

Abstract
Ferroptosis is a non-apoptotic form of regulated cell death caused by the failure of the glutathione-dependent lipid-peroxide-scavenging network. FINO2 is an endoperoxide-containing 1,2-dioxolane that can initiate ferroptosis selectively in engineered cancer cells. We investigated the mechanism and structural features necessary for ferroptosis initiation by FINO2. We found that FINO2 requires both an endoperoxide moiety and a nearby hydroxyl head group to initiate ferroptosis. In contrast to previously described ferroptosis inducers, FINO2 does not inhibit system xc− or directly target GPX4, as do erastin and RSL3, respectively, or deplete GPX4 protein, as does FIN56. Instead, FINO2 causes both indirect loss of GPX4 enzymatic function and directly oxidizes iron, ultimately causing widespread lipid peroxidation. These findings suggest that endoperoxides such as FINO2 can initiate a multi-pronged mechanism of ferroptosis.
Introduction
Regulated cell death includes several processes that lead to cell death through specific mechanisms that can be modulated with pharmacological and genetic tools. The recognition of cell death as a regulated process began with the discovery and characterization of apoptosis.1, 2 Ongoing work has since uncovered several other regulated cell death processes, including ferroptosis. Ferroptosis is an iron-dependent, oxidative form of regulated cell death that is distinct from apoptosis, and characterized by the failure of the glutathione-(GSH)-dependent lipid peroxide defense network.3–5 Consequently, cells undergoing ferroptotic cell death exhibit an increased accumulation of lipid peroxides and cannot be rescued by inhibitors of apoptosis or other cell death processes.6
Organic peroxides, such as artemisinin (1) and artesunate (2), are used therapeutically as cytotoxic agents for the treatment of cancers.7–9 Recently, development of analogues based on the plakinic acid natural products (3) identified the 1,2-dioxolane FINO2 (4), which triggers ferroptosis (Figure 1A).10 Further evaluation of ferroptosis induction by FINO2 against multiple cancer lines revealed that FINO2 selectively initiates ferroptosis in BJ-eLR cancer cells compared to the isogenic, non-cancerous BJ-hTERT cell line.10 Since evasion of apoptotic signaling is a hallmark of cancer,11 the ability of FINO2 to initiate a non-apoptotic programmed cell death process selectively in tumorigenic cells makes it an attractive target for further study.
Figure 1. FINO2 induces ferroptotic cell death.
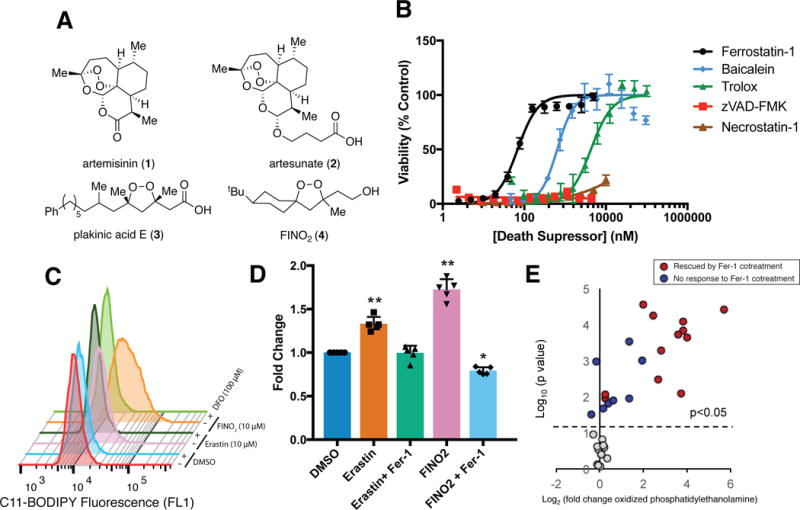
(A) Organic peroxides and FINO2 (B) The dose-dependent effect of cell death-suppressing compounds on ferroptosis triggered by FINO2 (10 μM) in HT-1080 cells. Viability measured 24 h after compound treatment. Experiments were performed with triplicate cell cultures. Data are plotted as the mean ± s.d., n=3. (C) Ability of iron chelator deferoxamine (DFO) to prevent ferroptosis-dependent C11-BODIPY oxidation when incubated together for 6 h. Three independent experiments were performed with similar results. (D) Ability of ferrostatin-1 (Fer-1) (2 μM) to prevent accumulation of thiobarbituric acid reactive substances (TBARS) when co-treated with erastin (5 μM) or FINO2 (10 μM) for 6 h. Data are plotted as the mean ± s.d., n=5. P values were determined using one-way ANOVA; *P=0.003, **P < 0.001 versus DMSO control. (E) Changes in oxidized phosphatidylethanolamine abundance as detected by LC-MS after treatment with FINO2 (10 μM) for 6 h. Individual lipid species are plotted based on their Log2 fold change in abundance (horizontal axis) and the statistical significance of the change (Log10 P-value) on the vertical axis. P values were determined using two-sided t test. Lipid species with significant change upon FINO2 treatment were plotted above the dot line (p<0.05). Experiments were performed in triplicate with biologically independent samples.
Here, we sought to define the mechanism by which FINO2 induces ferroptosis, and which structural features of FINO2 are necessary for its function. These experiments demonstrate that, in contrast to other ferroptosis-inducing compounds, such as erastin, FINO2 does not deplete GSH through inhibiting system xc−. FINO2 instead bypasses GSH depletion to cause iron oxidation, as well as loss of activity of the lipid-peroxide-reducing enzyme GPX4 indirectly, by a mechanism that is distinct from other GPX4 inhibitors. Exploration of the structure-activity relationship around the FINO2 scaffold revealed that both the endoperoxide moiety and the pendant hydroxyethyl group are necessary to induce oxidative events leading to ferroptotic cell death. We found that FINO2 exerts dual effects involving iron oxidation and loss of GPX4 enzymatic activity to induce ferroptosis, and therefore represents a novel class of ferroptosis inducers.
Results
FINO2 induces ferroptosis
We initially sought to evaluate the lethality of FINO2 in a cell line in which ferroptosis had been previously examined. Ferroptosis-sensitive HT-1080 fibrosarcoma cells3 were treated with a lethal concentration of FINO2 (10 μM) (Supplementary Figure 1A) alone, or co-treated with a panel of death-suppressing compounds at varied concentrations (Figure 1B). The lethality of FINO2 was suppressed by the ferroptosis inhibitor ferrostatin-1, which prevents the accumulation of lipid peroxides, likely through a radical trapping mechanism.12, 13 Baicalein and Trolox, which have been reported to inhibit ferroptosis,6 both suppressed FINO2 lethality (Figure 1B). The apoptosis inhibitor zVAD-FMK was unable to suppress cell death. Necrostatin-1, an inhibitor of necroptosis, an alternative form of regulated cell death, was similarly unable to prevent FINO2-induced death. We also evaluated the ability of nitroxide antioxidants XJB-5-131 and JP4-039 to suppress FINO2 lethality, which were previously found to suppress ferroptosis.14 The mitochondria-targeted nitroxide XJB-5-131 was 39-fold more potent than the cytosolic JP4-039, highlighting the potential importance of mitochondrial lipid peroxidation in mediating FINO2 lethality (Supplementary Figure 1).
We next aimed to validate that FINO2 causes lipid peroxidation, a defining event in ferroptosis. HT-1080 cells were treated with either FINO2 or the ferroptosis inducer erastin, and the increase in fluorescence intensity of the fluorescent lipid peroxidation probe C-11 BODIPY was monitored by flow cytometry (Figure 1C).3 Both erastin and FINO2 showed an increase in fluorescence 6 hours after treatment, with FINO2 showing a much larger shift, suggesting FINO2 causes a more rapid onset or overall greater quantity of lipid peroxidation. This increase was suppressed by co-treatment with deferoxamine (DFO), an iron chelator, confirming the iron dependence of lipid peroxidation induced by both FINO2 and erastin. We examined the oxidation of endogenous lipids by measuring the accumulation of thiobarbituric-acid-reactive substances (TBARS) in cells treated with erastin or FINO2 (Figure 1D). This assay detects malondialdehyde, a common product from the degradation of multiple lipid species.15 FINO2-treated cells showed a greater increase in TBARS than did erastin. In both cases, co-treatment with ferrostatin-1 suppressed TBARS accumulation, indicating the presence of lipid peroxidation suppressible by a specific ferroptosis inhibitor.
To gain insight into the type of lipid oxidation caused by FINO2, we used liquid chromatography-mass spectrometry (LC-MS) analysis to measure the oxidation of phosphatidylethanolamine (PE), a lipid critical to propagating ferroptosis (Figure 1E and Supplementary Figure 2).16 FINO2 treatment caused a large increase in a diverse set of oxidized PE species. Not all PEs that were oxidized in response to FINO2 were suppressed by co-treatment with ferrostatin-1, suggesting that only a specific set of oxidized PE species contribute to ferroptosis.
