

Abstract
The bacterial chaperonins are highly sophisticated molecular nanomachines, controlled by the hydrolysis of ATP to dynamically trap and remove from the environment unstable protein molecules that are susceptible to denaturation and aggregation. Chaperonins also act to assist in the refolding of these unstable proteins, providing a means by which these proteins may return in active form to the complex environment of the cell. The Escherichia coli GroE chaperonin system is one of the largest protein supramolecular complexes known, whose quaternary structure is required for segregating aggregation-prone proteins. Over the course of more than two decades of research on GroE, it has become accepted that GroE, more specifically the GroEL subunit, is a “high-tolerance” molecular system, capable of accommodating numerous mutations, while retaining its molecular integrity. In some cases, a given site of mutation was revealed to be absolutely required for GroEL function, providing hints regarding the network of signals and triggers that propel this unique system. In other instances, however, a mutation has produced a more delicate response, altering only part of, or in some cases, only a single facet of, the molecular mechanism, and these mutants have often provided invaluable hints on the extent of the complexity underlying chaperonin-assisted protein folding. In this review, we highlight some examples of the latter type of GroEL mutants which compose the unique “mutational repertoire” of GroEL and touch upon the important clues that each mutant provided to the overall effort to elucidate the details of GroE action.
Keywords: Chaperonin GroEL, Molecular nanomachine, Versatile mutation
Introduction
The process by which proteins attain and retain their functional structure in the cellular environment is truly complex. Governed by the fundamental thermodynamic principles elucidated by Anfinsen, protein folding in the cell is highly sensitive to various changes in the cell and the environment surrounding it. The structural and functional integrity of cellular proteins is protected by a cohort of chaperone proteins, whose function is to sense and rescue various proteins that are rendered unstable by these numerous changes that occur in the cell (Hartl et al. 2011; Kim et al. 2013). Studies regarding the overall integrity of the cellular proteome, the “proteostasis” of a given cell, have revealed a multitude of important mechanisms, factors, and interactions that must proceed smoothly in order to assure the normal function of a typical cell.
Chaperonins are a crucial component of the proteomic system that maintains proteostasis in cells. Chaperonins are ubiquitous and found in all three branches of the tree of life, and examples of life forms that do not possess these molecular machines are extremely rare (Williams and Fares 2010). Members of the chaperonin family may be grouped into two groups, Group I and Group II (Kim et al. 1994; Yébenes et al. 2011). Group I chaperonins, which include most of the bacterial chaperonins and similar proteins from mitochondria and chloroplasts, are typically composed of a large double ring-like complex containing 14 homogeneous protein subunits of average Mr ~ 57 kDa (the Hsp60 subunit). This double ring is accompanied by a smaller accessory protein, Hsp10 (Mr ~ 10 kDa). Seven subunits of Hsp10 also form a ring-like structure that conditionally binds to one ring of Hsp60 to act as a “lid” on the circular cavity formed by the larger protein. Group II chaperonins, which include the archaeal and cytoplasmic chaperonins, are typically much more diverse in composition and structure, consisting of 2 to 8 highly similar but heterogeneous subunits that also associate to form a double ring-like quaternary structure. In the case of Group II chaperonins, however, a companion subunit such as Hsp10 is frequently missing. A unique helical structure, known as the “helical protrusion”, is found in the subunits of Group II chaperonins, and this structure is postulated to act as a substitute for Hsp10 in members of this group (Yébenes et al. 2011).
The GroE protein from Escherichia coli is the representative member of the Group I chaperonins (Georgopoulos et al. 1972; Takano and Kakefuda 1972), and much of the work done to understand the details regarding chaperonin-assisted folding have been performed using this protein (Fig. 1a). The basic mechanism of E. coli GroE in assisting the folding of proteins roughly consists of three distinct stages. In the first stage of the mechanism, the larger GroEL subunit (Mr 57 kDa) utilizes hydrophobic interactions to sense and bind to protein molecules that have lost their active, native structures due to various stresses (Elad et al. 2007; Farr et al. 2000). After recognition and binding, the mechanism moves on to the second stage, where the bound protein is guided into the capsule-like quaternary structure formed by the GroEL ring and the GroES (Mr 10 kDa) lid (Mayhew et al. 1996). This construct, the “central cavity”, segregates the protein molecule from solution and shields them from irreversible aggregation and precipitation. After a predetermined interval has passed, the mechanism moves on to the final stage of the cycle, where the lid of the central cavity is dissociated, and the protein molecule is released into the solution. The chaperonin is also primed during this final stage for further cycles of protein recognition, segregation, and assistance. The complete repetitive mechanism of GroEL and GroES-assisted protein folding is coordinated by the binding of ATP to the GroEL subunit and its subsequent hydrolysis, which act as triggers to initiate additional binding/dissociation events and propel the molecular mechanism forward.
Fig. 1.

