

Abstract
Most genome-wide association studies have been conducted in European individuals, even though most genetic variation in humans is seen only in non-European samples. To search for novel loci associated with blood lipid levels and clarify the mechanism of action at previously identified lipid loci, we examined protein-coding genetic variants in 47,532 East Asian individuals using an exome array. We identified 255 variants at 41 loci reaching chip-wide significance, including 3 novel loci and 14 East Asian-specific coding variant associations. After meta-analysis with > 300,000 European samples, we identified an additional 9 novel loci. The same 16 genes were identified by the protein-altering variants in both East Asians and Europeans, likely pointing to the functional genes. Our data demonstrate that most of the low-frequency or rare coding variants associated with lipids are population-specific, and that examining genomic data across diverse ancestries may facilitate the identification of functional genes at associated loci.
Introduction
Genome-wide association studies (GWAS) have revealed over 175 genetic loci contributing to lipid levels1–6, which are heritable risk factors for cardiovascular disease, fatty liver disease, age-related macular degeneration and type 2 diabetes7–9. However, most of the published lipid-associated variants fall in non-protein-coding regions of the genome, are without obvious biological significance, and explain only a small fraction of the heritability of lipid levels. The examination of low frequency and potentially functional variants, poorly captured by standard GWAS arrays, has the potential to pinpoint causal variants and genes for follow-up and functional analyses, therefore promoting translation of the finding of genetic studies into new therapeutic targets. For example, low-frequency coding variants in PCSK9 lower plasma low density lipoprotein cholesterol (LDL-C), reduce risk of coronary artery disease (CAD), and have prompted the development of a new class of therapeutics10. Thus, we investigated the effect on lipid levels of the rare and low-frequency variants in the coding portion of the genome in an East Asian population, which has not been as extensively studied as the European population11–13.
We performed a meta-analysis of exome-wide association studies of blood lipid levels (high density lipoprotein cholesterol [HDL-C], LDL-C, triglycerides [TG], and total cholesterol [TC]) in a total of 47,532 East Asian samples that were genotyped using an exome array. We further integrated the exome array data for plasma lipids in over 300,000 individuals, primarily European ancestry (84%), conducted by the Global Lipids Genetics Consortium (GLGC). We aimed to determine whether novel or population-specific variants and genes influencing lipid levels could be identified in East Asian and multi-ancestry meta-analysis. Secondly, we aimed to determine if the protein-altering variants located in known lipid loci explained the association signal or were independent evidence of functional genes. And lastly, we examined whether exome data would implicate the same putatively functional genes in European and East Asian ancestries at lipid loci.
Results
To improve the coverage for the low frequency variants in Asian populations and follow up various GWAS variants, approximately 60K custom content variants were added to the standard exome array. Among 319,272 variants passing quality control, 204,408 (64.0%) were polymorphic in the East Asian individuals, of which about 25% (n = 50,126) were from the custom content. Approximately 76.1% (n=155,566) of the polymorphic variants are annotated as nonsynonymous or loss of function (stop-gain, stop-loss and splice variants) (Supplementary Table 1). By determining the proportion of variants observed in ExAC East Asian samples (n = 4,327 individuals) that were successfully genotyped by the array, we estimated that the exome array captured a large fraction of common and low-frequency coding variants (71.15% and 72.59% for variants with minor allele frequency (MAF) >5% and MAF = 1–5%, respectively). Among rare coding variants identified in ExAC sequenced individuals, 59.91% (MAF = 0.1–1%) and 19.92% (two or more copies) were captured by the array. Therefore, the array provided good coverage for low-frequency and moderate coverage for rare coding variants in East Asians. In addition, we examined 76K polymorphic coding variants that were not available or monomorphic in ExAC East Asian samples.
Discovery of novel variants associated with lipid levels
Our analysis identified three study-wide significant variants in three novel loci in East Asians, located at least 1 megabase from previously reported GWAS signals of lipid levels (Table 1). These include rs4377290 in ACVR1C (TC, P = 4.69 × 10−8), rs7901016 in MCU (LDL-C, P = 5.12 × 10−9), and the missense variant rs4883263 (encoding p.Ile342Val) in CD163 (HDL-C, P = 5.24 × 10−11). Each of these three variants demonstrated evidence for association (P = 1.80 × 10−3~ 6.68 × 10−5) in over 300,000 GLGC individuals.