FINO2 does not alter glutathione homeostasis
Having validated that FINO2 causes ferroptosis in HT-1080 cells, we sought to define the mechanism by which FINO2 induces ferroptosis in these cells. Originally, ferroptosis-inducing compounds were divided into two classes. Class 1 ferroptosis inducers decrease intracellular levels of GSH, a necessary cofactor for the lipid-peroxide-reducing enzyme GPX4. Class 2 ferroptosis inducers inhibit GPX4 directly through active site inhibition. More recently, the small molecule FIN56 was reported to deplete GPX4 protein, and also deplete coenzyme Q10, an endogenous inhibitor of lipid peroxidation, through modulation of the mevalonate pathway.17
A major source of cysteine for GSH synthesis is the cystine/glutamate antiporter, system xc− (Supplementary Figure 3). Inhibition of system xc− by erastin, sulfasalazine, sorafenib, or glutamate depletes intracellular GSH and induces ferroptosis.18 To test whether FINO2 acts via system xc− inhibition and GSH depletion, we examined the ability of FINO2 to inhibit system xc− function using a fluorescent enzymatic assay that quantifies the amount of glutamate released by cells into glutamate-free medium (Figure 2A).18 Both erastin and sulfasalazine were able to inhibit glutamate release compared to vehicle-treated cells. FINO2 showed minimal inhibition of glutamate release, suggesting that inhibition of system xc− is not a primary mechanism of FINO2 lethality. Glutathione levels were also quantified in cells undergoing ferroptosis using a reactive fluorescent reporter of free thiols in cell lysates (Figure 2B). To eliminate non-specific reactivity with accessible cysteine residues on proteins, we precipitated the protein fraction of lysates, leaving GSH as the major thiol in solution.19 Cells treated with erastin exhibited a three-fold decrease in GSH content compared to cells treated with vehicle or the GPX4 inhibitor (1S, 3R)-RSL3, hereafter referred to as RSL3. FINO2-treated cells showed no decrease in thiol content, indicating that FINO2 does not deplete GSH. These results suggest that FINO2 is not a system xc− inhibitor and does not deplete GSH through other mechanisms.
Figure 2. FINO2 does not alter glutathione homeostasis.
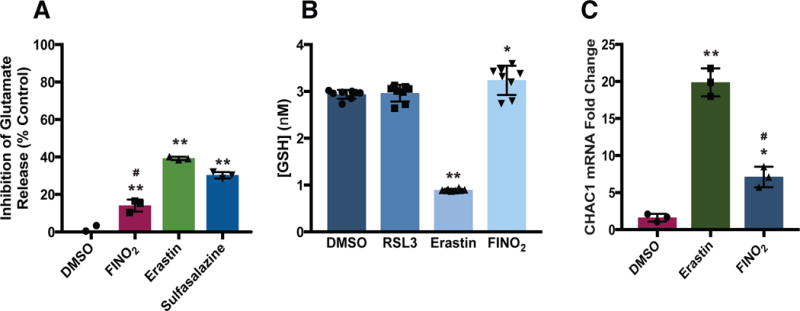
(A) Effect of FINO2 (10 μM) and system xc− inhibitors erastin (10 μM) and sulfasalazine (1 mM) on glutamate release after 1 h incubation. Data are plotted as the mean ± s.d., n=3 biologically independent samples. **P and #P < 0.001 versus DMSO control and Erastin, respectively. (B) Intracellular GSH levels in HT-1080 cells treated with ferroptosis inducers RSL3 (0.5 μM) for 90 m or erastin (5 μM), and FINO2 (10 μM) for 6 h, data are plotted as the mean ± s.d., n=8 biologically independent samples. *P=0.016, **P<0.001 versus DMSO control. (C) CHAC1 mRNA levels following erastin (10 μM) or FINO2 (10 μM) treatment for 6 h. Data are plotted as the mean ± s.d., n=3 biologically independent samples. *P=0.007, **P<0.001 versus DMSO control; #P < 0.001 versus erastin. All P values were determined using one-way ANOVA.
The oxidative stress caused by erastin upregulates components of the endoplasmic reticulum (ER) stress response pathway. The GSH-specific gamma-glutamylcyclotransferase enzyme encoded by the CHAC1 gene has been observed as a pharmacodynamic marker of exposure to erastin.18 To test whether FINO2 induces transcriptional changes similar to other ferroptosis inducers, we performed RT-qPCR to quantify the amount of CHAC1 mRNA in cells treated with erastin or FINO2 (Figure 2C). Cells treated with FINO2 showed a 7-fold increase in CHAC1 mRNA levels compared to vehicle. This upregulation was nearly 3-fold less than the upregulation in cells treated with erastin. This more modest upregulation indicates that FINO2 different transcriptional responses than an equivalent concentration of erastin.
FINO2 indirectly inhibits GPX4 activity in cells
Because FINO2 does not display the characteristic functional or genetic hallmarks of a class 1 ferroptosis inducer, we next evaluated the ability of FINO2 to act as a class 2 ferroptosis inducer by inhibiting GPX4 enzymatic activity. GPX4 is a selenocysteine-containing enzyme responsible for reducing lipid hydroperoxides to lipid alcohols, making it a master regulator of ferroptotic signaling (Supplementary Figure 3). To understand the impact of FINO2 on GPX4 activity, we used an LC-MS-based assay to monitor the ability of GPX4-containing lysates collected from cells treated with vehicle or ferroptosis inducers to reduce the GPX4-specific substrate phosphatidylcholine hydroperoxide (PCOOH) (Supplementary Figure 4).4, 17 Whereas erastin-treated cells did not show reduced GPX4 activity in this assay, treatment of cells with FINO2, FIN56, or RSL3 decreased the activity of GPX4 in the resulting lysates to a similar extent. We next monitored the kinetics of GPX4 inhibition by RSL3 and FINO2 by varying the amount of time cells were incubated with either compound. We observed that RSL3 causes a more rapid inhibition of GPX4 activity than does FINO2 (Supplementary Figure 5), suggesting that FINO2 might have a distinct mechanism of action from RSL3.
The observation that FINO2 caused decreased GPX4 activity suggested that, like RSL3, FINO2 might be a direct inhibitor of GPX44, 6, or that FINO2 could deplete GPX4 protein, similar to the ferroptosis inducer FIN56.17 To test whether FINO2 is a direct inhibitor of GPX4, we again used the LCMS-based PCOOH reduction assay to monitor the reduction of PCOOH in GPX4-containing cell lysates treated with ferroptosis inducers after cell lysis (Figure 3A). Because ferroptosis inducers are added after cell lysis, only molecules capable of directly inhibiting active GPX4 can prevent PCOOH reduction. As expected, the direct GPX4 inhibitor RSL3 was able to prevent PCOOH reduction in this assay (Figure 3A). FINO2 was unable to prevent PCOOH reduction, even at elevated concentrations, suggesting that FINO2 is not a direct inhibitor of GPX4. To support our hypothesis that FINO2 does not directly interact with GPX4, we performed 1H-15N heteronuclear single quantum coherence (HSQC) NMR on GPX4U46C in the presence and absence of FINO2. Unlike RSL3, FINO2 did not cause a change in the HSQC spectrum of GPX4, indicating that FINO2 is neither an allosteric nor active site ligand of GPX4 (Figure 3B and Supplementary Figure 6).
Figure 3. FINO2 indirectly inhibits GPX4 activity.
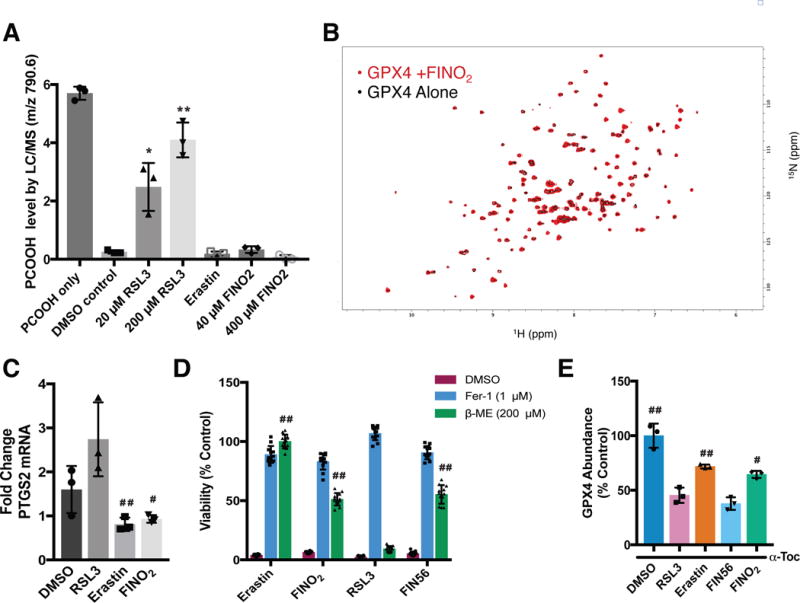
(A) Effect of ferroptosis inducers on GPX4 activity within the GPX4-containing cell lysates. Cell lysates were treated with PCOOH and GSH and the abundance of PCOOH was measured by LC-MS. Data are plotted as the mean ± s.d., n=3 biologically independent samples. *P=0.009, **P < 0.001 versus DMSO control (B) HSQC spectrum of GPX4 protein (black; 50 μM) overlaid with the spectrum of GPX4 incubated with FINO2 for 6 h (red; 50 μM and 500 μM, respectively) (C) PTGS2 mRNA levels following treatment with RSL3 (0.5 μM), erastin (10 μM), and FINO2 (10μM) for 6 h. Data are plotted as the mean ± s.d., n=3 biologically independent samples. #P=0.021, ##P=0.017 versus RSL3. (D) Ability of β-mercaptoethanol to prevent ferroptosis initiated by different ferroptosis inducers. Viability measured 24 h after co-treatment. Data are plotted as the mean ± s.d., n=12 biologically independent samples. ##P < 0.001 versus RSL3. (E) GPX4 protein abundance in HT-1080 cells co-treated with RLS3 (1 μM), erastin (10 μM), FIN56 (5 μM), or FINO2 (10 μM) and 100 μM α-tocopherol for 10 h, data are plotted as the mean ± s.d., n=3 biologically independent samples. #P=0.002, ##P<0.001 versus FIN56. Representative blot image is shown in Supplementary Figure 7. All P values were determined using unpaired two-tailed Student’s t-test.
