.
A more vacation-oriented strategy can control the tumor at the expense of higher doses
The AT schedule used so far, which we now refer to as AT1 (α=0.25, β=0.05), was chosen somewhat arbitrarily. Changing α and β changes the amount the dose is modulated (α) if the population changes by a threshold amount (β), therefore these values can have a direct impact on AT control. We tested another schedule that boosts both values, requiring a larger change in population size (10%) to produce a larger change in the dose modulation (50%), which we call AT2 (α=0.50, β=0.10).
We compared all treatment strategies (CT, AT1, and AT2) in Fig. 4 using a tumor with a pre-growth distribution that centered around a sensitive phenotype with a large degree of heterogeneity (s=100%, σs=25%). At the start of treatment the resistant cells were trapped in the interior by sensitive cells. The tumor recurred with the CT strategy after a year while both AT strategies controlled the tumor for the full 2 years (Fig. 4A). We found that the AT1 dose lowered initally then incorporated both vacations and dose adjustments to control the tumor using an average of 35% of the MTD. In the AT2 schedule, however, we found that the population did not change sufficiently fast to invoke a dose change, so control was achieved solely by having treatment vacations using an average of 76% of the MTD. However, since the proliferating cells are very sensitive to the drug, a large dose was not needed to control the tumor, so the more modulating AT1 dose schedule gave less drug to the patient while achieving a similar effect. The average sensitivity vs. the average dose for each schedule is plotted in Fig. 4B against a background heat map of the expected net proliferation rate of the tumor: . With CT, the mean sensitivity decreased over time as the most sensitive cells were eliminated and the more resistant cells took over. AT schedules, however, keep the cells, on average, in the sensitive region. The AT1 strategy shifted to the lowest dose that still kills the most sensitive phenotypes while the AT2 dose remained high.
Figure 4.
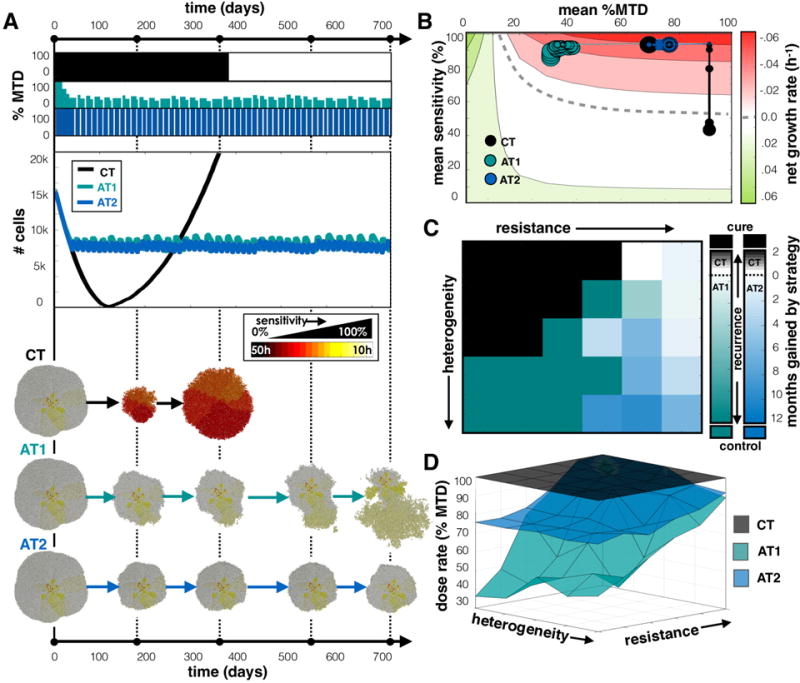
Comparing CT, AT1 (α=0.25, β=0.05), and AT2 (α=0.50, β=0.10) treatment schedules. A) Dose schedules, population dynamics, and spatial layout at various time points are shown for the treatment of a tumor with a pre-growth normal distribution of s=100% and σs=25%. See the Supplement for animations comparing treatment strategies for several tumor compositions (Movies 1–4). B) Trajectories for average sensitivity vs. average dose plotted every month with increasing point size. The background color indicates the expected growth rate of a tumor with a given cell cycle time, T, receiving a drug dose D (see text). The dashed line indicates where net growth is expected to be zero. C) Heat map indicates the strategy that gives the best outcome averaged from 3 simulations for different tumor compositions. Outcomes are favored in following order: cure, control, and longest time to recurrence. If recurrence is most likely, color indicates the average time gained by the winning strategy. D) Average dose given for each treatment strategy.
