

Abstract
The bacterial flagellum is a biological nanomachine for the locomotion of bacteria, and is seen in organisms like Salmonella and Escherichia coli. The flagellum consists of tens of thousands of protein molecules and more than 30 different kinds of proteins. The basal body of the flagellum contains a protein export apparatus and a rotary motor that is powered by ion motive force across the cytoplasmic membrane. The filament functions as a propeller whose helicity is controlled by the direction of the torque. The hook that connects the motor and filament acts as a universal joint, transmitting torque generated by the motor to different directions. This report describes the use of molecular dynamics to study the bacterial flagellum. Molecular dynamics simulation is a powerful method that permits the investigation, at atomic resolution, of the molecular mechanisms of biomolecular systems containing many proteins and solvent. When applied to the flagellum, these studies successfully unveiled the polymorphic supercoiling and transportation mechanism of the filament, the universal joint mechanism of the hook, the ion transfer mechanism of the motor stator, the flexible nature of the transport apparatus proteins, and activation of proteins involved in chemotaxis.
Keywords: Molecular dynamics, Polymorphic supercoiling, Universal joint, Protein export, Ion transport, Chemotaxis
Introduction
The bacterial flagellum, a biological nanomachine for the locomotion of bacteria, is found in both Gram-positive and -negative bacteria. The flagellum contains tens of thousands of protein molecules and more than 30 different kinds of proteins (Namba and Vonderviszt 1997). Bacteria, such as Escherichia coli and Salmonella, swim towards favorable conditions in their living environments by rotating the bacterial flagellar filaments. These microbes run straight by rotating the filaments for a few seconds in the counter-clockwise (CCW) direction as viewed from the outside of the cell (namely, swimming or run mode). Alternatively, in the tumble mode, the bacteria tumble their bodies upon the clockwise (CW) rotation of the filaments for ~ 0.1 s (Berg and Anderson 1973; Larsen et al. 1974b; Silverman and Simon 1974). In the run mode of E. coli and Salmonella, several flagellar filaments of a left-handed helical structure form a bundle and act as a screw, whereas in the tumble mode, as a transition of the filament structure into a right-handed helix is induced, the bundle is untangled, and the bacterium changes the direction of movement (Larsen et al. 1974b). The bacterial flagellum comprises the basal body (consisting of a rotary motor and export apparatus), the hook (which serves as a universal joint), and the filament (which serves as a propeller) (Fig. 1) (Berg 2003; Macnab 2003). The motor is powered by proton- or other ion-motive force across the cytoplasmic membrane, and the motor torque is transmitted to the filament through the hook. Bacterial flagella are paralogous to a protein export apparatus called a type III secretion system (T3SS) or type III injectisome (Aizawa 2001; Cornelis 2006). Interestingly, the archaeal flagellum, which is orthologous to a bacterial type IV pilus, is evolutionally different from the bacterial flagellum, although both flagella are used for swimming motility (Thomas et al. 2001; Ghosh and Albers 2011).
Fig. 1.

Overall structure of the flagellum of Salmonella typhimurium
The molecular structure of the bacterial flagellum has been investigated for many years. Atomic-resolution structures have been determined for many parts of the flagellum, including the filament, the hook, the rod, a part of the motor proteins, the soluble part of the export apparatus, and chemotactic proteins. However, the molecular mechanisms of bacterial flagella are still not yet fully understood. One reason for this is that it has been difficult to experimentally determine atomic-resolution structures for some segments of flagellar proteins, such as the motor proteins and membrane domains of the export apparatus proteins. Another reason is that the bacterial flagellum constitutes a highly flexible system, whose functional mechanism needs to be investigated from the point of view of dynamics.
Molecular dynamics (MD) simulation is a powerful computer simulation method that can be used to investigate the structure, dynamics, and function of proteins and other biomolecules, as well as their assembly. Many parts of the bacterial flagellum have been elucidated by MD, as shown below, although a whole flagellum is still too large for atomic-resolution MD. These MD results summarized here provide essential insights into the molecular mechanisms of the bacterial flagellar system.
Polymorphic supercoiling of the filament
The flagellar filament of Salmonella is a tubular supercoil structure that consists of subunits composed of a single protein, FliC or flagellin (Fig. 1). The filament grows up to 10 μm long, which contains 30,000 flagellin molecules. The structure of the filament can be described as stacked helical units, each consisting of 11 subunits, or as 11 circularly arranged protofilaments, and each filament forms nearly longitudinal helical arrays of subunits (O’Brien and Bennett 1972). The filament can be transformed into various distinct supercoil forms by changes in the chemical environment (Kamiya and Asakura 1976a, b; Hotani 1980), single amino acid mutations (Kamiya et al. 1982; Kanto et al. 1991), or mechanical forces (Macnab and Ornston 1977; Hotani 1982). The filament state in the run mode is called “normal” and the state in the tumble mode is either in “semicoil”, “curly I”, or “curly II” (Macnab and Ornston 1977; Turner et al. 2000). From a static point of view, the polymorphism of supercoils is reasonably well understood as a bi-stable protofilament model (Asakura 1970; Calladine 1975, 1976, 1978). In this model, it is assumed that subunit conformations are the same within each protofilament, and that protofilament conformation which includes the interactions with neighboring protofilaments is classified into two distinct forms, R-type and L-type. The assumed intrinsic bi-stable character of the protofilament and geometrical constraints to maintain a tubular structure result in discrete polymorphic states of the flagellar filament, as characterized by combination of the R- and L-type protofilaments, i.e., normal (2R/9L: 2 R-type and 9 L-type protofilaments), semicoil (4R/7L), curly I (5R/6L), and curly II (6R/5L) (Hasegawa et al. 1998; Yamashita et al. 1998). Although the bi-stable protofilament model gives a reasonable static and mechanical view of polymorphic supercoiling from a macroscopic point of view, the underlying molecular mechanisms should be understood based on atomic-resolution structures.
The A449V flagellin mutant from the SJW1655 strain of S. typhimurium forms a straight filament, whose protofilaments are all in the R-type. Its atomic structure was first solved for the F41 fragment (41 kDa) of flagellin by X-ray crystallography (Samatey et al. 2001) and for the whole R-type straight filament by electron cryoelectron microscopy (cryoEM) at 4 Å resolution (Yonekura et al. 2003). Later, the L-type straight filament structure (G426A mutant from the SJW1660 strain) was also determined by cryoEM at a similar resolution (Maki-Yonekura et al. 2010).
All-atom MD simulation of the filament in explicit solvent molecules was successful in investigating the molecular mechanism of polymorphic supercoiling (Kitao et al. 2006). Starting from the R-type straight filament structure consisting of 44 flagellin subunits, 2.4-million-atom MD simulations for 20 ns were conducted and the polymorphic supercoiling mechanism was investigated. In addition to the R-type straight filament, the wild-type (SJW1103) and G426A mutant (SJW1160) which form “normal (2R/9L)” and L-type straight (0R/11L) filaments in the native state, respectively, were also investigated. It was found that an MD simulation was successful in constructing the atomic-level supercoil structures consistent with various experimental data and, further, in elucidating the detailed underlying molecular mechanisms of the polymorphic supercoiling (Fig. 2). It was also identified that the following three types of interactions are identified as keys to understanding the supercoiling mechanism. “Permanent” interactions are always maintained between subunits in the various supercoil structures. This is consistent with the experimental result that filaments are severely destabilized if the first residues involved with permanent interactions in N-terminal regions (ASP42 and ASP43) are deleted (Vonderviszt et al. 1991). “Sliding” interactions are formed between variable hydrophilic or hydrophobic residue pairs, allowing intersubunit shear without large changes in energy. The formation of “switch” interactions (salt bridges and hydrogen bonds) stabilize intersubunit interactions, which drive the subunit to take the L-type conformation. In contrast, the breakage of switch interactions leads to the R-type conformation, which is similar to the monomer structure of flagellin. In other words, flagellin intrasubunit interactions prefer the R-type conformation. To construct the filament structure, part of the protofilament interface takes the L-type form and others take the R-type form. Since the L- and R-type interactions cannot be satisfied at the same time, the filament is energetically frustrated, which causes multiple energy minima. Since conformational changes among multiple energy minima can be caused by relatively low energy costs (Kitao and Takemura 2017), different types of perturbations, e.g., reversal of torque direction, pH and ionic strength change, and mutations, can easily induce conformational change. Therefore, polymorphic supercoiling is caused by the energy frustration between intra- and intersubunit interactions. This mechanism explains the phase diagram of the supercoil structure versus pH (Kamiya and Asakura 1976a). Switch interaction also explains the mutation D107E, which results in the L-type straight filament (Kamiya et al. 1982; Kanto et al. 1991). Since substitution of the aspartic acid (D) side chain with a longer one of glutamic acid (E) promotes the formation of salt bridges, D107E should prefer the L-type form. In contrast to the bi-stable protofilament model, the results of MD provide a microscopic view of supercoiling.