Gene PTGS2, which encodes prostaglandin synthase, is upregulated following RSL3 treatment.4 We determined whether cells experienced a similar upregulation following treatment with FINO2. In contrast to RSL3, neither erastin-treated cells nor FINO2-treated cells showed significant upregulation of PTGS2, indicating that FINO2 does not cause the same transcriptional changes as the class 2 ferroptosis inducer RSL3 (Figure 3C).
To evaluate whether FINO2 acts as an irreversible covalent inhibitor of GPX4, HT-1080 cells were treated with a lethal dose of each ferroptosis inducer and either vehicle, ferrostatin-1, or β-mercaptoethanol (β-ME) (Figure 3D). In the extracellular medium, β-ME reacts with cystine to form a disulfide bond with cysteine. Neutral amino acid transporters import this mixed disulfide, bypassing system xc− and increasing intracellular cysteine availability.20 When cells are co-treated with RSL3 and β-ME, the increase in cysteine availability for GSH synthesis is unable to rescue cells because GPX4 is irreversibly and covalently inhibited. Conversely, the lethality of erastin is fully suppressed by β-ME, as cysteine availability is no longer dependent on system xc− function. β-ME also rescues ferroptosis induced by FIN56, which depletes GPX4 protein abundance (Figure 3D)17: β-ME may prevent FIN56 lethality by increasing the cysteine available for GSH synthesis, thereby improving the activity of the remaining GPX4.21 Ferroptosis induced by FINO2 was partially rescued by β-ME, further indicating that FINO2 is not an irreversible covalent inhibitor of GPX4. Since FINO2 does not decrease intracellular glutathione levels (Figure 2B), we reasoned that β-ME supplementation might rescue FINO2-treated cells by enhancing GPX4 activity, similar to the situation with FIN56.
To test whether FINO2 causes GPX4 protein depletion similar to FIN56, we quantified the abundance of GPX4 protein in cells undergoing ferroptosis by western blotting (Figures 3E and Supplementary Figure 7). As expected, both RSL3 and FIN56 caused a large decrease in the abundance of GPX4 at the protein level.17 Erastin and FINO2 caused only a minor decrease in GPX4 protein abundance, suggesting that FINO2 is unlike FIN56.
The endoperoxide moiety is required for FINO2 lethality
Since FINO2 does not display behavior characteristic of a class 1 or class 2 ferroptosis inducer, we sought to gain insight into its mechanism by determining which functional groups in FINO2 are required for inducing ferroptosis. Earlier reports indicated that the ether linkage in artemisinin dimers may be more critical for cytotoxic activity than the endoperoxide component.22, 23 Therefore, a series of non-peroxide derivatives of FINO2 were prepared.
The first derivatives synthesized possessed a spirocyclic oxetane unit. These derivatives can be viewed as analogues in which an oxygen atom was removed from the peroxide functional group. Earlier studies indicate that spirocyclic oxetanes, which are emerging as effective drug candidates, are cytotoxic towards cancer cells.24, 25
The spirocyclic oxetanes were synthesized from cyclohexanones 5-7 in three steps. Addition of allylmagnesium chloride afforded alkenes 8-10.26, 27 Treatment of these alkenes with mCPBA yielded epoxides 11-13. These epoxides were then heated under basic conditions to afford oxetanes 14-16.28 During attempts to synthesize the O-axial diastereomers, however, rapid elimination of water occurred under the strongly basic conditions required for cyclization.29
Similar methodology was used to prepared tetrahydrofuran derivatives of FINO2. These furans were selected because they maintain the same sized rings as found in FINO2, while removing the peroxide functional group. Terpenoid-derived spirocyclic furans are reported to induce apoptosis in a range of cancers, with IC50 values in the low micromolar range.30 Other fused-ring furans have shown biological activity for a range of diseases.31
The spirocyclic furan derivatives were prepared from cyclohexanone 5 in two steps. Homoallylation of ketone 5 afforded alkenes 17a and 17b. Upon exposure to the epoxidation conditions, tetrahydrofurans 18a and 18b were formed. This intramolecular cyclization is likely to occur by cyclization of the hydroxyl group onto a protonated epoxide intermediate.32 Avoiding the basic cyclization step allowed us to prepare a FINO2 derivative with an axial oxygen atom (18a).
Another derivative of FINO2 was synthesized by ring expansion. Hydrogenation of the oxygen−oxygen bond in FINO2 yielded triol 19. Subsequent protection of this triol with benzyl bromide occurred only at the primary hydroxyl group, forming 1,3-diol 20. Protection of this 1,3-diol with dibromomethane afforded 1,3-dioxane 21. Deprotection of the benzyl group yielded the desired 1,3-dioxane analogue 22.
Triol 19 was also considered to be an analogue of FINO2 that warranted cell studies. Considering the reducing environment of the cell,33 FINO2 could act as a pro-drug, forming triol 19. Thus, the activity of FINO2 could be due to the reduced product, as observed for some other peroxide-based drugs.34, 35
The activities of FINO2 and its analogues were measured in several cell lines. These cells include the renal cancer cell line, CAKI-1, and two immortalized fibroblast cell lines, BJ-hTERT, and its tumorigenic counterpart, BJ-eLR. The activities of FINO2 and its analogues were measured using the CellTiter-Glo® assay (Figure 4A).
Figure 4. Potency of non-peroxide analogs (A) and peroxide analogs (B) of FINO2.
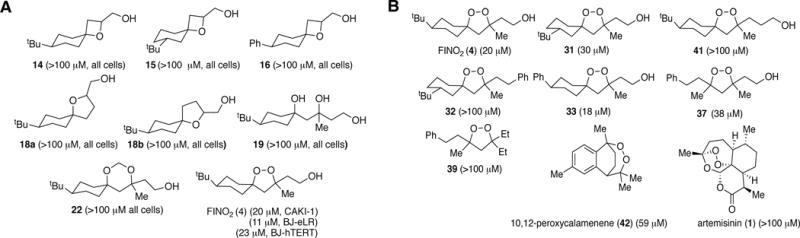
BJ-eLR, BJ-hTERT, or CAKI-1 cells were treated with either vehicle (DMSO), FINO2, or a FINO2 analogue at 5, 10, 25, 50, or 100 μM concentrations for 48 h. Cell viability was then measured and normalized to the DMSO vehicle to extract EC50 values. EC50 values are shown in parentheses. Compounds 15, 31, 32 and 37 were tested as a mixture of diastereomers
In all cases, the non-peroxide compounds displayed essentially no activity. FINO2, however, was able to kill oncogenic BJ-eLR and CAKI-1 cells selectively compared to BJ-hTERT cells. This result suggested that the peroxide moiety was essential to induce ferroptosis.
Earlier studies have also reported biological activity of 1,2-dioxolane-containing natural products, such as the plakinic acids and plakortides.36, 37 To determine the general ability of 1,2-dioxolanes to initiate ferroptosis, a series of peroxide-containing FINO2 analogues were synthesized. These synthetic analogues had modifications of both the hydrophobic and hydrophilic moieties, while retaining the 1,2-dioxolane core.