We compared these 3 strategies over a range of different tumor compositions to determine which is most efficacious in each case. Specifically, we grow and test treatments on an array of tumors with different starting distributions with unique sensitivity and standard deviation (s=50-100%, σs=5-25%). Presuming that the descending order of desired outcomes is to: cure, maintain, then gain the most time before recurrence, we obtained the best choice of treatment using 3 trials for each tumor composition in the array (Fig. 4C). When several strategies gave the same results, preference went to the lowest average dose. We found that the most sensitive and homogeneous tumors were cured by CT, but AT strategies worked better for heterogeneous tumors. The AT1 schedule provided a lower dose when the cells are more sensitive and controllable, the AT2 schedule extended the time to recurrence when recurrence was inevitable, and the more resistant, less heterogeneous tumors recurred similarly regardless of treatment strategy. The time to recurrence using each treatment schedule over the array of tumors is plotted in Fig. S4A. The average doses over the array of tumor compositions are given in Fig. 4D for each treatment. We found that we can control tumors bounded by drug-sensitive cells either by shifting to lower doses (AT1) or applying vacations between high doses (AT2). They both work because very sensitive proliferating cells do not need a full dose to be killed. However, when the tumor contains more resistant phenotypes, not all cells will respond to lower doses, so high doses and vacations grant better control.
More treatment vacations are needed to control heterogeneous invasive tumors
For the adaptive schedules to work, the sensitive cells must impede the proliferation of the resistant cells by competing for space and trapping the resistant cells inside the tumor. However, if the cells can move, the spatial structure that keeps the cells quiescent and hidden from the drug, is disturbed. We next examined the effect of cell migration by allowing cells to move in a persistent random walk at a modest speed of 5 μm/h (see methods for details).
Figure 5A shows an example with the same initial conditions as the previous example (s=100% and σs=25%), but with migrating cells. The tumor composition appears similar to the previous example, but the cells are more spatially mixed. In this case, both the CT and AT1 schedules led to recurrence while the AT2 schedule maintained control for the full 2 years (not fully shown). We found that for the CT case, compared to the case without migration (Fig. 4A), a quicker decline in the population was followed by faster relapse at 153 days. Due to the spatial spreading from migration, less cells were quiescent so the drug was able to affect more cells. This also allowed resistant cells to escape confinement sooner, leading to a faster recurrence. For AT1, dose modulation occurred, but the tumor population did not get small enough to trigger a vacation. The dose reduced substantially, and then rose again when resistant, unresponsive cells took over. The AT2 dose schedule incorporated treatment vacations, but also modulated because the larger proliferating fraction of cells (that are susceptible to the drug) caused larger fluctuations in the population size during both growth and treatment phases, triggering dose changes. Importantly, by employing the quicker switching between fast growth (during the vacations) and fast death (many cells susceptible and higher doses), AT2 kept the sensitive population at the invasive edge and the resistant cells suppressed. The AT2 schedule gave a larger average dose (67% of the MTD compared to 30% of the MTD for AT1), but did not recur over the monitored 2 year time period. Average sensitivity vs. average dose for each schedule is plotted in Fig. 5B. The CT strategy, again, resulted in selection for resistant cells over time, but the AT1 strategy, on the other hand, shifted to the lowest dose that still kills the most sensitive phenotypes. After the most sensitive cells were killed, the next most proliferative cells had a net positive proliferation rate and outgrew due to the low dose. This increased growth then caused a dose increase to keep proliferation in check. So sensitive phenotypes were essentially killed off sequentially with ever increasing doses until the resistant cells took ahold of the invasive edge. At that point, the resistant cells had a small (albeit positive) growth rate with limited chance of control. With the AT2 strategy there is a sharp transition between quick drug-free expansions and MTD-induced contractions. Because mostly sensitive cells remained on the outer rim, the net decline during treatment was approximately equal to the net increase when the dose was zero, and the tumor was controlled.
Figure 5.