Fig. 2.

Polymorphic supercoiling of flagellar filament (Kitao et al. 2006). Simulated models of flagellar filaments and helical parameters of wild-type filaments in the curvature-twist diagram. a Initial structure for simulation. Snapshots of normal (b) and semicoil (c) during simulation. d Helical parameters of meta-stable structures of ten distinct molecular dynamics (MD) trajectories (last 150 ps). Red “+” connected by lines: parameters expected from the bi-stable protofilament model. Long filaments consisting of 10,680 subunits are also shown. e Structures of randomly generated long filaments. f Results of force-bias MD starting from right- towards left-handed supercoil. The magenta and yellow lines represent the results obtained for a torque of 5.0 and 2.0 × 102 pN nm, respectively. Density map: probability determined from 11 trajectories that reached near 2R/9L or 1R/10L. White circle: initial structure. Black circles: final structures. Black line: probable position of the energy barrier. White arrow: expected relaxation pathway. g same as f but in the opposite direction. The magenta and yellow lines represent the results for a torque of 4.0 and 2.0 × 102 pN nm, respectively. The density map was determined from 34 trajectories that reached near 4R/7L or 5R/6L. White triangle and square: transient structures experimentally observed (Hotani 1982). Reproduction from the original paper (Kitao et al. 2006). Copyright (2006) National Academy of Sciences
Kitao et al. (2006) also found that the transition between distinct supercoils is achieved by a “transform and relax” mechanism, in which the filament structure is geometrically transformed rapidly and then slowly relaxes to energetically meta-stable states by rearranging interactions (Fig. 2f, g). The supercoil transformations were observed in MD when a torque of 3.0 × 102 pN nm or greater was applied, which agrees with the experimental value (Hotani 1982).
To investigate the mechanism responsible for inducing polymorphic transitions in the protofilament arrangement between the run and tumble modes, coarse-grained MD simulations of a 0.5-μm-long flagellar filament were performed for tens of microseconds with varying torque and solvent conditions (Arkhipov et al. 2006). It is concluded that rearrangement of protofilaments and supercoiling do not arise without solvent, which shows that the behavior of the flagellum is determined not only by interprotein interactions, but also by hydrodynamic effects.
As seen in these studies, all-atom and coarse-grained MD simulations provided important insights for the supercoiling mechanism.
Universal joint mechanism of the hook
The flagellar hook forms the tubular supercoil structure that is made of 11 circularly arranged protofilaments, and each protofilament constructs nearly longitudinal helical arrays of subunits (Wagenknecht et al. 1982; Morgan et al. 1993; Shaikh et al. 2005). The hook is similar to the filament in this regard; however, it is much more flexible (Kato et al. 1984). The hook is a helical assembly of ~ 130 copies of a single hook protein, FlgE, which forms a short, highly curved tube (Fig. 1). The length of the hook is relatively well regulated to ~ 55 nm (Hirano et al. 1994), but it grows much longer if FliK or FlhB proteins, which are involved with protein export and flagellar assembly, are mutated (Patterson-Delafield et al. 1973; Kutsukake et al. 1994; Williams et al. 1996). This elongated hook, called a polyhook, clearly shows a right-handed helical structure with a helical pitch of about 0.1 μm. The polyhook can be reversely transformed into various helical forms, such as a left-handed helix, “coiled” with almost no helical pitch, and also straight form. The radius and pitch of the polyhook supercoils were measured under different pH and temperature conditions (Kato et al. 1984). The right-handed supercoil, called “normal”, which is stable at neutral pH and room temperature, has a diameter of 35 nm and a pitch of 95 nm. The atomic structure of a 31-kDa hook protein fragment of S. typhimurium (FlgE31), which contains D1 and D2 domains, has been determined by the X-ray crystallography (Samatey et al. 2004) and fitted to the density map of the straight hook reconstructed from 9-Å resolution images obtained by cryoEM (Shaikh et al. 2005). Fujii et al. (2009) determined a more complete atomic structure of hook of Salmonella enterica serovar typhimurium (S. enterica) by cryoEM image of 7.1 Å, except for the Dc domain situated between the D0 and D1 domains. Matsunami et al. (2016) recently determined a full atomic structure of Campylobacter jejuni hook, which includes extra D3 and D4 domains, at 3.5 Å resolution.
Samatey et al. (2004) performed MD simulation of the protofilament extension and compression along the protofilament axis (11-start direction) using hook dimer and trimer. The simulation was conducted by shifting the reference coordinates of domain D2 of the subunit at the top and domain D1 of the subunit at the bottom in opposite directions to compress by 5 Å or extend by 15 Å while applying positional restraints to the main-chain atoms of these domains. They found that hydrogen bonding interactions between amino acid residues of the triangular loop and the inner face of D2 show multiple steps of changes in bonding partners, which realizes a large sliding at this D1–D2 interface. This “sliding” was interpreted as a mechanism to enable low energy structural change, as the intersubunit hydrogen bonds can be rearranged between different residue pairs, as also seen the filament (Kitao et al. 2006). Certain flexibility in the relative domain orientation between D1 and D2 also plays a role in the changes in bonding partners.
Furuta et al. (2007) further investigated the intersubunit interactions in the flagellar hook structure using 44 subunits of FlgE31 by two-million-atom MD. Energetically stable models for the St-type (straight form), Lh-coil (left-handed supercoil), and Rh-coil (right-handed supercoil) were generated at atomic detail and intersubunit interactions were investigated. They showed that the intersubunit distance can be compressed or extended totally up to ~ 20 Å along the direction of the hook protofilament axis (11-start) (Fig. 3). On the innermost side of the supercoil where the intersubunit distance is minimized, subunit pairs along 11-start are highly packed and no gap is left for further compression, while on the outermost side, subunits are slightly separated and make indirect interactions through water molecules. Similar tendencies were observed in the intersubunit interactions between neighboring protofilaments. The hook supercoil structures of the two extreme cases, the Lh- and Rh-coils, make maximal use of the gaps between subunits. This mechanism is called the “gap compression/extension mechanism”. Two underlying mechanisms play important roles to allow such conformational change of the hook with relatively low energy costs. One mechanism is mutual subunit sliding on the intersubunit interface with accompanying rearrangements of intersubunit hydrogen bonds, as also seen in the filament. Another mechanism is relative domain motions between domains D1 and D2. Domain motions are widely recognized as a mechanism to allow large conformational change in many proteins (Qi et al. 2005; Nishima et al. 2009). The hook also adopts these general mechanisms to act as a universal joint.
Fig. 3.