The 1,2-dioxolanes were synthesized from ketones 5−7 and 34. Treatment with LiNiPr2 yielded silyl enol ethers 23−25 and 35.38 Regioselective cobalt-catalyzed peroxidation of these products furnished mixed peroxyketals 26−28 and 36.38 Treatment of these compounds with 5 mol % of SnCl4 and alkene 29, 30, 38, or 40 followed by immediate deprotection yielded FINO2 (4) and derivatives 31−33, 37, 39, and 41.38
These 1,2-dioxolanes were tested in the renal cancer cell line (CAKI-1) along with both 10,12-peroxycalamenene (42)37 and artemisinin (1) (Figure 4B). These natural products were selected because 10,12-peroxycalamenene has been shown to induce apoptosis in human breast carcinoma cells,39 and several studies have reported on artemisinin’s activity against cancer.7–9, 40–42
The structure-activity relationship studies suggest that FINO2 belongs to a class of ferroptosis-inducing compounds that are somewhat tolerant of modifications, while retaining biological activity, unlike RSL3. Moving the tert-butyl group of FINO2 from C-4 to C-3 (31) resulted in a small decrease in potency. Removing the polar functionality (32) reveals that, although a peroxide moiety is necessary for initiating ferroptosis, it is not sufficient. Increasing the distance between the peroxide bond and hydroxyl group (41) also resulted in a decrease in activity. These data demonstrate that the hydroxyl portion of FINO2 must be present and have a specific spatial relationship to the peroxide, suggesting a specificity of mechanism for FINO2. Observations of cytotoxic activity of naturally occurring peroxides noted a similar requirement.37
The tert-butyl group, which can be metabolically unstable,43 is not required. When that group was replaced with an aromatic ring, 1,2-dioxolane 33 was similarly potent. Retaining the spirobicyclic core structure of FINO2 was also not necessary. Monocyclic peroxide 37, analogous to plakinic acid J (3) (Figure 1A), displayed activity similar to FINO2. The activity of 37 suggests that the cytotoxicity of 3 may be due to ferroptosis, not apoptosis.36,37 As observed for peroxide natural products,37 hydrophobic analogue 39 was also inactive.
FINO2 is a stable oxidant that oxidizes ferrous iron
Since the peroxide moiety in FINO2 is necessary for inducing ferroptosis, we hypothesized that FINO2 might be a selective pro-oxidant molecule that initiates ferroptosis through specific oxidation of ferroptosis-relevant substrates. Consistent with this hypothesis of relative inertness to most substrates, FINO2 was previously determined to be stable to 150 °C by thermo-gravimetric analysis.10 Similar 1,2-dioxolanes have also shown stability to LiBH4 and LiEt3BH at room temperature.44
Subjection to these high temperatures and strong reducing conditions, however, are not necessarily biologically relevant. FINO2 was therefore subjected to a variety of biologically relevant redox conditions in vitro (Supplementary Figure 8) and monitored for reaction by 1H-NMR spectroscopy. Heating FINO2 at 37 °C with a thiol, a class of compounds that are typically found between 0.5−10 mM in human cells,19 showed no decomposition. FINO2 was found to be stable to GSH, in agreement with the observation that FINO2 does not deplete GSH (vide supra). Arachidonic acid, a ubiquitous PUFA in cell membranes, showed no degradation following treatment with FINO2. Selenium-containing compounds such as selenocysteine and ebselen, the latter of which is model for GPX4,45 also did not reduce the peroxide bond. The inability of ebselen to reduce FINO2 is consistent with the observation that GPX4 cannot reduce endoperoxides.46 Ebselen was only able to reduce FINO2 at 90 °C, which suggests that FINO2 does not readily degrade to its reduced form 19 or to reactive oxygen species to confer biological activity. These results, and the lack of reactivity toward most reducing agents, suggest that the intact endoperoxide form is responsible for activity.
FINO2 was also stable at varying pH levels. No reaction was observed with amines or strong bases, even under conditions beyond the basicity found in peroxisomes.47 FINO2 was also found to be stable under acidic conditions that mimic conditions found in lysosomes.48 To examine the stability of FINO2 in biological systems, we measured the stability of FINO2 in human and mouse plasma and liver microsomes. We found that FINO2 had high stability in plasma, and human microsomes (Supplementary Figure 9).
Analogous to Fenton chemistry, 1,2-dioxolanes and ferrous iron can generate oxygen-centered radicals that initiate oxidative damage in biological systems.44 We hypothesized that FINO2 might initiate Fenton-type chemistry selectively over other organic peroxides.3, 12 Using an in vitro colorimetric assay, we monitored the oxidation state of iron treated with oxidizing compounds (Figure 5A). Out of all tested organic peroxides, only FINO2 and tert-butyl hydroperoxide (tBuOOH) were able to oxidize ferrous iron in vitro (Figure 5A). Cell death initiated by tBuOOH cannot be suppressed by ferrostatin-1 or β-ME (Supplementary Figure 10), indicating that the lethality of tBuOOH cannot be attributed to ferroptosis.
Figure 5. FINO2 directly oxidizes ferrous ion.
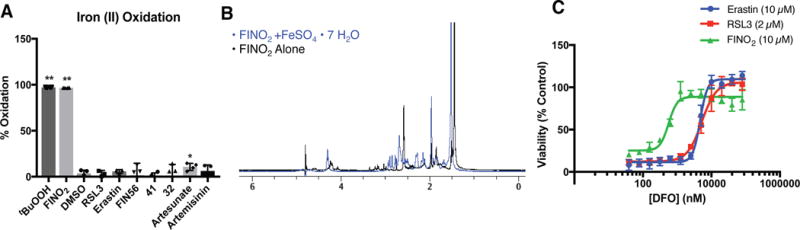
(A) Oxidation of ferrous iron (500 μM) in the presence of different oxidizing agents (500 μM). Data are plotted as the mean ± s.d., n=4 biologically independent samples. P values were determined using unpaired two-tailed Student’s t-test. *P = 0.035, **P<0.001 versus DMSO control. (B) 1H-NMR showing degradation of FINO2 following incubation with FeSO4•7H2O in CD3CN/D2O (1:1) for 12 h. (C) Ability of iron chelator deferoxamine (DFO) to inhibit ferroptosis initiated by different ferroptosis inducers. Experiments were performed in triplicate with three biologically independent samples. Data are plotted as the mean ± s.d..
The iron oxidation by FINO2 is accompanied by a degradation of the endoperoxide. Incubation of FINO2 at 37 °C for 12 hours reveals reduction of the peroxide bond by FeSO4•7H2O, as seen in similar systems (Figure 5B).49 This reduction is consistent with studies on 1,2,4-trioxolanes and plakortin derivatives that suggest rapid reduction of the peroxide functionality.34, 35, 49, 50 Consistent with previous studies, artesunate (2) also decomposed after exposure to iron (Supplementary Figure 11).7 Inactive analogues 32 and 41 were only partially decomposed after iron exposure (Supplementary Figure 11). These results suggest that the complete breakdown of the peroxide moiety by iron is required to induce ferroptosis.
The selectivity of iron oxidation by FINO2 compared to analogues 32 and 41 suggested that iron oxidation might be important to ferroptosis initiation by FINO2. The iron chelator DFO is able to suppress the lethality of FINO2 ten-fold more potently than for erastin or RSL3 (Figure 5C). Similarly, supplementation of cell media with ferrous or ferric salts sensitizes cells to FINO2 initiated ferroptosis, but not to RSL3 initiated ferroptosis (Supplementary Figure 12). Despite the apparent importance of iron to FINO2 lethality, FINO2 treated cells did not show changes in the abundance of the iron regulatory proteins IRP2, FTL1, or TFR (Supplementary Figure 13), suggesting that FINO2 may engage labile cellular iron preferentially over iron-containing proteins.
FINO2 oxidizes the lipidome independent of ALOX activity
Previously, the family of arachidonic acid lipoxygenases (ALOX) was shown to be critical in generating lipid peroxides during ferroptosis, particularly in response to erastin or RSL3.6 siRNA knockdown of either the ALOX15B or ALOXE3 genes suppressed the lethality of erastin. Similarly, a GFP-ALOX5 fusion protein expressed in ferroptosis-sensitive cells was activated and translocated to the nuclear membrane following erastin treatment. Because FINO2 is able to oxidize ferrous iron directly, we sought to evaluate whether ferroptosis initiated by FINO2 required enzymatic lipid peroxidation, similar to erastin and RSL3. We assembled a collection of arachidonic acid derivatives deuterated at the bis-allylic positions. Deuteration at the bis-allylic position creates a strong kinetic isotope effect in lipoxygenase enzymes, making deuterated arachidonic acids effective and highly specific inhibitors of lipoxygenases (Figure 6A).
Figure 6. Ferroptosis initiated by FINO2 oxidizes a large subset of the lipidome independent of lipoxygenase activity.