Table 1.
Genetic variants at novel loci associated with lipid levels in East Asian samples
| Gene | rsID | Position | Alleles | Variants | Trait | East Asian | GLGC | Combined | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||||||
| AAF | BETA | S.E. | P | N | AAF | P | AAF | P | I2 | ||||||
| ACVR1C | rs4377290 | 2:158437683 | C/T | TC | 0.33 | −0.039 | 0.007 | 4.69×10−8 | 46,025 | 0.46 | 1.59×10−4 | 0.44 | 6.06×10−8 | 16.2% | |
| MCU | rs7901016 | 10:74637326 | C/T | LDL-C | 0.27 | −0.044 | 0.008 | 5.12×10−9 | 44,985 | 0.09 | 1.80×10−3 | 0.12 | 2.21×10−9 | 18.2% | |
| CD163 | rs4883263 | 12:7649484 | C/T | p.Ile342Val | HDL-C | 0.69 | −0.047 | 0.007 | 5.24×10−11 | 47,456 | 0.94 | 6.68×10−5 | 0.90 | 6.30×10−13 | 2.38% |
AAF, alternative allele frequency.
Position is reported in human genome build hg19.
Alleles are listed as alternative/reference allele on the forward strand of the reference genome.
Summary of association results
We assessed association of 110,986 polymorphic variants that had at least 20 minor alleles in 47,532 East Asian samples. Overall, we detected 255 variants (including 51 coding variants) at 41 loci that reached exome-wide significant association with one or more lipid trait (P < 4.5× 10−7), of which 3 loci have not been previously reported (Figure 1). Collectively, the overall variance in each lipid trait explained by exome-wide significant variants in East Asian samples was 5.97% for TC, 6.20% for LDL-C, 6.93 % for HDL-C, and 6.89% for TG levels, respectively, of which 3.22 %, 4.77%, 3.35% and 3.86% can be attributed to coding variants (Figure 2). Our results also showed that additional 7 known loci were associated with lipid levels at suggestive significance (P < 4.46 × 10−6, Bonferroni correction of 11,215 variants) (Supplementary table 2), and that, taken together they increased the trait variance explained to 6.08%~7.20%.
Figure 1. Manhattan plot of exome-wide association results in 47,532 East Asians.

Manhattan plot showing −log10 P of the variants for LDL-C, HDL-C, TC and TG. Signals with exome-wide levels of significance (horizontal dash line; P < 4.5×10−7) are highlighted and the previously reported GWAS lead variant of each region are labeled separately in diamond. East Asian-specific variants are defined as the variants with conditional P values reaching exome-wide significance after conditioning on all independent variants in the corresponding loci identified by GLGC exome-wide association studies.
Figure 2. Proportion of total trait variance explained by the significant and coding variants.
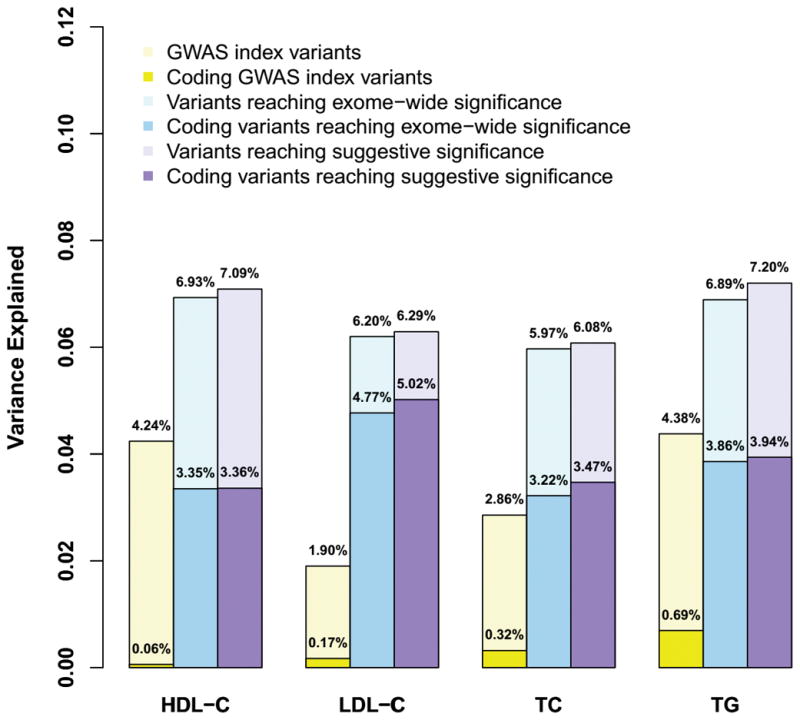
The variances explained by all the variants reaching exome-wide significance (P < 4.5 × 10−7), and together with the variants at suggestive significance (P < 4.46 × 10−6) are presented with light blue and purple bars, respectively. The proportions of variance explained by the corresponding protein-altering variants are represented by dark blue and purple bars, respectively. The proportions of variance explained by GWAS index variants are represented by yellow bars.