Arrangements of neighboring three subunits along different helical lines in the inner, intermediate, and outer sides of two different supercoils (Furuta et al. 2007). a–c Lh-coil (left-handed supercoil) and d–f Rh-coil (right-handed supercoil). a, d Along the protofilament direction; b, e between neighboring protofilaments viewed from inside; c, f as b, e but viewed from outside. In each panel, three sets of trimers are superimposed by best-fitting the subunits in white. Red, green, and blue represent the inner, intermediate, and outer sides of the supercoils, respectively. The reference subunits numbered “0” are shown as the Cα ribbon model and the others by surface representation. Snapshots at 0.5 ns are shown. Reproduction from the original paper with permission (Furuta et al. 2007)
Structural change and ion transportation mechanisms of the motor stator
The flagellar motor utilizes the motive force of protons and other ions as an energy source (Larsen et al. 1974a; Hirota et al. 1981; Dean et al. 1984; Stader et al. 1986; Chun and Parkinson 1988; Khan et al. 1988; Terahara et al. 2012). The flagellar motor can rotate in both the CW and CCW directions, which regulates the swimming pattern of bacteria (Yamaguchi et al. 1986). Escherichia coli has several proton-driven flagellar motors. The FliG, FliM, FliN, MotA, and MotB proteins in E. coli are involved in torque generation (Enomoto 1966; Yamaguchi et al. 1986). Among them, FliG, FliM, and FliN constitute the flagellar rotor and are also involved in CW/CCW switching. Each rotor is typically surrounded by ten stators that comprise MotA and MotB (Fig. 4a).
Fig. 4.

Overall structure of MotAB (Nishihara and Kitao 2015). a Schematic view of the flagellar motors of Escherichia coli (left) and Vibrio alginolyticus (right), b Transmembrane (TM) regions modeled, and c TM helix arrangement in the initial modeling of MotAB in E. coli. The obtained atomic model structure viewed parallel to the membrane (d) and from the periplasmic side (e). Spheres: P atoms in the lipid head groups. MotAB cross-sections of the area enclosed by magenta in e around a channel at the levels of Leu46 (f) and Asp32 (g). Reproduction from the original work (Nishima et al. 2009)
This motor can be considered as multifuel engines that convert the motive force of distinct ions to molecular motor rotation. The stator complex of the flagellar motor is embedded in the bacterial inner membrane (Fig. 4b). The stators of E. coli, MotAB complex (Fig. 1), act as a proton channel (Larsen et al. 1974a; Dean et al. 1984; Stader et al. 1986; Chun and Parkinson 1988; Khan et al. 1988), but Vibrio alginolyticus has a polar flagellum powered by sodium ions (Hirota et al. 1981), which uses PomAB as the stator (Fig. 4a). The chimera protein study suggested that the transmembrane (TM) region of MotAB utilizes both protons and sodium ions as energy sources (Asai et al. 2003). In Bacillus alcalophilus, the stator consists of MotPS and can be driven by rubidium (Rb+), potassium (K+), and sodium ions (Na+) (Terahara et al. 2012). The MotPS stator can be converted to an Na+-driven motor by a single mutation.
The MotAB complex of E. coli consists of four MotA and two MotB proteins (Fig. 4b, c). Each MotA protein contains four TM alpha-helical segments (A1–A4), two short loops in the periplasm, and two long segments in the cytoplasm (Dean et al. 1984; Zhou et al. 1995). The MotB protein is composed of a short N-terminal cytoplasmic segment, one TM helix (B), and a large C-terminal periplasmic domain (Stader et al. 1986; Chun and Parkinson 1988). The B segment is expected to form a proton channel together with A3 and A4 (Braun et al. 2004). Since only a few polar residues have been identified in the predicted TM segments of MotAB (Sharp et al. 1995a), the channel surface is expected to be relatively hydrophobic. Asp32 of MotB is conserved across species and considered to be the most plausible proton binding site (Zhou et al. 1995).
The generation of torque is hypothesized to originate from conformational changes in the MotA cytoplasmic domain upon proton association/dissociation at the carboxyl group of Asp32 in MotB and by interaction with FliG in the rotor (Berry 1993; Elston and Oster 1997; Walz and Caplan 2000; Kojima and Blair 2001). The periplasmic region of MotB contains a peptidoglycan-binding motif that acts to anchor the stator complex to the peptidoglycan (PG) layer around the rotor (Chun and Parkinson 1988; De Mot and Vanderleyden 1994).
The atomic structure of the periplasmic domain was determined for MotB of Helicobacter pylori (Roujeinikova 2008; O’Neill et al. 2011; Reboul et al. 2011) and S. typhimurium (Kojima et al. 2009) and PomB of V. alginolyticus (Zhu et al. 2014). To understand the insertion mechanism of the anchor domain of MotB into the PG mesh, all-atom MD simulations of the MotB periplasmic domain from H. pylori were performed (Reboul et al. 2011). These simulations and crystallographic data provided evidence that three loops in the domain move in a concerted fashion, which unmasks the PG binding site.
Systematic mutagenesis studies of E. coli MotAB conducted by Blair and coworkers have provided essential information on the structure and function of the flagellar motor, although the atomic structure of the TM region of the MotAB complex has not been determined experimentally. Trp scanning mutagenesis of MotA (Sharp et al. 1995b) and MotB (Sharp et al. 1995a) showed the effects of mutations on relative swarming rates and identified the lipid-facing regions of the helices and their arrangements. Disulfide cross-linking analyses of MotB (Braun and Blair 2001) revealed the arrangement of the MotB dimer in the MotAB complex. Subsequent cross-linking studies of the A3 and A4 segments of MotA and the B segment of MotB (Braun et al. 2004) revealed the helical arrangement of the complex core. Cys mutagenesis of A1 and A2 (Kim et al. 2008) led to the determination of the overall arrangement of the MotAB complex.
The atomic structure of the TM region of the MotAB complex was constructed and the mechanism of proton permeation was investigated (Fig. 4d–g) (Nishihara and Kitao 2015) based on these experimental data (Sharp et al. 1995a, b; Braun and Blair 2001; Braun et al. 2004; Kim et al. 2008). The steered molecular dynamics (SMD) simulations (Isralewitz et al. 2001a, b; Jensen et al. 2002) applied to ions and a water molecule showed that Leu46 of MotB acts as the gate for hydronium ion permeation, which induces the formation of a water wire that may mediate the transfer of a proton to Asp32 of MotB. The free energy barrier for H3O+ permeation is consistent with the proton transfer rate deduced from the flagellar rotational speed and number of protons transferred per rotation (Block and Berg 1984; Meister et al. 1987; Samuel and Berg 1996). This agreement indicates that gating is the rate-limiting step. A narrowing of the channel shown by the MD simulations of MotB mutants is consistent with size-dependent ion selectivity (Terahara et al. 2012). It was also shown that the A3 segment of MotA maintained a kink with non-protonated Asp32 of MotB, whereas protonation of Asp32 induced a straighter conformation. This movement suggests that the protonation and deprotonation cycle of Asp32 induces a ratchet-like motion of the cytoplasmic domain, which may be correlated with the motion of the flagellar rotor.
Flexibility of proteins in the export apparatus
The protein export apparatus of bacterial flagella in the basal body is similar to T3SS, which comprises a cytoplasmic complex and an export gate embedded in the inner membrane (Cornelis 2006; Kawamoto et al. 2013). The export gate of the flagellar protein export apparatus is composed of six membrane proteins, FlhA, FlhB, FliO, FliP, FliQ, and FliR (Fig. 1). As the cytoplasmic complex, FliI ATPase forms a cytoplasmic hexamer ring complex with FliH and FliJ.