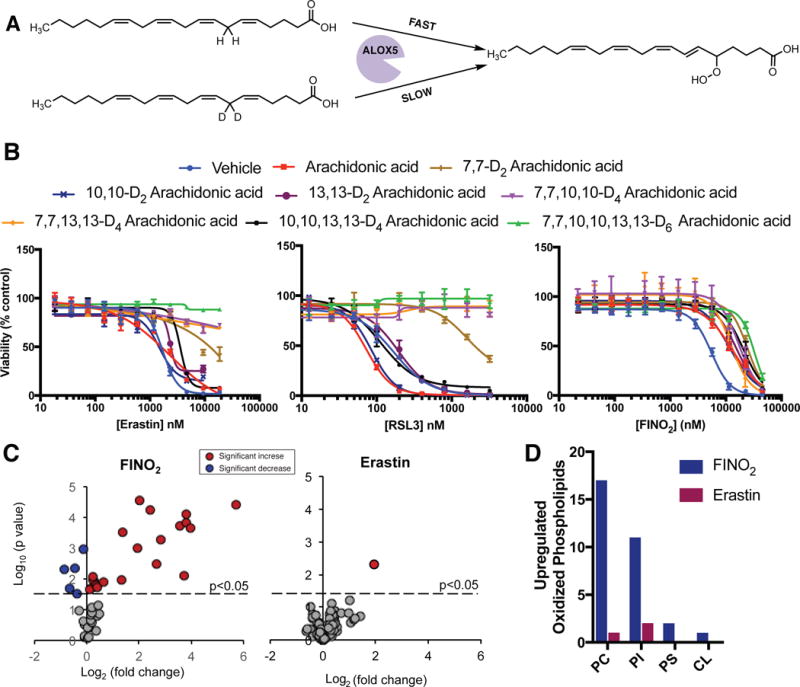
(A) Schematic of lipoxygenase inhibition by deuterated arachidonic acid. (B) Effect of deuterated arachidonic acid incubation (80 μM for 24 h) on HT-1080 sensitivity to ferroptosis inducers. Experiments were performed in triplicate with biologically independent samples. Data are plotted as the mean ± s.d.. (C) Volcano plots showing the change in abundance of oxidized phosphatidylethanolamine species in HT-1080 cells following incubation with FINO2 (10 μM) or erastin (5 μM) for 6 h. Red circles indicate significant increase in abundance, blue circles indicate a significant depletion following treatment. Experiments were performed in triplicate with biologically independent samples. P values were determined using two-sided t test. Lipid species with significant change upon FINO2 treatment were plotted above the dot line (p<0.05). (D) Number of oxidized phosphatidylcholine (PC), phosphatidylinositol (PI), phosphatidylserine (PS), and cardiolipin (CL) species upregulated in HT-1080 cells following treatment with FINO2 (10 μM) or erastin (5 μM) for 6 h.
We incubated HT-1080 cells with vehicle only (0.1% ethanol), arachidonic acid, or a deuterated arachidonic acid overnight to allow incorporation of the fatty acid into cellular membranes. Cells were then treated with erastin, RSL3, or FINO2 (Figure 6B). Deuteration at the 7 position was strongly protective against cell death initiated by erastin and RSL3, confirming the importance of ALOX5 in initiating ferroptosis for these molecules. Ferroptosis induced by FINO2 was only weakly rescued by deuteration at the 7 position, suggesting that FINO2 either activates an ALOX isoform with different regioselectivity or preferentially oxidizes fatty acid groups other than arachidonic acid. To evaluate whether FINO2 activates a different lipoxygenase isoform than erastin or FINO2, we tested all combinations of deuterated arachidonic acid analogues against all ferroptosis inducers. Additional deuteration increased the amount of death suppression for erastin and RSL3, and the fully deuterated D6-arachidonic acid completely suppressed the lethality of both of these ferroptosis inducers. No significant suppression of ferroptosis initiated by FINO2 was observed for any deuterated arachidonic acid tested (Figure 6B), suggesting that FINO2 does not require ALOX-mediated peroxidation for ferroptosis induction.
To compare the global lipidomic changes following treatment with FINO2 or erastin, we used LC-MS to identify oxidized esterified phospholipids extracted from HT-1080 cells treated with vehicle (DMSO), FINO2, or erastin with and without ferrostatin-1. Because of its importance to ferroptosis, we first compared the changes in oxidized PE following treatment with either ferroptosis inducer at equivalent time points (Figure 6C). Whereas erastin caused an increase in 1 PE species after treatment, FINO2 caused an increase in 21 PE species (Supplementary Figure 2). This pattern was consistent for other phospholipids, including phosphatidylcholine (PC), phosphatidylinositol (PI), phospatidylserine (PS), and cardiolipin (CL) (Figure 6D). The increases in oxidized phosphatidylserine and cardiolipin were unresponsive to ferrostatin treatment, suggesting they do not contribute to ferroptosis (Supplementary Figure 2B). MS analysis allowed us to identify the acyl tail portion of the oxidized phospholipids. As expected, all acyl tails contained some degree of polyunsaturation. Many of the oxidized phospholipids contained linoleic (18:2), arachidonic (20:4), and other polyunsaturated moieties. Cumulatively, these data show that FINO2 causes the oxidation of a much more diverse set of phospholipids than does erastin, and does not have strong substrate selectivity within the set of polyunsaturated lipids.
Discussion
A common feature of all ferroptosis inducers is the ability to overcome the endogenous GSH-dependent lipid peroxidation defense network. In this study, we investigated the mechanism by which the 1,2-dioxolane FINO2 was able to induce ferroptosis in cells. FINO2 does not elicit the same transcriptional, translational, and phenotypic responses expected from previously described ferroptosis inducers. We hypothesize that FINO2 is able to initiate ferroptosis through a combination of its ability to directly oxidize labile iron and inactivate GPX4.
In support of this hypothesis, we have demonstrated that the highly oxidizing peroxide group and a nearby polar head group are both required for lethality and for oxidation of ferroptosis-relevant substrates. Both non-peroxide derivatives and non-polar derivatives of FINO2 were not lethal to cells. Elongation of the polar head group led to a loss in activity as well as an inability to oxidize iron, suggesting that the spatial relationship between the peroxide and hydroxyl moieties in FINO2 is required to engage labile iron or may facilitate the reduction of the oxygen-oxygen bond. Ferroptosis initiation by FINO2 is highly sensitive to iron availability, and oxidizes a broad set of polyunsaturated lipids. Unlike other peroxide-containing compounds, FINO2 is able to initiate ferroptosis preferentially over other forms of cell death. Multiple factors could account for this remarkable selectivity, including inactivation of GPX4, or the lipophilicity of FINO2. The predicted octanol/water partition coefficient (sLogP) for FINO2 is 3.54, whereas the value for artemisinin is 2.39, making FINO2 more than an order of magnitude more lipophilic than artemisinin. Because of its high lipophilicity, FINO2 may accumulate in the appropriate lipid bilayers of cell membranes, allowing it to oxidize PUFAs directly in the locations that trigger ferroptosis.
In summary, this study provides insight into the mechanism of FINO2 and offers new perspectives on how ferroptosis can be initiated. Structural exploration of FINO2 and its analogues reveal that this new class of ferroptosis inducer can be modified and retain biological activity, but requires the endoperoxide and hydrophilic head to be present. In addition, whereas previous ferroptosis initiators inhibited components of the endogenous lipid-peroxide-scavenging network in cells, FINO2 induces ferroptosis through a combination of direct oxidation of ferroptosis-relevant substrates and indirect GPX4 inactivation. The mechanism of GPX4 inactivation by FINO2 remains unclear, since FINO2 does not decrease the protein level of GPX4 and is not a GPX4 ligand. Nonetheless, we hypothesize that this specific combination of pro-oxidant functionality and GPX4 inactivation is required to navigate cells to ferroptosis in response to this class of compounds.
Online Methods
Cell lines and media
HT-1080 cells were obtained from ATCC and grown in DMEM with glutamine and sodium pyruvate (Corning 10-013) supplemented with 10% Heat-Inactivated FBS, 1% non-essential amino acids (Invitrogen), and 1% penicillin-streptomycin mix (Invitrogen). BJ-5ta (BJ-hTERT) cells were obtained from ATCC and grown in DMEM media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. BJ-eLR cells were donated by the laboratory of William Han at the Dana-Farber Cancer Institute and grown in the same media as BJ-hTERT cells. CAKI-1 cells were purchased from ATCC and grown in RPMI media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. All cells were maintained in a humidified environment at 37 °C and 5% CO2 in a tissue incubator.
Chemical synthesis of FINO2 analogues
Synthetic protocols are described in the Supplementary Note.
FINO2 analogue potency measurement
BJ-eLR, BJ-hTERT, or CAKI-1 cells were plated at 10,000 cells per well in white 96-well plates in technical triplicates and incubated for 24 h to allow for cell adhesion. The cells were then treated with either vehicle (DMSO), FINO2, or a FINO2 analog at 5, 10, 25, 50, or 100 μM concentrations. The cells were returned to the cell culture incubator for 48 h. CellTiter-Glo® (Promega) was used according to the manufacturer’s protocol. Luminescence was read on a Bioteck Microplate Reader. All cell viability data were normalized to the DMSO vehicle. Experiments were performed three independent times with different passages for each cell line. EC50 values and error values were computed using Prism 7.0 (GraphPad).