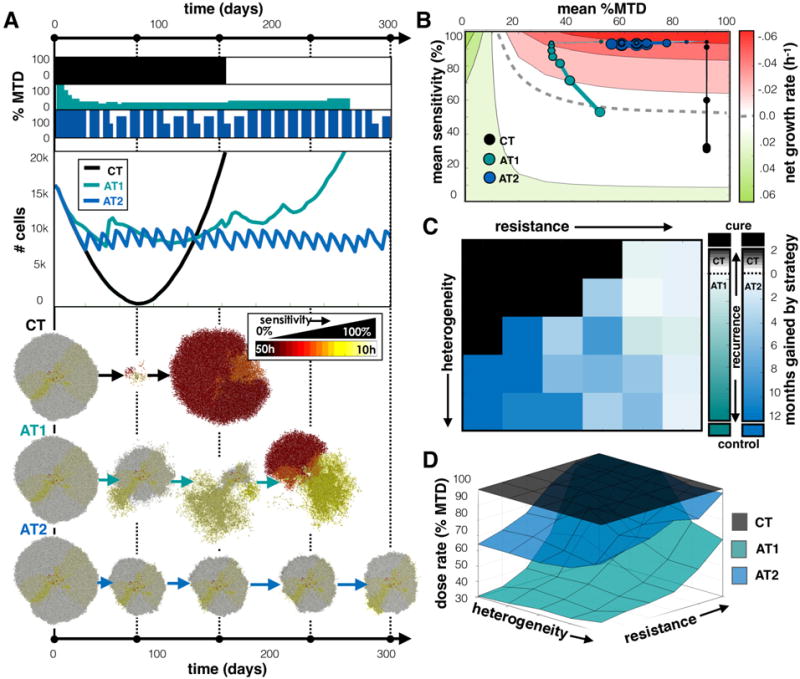
Comparing treatments when tumor cells migrate. A) Comparing CT, AT1, and AT2 treatments using a tumor from a pre-growth normal distribution of s=100% and σs=25%. Dose schedules, population dynamics, and spatial layout at various time points are shown for each treatment. See the Supplement for animation (Movie 5). B) Trajectories for average sensitivity vs. average dose over time. C) Winning strategies, and D) Average dose for different initial tumor compositions. See Figure 4 caption for more details on each panel.
A full sweep comparing CT, AT1, and A2 over many tumor compositions is shown in Fig. 5C. We found that while most of the original region cured by CT remains, allowing migration has destroyed the control by AT1 while the regions previously controlled with AT2 are somewhat preserved. The average doses for each strategy are shown in Fig. 5D. The doses were lower for both AT1 and AT2 in the sensitive region, which was previously controlled (in non-migrating tumors) by both adaptive strategies. However, control was only achieved with the AT2 strategy. As we increased the migration rate further to 10 μm/h, neither adaptive schedule successfully controlled tumors. The time to recurrence using each treatment schedule over the array of tumors is plotted in Fig. S4B-C for migration rates of 5 and 10 μm/h. A more extensive exploration of intermediate migration speeds and their outcomes is shown in Fig. S5, and is consistent with the result that AT2 is the best strategy for heterogeneous tumors with higher migration speeds.
Treatment vacations help delay recurrence of heterogeneous tumors with phenotypic drift
We have considered treatment responses to tumors that have pre-existing heterogeneity but assumed that progeny directly inherit the same phenotypes as the parental cell. However, this ignores the potential impact of subsequent mutations or epigenetic changes that may alter the cell phenotype over generations. Here, we tested how phenotypic drift affects the response to treatment strategy, i.e. we allowed a cell the opportunity to increase or decrease its proliferation rate upon division (see methods for details).
Starting with a population that is slightly less sensitive but heterogeneous (s=80% and σs=25%), we grew and treated a tumor that can change its proliferation rate (and therefore sensitivity) upon division (Fig. 6A). We found that each treatment strategy eventually failed because sensitive cells eventually became more resistant over generations. However, while CT and AT1 recurred at nearly the same time, AT2 was able to control the tumor for around 3 months longer. For the adaptive strategies, resistant regions started to emerge on the tumor’s edge, but under the AT2 schedule, the most sensitive cells could regrow during vacation periods and suppress the outgrowth of resistant cells. The cells eventually drifted toward more resistant phenotypes, while the sensitive cells were killed, causing the mass to separate into individual clumps where the resistant regions existed. We plot the average sensitivity vs. the average dose for each schedule in Fig. 6B, where movement along the y-axis occurs not just due to selection but also due to inheritance of evolving traits. Like all treatment strategies, the drug killed off the most sensitive cells, so the phenotypes eventually ended up further into the resistant region, but for CT the recurrent tumor returned almost completely resistant. For AT1, the resistant phenotype emerged faster than the dose modulations can keep up with, but the AT2 schedule, that shifts between extreme doses, better controlled both the faster proliferators during treatment and selected against the more resistant cells during vacations. The effect was not sustained for very long.
Figure 6.