The GroE chaperonin system from Escherichia coli. a (Left) The architecture of the GroE system. Fourteen subunits of GroEL form two heptameric rings (GroEL7) that are associated back to back. In the presence of nucleotides such as ATP, the cochaperonin GroES (GroES7) binds to GroEL to form the cis–GroEL–GroES complex depicted in the figure. Four subunits in the foreground of the GroEL 14-mer are colored by domain; the equatorial domain in light blue, the intermediate domain in red, and the apical domain in dark green. One subunit of GroES is colored magenta. (Right) The two GroEL subunits depicted show the two main conformations of GroEL (Closed and Open) that are observable in the crystal structure shown here (1SVT, Chaudhry et al. 2004). Crystal structures in this and subsequent figures were drawn using UCSF Chimera (Pettersen et al. 2004). b (Without Substrate Protein) The basic functional cycle of GroE. Binding and hydrolysis of ATP triggers various conformational changes that trigger GroES binding, formation of the encapsulating complex, and cycling. In the absence of unfolded polypeptide (the substrate protein), allosteric mechanisms cause the cycle to favor an asymmetric form where ATP binding and hydrolysis occur on alternating GroEL heptamers. (With Substrate Protein) The presence of unfolded substrate proteins mainly affects two facets of the basic cycle outlined above; first, the substrate proteins bind multivalently to the apical domains of GroEL to apply a “load” on the molecule, which must be overcome by the conformational changes of GroEL, as well as the binding of GroES (“Substrate Protein Load”). Substrate binding also confers an additional effect where the release of hydrolyzed ADP and bound GroES is enhanced by the binding of unfolded peptide to the trans ring (“Enhanced Release”)
The subunit structure of GroEL is composed of three clearly defined domains; each of which provides a distinct functional role in the mechanism outlined above (Fig. 1a) (Braig et al. 1994). The equatorial domain, containing the inter-ring interface and the ATPase active site that times the overall functional cycle, the apical domain, containing the recognition site for denatured proteins and GroES, and the intermediate domain, which links these two domains together via two “hinge” sites that connect the equatorial/intermediate domains (Hinge 1) and the intermediate/apical domains (Hinge 2). When ATP binds to GroEL, these three domains are rearranged in a highly dynamic fashion, and these dynamic conformations comprise the structural basis of the chaperonin functional mechanism.
X-ray crystal structure analysis of the E. coli GroEL–GroES–ADP complex (Xu et al. 1997) revealed two major conformations of the larger GroEL subunit: the closed conformation formed in the absence of bound nucleotide and the open conformation, which was visualized in complex with GroES and formed as a result of ATP binding and hydrolysis (Fig. 1a). More detailed characterizations using both X-ray crystallography (Chaudhry et al. 2004) and cryoelectron microscopy (Clare et al. 2012; Ranson et al. 2006) has revealed a number of additional conformations that are formed in the course of completing the GroE functional cycle. Combined with the results from numerous functional studies, as well as detailed kinetic analyses of both the ATPase activity of GroEL and the transient conformational changes that are triggered in GroEL as a result of ATP binding and hydrolysis, these conformational characterizations provide an outline of the process through which GroEL and GroES facilitates the refolding of numerous unfolded polypeptides (Fig. 1b).
Cryoelectron microscopy of an ATP-hydrolysis deficient mutant of GroEL, D398A (Clare et al. 2012), revealed after binding ATP to its equatorial domain, the GroEL subunit forms a total of two distinct conformations (Rs1, RS2, RSopen) before forming the “open” conformation of GroEL with bound GroES (R-ES) that resembles structures visualized in X-ray crystallography studies (Chaudhry et al. 2004). In this conformation, the GroEL ring and the GroES lid forms a large chamber in which unfolded polypeptides may be enclosed and segregated from solution (the cis–GroEL–GroES complex). The inner surface of the chamber is lined with hydrophilic amino acid residues (Xu et al. 1997), and this characteristic is conducive to the structural recovery of the segregated peptide. All of the conformational changes within the GroEL subunit outlined above consist of domain-level movements arranged around the two hinges in the intermediate domain. These rearrangements of domains in GroEL, therefore, give the impression that a highly “mechanical” scheme underlies chaperonin function, and, so, this protein may very aptly be described as a “protein nanomachine”.
The relatively weak ATPase activity of the GroEL subunit (rate of turnover ~ 0.1 s−1; Viitanen et al. 1990) dictates that the cis–GroEL–GroES complex will persist for a duration of several (8 to 10) seconds (Rye et al. 1999). Conversion of all seven of the ATP molecules to ADP will result in a rearrangement that will first weaken the affinity of GroES toward this cis ring (Rye et al. 1997) and also prime the GroEL heptameric ring that is distal from the bound GroES (the trans) ring so that this ring now displays a tendency to bind unfolded proteins and ATP (Ranson et al. 2006). This “transfer” of activities between the two GroEL rings is made possible by a highly sophisticated, nested allosteric mechanism within the GroEL 14-mer (Yifrach and Horovitz 1995). ATP binding to the seven GroEL subunits in a given heptameric ring is controlled by a positively cooperative, concerted allosteric mechanism. This results in the preferential binding of seven ATP molecules to one of the two rings in the GroEL oligomer. Contrastingly, the two heptameric rings in GroEL are controlled by a sequential, negatively cooperative allosteric mechanism. This mechanism guides the GroEL oligomer toward an asymmetrical mechanism; in other words, there is an inherent tendency for only one of the two rings of GroEL to be active at any given time. As a result, the GroE system functions in a manner similar to a reciprocating engine, where the two rings alternately undergo cycles of ATP/GroES binding and nucleotide hydrolysis (Fig. 1b, Without Substrate Protein).
In the actual act of facilitating the folding of proteins, the presence of unfolded polypeptide confers a number of additional effects to this system (Fig. 1b, With Substrate Protein). First, the affinity of GroEL toward unfolded polypeptide is higher in GroEL heptamers that do not possess bound nucleotide (i.e., the apo GroEL 14-mer, or the trans GroEL heptamer in the above scheme) and binding of ATP and other nucleotides to GroEL weakens this affinity. Multiple GroEL subunits in the heptameric ring interact with the unfolded substrate protein during this process, and, so, binding results in a “load” to be applied to this ring which must be overcome by the concerted rearrangement of apical domains within this ring, and the binding of GroES (Motojima et al. 2004). When peptide binding occurs at the trans ring of a preformed GroEL–ADP–GroES complex, this results in a decrease in affinity toward the hydrolysis product ADP and the bound co-chaperonin GroES in the cis GroEL ring (Rye et al. 1999). This latter effect partially aids the functional cycling between the two GroEL rings.
In this manner, GroEL and GroES cooperate to identify, isolate, and prevent the irreversible aggregation of multiple proteins in the cell. The binding and subsequent hydrolysis of ATP sends signals through the modular domain architecture of the GroEL subunit, which is translated into conformational changes via several important structural foci to achieve the desired result. A much more detailed overview of the individual steps that govern chaperonin action has been the subject of several excellent review articles which also highlight the points of further discussion (Hayer-Hartl et al. 2016; Saibil et al. 2013), and the complex subject of allostery in GroEL has been described in great detail by Gruber and Horovitz (2016); readers are referred to these reviews for more details on this system.