Ferroptosis rescue and sensitivity modulation
3,000 cells were seeded per well in black, clear bottom 384-well plates (Corning) and allowed to adhere overnight. For ferroptoisis rescue assays, the medium was replaced on the following day with 50 μL of growth medium and 5 μL medium containing a ferroptosis inducer and a dilution series of protective molecule. The final concentration of ferroptosis inducers was 10 μM erastin, 10 μM FINO2, 2 μM RSL3, and 5 μM FIN56 unless otherwise noted. For sensitivity modulation experiments, cells were co-treated with a fixed concentration of the modulating compound and a dilution series of the ferroptosis inducer. For deuterated arachidonic acids, cells were incubated in medium containing 80 μM of a deuterated arachidonic acid overnight to allow for incorporation into cellular membranes. The following day cells were treated with a dilution series of ferroptosis inducer. In all cases, after 24 h incubation with ferroptosis inducer, 6.1 μL of Presto Blue (Thermo-Fisher) were added. Cells were incubated for an additional 3 hours and the Presto Blue fluorescence intensity was measured using a Victor X5 plate reader (PerkinElmer)(ex/em 530/590). Background (no cells) fluorescence was subtracted and the resulting fluorescence intensities were averaged between biological replicates. From these data, dose-response curves and EC50 values were computed using Prism 7.0 (GraphPad).
C11-BODIPY lipid peroxidation measurement
The day before the experiment, 600,000 HT-1080 cells were seeded in 6 well plates and allowed to adhere overnight at 37 °C. On the day of the experiment, cells were treated with DMSO, erastin (10 μM), or FINO2 (10 μM) with or without DFO (100 μM) and allowed to incubate for 6 h at 37 °C. Cells were trypsinized, washed, and suspended in HBSS containing 1.5 μM C11-BODIPY (ThermoFisher) and incubated at 37 °C for 15 min. Cells were pelleted and resuspended in HBSS. Fluorescence intensity was measured on the FL1 channel with gating to record live cells only (gate constructed from DMSO treatment group). A minimum of 10,000 cells was analyzed per condition.
Reduced glutathione measurement
10 million HT-1080 cells were treated with 5 μM erastin, 10 μM FINO2, 500 nM RSL3, or vehicle. Cells were harvested after 1.5 h (RSL3) or 6 h (vehicle, erastin, FINO2), and lysed using ice-cold PBS/0.5% Nonidet P-40. Samples were centrifuged for 15 min at 4 °C at top speed. The resulting supernatant was deproteinized using a Deproteinizing Kit (ab2047080) and kept on ice. Reduced GSH levels were determined using GSH/GSSG Ratio Detection Assay Kit (Fluorometric – Green) (ab138881) following the manufacturer’s protocol.
TBARS measurement
20 million HT-1080 cells were treated with 5 μM erastin, or 10 μM FINO2 with and without 2 μM ferrostatin-1, harvested after 6 h into 1 mL PBS. Samples were sonicated and the homogenate was stored on ice. TBARS were measured using a TBARS (TCA Method) Assay Kit – (Cayman 700870) following the manufacturer’s protocol.
qPCR
HT-1080 cells were treated with either 10 μM erastin, 10 μM FINO2, or 2 μM RSL3 for 5 h. RNA was extracted using the Qiashredder and Qiagen RNeasy Mini kits (Qiagen) according to the manufacturer's protocol. For each sample 2 μg of RNA were used as input for each reverse transcription reaction, performed using the TaqMan RT kit (Applied Biosystems/Life Technologies Corp., Foster City, CA). Quantitative PCR reactions were performed using the Power SYBR Green PCR Master Mix (Applied Biosystems). Triplicate samples per condition were analyzed on a ViiA 7 qPCR instrument (Thermo Fischer) using absolute quantification settings. Differences in mRNA levels compared to ACTB internal reference control were computed between control and experimental conditions using the ΔΔCt method.
HSQC NMR
Uniformly 15N-labeled GPX4U46C was expressed in and purified from E. Coli. For HSQC of GPX4 with FINO2, 50 μM 15N-labeled GPX4 was pre-incubated with 500 μM FINO2 for 6 hours at room temperature in buffer (100 mM MES, 5 mM TCEP, pH 6.5). For HSQC of GPX4 with RSL3, since solubility of RSL3 in aqueous solution is low, 10 μM 15N-labeled GPX4 was pre-incubated with 100 μM RSL3 for 12 hours at 4°C. Then the protein solution was concentrated to 50 μM before testing. 10 % D2O was added for the field frequency lock. The 1H-15N HSQC spectra were collected on Bruker Avance III 500 Ascend (500 MHz) spectrometers at ambient temperature. The 1H carrier frequency was positioned at the water resonance. The 15N carrier frequency was positioned at 115 ppm. The spectral width in the 1H dimension was 7500 Hz and the width in 15N dimension was 1824.6 Hz. Suppression of water signal was accomplished using the WATERGATE sequence. Heteronuclear decoupling was accomplished using GARP decoupling scheme.
Western blotting
0.8 million HT-1080 cells were seeded per well in a 60 mm plate and allowed to adhere overnight. Cells were then co-treated with 100 μM α-Tocopherol and either 1 μM of RSL3, 10 μM of Erastin, 5 μM of FIN56, 10 μM of FINO2, or vehicle for 10 h. Cells were harvested with trypsin (Invitrogen, #25200-114), pelleted, and frozen at -80˚C. Cell pellets were thawed, lysed, blotted, and imaged as previously described.3 In particular, for this experiment, the set of 15 pellets was run on a single gel. Ultimately, two gels were run and quantified from a total of six biological replicates (30 pellets in total). For the other proteins of interest, experiments were performed in biological triplicate. Antibodies used were: GPX4 (abcam, #ab125066, 1:250 dilution), Ferritin Light Chain (Santa Cruz, #sc-390558, 1:1000 dilution), IRP2 (Nous Biological, #NB100-1798, 1:1000 dilution), Transferrin receptor 1 (CD71, TFRC, Cell Signaling, #13113, 1:1000 dilution), Actin (Cell signaling, #D18C11, 1:3000 dilution), alpha-tubulin (Santa Cruz, #sc-32293, 1:3000 dilution) and GAPDH (Santa Cruz, # sc-47724, 1:10,000 dilution). Results were quantified using a LI-COR Odyssey CLx IR scanner and GraphPad Prism 7.
Decomposition of FINO2 by FeSO4
An NMR tube was charged with FINO2 (5.0 mg, 0.020 mmol) and D2O:CD3CN (1:1, 0.04M per solvent). The NMR was preheated to 37 °C and the 1H spectrum was acquired. Then, FeSO4•7H2O (11 mg, 0.039 mmol) was added. After 12 h, a 1H spectrum was acquired at room temperature.
Colorimetric iron oxidation
In a clear 96 well plate, 4 μL of a 25 mM DMSO solution of test compound was added to 196 μL of a 2:1 solution of water and DMSO (v/v) containing 500 μM FeCl2. This mixture was incubated at 37 °C for 1 h. 5 μL of a 20 mM stock of FerroZine™ (Sigma Aldrich) was added. Absorbance was measured at 567 nm.
LC-MS GPX4 activity assay
For experiments performed on whole cells, 20 million HT-1080 cells were treated with vehicle (DMSO), erastin (10 μM), FINO2 (10 μM), FIN56 (5 μM) for 6 h, or RSL3 (0.5 μM) for 2 h. Cells were harvested by trypsinization and washed twice in PBS and resuspended in lysis buffer (25 mM sodium phosphate, 125 mM NaCl, 1 mM EDTA, 0.1 mM Deferoxamine, 25 μM butylated hydroxytoluene, 0.3% Triton-X 100, and protease inhibitor tablet [1 tab/10 mL]). Cells were lysed by sonication and cleared of insoluble components by centrifugation at 14,000 xg for 10 min at 4 °C. For cell-free assays, HT1080 cells were harvested by trypsinization and washed twice in PBS and re-suspended in ice-cold NP40 lysis buffer (10 mM Tris/Cl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 5mM TCEP and protease inhibitor [1 tab/10mL]). Cells were lysed by placing on ice for 30 min with extensively pipetting every 10 min and then centrifuging at 14,000g for 10 min at 4°C. The protein concentration of each sample was determined using the Bradford assay. For cell-free experiments, cell lysate containing 200μg protein was separately pre-incubated with vehicle (DMSO, 0.2%), erastin (10 μM), RSL3 (20/200 μM) or FINO2 (40/400 μM) for 10 min at 37°C, in 500 μL GXP4 reaction buffer(25mM sodium phosphate, 125 mM NaCl, 1mM EDTA, 0.1 mM deferoxamine, 0.1% Triton X-100 and 5mM TCEP). Otherwise, PCOOH reduction reactions contained 200 μg of protein from treated groups, 5 mM reduced glutathione, 10 μM PCOOH, and sufficient reaction buffer to raise the volume to 500 μL (Reaction buffer: 25 mM sodium phosphate, 125 mM NaCl, 1 mM EDTA, 0.1 mM deferoxamine, 0.1% TritonX-100). Reactions were incubated at 37 °C for 30 min and then extracted with 1 mL of chloroform/methanol mixture (2:1 v/v). The organic portion was isolated and evaporated. Dry extracted materials were reconstituted in methanol. The samples were analyzed by LC-MS for PCOOH content as described previously.4, 17
MS analysis of phospholipids
Lipids were extracted by using Folch’s procedure.51 Lipid phosphorus was determined by a micro-method.52 MS analysis of PLs was performed on a Q-Exactive hybrid-quadrupole-orbitrap mass spectrometer (ThermoFisher Scientific) as previously described.53 Phospholipids were separated on a normal phase column (Silica Luna 3 μm, 100A, 150x2 mm, (Phenomenex, Torrance CA)) at 35 °C using gradient solvents containing 5 mM ammonium acetate (A – n-hexane:2-propanol:water, 43:57:1 (v/v/v) and B – n-hexane:2-propanol:water, 43:57:8 (v/v/v). The gradient conditions (all linear) were as follows: 0-23 min (10% B to 32% B); 23-32 min (32% B to 65% B); 32-35 min (65% B to 100% B) 35-62 min (hold at 100% B); 62-64 min (100% B to 10% B); 64-80 min (10% B). Flow rate was maintained at 200 μl/min except for the 35-62 min time frame where the flow rate was increased to 225 μl/min. MS analysis was performed in negative ion mode at a resolution of 140,000 for the full MS scan in a data-dependent mode. The scan range for MS analysis was 400-1800 m/z with a maximum injection time of 128 ms using 1 microscan. An isolation window of 1.0 Da was set for the MS and MS2 scans. Capillary spray voltage was set at 3.5 kV, and capillary temperature was 320 °C. The S-lens Rf level was set to 60. Analysis of LC/MS data was performed using the software package Compound Discoverer™ (ThermoFisher Scientific, San Jose, CA) with an in-house generated analysis workflow and oxidized phospholipid database.