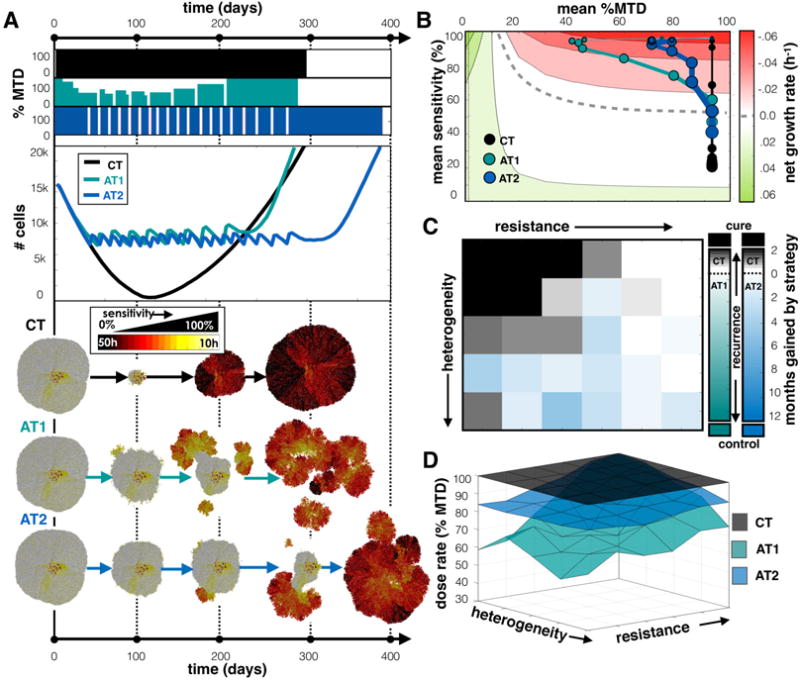
Comparing treatments when tumor cell phenotypes can drift. A) Comparing CT, AT1, and AT2 treatments using a tumor from a pre-growth normal distribution of s=80% and σs=25%. Dose schedules, population dynamics, and spatial layout at various time points are shown for each treatment. See the Supplement for animation (Movie 6). B) Trajectories for average sensitivity vs. average dose over time. C) Winning strategies, and D) Average dose for different initial tumor compositions. See Figure 4 caption for details on each panel.
The array of different tumor compositions was evaluated to compare CT, AT1, and AT2 (Fig. 6C). The region originally cured by CT was significantly reduced, and the ability to control the tumor through adaptive means was completely eliminated. CT actually did a better job with just a few months gain over AT1 in many regions. But again, the AT2 schedule delayed recurrence for longer over the CT schedule for heterogeneous tumor compositions. Regardless, neither AT treatment sustained control for long, as the net gain with AT2 was at the most 6 months. The average doses for CT, AT1, and AT2 are shown in Fig. 6D. In the sensitive region, which was controlled when there was no drift, the doses were found to be higher for both AT1 and AT2. Increasing the drift rate further destroyed any control whatsoever using CT, AT1, or AT2. See Fig. S6A-C for the time to recurrence plots using each treatment schedule over the array of tumors for drift rates of 0%, 10%, and 100%.
Discussion
Our simulations demonstrate the necessity of matching any cancer treatment regimen to the corresponding intratumoral evolutionary dynamics. The term “precision medicine” is often applied to strategies that match tumor treatment to specific predictive biomarkers. While this approach increases the probability of success it neglects the reality that virtually all tumor responses are followed by evolution of resistance, and thus, treatment failure. Our results indicate that precision medicine also needs to encompass the complex evolutionary dynamics that govern emergence and proliferation of resistant phenotypes. Here we present the diverse Darwinian interactions that can lead to resistance but also corresponding treatment strategies that can exploit these dynamics to delay the time to progression.
It is clear from our analysis that there is no one-size-fits-all evolutionary strategy. Using a modified off-lattice agent based model, we found conventional MTD application of cancer therapy can cure homogeneous tumors that consist entirely or almost entirely of cells that are sensitive to the applied treatment. While this is improbable at advanced stages, it is observed clinically as some tumors that histologically appear homogeneous, such as testicular cancer and some lymphomas, are frequently cured by conventional chemotherapy. However, decades of clinical experience, consistent with our model predictions, have found cure is not typically achievable in cancers that are highly heterogeneous (e.g. melanoma, lung, breast), since they already contain cells that are therapy resistant due to genetic, epigenetic or phenotypic properties or environmentally-mediated mechanisms (30).