Among them, FlhA is the largest protein, which consists of the N-terminal TM region (FlhATM), C-terminal cytoplasmic region (FlhAC), and a linker between them. FlhA is required for the export of the hook (FlgE), hook-capping protein (FlgD), and flagellin (FliC) (Minamino and Macnab 1999). FlhATM is expected to have eight TM helices. The structure of FlhAC shows that it comprises four domains, D1, D2, D3, and D4 (Saijo-Hamano et al. 2010). Interestingly, a large gap is seen between D2 and D4. A 20-ns MD simulation showed that the interdomain distance between D2 and D4 fluctuates around 10 Å multiple times within this timescale, which indicates the highly flexible nature of this protein (Fig. 5). Dynamic domain analysis (Hayward and Berendsen 1998; Hayward 1999) on the MD trajectory indicated that subdomain interfaces shown by green act as hinges of domain motion. Six mutations of FlhA, which restrict protein export at 42 °C, namely temperature-sensitive (ts) mutations, are all situated in FlhAC. Half of the ts mutations are located at subdomain interfaces. In conclusion, the dynamic domain motion of FlhAC is suggested to be essential for protein export.
Fig. 5.
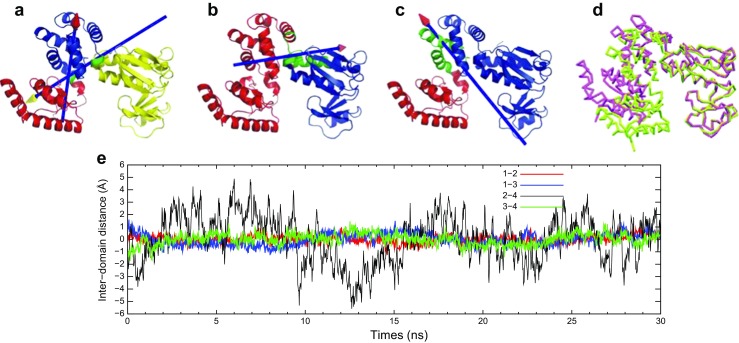
“Dynamic” domains identified by MD simulation and DynDom analysis (Saijo-Hamano et al. 2010). a The first, b second, and c third principal modes of the last 20-ns MD trajectory. Dynamics domains consist of the residues a 363–502 (yellow), 503–585 (blue), and 586–684 (red), b 363–551 (blue) and 571–684 (red), c 363–393, 396, 401–401 (blue) and 394–395, 397–400, 507–684 (red). The residues shown by green represent the bending residues that connect dynamics domains. In each case, the red and yellow domains can be considered to rotate around the axes with the arrow heads of the corresponding colors with the fixed blue domain. d Open and closed forms of FlhAC in magenta and green, respectively, identified by MD simulation. e Fluctuation of interdomain distance between four domains in FlhAC from the average as a function of simulation time. Center of mass distance between domains D1–D2 (magenta), D1–D3 (blue), D2–D4 (red), and D3–D4 (green). Reproduction from the original paper with permission (Saijo-Hamano et al. 2010)
FlhB also plays important roles for protein export. Similar to FlhA, FlhB comprises an N-terminal transmembrane region containing four helices (FlhBTM), C-terminal cytoplasmic region (FlhBC), and a linker between them; however, the protein size is smaller. FlhB permits export of rod- and hook-type proteins before hook completion; however, after cleaving between Asn269 and Pro270 in FlhBC, it specifically recognized filament-type proteins (Fraser et al. 2003). Meshcheryakov et al. (2013) determined the crystal structures of FlhBC from S. typhimurium and Aquifex aeolicus, which shows high structure similarity to T3SS proteins, EscUC from enteropathogenic E. coli and SpaSC from S. typhimurium (Zarivach et al. 2008), and YscUC from Yersinia pestis (Lountos et al. 2009). Meshcheryakov et al. also found that deletion of a short flexible loop in a globular part of S. typhimurium FlhBC (residues 281–285) leads to complete inhibition of secretion by the flagellar secretion system. MD simulation showed that N-terminal helices (in S. typhimurium, α1a and α1b) are highly flexible, but that the deletion of the loop reduces this flexibility (Fig. 6). The importance of such flexibility has previously been shown for EscU, suggesting a general feature for export function.
Fig. 6.

Flexibility of the N-terminal α-helix of Salmonella FlhBC observed in MD simulations (Meshcheryakov et al. 2013). a Key residues and vectors used for MD analysis. The vectors connecting residues 229–238, 238–256, 256–264, 265–269, and 238–264 are defined as V1–V5, respectively. b–e Structural variations of the N-terminal α-helix during MD simulations when the globular domain is superimposed for: b the wild-type FlhBC, c FlhBC Δ(281–285), d FlhBC AAAAA281–285, and e FlhBC PPPPP281–285. Reproduction from the original paper (Meshcheryakov et al. 2013)
Export mechanism of the flagellar filament protein
Extracellular components of the flagellum are exported by the flagellar export apparatus after synthesis and are assembled (Fig. 1) (Macnab 2003). For filament construction, each flagellin subunit is transported through a central channel of the filament and reaches the filament tip, where a capping protein complex assists in flagellin polymerization and prevents flagellin loss (Fig. 7) (Ikeda et al. 1993). Flagellin of S. typhimurium comprises four domains, D0, D1, D2, and D3. Domains D0 and D1 make intersubunit contacts and compose the filament core. Domain fragment Df1 is the proteolysis-resistant portion of D1, which includes residues from the N-terminal side as originally defined (Yonekura et al. 2000) and those from the C-terminal side. Domains D2 and D3, which contain two hypervariable regions (HVRs), protrude into solvent and have almost no contribution to intersubunit contacts. D2 can be separated into subdomains D2a and D2b. The flagellar channel diameter of 20 Å may be too narrow for a folded multidomain flagellin to pass through each flagellin subunit, as the filament has a cross-section larger than 20 Å. Flagellin and possibly other flagellar export proteins are expected to be transported in at least partially unfolded form (Yonekura et al. 2003).
Fig. 7.

Bacterial flagellin export (Chng and Kitao 2008). A flagellum with a few assembled flagellin near the tip of the filament is shown in this schematic diagram. The conformation of flagellin during transport through the channel is suggested to be highly unfolded due to the narrow channel cross-section. Refolding then takes place in the “chamber” before assembly with the help of HAP2 chaperone. The newly added flagellin shown still has disordered termini helices, with the filled circle representing the N-terminal. Reproduction from the original paper with permission (Chng and Kitao 2008)
For flagellar proteins to be largely or completely unfolded during transport, they could have been either (i) maintained in an unfolded state after their synthesis or (ii) actively unfolded before transport. The chaperone of flagellin, FliS, binds to the partially structured C-terminal segment to inhibit cytoplasmic polymerization of flagellin while leaving the rest of the molecule folded (Ozin et al. 2003; Muskotál et al. 2006). Hence, flagellar proteins are likely to be largely folded in the cytoplasm while waiting to be exported.
For efficient transport and assembly, the multidomain flagellin may have been evolutionarily designed to unfold easily, yet quick to refold from the denatured state. Thermal denaturation using differential scanning calorimetry (DSC) and circular dichroism (CD) melting measurements showed that flagellin unfolds in stages in accord to its multidomain nature (Honda et al. 1999). Based on a multistate transition model, the authors obtained melting curves for each thermodynamic domain that is subsequently assigned to a unique structural domain via CD measurements. An unfolding order for the domains was suggested based on the melting temperatures.
Chng and Kitao (2008) conducted a thermal unfolding MD simulation in aqueous solution as an attempt to gain atomic-level insight into the refolding process. They found a similar unfolding order of the domains as reported in experimental thermal denaturation (Honda et al. 1999). D2a and D3 exhibited high thermal stability and contained persistent three-stranded beta-sheets in the denatured state, which could serve as folding cores to guide refolding. If a transported flagellin subunit refolds in a “chamber” formed by a tip of the flagellar filament and the cap protein of the filament HAP2 (Fig. 7), the chamber is large enough for either denatured HVR domains or filament-core domains, but not the whole flagellin. This suggests a two-stage refolding process: filament-core domains become folded first and then docking of Df1 to folded D2 would complete the refolding process for flagellin. In bacterial species with short or negligible HVR segment, refolding of filament-core domains is suggested to be the major step.