NADPH quantification
NADPH levels were analyzed using Amplite fluorimetric NADP/NADPH ratio assay kit (AAT bioquest). 1 million HT1080 cells were seeded in 100-mm tissue culture dishes for 16 h. Cells were treated with either vehicle or a ferroptosis inducer (0.1% DMSO for 8 h, 5 μM FIN56 for 8 hr, 10 μM Erastin for 8 h, 0.5 μM RSL3 for 3 h, 10 μM FINO2 for 8 h). After treatment, the cells were trypsinized, pelleted, lysed and analyzed for NADPH level in the same way as described before.54 The concentrations of these metabolites were normalized to the amount of total protein. Measurements were done in biological triplicates. NADPH levels of all samples were normalized to DMSO vehicle control.
In vitro microsomal stability assay
To a 96 well round bottom polypropylene plate (PerkinElmer #6008190) was added phosphate buffer (182.2 μL, pH 7.4, 100 mM) followed by addition of NADPH-regenerating system solution A (10 μL), and NADPH regenerating system solution B (2 μL) (Corning Gentest 3P NADPH regenerating system solution A (#451220) and B (#451200)). A stock solution of FINO2 (0.8 μL, 5 mM) or ferrostatin-1 (Fer-1) (positive control, 0.8 μL, 5 mM) in methanol was added and the mixture was warmed to 37 °C for 5 min. Mouse microsomes (CD-1, 20 mg/mL, Life Technologies) or human microsomes (pooled 50 donors, 20 mg/mL, Thermo Fisher Scientific) (5 μL, thawed in 37 °C bead bath before use) were added. At selected time points (0, 1, 5, 10, 20, 30, 60 and 120 min) aliquots (15 μL) were withdrawn from the plate and quenched upon addition to cold methanol (60 μL), containing a terfenadine internal standard (1.25 mM) in a 96 well polypropylene plate kept on ice. The samples were centrifuged at 4,000 rpm for 5 min at 4 °C. The supernatant (40 μL) was withdrawn and transferred to a sample vial with insert.
In vitro plasma stability assay
Mouse and human plasma from GeneTex (mouse: #GTX73236, human: #GTX73265) aliquoted and stored at -80 °C were thawed in a 37 °C bead path and centrifuged at 3,000 rpm for 10 min at 10 °C to remove any particulates. The particulate-free plasma was diluted in phosphate buffer (pH 7.4, 100 mM) to a final plasma concentration of 50%. To a 96 well round bottom polypropylene plate (PerkinElmer #6008190) was added the 50% plasma solution (195 μL) and the mixture was warmed to 37 °C for 5 min. A stock solution of FINO2 (5 μL, 800 μM), ferrostatin-1 (Fer-1) (negative control 5 μL, 800 μM), or lovastatin (positive control, 5 μL, 800 μM) DMSO was added. At selected time points (0, 5, 15, 30, 60, 120, 180, and 360 min) aliquots (15 μL) were withdrawn from the plate and quenched upon addition to cold acetonitrile (60 μL), containing a terfenadine internal standard (1.25 mM) in a 96 well polypropylene plate kept on ice. The samples were centrifuged at 4,000 rpm for 5 min at 4 °C. The supernatant (40 μL) was withdrawn and transferred to a sample vial with insert.
LC-MS analysis of in vitro Pharmacokinetic studies
The samples were analyzed by LC-MS. LC-MS analysis was performed on a platform comprising a Thermo Scientific Dionex Ultimate 3000 and a Bruker amaZon SL equipped with an electrospray ionization source controlled by Bruker Hystar 3.2. Chromatographic separation was performed by injecting 5 mL of the sample onto an Agilent Eclipse Plus C18 column (2.1 3 50 mm, 3.5 mm) maintained at 20 °C. The flow rate was maintained at 400 mL/min. The initial flow conditions were 80% solvent A (95:5:0.1 water:methanol:acetic acid with 10 mM ammonium acetate) and 20% solvent B (methanol containing 0.1% acetic acid). Solvent B was maintained at 20% for the first minute of the run and was raised to 80% over 0.50 min. Solvent B was raised to 100% by 5 min and was lowered back to initial conditions (20%) by 8.750 min with a total run time of 12.0 min. The retention times and m/z were summarized in Supplementary table 1.
The area of the base peak chromatogram for each compound was measured at each time-point and compared against the zero minute time-point to calculate the percentage of compound remaining at each time-point. For samples where the percent of compound remaining was greater than 100% at the second time-point, the second time-point used to calculate the percent of compound remaining. The data were plotted in Prism 7 and the data fit with a one-phase linear decay model with y0 value set to 100 to calculate the half-life. For compounds with half-lives greater than two hours, the percent of compound remaining after 120 min is reported.
Statistical analysis
The data reported in this study represent the mean and s.d. of at least three independent experiments measured in triplicate, unless otherwise stated in the figure legend. For statistical analyses, ANOVA and two-tailed Student’s t-tests were conducted to assess whether a significant difference exists between two groups of samples. A P value of less than 0.05 was considered as statistically significant. All statistical tests were carried out using Prism 7 software (GraphPad).
Data availability
The data that support the findings of this study are available from the corresponding authors upon request.
Supplementary Material
Acknowledgments
This research was supported by the Training Program in Molecular Biophysics Grant (T32GM008281 to M.M.G.), the National Cancer Institute (R35CA209896 and P01CA087497 to B.R.S), the National Institute of General Medical Sciences (1RO1GM118730 to K. A. W.), the National Heart, Lung, and Blood Institute (HL114453 to V.E.K. and Y.Y.T.), the National Institute of Allergy and Infectious Diseases (U19AI068021 to V.E.K. and Y.Y.T.), and the MRSEC Program of the National Science Foundation (DMR-1420073 to E.P.-P.). The Bruker Avance-400, 500, and 600 MHz Spectrometers (NYU) were acquired through the support of the National Science Foundation (CHE-01162222). We thank Dr. C. Lin for assistance with NMR spectroscopy and mass spectrometry, Dr. C. Hu for X-ray analysis and the Materials Research Science and Engineering Center (MRSEC) program of the National Science Foundation (NSF) under Award Numbers DMR-0820341 and DMR-1420073, and J. Chung for his assistance with cell culture.
Footnotes
Author Contributions
M.M.G., A.A.A., H.L., B.R.S., and K.A.W. contributed to the writing of the manuscript. M.M.G., A.A.A., L.K.M, V.E.K., B.R.S., and K.A.W. designed and planned research. M.M.G., A.A.A., H.L., J.M.C., D.W.H., D.S.Z., P.H.B., Y.Y.T., and J.D.D. conducted in vitro biochemical and metabolomic assays. M.M.G, A.A.A., B.H., C.A.V., D.W.H., and A.J.L., collected and analyzed cell viability data. L.Y. performed qPCR. A.A.A. and H.L. conducted NMR studies. A.A.A. conducted stability studies. H.L. and E.R. conducted western blotting experiments. A.A.A., B.H., and D.S.Z. synthesized FINO2 and all structural analogues. A.Y.C. aided in furan synthesis. E.P.-P. aided in oxetane synthesis. M.A.F., A.V.B., V.V.S., A.J.L., and M.S.S., synthesized deuterated arachidonic acids. All authors have given their approval of the final version of the manuscript.
Conflict of Interest
M.S. Shchepinov declares a competing financial interest as the Chief Scientific Officer of Retrotrope, Inc., all other authors declare no competing financial interests.