In tumors that are not curable by standard MTD therapy, our models found that evolution-based strategies that exploit the fitness cost of resistance can delay treatment failure and tumor progression. Recent experimental studies have shown that adaptive therapy can enforce prolonged tumor control (9), however, none of the prior theoretical models have included spatial dynamics and thus assume that the different tumor subpopulations are well mixed (4,5,17). This is in contrast to radiological and pathological images of tumor that show marked spatial heterogeneity (31–34). Spatial structure can affect the emergence of resistant phenotypes due to space limitation (1,35) and limited drug perfusion (36,37). We present some insight into how spatial context might influence disease control, which reflects similar ideas presented in a recent study with growing bacterial colonies of E. Coli (35). In this study, less fit resistant clones were trapped due to spatial constraints, but high intensity drug exposure led to the competitive release of dormant mutants. Our model showed the same nature of competition, and we were able to test multiple treatment strategies on different tumor phenotypic compositions.
Previous modeling work has established the importance of genetic (38–40) and phenotypic heterogeneity (1,41,42) as well as mapping between them (43) in tumor progression. There have also been models that consider the evolution of drug sensitivity in low dose or metronomic treatments, but either ignore space (17) or heterogeneity in drug sensitivity (18), considering sensitive and resistant populations in a binary manner. We considered phenotypic heterogeneity over a wide spectrum of tumor compositions in space. By systematically testing an array of different initial phenotypic distributions we were able to delineate different regions where some schedules work better than others. We also found that some AT strategies are better than others for different situations. For example, with cell migration and phenotypic evolution, we can apply a strategy with less dose modulation and more emphasis on treatment vacations to keep the population sensitive. The extreme dose changes keep the tumor responding quickly during both the growth phase and the treatment phase to either maintain the spatial structure in the case of migration or to prevent the evolution of less sensitive cells in the case of phenotypic drift.
Comparing widely disseminated cancer, where the distribution of tumor cell subpopulations will likely vary among the metastatic sites, to our model, which only considers the response of single tumors, may seem disconnected. It is thus important to ask how much variation in clinical response to CT and AT should be expected. We tested the treatments on a set of 6 tumor metastases with mixed compositions using the total number of cells across all tumors to represent a systemic measure of burden and showed that control can still be achieved (Fig. 7 left). We found that with the CT strategy the more sensitive tumors responded with complete eradication, while the less sensitive tumors eventually recurred. In contrast, both AT schedules could still control the disease. While the more sensitive tumors shrank, the less sensitive ones grew, keeping the total population relatively constant. The full dose schedules and population dynamics are shown in Fig. S7A-B along with a case where the set of metastases all have the same heterogeneous composition. In the latter situation, the set of tumors responded as if they were single independent tumors.
Figure 7.
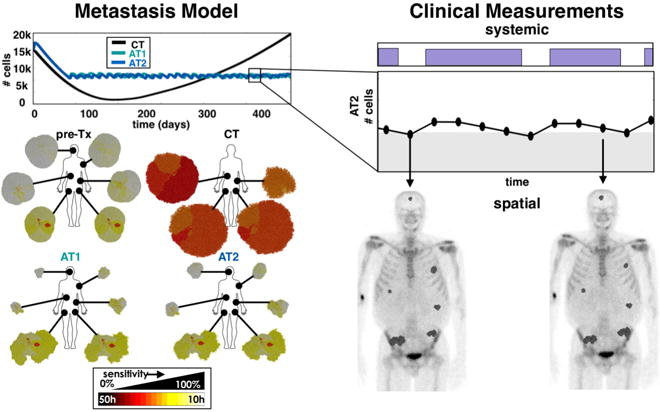
Connecting a set of tumors at the tissue scale to a systemic measure of tumor burden. A set of tumors with dissimilar compositions are treated using CT, AT1 (α=0.25, β=0.05), and AT2 (α=0.50, β=0.10) schedules using the change in the total tumor burden (the sum of tumor cells from all metastatic sites) to adjust treatment doses. The population dynamics (upper left), and the individual spatial compositions at the end of the simulation are shown (lower left). This model can be used to couple the total systemic tumor burden to the individual spatial distributions found from imaging (right).
To translate this to real patients, ideally we would to connect the tissue scale heterogeneity with a systemic biomarker of tumor burden (Fig. 7 right). A measure of variation in drug sensitivity could be done prior to treatment using immunohistochemistry. Further monitoring and assessment of disease burden is then needed to make treatment decisions over time, which could be done using systemic biomarkers, circulating tumor cells, cell-free DNA, or imaging (44). Our model represents a generic solid tumor that assumes that we can perfectly measure the total number of tumor cells periodically instead of a surrogate biomarker. The methods for actually measuring tumor burden non-invasively and frequent enough to direct treatment decisions are not well developed for all cancers. In the ongoing adaptive therapy trial on prostate cancer (26), PSA is used as a marker for disease burden. PSA may not be a perfect indicator, but it is readily available, standardized, and utilized (45). Other biomarkers are used for surveillance of various cancers with degrees of specificity and sensitivity (46), such as CA-125 for ovarian cancer and LDH for melanoma. As technologies for serum biomarkers, circulating tumor cells, and cell-free DNA continue to be developed (44), we hope this challenge can be addressed to better measure burden in advanced and disseminated cancers through periodic blood draws.