Evaluation of known lipid signals
Among the 38 previously established lipid loci that reached significance, we identified a more significant candidate variant at 14 loci (Supplementary Table 3 and Figure 1), where the initially reported GWAS index variants showed no significant associations or were independent of previously identified associations in European populations (r2 < 0.02) (APOB and APOE), demonstrating allelic heterogeneity between East Asian and European ancestries. The lead variants in the remaining 24 loci were the same as or strongly related (r2 > 0.69) to the reported GWAS index variants from previous studies in primarily European samples. Sequential conditional analyses revealed that 12 loci with evidence of association exhibited two or more significant signals (Supplementary Table 4). For example, a novel missense variant (rs2075260, encoding p.Val2141Ile) at ACACB was detected and largely independent of the originally reported GWAS index rs7134594 at MVK (r2 = 0.01)2, representing novel association not previously reported. The GWAS index rs7134594 could be explained by another missense variant (rs9593 p.Met239Lys) at MMAB (conditional P = 0.73).
For gene-base analysis, nine genes (PCSK9, EVI5, HMGCR, CD36, APOA1, PCSK7, CETP, LDLR and PPARA) reached gene-based significance (P < 2.8 × 10−6) with lipid levels (Supplementary Figure 1 and Supplementary Table 5), however, no new genes were identified by gene-based analyses that weren’t already highlighted by single variant tests.
Putative functional coding variants at the known loci
Identifying coding variants in known loci has the potential to help pinpoint causal genes. We observed that the protein-altering variants are more likely to have strong effect sizes on lipid levels (Figure 3 and Supplementary Table 6), compared to the non-coding variants significantly associated with lipid levels. Ten coding variants in eight genes showed strong effects on lipid levels (beta range from 0.20 to 1.17 SD units), and 8 were low-frequency or rare variants (MAF < 3%). We next sought to quantify what proportion of GWAS loci might be due to a protein-altering variant, implicating a candidate functional gene. We make the reasonably well-supported assumption that a protein-altering variant, if the top signal, explains the signal, or is independent of the original signal, is the most likely causal variant for each region14–16. Among the 38 known loci showing association evidence at study-wide significance, 12 loci harbored a protein-altering variant that exhibited strongest association with lipid levels, while 4 loci have a protein-altering variant that was not the top signal but could explain the association of the reported index variant (Supplementary Table 7 and Figure 1). In 8 of these 16 loci (PCSK9, EVI5, CD36, MMAB, ALDH2, SLC12A4, LDLR, and PPARA), the previously identified lead variants in European populations did not reach exome-wide significance. In the remaining 8 loci (GCKR, MLXIPL, HNF1A, LPL, ABO, GPAM, PMFBP1, and TM6SF2), the GWAS index variant in each locus (P values range from 4.86×10−8 to 1.26×10−62) is in strong LD with the corresponding protein-altering variant (r2 > 0.68) and does not remain significant after accounting for the effect of the protein-altering variant (conditional P values > 0.01), suggesting that the index variant might act as a proxy for the functional protein-altering variant. Together, 42.1% (16/38) of loci appear to have a protein-altering variant that could account for the original association signal. In addition, we identified 15 protein-altering variants in 9 genes (APOB, HMGCR, ABCA1, APOA1-APOA5, ACACB, CETP, PKD1L3, LIPG, and APOE) that were independent of the original signal but may highlight functional genes in the region. All of these putative functional variants may point to functional candidate genes: either well-established causal genes (such as the genes that cause Mendelian dyslipidemias (Supplementary Table 8)) or to potential new candidate genes (MMAB, ACACB, SLC12A4, and PMFBP1). In total, the 31 protein-altering variants in the known loci may point to 25 candidate functional lipid genes.