Notable examples of the GroE mutational repertoire (Fig. 2)
Fig. 2.

Examples of GroEL mutants where the functional mechanism has been “reprogrammed”. Depicted at center is the cis–GroEL–GroES complex from 1SVT; arranged around this depiction are four panels which show the locations of various amino acid residues that were mutated to induce functional modifications to GroEL. The Inter-ring residues panel shows the four amino acid residues that are altered to form the single-ring GroEL mutant SR-1 in green and turquoise, and Arg13 and Ala126, which were altered to negate the negative cooperativity between GroEL rings, are shown in magenta and purple in their respective subunits. In the Asp398 panel, Helix M, which orients this residue toward the ATP binding site upon nucleotide binding, is depicted in green for the closed form and yellow in the open form. Asp398 is depicted in ball and stick form. This panel was generated by fitting the equatorial domains of the open and closed forms of the GroEL subunits in PDBID 1SVT using the MatchMaker tool in UCSF Chimera (Pettersen et al. 2004). The Intra-ring residues panel shows the location of Arg197 (orange), Glu386 (yellow), Asp155 (blue), and Arg395 (light blue). The Hinges panel shows the two residues that were modified to confer temperature sensitivity (Cys138, cyan) and substrate binding affinity (Gly192, dark blue)
Since the elucidation of the first X-ray crystal structure of E. coli GroEL 14-mer by Braig et al. (1994), many research groups have constructed various mutants of GroEL in order to map the relationship between the subunit structure of GroEL and its functional mechanism. A very comprehensive initial characterization of the GroE architecture was performed by Fenton et al. (1994) at the time the above crystal structure was reported, and, subsequently, many other research groups followed with their own efforts. As new findings continued to be reported, the general consensus was that the modular architecture of GroE proved to be especially conducive to analysis through mutation (in other words, mutations in GroEL caused extensive aggregation and misfolding of GroEL only in very rare cases, and recovery of mutants in soluble, analyzable form was the norm rather than the exception). In many cases, mutation of a single residue completely abolished the chaperonin activity of GroE (as characterized by an inability to support E. coli growth in the absence of wild-type GroEL). These mutants could invariably be correlated with an essential function of the GroEL mechanism; for example, the mutation of Asp 87 to Lys (abolishes ATPase activity) or Tyr 199 to Glu (abolishes recognition and binding of unfolded polypeptide substrate) (Fenton et al. 1994). In other cases, however, the results were more ambiguous, resulting in qualitative changes in the ATPase, peptide binding, and cumulative folding assistance abilities of GroEL. The availability of the X-ray crystal structure of GroE allowed researchers to target localized areas of the GroE architecture with greater and greater precision, and, in certain cases, resulted in changes to the GroE functional mechanism that may be termed as a “reprogramming” of the original functional mechanism.
Below, we give some examples of GroEL mutants which display a qualitative change in the chaperonin mechanism (a “reprogramming”), to highlight the contributions that each mutant has provided in our efforts to understand the details regarding GroE-facilitated protein folding, and also to put forth an idea regarding the versatility and applicability of this system in various fields in addition to protein folding. We hasten to add that the list below is hardly extensive; we apologize in advance for the omission of numerous important mutations that contributed greatly to our understanding of GroE-facilitated protein folding.
Quaternary structure and function
GroEL SR-1 (Weissman et al. 1995, 1996)
GroEL SR-1 is a single-ring variant of GroEL which was created in order to determine details regarding the fate of polypeptide that undergoes a single binding–release cycle into the GroEL central cavity. Four amino acid residues located at the base of the GroEL equatorial domain (Arg452, Glu461, Ser463, and Val464) were all replaced by alanine residues in order to create GroEL SR-1. These amino acid replacements disrupt the major interface that allows contacts to be formed between the two rings of GroEL, and, true to design, GroEL SR-1 was isolated as a single-ring version of the original GroEL oligomer. Importantly, it was found that GroEL SR-1 retained most of the functional abilities of GroEL, specifically ATP binding/hydrolysis (albeit with a reduced rate), and substrate protein binding and encapsulation (in concert with GroES).
The absence of the second ring in GroEL SR-1 induced many subtle changes to its functional characteristics, and these changes proved to be vital hints toward understanding the importance of inter-ring communication in the chaperonin system (Rye et al. 1997). Proteins bound to SR-1 are encapsulated within the central chamber upon addition of GroES and ATP; however, the chaperonin cycle is arrested at this point, and the protein is entrapped within this complex until artificial measures, such as a reduction in experimental temperature to 4 °C, is applied (Rye et al. 1997). The reason for this arrest is the absence of inter-ring signals from the missing ring, which, in the native GroEL oligomer, is triggered by the binding of unfolded proteins and/or ATP to this second ring. In addition to being a useful trapping molecule to rapidly remove GroES from experiments (Weissman et al. 1995), SR-1 was used to demonstrate the folding of proteins that were trapped inside the closed central cavity (Rye et al. 1997; Weissman et al. 1996) and in determining the importance of ATP binding, as compared to ATP hydrolysis, in assisting the folding of denatured polypeptide.