The tumor model presented here is greatly simplified and abstracted to study the impact of spatial intratumoral heterogeneity on tumor progression and how it might be treated using adaptive therapy. We have ignored pharmacokinetics (47), spatially-weighted dose dependence (48,49), microenvironmental influence (50), therapy-induced drug resistance (50), and of course, the 3rd dimension (51). All of these are possible extensions of this work, and the model can be modified to represent specific cancers, resistance mechanisms, and associated biomarkers, but we have chosen a simple starting point to understand how a spectrum of cell phenotypes compete for space under different drug strategies. Our work illustrates clearly the importance of using treatment response as a key driver of treatment decisions, rather than fixed strategies. We strongly believe that the future of precision medicine should not only be focused on the development of new drugs but also in the smarter evolutionary enlightened application of preexisting ones.
Supplementary Material
Major findings.
Recurrence is inevitable when applying a continuous treatment schedule for a tumor with any pre-existing resistance. However, adaptive strategies can control such heterogeneous tumors using either dose modulation or treatment vacations. Further, a treatment vacation approach seems to be the best strategy for dealing with more invasive and evolving tumors.
Acknowledgments
The authors gratefully acknowledge funding from both the Cancer Systems Biology Consortium (CSBC) and the Physical Sciences Oncology Network (PSON) at the National Cancer Institute, through grants U01CA151924 (supporting A. Anderson and J. Gallaher) and U54CA193489 (supporting A. Anderson, P. Enriquez-Navas, K. Luddy, and R. Gatenby).
Footnotes
Authors report no conflicts of interest.
References
- 1.Gallaher JA, Anderson ARA. Evolution of intratumoral phenotypic heterogeneity: the role of trait inheritance. 2013 Aug 6;3(4):20130016. doi: 10.1098/rsfs.2013.0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreso A, O’Brien CA, van Galen P, Gan OI, Notta F, Brown AMK, et al. Variable Clonal Repopulation Dynamics Influence Chemotherapy Response in Colorectal Cancer. Science. 2013 Jan 31;339(6119):543–8. doi: 10.1126/science.1227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chmielecki J, Foo J, Oxnard GR, Hutchinson K, Ohashi K, Somwar R, et al. Science Translational Medicine. 90. Vol. 3. American Association for the Advancement of Science; 2011. Jul 6, Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling; pp. 90ra59–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silva AS, Kam Y, Khin ZP, Minton SE, Gillies RJ, Gatenby RA. Evolutionary Approaches to Prolong Progression-Free Survival in Breast Cancer. Cancer Research. 2012 Dec 16;72(24):6362–70. doi: 10.1158/0008-5472.CAN-12-2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Silva AS, Durand A, Ribeiro MC, Alsina M, Shain K, Baz R. An Evolutionary Approach for Personalized Therapy in Multiple Myeloma. AM. 2016;07(02):159–69. [Google Scholar]
- 6.Moore N, Houghton JM, Lyle S. Slow-cycling therapy-resistant cancer cells. Stem cells and development. 2011 doi: 10.1089/scd.2011.0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gesztelyi R, Zsuga J, Kemeny-Beke A, Varga B, Juhasz B, Tosaki A. The Hill equation and the origin of quantitative pharmacology. Arch Hist Exact Sci. 2012;66(4):427–38. [Google Scholar]
- 8.Gardner SN. A mechanistic, predictive model of dose-response curves for cell cycle phase-specific and -nonspecific drugs. Cancer Research. 2000 Mar 1;60(5):1417–25. [PubMed] [Google Scholar]
- 9.Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva AS, Foroutan P, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Science Translational Medicine. 2016 Feb 24;8(327):327ra24–4. doi: 10.1126/scitranslmed.aad7842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gelles JD, Chipuk JE. Robust high-throughput kinetic analysis of apoptosis with real-time high-content live-cell imaging. Cell Death Dis. 2016 doi: 10.1038/cddis.2016.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Nieuwenhuijze AEM, Van Lopik T, Smeenk RJT, Aarden LA. Annals of the Rheumatic Diseases. 1. Vol. 62. BMJ Publishing Group Ltd; 2003. Jan 1, Time between onset of apoptosis and release of nucleosomes from apoptotic cells: putative implications for systemic lupus erythematosus; pp. 10–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncology. 2012 Aug;8(8):999–1014. doi: 10.2217/fon.12.86. [DOI] [PubMed] [Google Scholar]