Figure 3. Effect Size vs. Allele Frequency for variants associated with blood lipids at exome-wide significance.

The protein-altering variants are shown in red in comparison to the non-coding variants shown in black. East Asian-specific protein-altering variants are labeled in diamond. The variants shown in triangle, PCSK9 (p.Arg93Cys) and APOA5 (p.Gly185Cys), have extremely rare minor allele frequencies in Europeans, although they do not display population-specific association. The protein-altering variants show strong effects on lipid levels (beta > 0.20 SD units) are highlighted. Estimated power curves are shown (as dashed lines) for the minimum standardized effect sizes (in s.d. units) that could be identified for a given effect-allele frequency with 10% (purple), 50% (green) and 80% (blue) power, assuming sample size 47,532 and alpha level 4.5 × 10−7.
Association with coronary artery disease
To further evaluate whether the novel variants and putative functional variants in known regions identified in our samples also influenced CAD risk, we tested for association in 28,899 Chinese individuals with and without coronary disease (9,661 CAD cases and 18,558 controls) and in the largest publicly available CAD GWAS analyses (CARDIoGRAMplusC4D) of ~185,000 CAD cases and controls17 (Supplementary Table 9). For the novel non-coding variant near MCU (rs7901016), the C allele associated with lower LDL-C was similarly associated with reduced risk for CAD in Chinese samples (OR = 0.94, 95% CI = 0.90–0.98, P = 2.8×10−3) and CARDIoGRAMplusC4D (OR = 0.94, 95% CI = 0.91–0.98, P = 4.55×10−4). Among the 31 putative functional coding variants in the known regions, all the 20 non-HDL-C related variants displayed a consistent direction of effect between lipid traits and CAD. 15 out of 20 showed nominal significance (P < 0.05) in Chinese or CARDIoGRAMplusC4D CAD data, whereas 7 variants in PCSK9, APOB, LDLR, APOE, HNF1A, and APOA5 displayed significant associations even after accounting for multiple testing (P-value range from 5.95×10−4 to 8.17×10−11 < 0.05/31). In particular, nearly all of the LDL-associated coding variants demonstrated association with CAD, and the strengths of effect on CAD risk and LDL-C were highly correlated (r2 = 0.78, P = 3.3× 10−4, Supplementary Figure 2).
Novel loci identified by East Asian and GLGC samples
An exome-wide association screen for plasma lipids in >300,000 individuals genotyped by the exome array was conducted in parallel by the Global Lipids Genetics Consortium. The majorities (84%) of the participants were of European ancestry, and only 2.3% were East Asian. We further carried out large-scale trans-ancestry meta-analysis in our East Asian and GLGC samples, being careful to include overlapping samples only once, to seek both novel and population-specific genetic variants for lipid levels.
In the combined GLGC and East Asian samples, 9 additional variants showed significant associations (P < 2.1 × 10−7, Bonferroni correction of 242,289 variants analyzed in GLGC) with at least one lipid trait, that were not significant in either the East Asian or GLGC analyses. All of them are common (MAF > 0.05 in both East Asian and GLGC), including 4 coding variants (Table 2 and Supplementary Figure 3): FAM114A2 (p.Gly122Ser, HDL-C, P = 1.74 × 10−7), MGAT1 (p.Leu435Pro, HDL-C, P = 9.36 × 10−8), ASCC3 (p.Leu146Phe, LDL-C, P = 5.84 × 10−8, TC, P = 5.22 × 10−9), PLCE1 (p.Arg1575Pro, TC, P = 9.92 × 10−8).
Table 2.