The same authors further explored how to mechanically deform flagellin so as to attain a conformation suitable for transport through the narrow flagellar channel by means of force-probe MD simulations (Chng and Kitao 2010). Two convenient pulling schemes were chosen. In the first scheme, termed “Unzip”, flagellin is unraveled from its adjacently placed termini into a linear polypeptide (Fig. 8a). In the second scheme, termed “Stretch”, flagellin is stretched along its major principal axis into a hairpin-like conformation (Fig. 8b). In the Stretch scheme, flagellin is stiffer and requires larger unfolding forces when stretched as the relative sliding of beta-strands requires the breaking of multiple hydrogen bonds at once. In contrast, unzipping requires lower unfolding forces as it mainly involves unraveling beta-sheets by breaking hydrogen bonds one by one. Therefore, Unzip is identified as the more likely scheme to produce a transport-capable flagellin.
Fig. 8.

Mechanical unfolding of flagellin under Unzip (a) and Stretch schemes (b) (Chng and Kitao 2010). The blue and red spheres represent the N- and C-terminal Cα atoms, denoted as A and B. The orange spheres represent atoms of the D3 surface three-residue aromatic cluster. The dark blue circle in a indicates the salt bridge interaction between the D1 N-terminal hairpin and D1 C-terminal. In b, the colored ovals indicate load-bearing interactions. Reproduction from the original paper with permission (Chng and Kitao 2010)
To view the behavior of translocating flagellin in its channel environment, an MD simulation of a 100-residue segment of flagellin translocated through the flagellar channel was performed by Tanner et al. (2011). Although the translocation rate in their simulation is much faster than the in vivo translocation rate, a series of salt bridge formations were observed during the 52-ns simulation. Based on the MD results, they constructed a mathematical model describing that the flagellum growth rate decreases exponentially with length, because protein compression and friction between translocating flagellin and the flagellar channel increase proportionally along the flagellum.
Rotational switching of the motor
Switching of the rotational direction of the flagella motor plays a central role for bacterial chemotaxis. The rotational switching is controlled by the signaling molecule phospho-CheY (CheY-P), whose binding onto the FliM motor protein promotes CW rotation (Welch et al. 1993). Phosphorylation of CheY is believed to promote binding to the FliM via a conformational change from an inactive conformation to an active form (CheY activation) (Welch et al. 1994).
Molecular features for interactions between CheY-P and FliM were computationally investigated by modeling and comparing four CheY-P homologs contained in V. cholera (Dasgupta and Dattagupta 2008). Among these homologs, only one of them (CheY3v) has been reported to directly switch flagella rotation by an in vivo study (Hyakutake et al. 2005). The simulations showed that only CheY3v possesses a suitable depth and hydrophobicity at the FliM binding pocket and the residues that form the hydrogen bonds with FliM are found in the correct position.
The CheY activation is also likely to be affected by acetylation of CheY (Fraiberg et al. 2015). This in vivo study showed that the acetylation of the K91 residue in CheY is responsible for acetate-dependent generation of CW rotation. Subsequent all-atom MD simulations revealed that the K91H CheY mutant has a higher flexibility in the protein regions surrounding position 91, and spends much more time in the active state than the wild-type CheY, which suggests that lysine exclusion from position 91 removes the repression of CheY activation.
Conclusion
In this article, we reviewed molecular dynamics (MD) studies in which various mechanisms of bacterial flagellar function were investigated. All-atom MD simulation showed that the molecular mechanisms were elucidated successfully, including quantitative comparison with experimental results and confirmation of mutation and deletion effects. In addition, MD simulation permitted the construction of reasonable atomic models of proteins, as shown in the example of MotAB. The results of coarse-grained MD indicated that a large portion of the flagellar structure can be simulated. As atomic structures of further components of the bacterial flagellum are solved, MD simulation is expected to play an increasingly important role in the investigation of molecular mechanisms. A possible goal might be MD simulation of the entire bacterial flagellum.
Acknowledgements
This research was supported by MEXT/JSPS KAKENHI (nos. 25104002 and 15H04357) to A.K. and by MEXT as “Priority Issue on Post-K Computer” (Building Innovative Drug Discovery Infrastructure Through Functional Control of Biomolecular Systems) to A.K. The computations were partly performed using the supercomputers at the RCCS, The National Institute of Natural Science, and ISSP, The University of Tokyo. This research also used computational resources of the K computer provided by the RIKEN Advanced Institute for Computational Science through the HPCI System Research project (project IDs: hp120223, hp140030, hp140031, hp150049, hp150270, hp160207, and hp170254).
Compliance with ethical standards
Conflict of interest
Akio Kitao declares that he has no conflict of interest. Hiroaki Hata declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Footnotes
This article is part of a Special Issue on ‘Biomolecules to Bio-nanomachines - Fumio Arisaka 70th Birthday’ edited by Damien Hall.
References
- Aizawa S (2001) Bacterial flagella and type III secretion systems. FEMS Microbiol Lett 202(2):157–164 [DOI] [PubMed]
- Arkhipov A, Freddolino PL, Imada K, Namba K, Schulten K. Coarse-grained molecular dynamics simulations of a rotating bacterial flagellum. Biophys J. 2006;91(12):4589–4597. doi: 10.1529/biophysj.106.093443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asai Y, Yakushi T, Kawagishi I, Homma M. Ion-coupling determinants of Na+-driven and H+-driven flagellar motors. J Mol Biol. 2003;327(2):453–463. doi: 10.1016/S0022-2836(03)00096-2. [DOI] [PubMed] [Google Scholar]
- Asakura S. Polymerization of flagellin and polymorphism of flagella. Adv Biophys. 1970;1:99–155. [PubMed] [Google Scholar]
- Berg HC. The rotary motor of bacterial flagella. Annu Rev Biochem. 2003;72(1):19–54. doi: 10.1146/annurev.biochem.72.121801.161737. [DOI] [PubMed] [Google Scholar]
- Berg HC, Anderson RA. Bacteria swim by rotating their flagellar filaments. Nature. 1973;245(5425):380–382. doi: 10.1038/245380a0. [DOI] [PubMed] [Google Scholar]
- Berry RM. Torque and switching in the bacterial flagellar motor. An electrostatic model. Biophys J. 1993;64(4):961–973. doi: 10.1016/S0006-3495(93)81462-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block SM, Berg HC. Successive incorporation of force-generating units in the bacterial rotary motor. Nature. 1984;309(5967):470–472. doi: 10.1038/309470a0. [DOI] [PubMed] [Google Scholar]
- Braun TF, Blair DF. Targeted disulfide cross-linking of the MotB protein of Escherichia coli: evidence for two H(+) channels in the stator complex. Biochemistry. 2001;40(43):13051–13059. doi: 10.1021/bi011264g. [DOI] [PubMed] [Google Scholar]