- 13.Saunders NA, Simpson F, Thompson EW, Hill MM, Endo-Munoz L, Leggatt G, et al. EMBO Mol Med. 8. Vol. 4. EMBO Press; 2012. Jun 25, Role of intratumoural heterogeneity in cancer drug resistance: molecular and clinical perspectives; pp. 675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kam Y, Das T, Tian H, Foroutan P, Ruiz E, Martinez G, et al. Sweat but no gain: Inhibiting proliferation of multidrug resistant cancer cells with “ersatzdroges”. Int J Cancer. 2014 Sep 2;136(4):E188–96. doi: 10.1002/ijc.29158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford S. Is it time for a new paradigm for systemic cancer treatment? Lessons from a century of cancer chemotherapy. 2013 Jun 24;4:1–18. doi: 10.3389/fphar.2013.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeilinger AR, Olson DM. Competition between stink bug and heliothine caterpillar pests on cotton at within-plant spatial scales. Entomolgia Experimentalis et Applicata. 2011 [Google Scholar]
- 17.Hansen E, Woods RJ, Read AF. How to Use a Chemotherapeutic Agent When Resistance to It Threatens the Patient. In: Schneider D, editor. Plos Biol. 2. Vol. 15. 2017. Feb 9, pp. e2001110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erren TC, Cullen P, Erren M, Bourne PE. Ten Simple Rules for Doing Your Best Research, According to Hamming. PLoS Computational Biology. Public Library of Science. 2007 Oct 26;3(10):e213–65. doi: 10.1371/journal.pcbi.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benzekry S, Hahnfeldt P. Maximum tolerated dose versus metronomic scheduling in the treatment of metastatic cancers. Journal of Theoretical Biology. 2013 doi: 10.1016/j.jtbi.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 20.Mpekris F, Baish JW, Stylianopoulos T, Jain RK. Role of vascular normalization in benefit from metronomic chemotherapy. Proceedings of the National Academy of Sciences. 2017 Feb 21;114(8):1994–9. doi: 10.1073/pnas.1700340114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banys-Paluchowski M, Schütz F, Ruckhäberle E, Krawczyk N, Fehm T. Metronomic Chemotherapy for Metastatic Breast Cancer - a Systematic Review of the Literature. Geburtshilfe Frauenheilkd. 2016 May;76(5):525–34. doi: 10.1055/s-0042-105871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hiley CT, Swanton C. Pruning Cancer’s Evolutionary Tree with Lesion-Directed Therapy. Cancer Discovery.American Association for Cancer Research. 2016 Feb 4;6(2):122–4. doi: 10.1158/2159-8290.CD-15-1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012 doi: 10.1038/nrc3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korolev KS, Xavier JB, Gore J. Nat Rev Cancer. 5. Vol. 14. Nature Publishing Group; 2014. Apr 17, Turning ecology and evolution against cancer; pp. 371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aktipis CA, Boddy AM, Gatenby RA, Brown JS, Maley CC. Nat Rev Cancer. 12. Vol. 13. Nature Publishing Group; 2013. Nov 11, Life history trade-offs in cancer evolution; pp. 883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moffitt Cancer Center. Adaptive Abiraterone Therapy for Metastatic Castration Resistant Prostate Cancer ( NCT02415621) [Google Scholar]
- 27.Zhang J, Cunningham JJ, Brown JS, Gatenby RA. Nature Communications. 1. Vol. 8. Nature Publishing Group; 2017. Nov 28, Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer; p. 1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melnyk AH, Wong A, Kassen R. The fitness costs of antibiotic resistance mutations. Evol Appl. 2015 doi: 10.1111/eva.12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid Y, Storts D, Riss T. L Minor - Bethesda: Eli Lilly & Company and the National …, 2004. Assay Guidance Manual. [PubMed] [Google Scholar]
- 30.Morris L, Riaz N, Desrichard A, Şenbabaoğlu Y. Pan-cancer analysis of intratumor heterogeneity as a prognostic determinant of survival. Oncotarget. 2016 doi: 10.18632/oncotarget.7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerlinger M, Rowan A, Horswell S. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New England Journal of Medicine. 2012 doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gillies RJ, Anderson ARA, Gatenby RA, Morse DL. Clinical Radiology. 7. Vol. 65. Elsevier; 2010. Jul 1, The biology underlying molecular imaging in oncology: from genome to anatome and back again; pp. 517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gatenby RA, Grove O, Gillies RJ. Quantitative Imaging in Cancer Evolution and Ecology. Radiology. 