Variants at novel loci associated with lipid levels identified from combined East Asian and GLGC samples
| Gene | rsID | Position | Alleles | Variants | Combined | GLGC | East Asian | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||||||
| Trait | AAF | BETA | S.E. | P | N | I2 | AAF | P | AAF | P | |||||
| PDGFC | rs4691380 | 4:157720124 | T/C | HDL-C | 0.35 | 0.014 | 0.003 | 1.07×10−7 | 335,481 | 0.54% | 0.36 | 2.80×10−7 | 0.31 | 0.14 | |
| FAM114A2 | rs2578377 | 5:153413390 | T/C | p.Gly122Ser | HDL-C | 0.67 | −0.014 | 0.003 | 1.74×10−7 | 335,481 | 4.25% | 0.63 | 2.35×10−7 | 0.87 | 0.36 |
| MGAT1 | rs634501 | 5:180218668 | G/A | p.Leu435Pro | HDL-C | 0.72 | −0.015 | 0.003 | 9.36×10−8 | 337,027 | 1.70% | 0.76 | 2.35×10−5 | 0.52 | 3.96×10−4 |
| ASCC3 | rs9390698 | 6:101296389 | A/G | p.Leu146Phe | LDL-C | 0.39 | 0.014 | 0.003 | 5.84×10−8 | 331,991 | 0.40% | 0.41 | 1.15×10−6 | 0.26 | 1.18×10−2 |
| TC | 0.39 | 0.015 | 0.003 | 5.22×10−9 | 358,251 | 0.70% | 0.41 | 1.89×10−7 | 0.26 | 5.05×10−3 | |||||
| LOC100996634 | rs884366 | 6:109574095 | A/G | HDL-C | 0.31 | −0.015 | 0.003 | 1.45×10−8 | 327,673 | 0.04% | 0.30 | 4.06×10−6 | 0.38 | 1.88×10−4 | |
| EEPD1 | rs4302748 | 7:36191699 | A/G | LDL-C | 0.18 | 0.018 | 0.003 | 2.10×10−8 | 333,359 | 4.30% | 0.20 | 5.55×10−7 | 0.09 | 3.82×10−3 | |
| PLCE1 | rs2274224 | 10:96039597 | C/G | p.Arg1575Pro | TC | 0.44 | −0.020 | 0.004 | 9.92×10−8 | 150,798 | 17.73% | 0.44 | 2.80×10−7 | 0.56 | 0.41 |
| EIF4B | rs7306523 | 12:53393964 | G/A | LDL-C | 0.70 | −0.017 | 0.003 | 1.38×10−7 | 313,750 | 1.10% | 0.77 | 1.75×10−5 | 0.29 | 1.27×10−3 | |
| TC | 0.70 | −0.017 | 0.003 | 5.36×10−8 | 338,266 | 0.00% | 0.77 | 1.42×10−6 | 0.29 | 1.15×10−2 | |||||
| SLC17A8 | rs7965082 | 12:100800193 | T/C | LDL-C | 0.52 | −0.013 | 0.002 | 9.21×10−8 | 333,359 | 0.00% | 0.54 | 1.89×10−6 | 0.41 | 1.31×10−2 | |
| TC | 0.52 | −0.014 | 0.002 | 8.28×10−9 | 358,251 | 0.00% | 0.54 | 1.47×10−6 | 0.41 | 3.86×10−4 | |||||
AAF, alternative allele frequency.
Position is reported in human genome build hg19.
Alleles are listed as alternative/reference allele on the forward strand of the reference genome.
Joint analysis of the novel signals with additional samples
To strengthen support for association, we performed in silico replication of significant variants in three additional independent genome-wide datasets, comprising a combined total of ~160,000 individuals from the Nord-Trøndelag Health Study (HUNT)18, GLGC GWAS samples2, and Chinese lipids GWAS study19. We found that the associations of 12 novel variants became more significant and reached genome-wide significance in the joint analysis (P values range from 2.99×10−8 to 7.62×10−15) (Supplementary Table 10).
Coding variants point to the same genes across ancestries
We further evaluated whether these variants identified in East Asian were also defined as putative functional variants in GLGC samples (Supplementary Table 11). We found that both East Asian and GLGC samples pointed to the same nine functional genes, but had different associated variants in each ancestry (Table 3). The eight coding variants (MAF range from 0.004% to 15.9%) at PCSK9, CD36, ABCA1, CETP, PMFBP1, LIPG, LDLR, and PPARA identified by GLGC showed lower minor allele frequencies (MAF range from 0 to 2.57%) in the East Asian samples and, thus, displayed no significance or only suggestive significance (CETP). Conversely, the coding variants at PCSK9, APOB, CD36, CETP, LDLR and PPARA identified in East Asian (MAF range from 0.094% to 12.45%) also had lower minor allele frequencies in GLGC (MAF range from 0.001% to 0.20%). In addition, the same putatively functional coding variants and genes at seven loci (GCKR, MLXIPL, LPL, GPAM, HNF1A, TM6SF2, and APOE) were identified in both East Asian and GLGC samples, with similar common minor allele frequencies (Table 4).