D398A (Rye et al. 1997)
In 1997, a refined crystal structure of the asymmetric GroEL–GroES–ADP complex (Xu et al. 1997), presumed to be the predominant form of GroE in E. coli cells, was determined. The structure revealed that Asp398 was a key amino acid residue in orienting the ATP molecule during hydrolysis, specifically through interactions with the coordinating Mg2+ ion to orient a water molecule to the γ-phosphate of ATP for subsequent use in hydrolysis. Asp398 also plays a role in orienting a long helical element (Helix M) toward the ATP binding site after this nucleotide is bound to the equatorial domain in the open GroEL conformation. When this residue was altered to an alanine, the resultant mutant could hydrolyze ATP at only 2% of the rate seen in the wild-type chaperonin (Rye et al. 1997). After further analysis, it was found that, although GroEL D398A indeed displays a greatly reduced ATPase activity, the other functional components of the GroE mechanism, such as the ability to bind GroES, the ability to encapsulate unfolded protein within the GroE central chamber, and the ability to release this trapped protein molecule into the chamber to allow refolding, were all conserved. GroEL D398A offered a unique opportunity for researchers to observe in detail a “cycle-arrested form” of GroEL, where refolding assistance reactions could be initiated, but would be stopped at a predetermined state, the asymmetric GroEL–GroES–ADP form containing the unfolded protein molecule within the cis encapsulated complex.
This arrested form of the chaperonin complex was incredibly useful in characterizing the details of functional transfer between the cis and trans rings during the chaperonin cycle, and also in determining the differences in substrate protein and nucleotide binding affinities between the two rings in this complex. Presently, this mutant is also vitally involved in clarifying an important question regarding the GroE functional mechanism: whether the chaperonin functions strictly as an “alternating” asymmetrical nanomachine (Hayer-Hartl et al. 2016) or is the folding assistance capacity of GroE expanded, under certain conditions of high client protein load, through the simultaneous use of both refolding chambers (Iizuka and Funatsu 2016; Yamamoto and Ando 2016).
Allosteric regulation and function
R197A, D155A
As outlined above, the complex molecular mechanism of GroE is supported by an intricate nested allosteric mechanism that coordinates intersubunit communications within the GroEL heptameric ring, and between the two rings of the GroEL 14-mer (Yifrach and Horovitz 1995). Intra-ring communication between GroEL subunits displays positive cooperativity, and is explained according to the Monod–Wyman–Changeux mechanism of concerted allostericity. The subunits, therefore, coordinate their molecular rearrangements so that the seven apical domains of the GroEL heptameric ring may coordinate their movements to dislodge the bound unfolded polypeptide prior to encapsulation.
Yifrach and Horovitz (1994) constructed a mutation, R197A, which displayed a great reduction in both the positive intra-ring cooperativity and the negative inter-ring cooperativity in the GroEL 14-mer, highlighting the importance of the R197–E386 salt bridge in the overall allosteric architecture of GroE. Subsequently, the importance of this residue was highlighted by the identification of two salt bridges that are formed between adjacent subunits in the GroEL ring (the first between Glu255 and Lys207, and the second between Arg197 and Glu386) (Clare et al. 2012; Piggot et al. 2012; Xu et al. 1997). Binding of ATP to the GroEL subunit disrupts these salt bridges, and the rearrangement of the domains to the GroEL open form causes new salt bridges to be created, between Glu255 and Lys245, and between Lys80 and Glu386 (Clare et al. 2012).
Taking these results one step further, Danziger et al. (2003) focused on another residue, Asp155, which was revealed in crystal structures to form an intrasubunit salt bridge with Arg395, which is, in turn, linked to Glu386 via the long helix motif Helix M. Strikingly, when Asp155 was replaced with an alanine, the resultant mutation caused a reprogramming of the intra-ring allosteric mechanism of GroEL, from a concerted mechanism to a sequential form (Danziger et al. 2003). This change in the allosteric mechanism was directly visualized in transition electron microscopy experiments, which showed that the apical domains of D155A displayed a break in symmetry in the orientation around the GroEL ring. This finding demonstrated the degree of precision that may be attained in manipulating the architecture of GroEL in a targeted fashion.
R13G/A126V
Separate experiments were also performed to analyze the inter-ring negative cooperativity of GroEL. The two rings of the GroEL 14-mer associate through interactions between multiple amino acid residues arranged at the base of the equatorial domain GroEL base. In addition to the four amino acids outlined above that were mutated in order to produce the single-ring SR-1 mutant, two amino acid residues, Arg13 and Ala126, were highlighted as potential candidates to disrupt the inter-ring communication network of GroEL. These two residues were targeted through an unconventional coincidence; both Arg13 and Ala126 were discovered in mutated from in the original crystal structure determined by Braig et al. (1994). Aharoni and Horovitz (1996) reasoned that these two mutations acted to reduce the number of attainable conformations in GroEL during crystallization, thereby facilitating crystal production and analysis. Their surmise turned out to be correct; the R13G/A126V double mutant was characterized by a selective disruption of the inter-ring negative cooperativity that coordinates the alternating chaperonin cycle in wild-type GroEL, while leaving the positive intra-ring mechanism intact (Aharoni and Horovitz 1996). The degree of selectivity in these mutations again demonstrates the precision with which researchers may manipulate the molecular architecture of GroEL to obtain a desired result.
Modular domain structure and function
C138W
Of the two hinge regions, the link between the three domains of GroEL, Hinge 1, which connects the equatorial domain with the intermediate domain, is situated in the vicinity of the ATP binding site and located next to the machinery that controls conformational rearrangements and allosteric transitions.
Taking into account previous chemical modification experiments that highlighted the importance of this region in interdomain communication (Martin 1998), we replaced a cysteine residue in Hinge 1, Cys138, with a larger, fluorescent tryptophan side chain. The resultant mutant, GroEL C138W, was, at first glance, indistinguishable from the wild type, with an ATPase activity comparable to wild-type GroEL that was sensitive to GroES binding (Kawata et al. 1999). However, this mutant was completely unable to assist in the folding of proteins such as rhodanese when we probed this ability under conditions that were routinely used (specifically, at 25 °C). A curious disappearance of the heretofore normal ATPase activity under these same conditions led us to probe further, where we found that the C138W mutant converted the chaperonin into a temperature-sensitive variant. At 37 °C, C138W was indistinguishable in all aspects from the wild type; however, at 25 °C, steric hindrances caused the chaperonin to be arrested in a “ternary complex” form, with both unfolded protein and GroES bound to the same ring of GroEL C138W (Kawata et al. 1999).