- Braun TF, Al-Mawsawi LQ, Kojima S, Blair DF. Arrangement of core membrane segments in the MotA/MotB proton-channel complex of Escherichia coli. Biochemistry. 2004;43(1):35–45. doi: 10.1021/bi035406d. [DOI] [PubMed] [Google Scholar]
- Calladine CR. Construction of bacterial flagella. Nature. 1975;255(5504):121–124. doi: 10.1038/255121a0. [DOI] [PubMed] [Google Scholar]
- Calladine CR. Design requirements for the construction of bacterial flagella. J Theor Biol. 1976;57(2):469–489. doi: 10.1016/0022-5193(76)90016-3. [DOI] [PubMed] [Google Scholar]
- Calladine CR. Change of waveform in bacterial flagella: the role of mechanics at the molecular level. J Mol Biol. 1978;118(4):457–479. doi: 10.1016/0022-2836(78)90285-1. [DOI] [Google Scholar]
- Chng C-P, Kitao A. Thermal unfolding simulations of bacterial flagellin: insight into its refolding before assembly. Biophys J. 2008;94(10):3858–3871. doi: 10.1529/biophysj.107.123927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng C-P, Kitao A. Mechanical unfolding of bacterial flagellar filament protein by molecular dynamics simulation. J Mol Graph Model. 2010;28(6):548–554. doi: 10.1016/j.jmgm.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Chun SY, Parkinson JS. Bacterial motility: membrane topology of the Escherichia coli MotB protein. Science. 1988;239(4837):276–278. doi: 10.1126/science.2447650. [DOI] [PubMed] [Google Scholar]
- Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4(11):811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- Dasgupta J, Dattagupta JK. Structural determinants of V. cholerae CheYs that discriminate them in FliM binding: comparative modeling and MD simulation studies. J Biomol Struct Dyn. 2008;25(5):495–503. doi: 10.1080/07391102.2008.10507196. [DOI] [PubMed] [Google Scholar]
- De Mot R, Vanderleyden J. The C-terminal sequence conservation between Ompa-related outer membrane proteins and Motb suggests a common function in both Gram-positive and Gram-negative bacteria, possibly in the interaction of these domains with peptidoglycan. Mol Microbiol. 1994;12(2):333–336. doi: 10.1111/j.1365-2958.1994.tb01021.x. [DOI] [PubMed] [Google Scholar]
- Dean GE, Macnab RM, Stader J, Matsumura P, Burks C. Gene sequence and predicted amino acid sequence of the motA protein, a membrane-associated protein required for flagellar rotation in Escherichia coli. J Bacteriol. 1984;159(3):991–999. doi: 10.1128/jb.159.3.991-999.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston TC, Oster G. Protein turbines. I: the bacterial flagellar motor. Biophys J. 1997;73(2):703–721. doi: 10.1016/S0006-3495(97)78104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto M. Genetic studies of paralyzed mutants in Salmonella. II. Mapping of three mot loci by linkage analysis. Genetics. 1966;54(5):1069–1076. doi: 10.1093/genetics/54.5.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraiberg M, Afanzar O, Cassidy CK, Gabashvili A, Schulten K, Levin Y, Eisenbach M. CheY’s acetylation sites responsible for generating clockwise flagellar rotation in Escherichia coli. Mol Microbiol. 2015;95(2):231–244. doi: 10.1111/mmi.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser GM, Hirano T, Ferris HU, Devgan LL, Kihara M, Macnab RM. Substrate specificity of type III flagellar protein export in Salmonella is controlled by subdomain interactions in FlhB. Mol Microbiol. 2003;48(4):1043–1057. doi: 10.1046/j.1365-2958.2003.03487.x. [DOI] [PubMed] [Google Scholar]
- Fujii T, Kato T, Namba K. Specific arrangement of alpha-helical coiled coils in the core domain of the bacterial flagellar hook for the universal joint function. Structure. 2009;17(11):1485–1493. doi: 10.1016/j.str.2009.08.017. [DOI] [PubMed] [Google Scholar]
- Furuta T, Samatey FA, Matsunami H, Imada K, Namba K, Kitao A. Gap compression/extension mechanism of bacterial flagellar hook as the molecular universal joint. J Struct Biol. 2007;157(3):481–490. doi: 10.1016/j.jsb.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Albers SV. Assembly and function of the archaeal flagellum. Biochem Soc Trans. 2011;39(1):64–69. doi: 10.1042/BST0390064. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Yamashita I, Namba K. Quasi- and nonequivalence in the structure of bacterial flagellar filament. Biophys J. 1998;74(1):569–575. doi: 10.1016/S0006-3495(98)77815-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward S. Structural principles governing domain motions in proteins. Proteins. 1999;36(4):425–435. doi: 10.1002/(SICI)1097-0134(19990901)36:4<425::AID-PROT6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hayward S, Berendsen HJ. Systematic analysis of domain motions in proteins from conformational change: new results on citrate synthase and T4 lysozyme. Proteins. 1998;30(2):144–154. doi: 10.1002/(SICI)1097-0134(19980201)30:2<144::AID-PROT4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Hirano T, Yamaguchi S, Oosawa K, Aizawa S (1994) Roles of FliK and FlhB in determination of flagellar hook length in Salmonella typhimurium. J Bacteriol 176(17):5439–5449 [DOI] [PMC free article] [PubMed]
- Hirota N, Kitada M, Imae Y. Flagellar motors of alkalophilic Bacillus are powered by an electrochemical potential gradient of Na+ FEBS Lett. 1981;132(2):278–280. doi: 10.1016/0014-5793(81)81178-7. [DOI] [Google Scholar]
- Honda S, Uedaira H, Vonderviszt F, Kidokoro S, Namba K. Folding energetics of a multidomain protein, flagellin. J Mol Biol. 1999;293(3):719–732. doi: 10.1006/jmbi.1999.3175. [DOI] [PubMed] [Google Scholar]
- Hotani H. Micro-video study of moving bacterial flagellar filaments II. Polymorphic transition in alcohol. Biosystems. 1980;12(3–4):325–330. doi: 10.1016/0303-2647(80)90030-1. [DOI] [PubMed] [Google Scholar]
- Hotani H. Micro-video study of moving bacterial flagellar filaments: III. Cyclic transformation induced by mechanical force. J Mol Biol. 1982;156(4):791–806. doi: 10.1016/0022-2836(82)90142-5. [DOI] [PubMed] [Google Scholar]
- Hyakutake A, Homma M, Austin MJ, Boin MA, Häse CC, Kawagishi I. Only one of the five CheY homologs in Vibrio cholerae directly switches flagellar rotation. J Bacteriol. 2005;187(24):8403–8410. doi: 10.1128/JB.187.24.8403-8410.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda T, Yamaguchi S, Hotani H. Flagellar growth in a filament-less Salmonella fliD mutant supplemented with purified hook-associated protein 2. J Biochem. 1993;114(1):39–44. doi: 10.1093/oxfordjournals.jbchem.a124136. [DOI] [PubMed] [Google Scholar]
- Isralewitz B, Baudry J, Gullingsrud J, Kosztin D, Schulten K. Steered molecular dynamics investigations of protein function. J Mol Graph Model. 2001;19(1):13–25. doi: 10.1016/S1093-3263(00)00133-9. [DOI] [PubMed] [Google Scholar]
- Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr Opin Struct Biol. 2001;11(2):224–230. doi: 10.1016/S0959-440X(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Jensen MØ, Park S, Tajkhorshid E, Schulten K. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc Natl Acad Sci U S A. 2002;99(10):6731–6736. doi: 10.1073/pnas.102649299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya R, Asakura S. Flagellar transformations at alkaline pH. J Mol Biol. 1976;108(2):513–518. doi: 10.1016/S0022-2836(76)80133-7. [DOI] [PubMed] [Google Scholar]