2013 Oct;269(1):8–14. doi: 10.1148/radiol.13122697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Connor JPB, Rose CJ, Waterton JC, Carano RAD, Parker GJM, Jackson A. Imaging intratumor heterogeneity: role in therapy response, resistance, and clinical outcome. Clinical Cancer Research. 2015 Jan 15;21(2):249–57. doi: 10.1158/1078-0432.CCR-14-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fusco D, Gralka M, Kayser J, Anderson A. Excess of mutational jackpot events in expanding populations revealed by spatial Luria-Delbruck experiments. Nature. 2016 doi: 10.1038/ncomms12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu F, Nowak MA, Bonhoeffer S. Spatial Heterogeneity in Drug Concentrations Can Facilitate the Emergence of Resistance to Cancer Therapy. In: Antia R, editor. PLoS Computational Biology. 3. Vol. 11. 2015. Mar 19, p. e1004142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shah AB, Rejniak KA, Gevertz JL. Limiting the development of anti-cancer drug resistance in a spatial model of micrometastases. MBE. 2016 Dec 1;13(6):1185–206. doi: 10.3934/mbe.2016038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durrett R, Foo J, Leder K, Mayberry J, Michor F. Intratumor heterogeneity in evolutionary models of tumor progression. Genetics. 2011 Jun;188(2):461–77. doi: 10.1534/genetics.110.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015 Aug 26;525(7568):261–4. doi: 10.1038/nature14971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McFarland CD, the KKPO Impact of deleterious passenger mutations on cancer progression. National Acad Sciences. 2013 doi: 10.1073/pnas.1213968110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robertson-Tessi M, Gillies RJ, Gatenby RA, Anderson ARA. Impact of metabolic heterogeneity on tumor growth, invasion, and treatment outcomes. Cancer Research. 2015 Apr 15;75(8):1567–79. doi: 10.1158/0008-5472.CAN-14-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carmona-Fontaine C, Bucci V. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. 2013 doi: 10.1073/pnas.1311939110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nichol D, Robertson-Tessi M, Jeavons P, Anderson ARA. Stochasticity in the Genotype-Phenotype Map: Implications for the Robustness and Persistence of Bet-Hedging. Genetics. 2016 Dec;204(4):1523–39. doi: 10.1534/genetics.116.193474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowes LE, Bratman SV, Dittamore R, Done S, Kelley SO, Mai S, et al. Circulating Tumor Cells (CTC) and Cell-Free DNA (cfDNA) Workshop 2016: Scientific Opportunities and Logistics for Cancer Clinical Trial Incorporation. 2016 doi: 10.3390/ijms17091505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallace TJ, Torre T, Grob M, Yu J, Avital I, Brücher B, et al. Current approaches, challenges and future directions for monitoring treatment response in prostate cancer. Journal of Cancer. 2014 doi: 10.7150/jca.7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Duffy MJ. Tumor markers in clinical practice: a review focusing on common solid cancers. Medical Principles and Practice. 2012 doi: 10.1159/000338393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Groh CM, Hubbard ME, Jones PF. Mathematical and computational models of drug transport in tumours. Journal of The Royal Society Interface. 2014 doi: 10.1098/rsif.2013.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rejniak KA, Estrella V, Chen T, Cohen AS, Lloyd MC, Morse DL. The role of tumor tissue architecture in treatment penetration and efficacy: an integrative study. Frontiers in Oncology. 2013;3:111. doi: 10.3389/fonc.2013.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim MJ, Gillies RJ, Rejniak KA. Current advances in mathematical modeling of anti-cancer drug penetration into tumor tissues. Frontiers in oncology. 2013 doi: 10.3389/fonc.2013.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun X, Bao J, Shao Y. Mathematical Modeling of Therapy-induced Cancer Drug Resistance: Connecting Cancer Mechanisms to Population Survival Rates. 1. Vol. 6. Nature Publishing Group; 2016. Mar 1, pp. 22498–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tang L, van de Ven AL, Guo D, Andasari V, Cristini V, Li KC, et al. Computational Modeling of 3D Tumor Growth and Angiogenesis for Chemotherapy Evaluation. In: Pappalardo F, editor. PLoS ONE. 1. Vol. 9. Public Library of Science; 2014. Jan 3, pp. e83962–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.