Upon further experimentation, the nature of the arrested C138W complex became clearer, and we concluded that this form was arrested at a stage of the cycle immediately after ATP hydrolysis had been completed, but prior to the release of bound protein into the central cavity, which would allow the folding of trapped polypeptide (Miyazaki et al. 2002). Characterization of this complex allowed us to propose a specific sequence of events that occur after binding of ATP to the target peptide–GroEL complex: after ATP is bound, GroES binds to the GroEL ring, and only after this event is the release of polypeptide from the apical domains initiated. Most interestingly, the nature of this arrested complex was reversible; simple increases in temperature to > 30 °C was sufficient to restart both target protein refolding and ATPase activity in GroEL C138W. This final characteristic argued strongly for the explanation that the arrested complex detected in GroEL C138W was not a side reaction that diverted GroEL from the main mechanistic pathway, but was, in fact, an “on-pathway” intermediate that provided vital hints to understanding the chaperonin mechanism. The ability to introduce a reversible, temperature-sensitive switch into GroEL also demonstrated again the degree of control that could potentially be incorporated into this system.
G192W
The second hinge region in GroEL, Hinge 2, was also an interesting target to probe regarding the extensive rearrangement of the apical domain during GroE function, and the potentially high dynamism of this site was inferred by the presence of no less than three glycine residues (Gly192, 374, and 375) situated in close proximity in this region. Each of these glycines was replaced by tryptophans, and, among them, Gly 192 proved to be an interesting site for further analysis due to the finding that inserting a bulky indole side chain into this site caused a tilting of the apical domain from its original position in the closed GroEL form (Machida et al. 2009). This change in apical domain tilt resulted in a number of interesting consequences for the structure and function of GroEL. G192W is capable of binding to GroES in the absence of ATP binding. Presumably, this novel ability of ATP-less GroES interaction was due to a change in the orientation of the apical domain that exposed the GroES binding site to solution. When ATP was added to GroEL G192W, experiments indicated that the double-ring structure was retained. Interestingly, the two rings of GroEL G192W could bind both unfolded protein or GroES, but not both at the same time, as was the case for the arrested complex in GroEL C138W. In other words, the addition of ATP and GroES to GroEL G192W simultaneously caused the forcible formation of a trans ternary complex, where GroES and refolding substrate protein were bound to opposite rings of the GroEL G192W tetradecamer.
Upon further study, it was found that mutation of Gly192 might result in a degree of GroEL manipulation that was much more precise than previously expected. Very recently, we constructed a series of substitution mutants of Gly192 in order to probe the possible utility of GroEL in controlling irreversible aggregation and fibril formation of various proteins implicated in molecular diseases (Fukui et al. 2016). Our findings indicated that, by selecting the amino acid to replace Gly192 according to the van der Waals volume of the substituting side chain, it was possible to change the orientation of the GroEL apical domain to a surprising degree of precision, and, in the process, modulate the affinity of this domain (and the GroEL oligomer) toward proteins such as α-synuclein. This “adjustable” affinity of the G192X series of mutants resulted in significant differences in both the ability of GroEL to suppress protein fibrillogenesis and, also, to alter the morphology of fibrils that are produced in the presence of chaperonin to a certain degree.
Radical alterations and expansions to the chaperonin repertoire (Fig. 3)
Fig. 3.
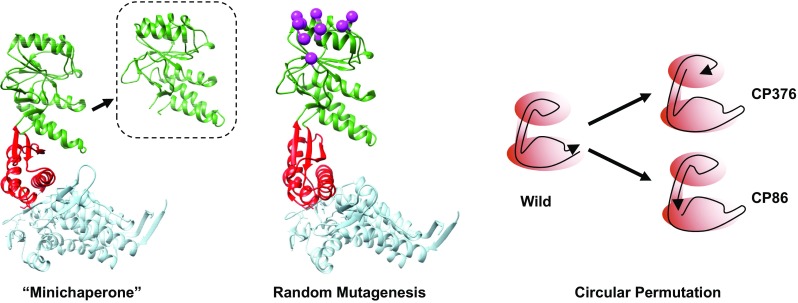
Unconventional modifications to the GroEL architecture. Left the “minichaperones”, isolated domains of GroEL that retain minimal chaperonin function; center random mutagenesis of selected apical domain residues (magenta) performed by Kawe and Plückthun (2006); right circular permutation to perturb the polypeptide backbone of GroEL
Minichaperones
An interesting characteristic of the GroEL subunit architecture was that the N- and C- termini of the polypeptide backbone are localized in roughly the same area of the subunit: at the base of the equatorial domain, inside the heptameric ring (Braig et al. 1994). The polypeptide backbone of GroEL starts at this position, proceeds to the apical domain, and returns to the equatorial domain, having transited through this domain and the intermediate domain twice (Fig. 1a, right). The apical domain, unlike the other two domains in GroEL, is, therefore, a self-contained domain. Zahn and coworkers (1996) focused on this characteristic of the GroEL apical domain and developed the concept of “minichaperones”, a soluble polypeptide derived from the GroEL apical domain that displayed a minimal mechanism of protein aggregate control and solubilization. These minichaperones bound to unfolded proteins, required no nucleotides to function, and were found to be active both in vitro and in vivo (Chatellier et al. 1998). Recently, we ourselves have found that the minichaperones are also effective in controlling the irreversible fibrillation of proteins such as α-synuclein and Alzheimer beta peptide 1-42 (Ojha et al. 2016). The relatively small size of this protein derivative makes the minichaperones an attractive tool in developing practical protein solubilization and anti-aggregation reagents, and some attempts to utilize them (Altamirano et al. 1997, 1999; Sharapova et al. 2016) and improve them for this role (Wang et al. 2000) have been reported.