- Kamiya R, Asakura S. Helical transformations of Salmonella flagella in vitro. J Mol Biol. 1976;106(1):167–186. doi: 10.1016/0022-2836(76)90306-5. [DOI] [PubMed] [Google Scholar]
- Kamiya R, Hotani H, Asakura S. Polymorphic transition in bacterial flagella. Symp Soc Exp Biol. 1982;35:53–76. [PubMed] [Google Scholar]
- Kanto S, Okino H, Aizawa S, Yamaguchi S. Amino acids responsible for flagellar shape are distributed in terminal regions of flagellin. J Mol Biol. 1991;219(3):471–480. doi: 10.1016/0022-2836(91)90187-B. [DOI] [PubMed] [Google Scholar]
- Kato S, Okamoto M, Asakura S. Polymorphic transition of the flagellar polyhook from Escherichia coli and Salmonella typhimurium. J Mol Biol. 1984;173(4):463–476. doi: 10.1016/0022-2836(84)90391-7. [DOI] [PubMed] [Google Scholar]
- Kawamoto A, Morimoto YV, Miyata T, Minamino T, Hughes KT, Kato T, Namba K. Common and distinct structural features of Salmonella injectisome and flagellar basal body. Sci Rep. 2013;3:3369. doi: 10.1038/srep03369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Dapice M, Reese TS. Effects of mot gene expression on the structure of the flagellar motor. J Mol Biol. 1988;202(3):575–584. doi: 10.1016/0022-2836(88)90287-2. [DOI] [PubMed] [Google Scholar]
- Kim EA, Price-Carter M, Carlquist WC, Blair DF. Membrane segment organization in the stator complex of the flagellar motor: implications for proton flow and proton-induced conformational change. Biochemistry. 2008;47(43):11332–11339. doi: 10.1021/bi801347a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitao A, Takemura K. High anisotropy and frustration: the keys to regulating protein function efficiently in crowded environments. Curr Opin Struct Biol. 2017;42:50–58. doi: 10.1016/j.sbi.2016.10.014. [DOI] [PubMed] [Google Scholar]
- Kitao A, Yonekura K, Maki-Yonekura S, Samatey FA, Imada K, Namba K, Go N. Switch interactions control energy frustration and multiple flagellar filament structures. Proc Natl Acad Sci U S A. 2006;103(13):4894–4899. doi: 10.1073/pnas.0510285103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Blair DF. Conformational change in the stator of the bacterial flagellar motor. Biochemistry. 2001;40(43):13041–13050. doi: 10.1021/bi011263o. [DOI] [PubMed] [Google Scholar]
- Kojima S, Imada K, Sakuma M, Sudo Y, Kojima C, Minamino T, Homma M, Namba K. Stator assembly and activation mechanism of the flagellar motor by the periplasmic region of MotB. Mol Microbiol. 2009;73(4):710–718. doi: 10.1111/j.1365-2958.2009.06802.x. [DOI] [PubMed] [Google Scholar]
- Kutsukake K, Minamino T, Yokoseki T. Isolation and characterization of FliK-independent flagellation mutants from Salmonella typhimurium. J Bacteriol. 1994;176(24):7625–7629. doi: 10.1128/jb.176.24.7625-7629.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SH, Adler J, Gargus JJ, Hogg RW. Chemomechanical coupling without ATP: the source of energy for motility and chemotaxis in bacteria. Proc Natl Acad Sci U S A. 1974;71(4):1239–1243. doi: 10.1073/pnas.71.4.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen SH, Reader RW, Kort EN, Tso WW, Adler J. Change in direction of flagellar rotation is the basis of the chemotactic response in Escherichia coli. Nature. 1974;249(5452):74–77. doi: 10.1038/249074a0. [DOI] [PubMed] [Google Scholar]
- Lountos GT, Austin BP, Nallamsetty S, Waugh DS. Atomic resolution structure of the cytoplasmic domain of Yersinia pestis YscU, a regulatory switch involved in type III secretion. Protein Sci. 2009;18(2):467–474. doi: 10.1002/pro.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnab RM. How bacteria assemble flagella. Annu Rev Microbiol. 2003;57:77–100. doi: 10.1146/annurev.micro.57.030502.090832. [DOI] [PubMed] [Google Scholar]
- Macnab RM, Ornston MK. Normal-to-curly flagellar transitions and their role in bacterial tumbling. Stabilization of an alternative quaternary structure by mechanical force. J Mol Biol. 1977;112(1):1–30. doi: 10.1016/S0022-2836(77)80153-8. [DOI] [PubMed] [Google Scholar]
- Maki-Yonekura S, Yonekura K, Namba K. Conformational change of flagellin for polymorphic supercoiling of the flagellar filament. Nat Struct Mol Biol. 2010;17(4):417–422. doi: 10.1038/nsmb.1774. [DOI] [PubMed] [Google Scholar]
- Matsunami H, Barker CS, Yoon YH, Wolf M, Samatey FA. Complete structure of the bacterial flagellar hook reveals extensive set of stabilizing interactions. Nat Commun. 2016;7:13425. doi: 10.1038/ncomms13425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister M, Lowe G, Berg HC. The proton flux through the bacterial flagellar motor. Cell. 1987;49(5):643–650. doi: 10.1016/0092-8674(87)90540-X. [DOI] [PubMed] [Google Scholar]
- Meshcheryakov VA, Kitao A, Matsunami H, Samatey FA. Inhibition of a type III secretion system by the deletion of a short loop in one of its membrane proteins. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 5):812–820. doi: 10.1107/S0907444913002102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamino T, Macnab RM. Components of the Salmonella flagellar export apparatus and classification of export substrates. J Bacteriol. 1999;181(5):1388–1394. doi: 10.1128/jb.181.5.1388-1394.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DG, Macnab RM, Francis NR, DeRosier DJ. Domain organization of the subunit of the Salmonella typhimurium flagellar hook. J Mol Biol. 1993;229(1):79–84. doi: 10.1006/jmbi.1993.1009. [DOI] [PubMed] [Google Scholar]
- Muskotál A, Király R, Sebestyén A, Gugolya Z, Végh BM, Vonderviszt F. Interaction of FliS flagellar chaperone with flagellin. FEBS Lett. 2006;580(16):3916–3920. doi: 10.1016/j.febslet.2006.06.024. [DOI] [PubMed] [Google Scholar]
- Namba K, Vonderviszt F. Molecular architecture of bacterial flagellum. Q Rev Biophys. 1997;30(1):1–65. doi: 10.1017/S0033583596003319. [DOI] [PubMed] [Google Scholar]
- Nishihara Y, Kitao A. Gate-controlled proton diffusion and protonation-induced ratchet motion in the stator of the bacterial flagellar motor. Proc Natl Acad Sci U S A. 2015;112(25):7737–7742. doi: 10.1073/pnas.1502991112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishima W, Qi G, Hayward S, Kitao A. DTA: dihedral transition analysis for characterization of the effects of large main-chain dihedral changes in proteins. Bioinformatics. 2009;25(5):628–635. doi: 10.1093/bioinformatics/btp032. [DOI] [PubMed] [Google Scholar]
- O’Brien EJ, Bennett PM. Structure of straight flagella from a mutant Salmonella. J Mol Biol. 1972;70(1):133–152. doi: 10.1016/0022-2836(72)90168-4. [DOI] [PubMed] [Google Scholar]
- O’Neill J, Xie M, Hijnen M, Roujeinikova A. Role of the MotB linker in the assembly and activation of the bacterial flagellar motor. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 12):1009–1016. doi: 10.1107/S0907444911041102. [DOI] [PubMed] [Google Scholar]
- Ozin AJ, Claret L, Auvray F, Hughes C. The FliS chaperone selectively binds the disordered flagellin C-terminal D0 domain central to polymerisation. FEMS Microbiol Lett. 2003;219(2):219–224. doi: 10.1016/S0378-1097(02)01208-9. [DOI] [PubMed] [Google Scholar]
- Patterson-Delafield J, Martinez RJ, Stocker BA, Yamaguchi S. A new fla gene in Salmonella typhimurium—flaR—and its mutant phenotype-superhooks. Arch Microbiol. 1973;90(2):107–120. doi: 10.1007/BF00414513. [DOI] [PubMed] [Google Scholar]