References
- 1.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Cell Biol. 2001;2:545–550. doi: 10.1038/35080097. [DOI] [PubMed] [Google Scholar]
- 3.Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang WS, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crespo-Ortiz MP, Wei MQ. Antitumor activity of artemisinin and Its derivatives: from a well-known antimalarial agent to a potential anticancer drug. J Biomed Biotechnol. 2012;2012:247597. doi: 10.1155/2012/247597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Li Q, Wu J, Wang MY, Yu JX. Artemisinin and its derivatives as a repurposing anticancer agent: what else do we need to do? Molecules. 2016;21 doi: 10.3390/molecules21101331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishna S, et al. A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. Ebiomedicine. 2015;2:82–90. doi: 10.1016/j.ebiom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrams RP, Carroll WL, Woerpel KA. Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol. 2016;11:1305–1312. doi: 10.1021/acschembio.5b00900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Skouta R, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136:4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zilka O, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3:232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krainz T, et al. A mitochondrial-targeted nitroxide is a potent inhibitor of ferroptosis. ACS Cent Sci. 2016;2:653–659. doi: 10.1021/acscentsci.6b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kagan VE, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimada K, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dixon SJ, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin J, et al. Cyanine-based fluorescent probe for highly selective detection of glutathione in cell cultures and live mouse tissues. J Am Chem Soc. 2014;136:5351–5358. doi: 10.1021/ja412628z. [DOI] [PubMed] [Google Scholar]
- 20.Ishii T, Bannai S, Sugita Y. Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. Role of the mixed disulfide of 2-mercaptoethanol and cysteine. J Biol Chem. 1981;256:12387–12392. [PubMed] [Google Scholar]
- 21.Toppo S, Flohe L, Ursini F, Vanin S, Maiorino M. Catalytic mechanisms and specificities of glutathione peroxidases: variations of a basic scheme. Biochim Biophys Acta. 2009;1790:1486–1500. doi: 10.1016/j.bbagen.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Beekman AC, et al. Stereochemistry-dependent cytotoxicity of some artemisinin derivatives. J Nat Prod. 1997;60:325–330. doi: 10.1021/np9605495. [DOI] [PubMed] [Google Scholar]
- 23.Beekman AC, et al. Artemisinin-derived sesquiterpene lactones as potential antitumour compounds: Cytotoxic action against bone marrow and tumour cells. Planta Med. 1998;64:615–619. doi: 10.1055/s-2006-957533. [DOI] [PubMed] [Google Scholar]
- 24.Burkhard JA, Wuitschik G, Rogers-Evans M, Muller K, Carreira EM. Oxetanes as versatile elements in drug discovery and synthesis. Angew Chem Int Ed. 2010;49:9052–9067. doi: 10.1002/anie.200907155. [DOI] [PubMed] [Google Scholar]
- 25.Wuitschik G, et al. Oxetanes in drug discovery: structural and synthetic insights. J Med Chem. 2010;53:3227–3246. doi: 10.1021/jm9018788. [DOI] [PubMed] [Google Scholar]
- 26.Trost BM, Bogdanow MJ. New synthetic reactions – versatile cyclobutanone (spiroannelation) and gamma-butyrolactone (lactone annelation) synthesis. J Am Chem Soc. 1973;95:5321–5334. [Google Scholar]
- 27.Fujioka H, et al. Reaction of the acetals with TESOTf-base combination; Speculation of the intermediates and efficient mixed acetal formation. J Am Chem Soc. 2006;128:5930–5938. doi: 10.1021/ja060328d. [DOI] [PubMed] [Google Scholar]
- 28.Murai A, Ono M, Masamune T. Intramolecular cyclization of 3,4-epoxy alcohols – oxetane formation. Bull Chem Soc Jpn. 1977;50:1226–1231. [Google Scholar]
- 29.Biemann K, Seibl J. Application of mass spectrometry to structure problems. 2. stereochemistry of epimeric, cyclic alcohols. J Am Chem Soc. 1959;81:3149–3150. [Google Scholar]
- 30.Liu Y, et al. Guanacastane-type diterpenoids with cytotoxic activity from Coprinus plicatilis. Bioorg Med Chem Lett. 2012;22:5059–5062. doi: 10.1016/j.bmcl.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Ki DW, et al. New antioxidant sesquiterpenes from a culture broth of Coprinus echinosporus. J Antibiot. 2015;68:351–353. doi: 10.1038/ja.2014.158. [DOI] [PubMed] [Google Scholar]
- 32.Vilotijevic I, Jamison TF. Epoxide-opening cascades in the synthesis of polycyclic polyether natural products. Angew Chem Int Ed. 2009;48:5250–5281. doi: 10.1002/anie.200900600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic-reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- 34.Spangler B, et al. A novel tumor-activated prodrug strategy targeting ferrous iron is effective in multiple preclinical cancer models. J Med Chem. 2016;59:11161–11170. doi: 10.1021/acs.jmedchem.6b01470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fontaine SD, DiPasquale AG, Renslo AR. Efficient and stereocontrolled synthesis of 1,2,4-trioxolanes useful for ferrous iron-dependent drug delivery. Org Lett. 2014;16:5776–5779. doi: 10.1021/ol5028392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidson BS. Cytotoxic 5-membered cyclic peroxides from a plakortis sponge. J Org Chem. 1991;56:6722–6724. [Google Scholar]
- 37.D'Ambrosio M, Guerriero A, Debitus C, Waikedre J, Pietra F. Relative contributions to antitumoral activity of lipophilic vs. polar reactive moieties in marine terpenoids. Tetrahedron Lett. 1997;38:6285–6288. [Google Scholar]
- 38.Hurlocker B, Miner MR, Woerpel KA. Synthesis of silyl monoperoxyketals by regioselective cobalt-catalyzed peroxidation of silyl enol ethers: application to the synthesis of 1,2-dioxolanes. Org Lett. 2014;16:4280–4283. doi: 10.1021/ol5020015. [DOI] [PubMed] [Google Scholar]
- 39.Park SE, et al. Induction of apoptosis in MDA-MB-231 human breast carcinoma cells with an ethanol extract of Cyperus rotundus L. by activating caspases. Oncol Rep. 2014;32:2461–2470. doi: 10.3892/or.2014.3507. [DOI] [PubMed] [Google Scholar]
- 40.Yu B, Reynisson J. Bond stability of the “undesirable” heteroatom-heteroatom molecular moieties for high-throughput screening libraries. Eur J Med Chem. 2011;46:5833–5837. doi: 10.1016/j.ejmech.2011.09.044. [DOI] [PubMed] [Google Scholar]
- 41.Nam W, et al. Effects of artemisinin and its derivatives on growth inhibition and apoptosis of oral cancer cells. Head Neck-J Sci Spec. 2007;29:335–340. doi: 10.1002/hed.20524. [DOI] [PubMed] [Google Scholar]
- 42.Ooko E, et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine. 2015;22:1045–1054. doi: 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Barnes-Seeman D, et al. Metabolically stable tert-butyl replacement. ACS Med Chem Lett. 2013;4:514–516. doi: 10.1021/ml400045j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang XF, et al. Spiro- and dispiro-1,2-dioxolanes: contribution of iron(II)-mediated one-electron vs two-electron reduction to the activity of antimalarial peroxides. J Med Chem. 2007;50:5840–5847. doi: 10.1021/jm0707673. [DOI] [PubMed] [Google Scholar]
- 45.Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Bio Med. 2003;34:496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 46.Hong Y, et al. The role of selenium-dependent and selenium-independent glutathione peroxidases in the formation of prostaglandin-F2-Alpha. J Biol Chem. 1989;264:13793–13800. [PubMed] [Google Scholar]
- 47.Dansen TB, Wirtz KWA, Wanders RJA, Pap EHW. Peroxisomes in human fibroblasts have a basic pH. Nat Cell Biol. 2000;2:51–53. doi: 10.1038/71375. [DOI] [PubMed] [Google Scholar]
- 48.Beasley DE, Koltz AM, Lambert JE, Fierer N, Dunn RR. The Evolution of Stomach Acidity and Its Relevance to the Human Microbiome. PLoS One. 2015;10:e0134116. doi: 10.1371/journal.pone.0134116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Creek DJ, Chiu FCK, Prankerd RJ, Charman SA, Charman WN. Kinetics of iron-mediated artemisinin degradation: effect of solvent composition and iron salt. J Pharm Sci-US. 2005;94:1820–1829. doi: 10.1002/jps.20400. [DOI] [PubMed] [Google Scholar]
- 50.Wu YK, Yue ZY, Wu YL. Interaction of Qinghaosu (artemisinin) with cysteine sulfhydryl mediated by traces of non-heme iron. Angew Chem Int Ed. 1999;38:2580–2582. doi: 10.1002/(sici)1521-3773(19990903)38:17<2580::aid-anie2580>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
Methods-only References
- 51.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 52.Boettcher C, Pries C, Vangent CM. A rapid and sensitive sub-micro phosphorus determination. Anal Chim Acta. 1961;24:203–204. [Google Scholar]
- 53.Tyurina YY, et al. A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem. 2014;6:542–552. doi: 10.1038/nchem.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shimada K, Hayano M, Pagano N, Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. 2016;23:225–235. doi: 10.1016/j.chembiol.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon request.