Random mutagenesis
Directed evolution using in vitro random mutagenesis is a powerful technique to probe the evolutionary potential of a given protein system. The E. coli GroE system has undergone two major efforts in this area; the first project involved using in vitro evolution to optimize the GroE system for the folding of green fluorescent protein (Wang et al. 2002) and the second effort localized the randomization to selected amino acid residues in the apical domain of GroEL in an attempt to modify the target specificity of the GroE mechanism (Kawe and Plückthun 2006). In the case of the GFP-optimizing experiments performed by Wang et al. (2002), the results of directed evolution were modest; GFP-optimized derivatives of GroE produced a modest increase in GFP refolding ability of up to 8-fold. However, this increase was accompanied by a correlating decrease in the efficiency toward other refolding proteins. It seemed that the mechanism of wild-type GroE was already strongly evolved toward a “general protein assistance chaperone”, and an unbiased effort at improving this system toward a certain protein target was, in effect, specializing this system toward that target at the expense of other target proteins. In the more localized survey performed by Kawe and Plückthun (2006), modifying selected amino acid residues in the GroEL apical domain that were implicated in substrate binding capability proved to be not as effective in obtaining an “improved” chaperonin. One major reason for this was that the binding region for unfolded polypeptide in the GroEL apical domain overlaps the binding site for the co-chaperonin GroES. In the cell, proteins that strictly require the assistance of GroE in order to attain its native structure invariably requires the participation of GroES during the functional cycle, and, so, any modifications to this site in directed evolution studies result in an immediate effect on co-chaperonin binding, thereby weakening the overall effect of GroE. The results of these two major efforts in GroE directed evolution highlighted the degree to which the GroE system was already tuned to support the folding of as many proteins as possible, by utilizing a highly complex, multistage protein nanomachine.
Circular permutation mutants
The fact that the polypeptide termini of GroEL are situated in close steric proximity allowed us to probe another method to perturb the subunit structure of GroEL, through circular permutation (Heinemann and Hahn 1995). By changing the location of the peptide termini in the GroEL subunit structure, it would be possible to perturb the domain linkage of GroEL by a method other than simple amino acid replacement. With this in mind, we constructed a random circular perturbation library of GroEL mutants with the polypeptide termini located at different locations of the subunit (Mizobata et al. 2011).
Two circularly permuted variants of GroEL, CP376 and CP86, were of particular interest in analyzing the domain structure of GroEL in relation to its functional mechanism. In GroEL CP376, the polypeptide termini were shifted to the Hinge 2 region, in effect cutting off one of the two polypeptide backbones that linked the apical and equatorial domains together (Mizobata et al. 2011). Experiments showed that, in this mutant, domain rearrangements that involved the apical domain were strongly affected. In particular, the large tilting of the apical domain in response to ATP binding, proposed to be the “power stroke” of the GroEL subunit that was responsible for dislodging unfolded polypeptide from this domain in order to initiate protein encapsulation, was selectively abolished. It should be noted, however, that, despite this radical departure from the original GroEL subunit architecture, GroEL CP376 was, nevertheless, capable of assisting the folding of proteins to a certain extent, demonstrating the tenacity of this protein in assisting the folding of unstable polypeptides.
The second circularly permuted mutant, CP86, has its polypeptide termini relocated to a site immediately adjacent to the ATP binding site in the equatorial domain (Mizuta et al. 2013). This mutant was characterized initially by a “reprogramming” of the functional mechanism of GroE in the form of a recalibrated substrate specificity; CP86 was capable of assisting the folding of malate dehydrogenase in vitro to the same extent as wild-type GroEL, while completely losing the ability to assist the folding of bovine rhodanese. What was more interesting, however, was that we were able to engineer an additional molecular circuit to this mutant in the form of a disulfide bond that linked the two polypeptide termini under oxidizing conditions. The engineering of a reversible redox switch into the GroEL architecture was an interesting feat that demonstrated the potential versatility of this molecule in the protein engineering field.
Final remarks
Although the structure of E. coli GroEL has proved to be a highly complex protein nanomachine system capable of assisting the folding of many proteins indiscriminately, the system has also been demonstrated to be robust and capable of accommodating many changes to its architecture to produce a variety of targeted modifications. Further studies will undoubtedly reveal even more intriguing details on the structure and function of the chaperonins, as well as provide opportunities to utilize the unique capabilities of this nanomachine.
Funding
Portions of the manuscript performed by the authors was funded by a Grant-in-Aid for Scientific Research (C) (no. 22570119 to T. M.) from the Japan Society for the Promotion of Science (JSPS) and by the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED).
Compliance with ethical standards
Conflict of interest
Tomohiro Mizobata declares that he has no conflict of interest. Yasushi Kawata declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines - Fumio Arisaka 70th Birthday’ edited by Damien Hall, Junichi Takagi and Haruki Nakamura.
Contributor Information
Tomohiro Mizobata, Email: mizobata@bio.tottori-u.ac.jp.
Yasushi Kawata, Email: kawata@bio.tottori-u.ac.jp.