- Qi G, Lee R, Hayward S. A comprehensive and non-redundant database of protein domain movements. Bioinformatics. 2005;21(12):2832–2838. doi: 10.1093/bioinformatics/bti420. [DOI] [PubMed] [Google Scholar]
- Reboul CF, Andrews DA, Nahar MF, Buckle AM, Roujeinikova A. Crystallographic and molecular dynamics analysis of loop motions unmasking the peptidoglycan-binding site in stator protein MotB of flagellar motor. PLoS One. 2011;6(4):e18981. doi: 10.1371/journal.pone.0018981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roujeinikova A. Crystal structure of the cell wall anchor domain of MotB, a stator component of the bacterial flagellar motor: implications for peptidoglycan recognition. Proc Natl Acad Sci U S A. 2008;105(30):10348–10353. doi: 10.1073/pnas.0803039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo-Hamano Y, Imada K, Minamino T, Kihara M, Shimada M, Kitao A, Namba K. Structure of the cytoplasmic domain of FlhA and implication for flagellar type III protein export. Mol Microbiol. 2010;76(1):260–268. doi: 10.1111/j.1365-2958.2010.07097.x. [DOI] [PubMed] [Google Scholar]
- Samatey FA, Imada K, Nagashima S, Vonderviszt F, Kumasaka T, Yamamoto M, Namba K. Structure of the bacterial flagellar protofilament and implications for a switch for supercoiling. Nature. 2001;410(6826):331–337. doi: 10.1038/35066504. [DOI] [PubMed] [Google Scholar]
- Samatey FA, Matsunami H, Imada K, Nagashima S, Shaikh TR, Thomas DR, Chen JZ, Derosier DJ, Kitao A, Namba K. Structure of the bacterial flagellar hook and implication for the molecular universal joint mechanism. Nature. 2004;431(7012):1062–1068. doi: 10.1038/nature02997. [DOI] [PubMed] [Google Scholar]
- Samuel AD, Berg HC. Torque-generating units of the bacterial flagellar motor step independently. Biophys J. 1996;71(2):918–923. doi: 10.1016/S0006-3495(96)79295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh TR, Thomas DR, Chen JZ, Samatey FA, Matsunami H, Imada K, Namba K, Derosier DJ. A partial atomic structure for the flagellar hook of Salmonella typhimurium. Proc Natl Acad Sci U S A. 2005;102(4):1023–1028. doi: 10.1073/pnas.0409020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp LL, Zhou J, Blair DF. Tryptophan-scanning mutagenesis of MotB, an integral membrane protein essential for flagellar rotation in Escherichia coli. Biochemistry. 1995;34(28):9166–9171. doi: 10.1021/bi00028a028. [DOI] [PubMed] [Google Scholar]
- Sharp LL, Zhou J, Blair DF. Features of MotA proton channel structure revealed by tryptophan-scanning mutagenesis. Proc Natl Acad Sci U S A. 1995;92(17):7946–7950. doi: 10.1073/pnas.92.17.7946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman M, Simon M. Characterization of Escherichia coli flagellar mutants that are insensitive to catabolite repression. J Bacteriol. 1974;120(3):1196–1203. doi: 10.1128/jb.120.3.1196-1203.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stader J, Matsumura P, Vacante D, Dean GE, Macnab RM. Nucleotide sequence of the Escherichia coli motB gene and site-limited incorporation of its product into the cytoplasmic membrane. J Bacteriol. 1986;166(1):244–252. doi: 10.1128/jb.166.1.244-252.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner DE, Ma W, Chen Z, Schulten K. Theoretical and computational investigation of flagellin translocation and bacterial flagellum growth. Biophys J. 2011;100(11):2548–2556. doi: 10.1016/j.bpj.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terahara N, Sano M, Ito M. A Bacillus flagellar motor that can use both Na+ and K+ as a coupling ion is converted by a single mutation to use only Na+ PLoS One. 2012;7(9):e46248. doi: 10.1371/journal.pone.0046248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NA, Bardy SL, Jarrell KF. The archaeal flagellum: a different kind of prokaryotic motility structure. FEMS Microbiol Rev. 2001;25(2):147–174. doi: 10.1111/j.1574-6976.2001.tb00575.x. [DOI] [PubMed] [Google Scholar]
- Turner L, Ryu WS, Berg HC. Real-time imaging of fluorescent flagellar filaments. J Bacteriol. 2000;182(10):2793–2801. doi: 10.1128/JB.182.10.2793-2801.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonderviszt F, Aizawa SI, Namba K. Role of the disordered terminal regions of flagellin in filament formation and stability. J Mol Biol. 1991;221(4):1461–1474. doi: 10.1016/0022-2836(91)90946-4. [DOI] [PubMed] [Google Scholar]
- Wagenknecht T, DeRosier DJ, Aizawa S, Macnab RM. Flagellar hook structures of Caulobacter and Salmonella and their relationship to filament structure. J Mol Biol. 1982;162(1):69–87. doi: 10.1016/0022-2836(82)90162-0. [DOI] [PubMed] [Google Scholar]
- Walz D, Caplan SR. An electrostatic mechanism closely reproducing observed behavior in the bacterial flagellar motor. Biophys J. 2000;78(2):626–651. doi: 10.1016/S0006-3495(00)76622-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Oosawa K, Aizawa S, Eisenbach M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc Natl Acad Sci U S A. 1993;90(19):8787–8791. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M, Oosawa K, Aizawa S, Eisenbach M (1994) Effects of phosphorylation, Mg2+, and conformation of the chemotaxis protein CheY on its binding to the flagellar switch protein FliM. Biochemistry 33(34):10470–10476 [DOI] [PubMed]
- Williams AW, Yamaguchi S, Togashi F, Aizawa S, Kawagishi I, Macnab RM (1996) Mutations in fliK and flhB affecting flagellar hook and filament assembly in Salmonella typhimurium. J Bacteriol 178(10):2960–2970 [DOI] [PMC free article] [PubMed]
- Yamaguchi S, Fujita H, Ishihara A, Aizawa S, Macnab RM. Subdivision of flagellar genes of Salmonella typhimurium into regions responsible for assembly, rotation, and switching. J Bacteriol. 1986;166(1):187–193. doi: 10.1128/jb.166.1.187-193.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita I, Hasegawa K, Suzuki H, Vonderviszt F, Mimori-Kiyosue Y, Namba K. Structure and switching of bacterial flagellar filaments studied by X-ray fiber diffraction. Nat Struct Mol Biol. 1998;5(2):125–132. doi: 10.1038/nsb0298-125. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Maki S, Morgan DG, DeRosier DJ, Vonderviszt F, Imada K, Namba K. The bacterial flagellar cap as the rotary promoter of flagellin self-assembly. Science. 2000;290(5499):2148–2152. doi: 10.1126/science.290.5499.2148. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Maki-Yonekura S, Namba K. Complete atomic model of the bacterial flagellar filament by electron cryomicroscopy. Nature. 2003;424(6949):643–650. doi: 10.1038/nature01830. [DOI] [PubMed] [Google Scholar]
- Zarivach R, Deng W, Vuckovic M, Felise HB, Nguyen HV, Miller SI, Finlay BB, Strynadka NC. Structural analysis of the essential self-cleaving type III secretion proteins EscU and SpaS. Nature. 2008;453(7191):124–127. doi: 10.1038/nature06832. [DOI] [PubMed] [Google Scholar]
- Zhou J, Fazzio RT, Blair DF. Membrane topology of the MotA protein of Escherichia coli. J Mol Biol. 1995;251(2):237–242. doi: 10.1006/jmbi.1995.0431. [DOI] [PubMed] [Google Scholar]
- Zhu S, Takao M, Li N, Sakuma M, Nishino Y, Homma M, Kojima S, Imada K. Conformational change in the periplamic region of the flagellar stator coupled with the assembly around the rotor. Proc Natl Acad Sci U S A. 2014;111(37):13523–13528. doi: 10.1073/pnas.1324201111. [DOI] [PMC free article] [PubMed] [Google Scholar]