References
- Aharoni A, Horovitz A. Inter-ring communication is disrupted in the GroEL mutant Arg13 → Gly; Ala126 → Val with known crystal structure. J Mol Biol. 1996;258:732–735. doi: 10.1006/jmbi.1996.0282. [DOI] [PubMed] [Google Scholar]
- Altamirano MM, Golbik R, Zahn R, Buckle AM, Fersht AR. Refolding chromatography with immobilized mini-chaperones. Proc Natl Acad Sci U S A. 1997;94:3576–3578. doi: 10.1073/pnas.94.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamirano MM, García C, Possani LD, Fersht AR. Oxidative refolding chromatography: folding of the scorpion toxin Cn5. Nat Biotechnol. 1999;17:187–191. doi: 10.1038/6192. [DOI] [PubMed] [Google Scholar]
- Braig K, Otwinowski Z, Hegde R, Boisvert DC, Joachimiak A, Horwich AL, Sigler PB. The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature. 1994;371:578–586. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- Chatellier J, Hill F, Lund PA, Fersht AR. In vivo activities of GroEL minichaperones. Proc Natl Acad Sci U S A. 1998;95:9861–9866. doi: 10.1073/pnas.95.17.9861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry C, Horwich AL, Brunger AT, Adams PD. Exploring the structural dynamics of the E. coli chaperonin GroEL using translation–libration–screw crystallographic refinement of intermediate states. J Mol Biol. 2004;342:229–245. doi: 10.1016/j.jmb.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Clare DK, Vasishtan D, Stagg S, Quispe J, Farr GW, Topf M, Horwich AL, Saibil HR. ATP-triggered conformational changes delineate substrate-binding and -folding mechanics of the GroEL chaperonin. Cell. 2012;149:113–123. doi: 10.1016/j.cell.2012.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danziger O, Rivenzon-Segal D, Wolf SG, Horovitz A. Conversion of the allosteric transition of GroEL from concerted to sequential by the single mutation Asp-155 → Ala. Proc Natl Acad Sci U S A. 2003;100:13797–13802. doi: 10.1073/pnas.2333925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elad N, Farr GW, Clare DK, Orlova EV, Horwich AL, Saibil HR. Topologies of a substrate protein bound to the chaperonin GroEL. Mol Cell. 2007;26:415–426. doi: 10.1016/j.molcel.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farr GW, Furtak K, Rowland MB, Ranson NA, Saibil HR, Kirchhausen T, Horwich AL. Multivalent binding of nonnative substrate proteins by the chaperonin GroEL. Cell. 2000;100:561–573. doi: 10.1016/S0092-8674(00)80692-3. [DOI] [PubMed] [Google Scholar]
- Fenton WA, Kashi Y, Furtak K, Horwich AL. Residues in chaperonin GroEL required for polypeptide binding and release. Nature. 1994;371:614–619. doi: 10.1038/371614a0. [DOI] [PubMed] [Google Scholar]
- Fukui N, Araki K, Hongo K, Mizobata T, Kawata Y. Modulating the effects of the bacterial chaperonin GroEL on fibrillogenic polypeptides through modification of domain hinge architecture. J Biol Chem. 2016;291:25217–25226. doi: 10.1074/jbc.M116.751925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgopoulos CP, Hendrix RW, Kaiser AD, Wood WB. Role of the host cell in bacteriophage morphogenesis: effects of a bacterial mutation on T4 head assembly. Nat New Biol. 1972;239:38–41. doi: 10.1038/newbio239038a0. [DOI] [PubMed] [Google Scholar]
- Gruber R, Horovitz A. Allosteric mechanisms in chaperonin machines. Chem Rev. 2016;116:6588–6606. doi: 10.1021/acs.chemrev.5b00556. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- Hayer-Hartl M, Bracher A, Hartl FU. The GroEL–GroES chaperonin machine: a nano-cage for protein folding. Trends Biochem Sci. 2016;41:62–76. doi: 10.1016/j.tibs.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Hahn M. Circular permutation of polypeptide chains: implications for protein folding and stability. Prog Biophys Mol Biol. 1995;64:121–143. doi: 10.1016/0079-6107(95)00013-5. [DOI] [PubMed] [Google Scholar]
- Iizuka R, Funatsu T. Chaperonin GroEL uses asymmetric and symmetric reaction cycles in response to the concentration of non-native substrate proteins. Biophys Physicobiol. 2016;13:63–69. doi: 10.2142/biophysico.13.0_63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawata Y, Kawagoe M, Hongo K, Miyazaki T, Higurashi T, Mizobata T, Nagai J. Functional communications between the apical and equatorial domains of GroEL through the intermediate domain. Biochemistry. 1999;38:15731–15740. doi: 10.1021/bi9909750. [DOI] [PubMed] [Google Scholar]
- Kawe M, Plückthun A. GroEL walks the fine line: the subtle balance of substrate and co-chaperonin binding by GroEL. A combinatorial investigation by design, selection and screening. J Mol Biol. 2006;357:411–426. doi: 10.1016/j.jmb.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Kim S, Willison KR, Horwich AL. Cystosolic chaperonin subunits have a conserved ATPase domain but diverged polypeptide-binding domains. Trends Biochem Sci. 1994;19:543–548. doi: 10.1016/0968-0004(94)90058-2. [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Machida K, Fujiwara R, Tanaka T, Sakane I, Hongo K, Mizobata T, Kawata Y. Gly192 at hinge 2 site in the chaperonin GroEL plays a pivotal role in the dynamic apical domain movement that leads to GroES binding and efficient encapsulation of substrate proteins. Biochim Biophys Acta. 2009;1794:1344–1354. doi: 10.1016/j.bbapap.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Martin J. Role of the GroEL chaperonin intermediate domain in coupling ATP hydrolysis to polypeptide release. J Biol Chem. 1998;273:7351–7357. doi: 10.1074/jbc.273.13.7351. [DOI] [PubMed] [Google Scholar]
- Mayhew M, da Silva AC, Martin J, Erdjument-Bromage H, Tempst P, Hartl FU. Protein folding in the central cavity of the GroEL–GroES chaperonin complex. Nature. 1996;379:420–426. doi: 10.1038/379420a0. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Yoshimi T, Furutsu Y, Hongo K, Mizobata T, Kanemori M, Kawata Y. GroEL–substrate–GroES ternary complexes are an important transient intermediate of the chaperonin cycle. J Biol Chem. 2002;277:50621–50628. doi: 10.1074/jbc.M209183200. [DOI] [PubMed] [Google Scholar]
- Mizobata T, Uemura T, Isaji K, Hirayama T, Hongo K, Kawata Y. Probing the functional mechanism of Escherichia coli GroEL using circular permutation. PLoS One. 2011;6:e26462. doi: 10.1371/journal.pone.0026462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuta T, Ando K, Uemura T, Kawata Y, Mizobata T. Probing the dynamic process of encapsulation in Escherichia coli GroEL. PLoS One. 2013;8:e78135. doi: 10.1371/journal.pone.0078135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motojima F, Chaudhry C, Fenton WA, Farr GW, Horwich AL. Substrate polypeptide presents a load on the apical domains of the chaperonin GroEL. Proc Natl Acad Sci U S A. 2004;101:15005–15012. doi: 10.1073/pnas.0406132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojha B, Fukui N, Hongo K, Mizobata T, Kawata Y. Suppression of amyloid fibrils using the GroEL apical domain. Sci Rep. 2016;6:31041. doi: 10.1038/srep31041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Piggot TJ, Sessions RB, Burston SG. Toward a detailed description of the pathways of allosteric communication in the GroEL chaperonin through atomistic simulation. Biochemistry. 2012;51:1707–1718. doi: 10.1021/bi201237a. [DOI] [PubMed] [Google Scholar]
- Ranson NA, Clare DK, Farr GW, Houldershaw D, Horwich AL, Saibil HR. Allosteric signaling of ATP hydrolysis in GroEL–GroES complexes. Nat Struct Mol Biol. 2006;13:147–152. doi: 10.1038/nsmb1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rye HS, Burston SG, Fenton WA, Beechem JM, Xu Z, Sigler PB, Horwich AL. Distinct actions of cis and trans ATP within the double ring of the chaperonin GroEL. Nature. 1997;388:792–798. doi: 10.1038/42047. [DOI] [PubMed] [Google Scholar]
- Rye HS, Roseman AM, Chen S, Furtak K, Fenton WA, Saibil HR, Horwich AL. GroEL–GroES cycling: ATP and nonnative polypeptide direct alternation of folding-active rings. Cell. 1999;97:325–338. doi: 10.1016/S0092-8674(00)80742-4. [DOI] [PubMed] [Google Scholar]
- Saibil HR, Fenton WA, Clare DK, Horwich AL. Structure and allostery of the chaperonin GroEL. J Mol Biol. 2013;425:1476–1487. doi: 10.1016/j.jmb.2012.11.028. [DOI] [PubMed] [Google Scholar]
- Sharapova OA, Yurkova MS, Fedorov AN. A minichaperone-based fusion system for producing insoluble proteins in soluble stable forms. Protein Eng Des Sel. 2016;29:57–64. doi: 10.1093/protein/gzv060. [DOI] [PubMed] [Google Scholar]
- Takano T, Kakefuda T. Involvement of a bacterial factor in morphogenesis of bacteriophage capsid. Nat New Biol. 1972;239:34–37. doi: 10.1038/newbio239034a0. [DOI] [PubMed] [Google Scholar]
- Viitanen PV, Lubben TH, Reed J, Goloubinoff P, O’Keefe DP, Lorimer GH. Chaperonin-facilitated refolding of ribulose bisphosphate carboxylase and ATP hydrolysis by chaperonin 60 (groEL) are potassium dependent. Biochemistry. 1990;29:5665–5671. doi: 10.1021/bi00476a003. [DOI] [PubMed] [Google Scholar]
- Wang Q, Buckle AM, Fersht AR. Stabilization of GroEL minichaperones by core and surface mutations. J Mol Biol. 2000;298:917–926. doi: 10.1006/jmbi.2000.3716. [DOI] [PubMed] [Google Scholar]
- Wang JD, Herman C, Tipton KA, Gross CA, Weissman JS. Directed evolution of substrate-optimized GroEL/S chaperonins. Cell. 2002;111:1027–1039. doi: 10.1016/S0092-8674(02)01198-4. [DOI] [PubMed] [Google Scholar]
- Weissman JS, Hohl CM, Kovalenko O, Kashi Y, Chen S, Braig K, Saibil HR, Fenton WA, Horwich AL (1995) Mechanism of GroEL action: productive release of polypeptide from a sequestered position under GroES. Cell 83:577–587 [DOI] [PubMed]
- Weissman JS, Rye HS, Fenton WA, Beechem JM, Horwich AL. Characterization of the active intermediate of a GroEL–GroES-mediated protein folding reaction. Cell. 1996;84:481–490. doi: 10.1016/S0092-8674(00)81293-3. [DOI] [PubMed] [Google Scholar]
- Williams TA, Fares MA. The effect of chaperonin buffering on protein evolution. Genome Biol Evol. 2010;2:609–619. doi: 10.1093/gbe/evq045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Horwich AL, Sigler PB. The crystal structure of the asymmetric GroEL–GroES–(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- Yamamoto D, Ando T. Chaperonin GroEL–GroES functions as both alternating and non-alternating engines. J Mol Biol. 2016;428:3090–3101. doi: 10.1016/j.jmb.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Yébenes H, Mesa P, Muñoz IG, Montoya G, Valpuesta JM. Chaperonins: two rings for folding. Trends Biochem Sci. 2011;36:424–432. doi: 10.1016/j.tibs.2011.05.003. [DOI] [PubMed] [Google Scholar]
- Yifrach O, Horovitz A. Two lines of allosteric communication in the oligomeric chaperonin GroEL are revealed by the single mutation Arg196→Ala. J Mol Biol. 1994;243:397–401. doi: 10.1006/jmbi.1994.1667. [DOI] [PubMed] [Google Scholar]
- Yifrach O, Horovitz A. Nested cooperativity in the ATPase activity of the oligomeric chaperonin GroEL. Biochemistry. 1995;34:5303–5308. doi: 10.1021/bi00016a001. [DOI] [PubMed] [Google Scholar]
- Zahn R, Buckle AM, Perrett S, Johnson CM, Corrales FJ, Golbik R, Fersht AR. Chaperone activity and structure of monomeric polypeptide binding domains of GroEL. Proc Natl Acad Sci U S A. 1996;93:15024–15029. doi: 10.1073/pnas.93.26.15024. [DOI] [PMC free article] [PubMed] [Google Scholar]