Table 3.
Inter-ancestry allelic heterogeneity at lipid genes.
| Protein-altering variants | Look-up in the other sample | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||
| Gene | Study | rsID | Note* | Position | Variants | Alleles | Trait | BETA | S.E. | P | AAF | Variance Explained | AAF | P |
| PCSK9 | GLGC | rs11591147 | Protein-altering is top | 1:55505647 | p.Arg46Leu | T/G | LDL-C | −0.475 | 0.011 | 0.00 | 1.48% | 0.70% | 0.01% | 0.26 |
| Asian | rs151193009 | Protein-altering is top | 1:55509585 | p.Arg93Cys | T/C | LDL-C | −0.542 | 0.029 | 7.62×10−77 | 1.32% | 0.77% | 0.01% | 7.62×10−9 | |
| APOB | GLGC | rs1367117 | Explaining index | 2:21263900 | p.Thr98Ile | A/G | LDL-C | 0.105 | 0.003 | 3.61×10−278 | 28.44% | 0.43% | 13.01% | 4.26×10−10 |
| Asian | rs13306194 | Protein-altering is top | 2:21252534 | p.Arg532Trp | A/G | LDL-C | −0.098 | 0.010 | 9.53×10−22 | 12.45% | 0.20% | 0.20% | 8.13×10−3 | |
| CD36 | GLGC | rs3211938 | Protein-altering is top | 7:80300449 | p.Tyr325* | G/T | HDL-C | 0.181 | 0.021 | 1.43×10−18 | 0.47% | 0.03% | 0.001% | 0.87 |
| Asian | rs148910227 | Protein-altering is top | 7:80302116 | p.Arg386Trp | T/C | HDL-C | 0.342 | 0.058 | 3.17×10−9 | 0.31% | 0.07% | 0.02% | 0.01 | |
| ABCA1 | GLGC | rs146292819 | Independent of index | 9:107556776 | p.Asn1800His | G/T | HDL-C | −0.843 | 0.059 | 3.99×10−46 | 0.05% | 0.07% | 0.00% | NA |
| Asian | rs2230808 | Independent of index | 9:107562804 | p.Lys1587Arg | C/T | HDL-C | 0.047 | 0.007 | 2.49×10−12 | 60.97% | 0.10% | 72.96% | 9.78×10−19 | |
| CETP | GLGC | rs5880 | Independent of index | 16:57015091 | p.Ala330Pro | C/G | HDL-C | −0.258 | 0.007 | 4.08×10−321 | 4.81% | 0.60% | 0.64% | 6.90×10−7 |
| Asian | rs2303790 | Independent of index | 16:57017292 | p.Asp459Gly | G/A | HDL-C | 0.407 | 0.025 | 7.53×10−62 | 2.23% | 0.72% | 0.02% | 3.16×10−5 | |
| PMFBP1 | GLGC | rs34832584 | Independent of index | 16:72162966 | p.Thr505Lys | T/G | TC | 0.020 | 0.003 | 1.62×10−8 | 15.93% | 0.01% | 2.57% | 0.50 |
| Asian | rs16973716 | Explaining index | 16:72156842 | p.Lys768Asn | G/T | TC | 0.042 | 0.008 | 1.75×10−7 | 28.96% | 0.07% | 44.69% | 2.66×10−7 | |
| LIPG | GLGC | rs77960347 | Independent of index | 18:47109955 | p.Asn396Ser | G/A | HDL-C | 0.259 | 0.012 | 1.62×10−98 | 1.07% | 0.14% | 0.01% | 0.79 |
| Asian | rs2000813 | Independent of index | 18:47093864 | p.Thr111Ile | T/C | HDL-C | 0.043 | 0.007 | 1.04×10−9 | 31.06% | 0.08% | 28.61% | 1.76×10−41 | |
| LDLR | GLGC | rs139043155 | Independent of index | 19:11217344 | p.Asp225Glu | A/T | LDL-C | 1.644 | 0.214 | 1.53×10−14 | 0.004% | 0.02% | 0.00% | NA |
| Asian | rs200990725 | Protein-altering is top | 19:11217315 | p.Arg257Trp | T/C | LDL-C | 0.882 | 0.109 | 6.35×10−16 | 0.094% | 0.15% | 0.001% | 1.96×10−4 | |
| PPARA | GLGC | rs1042311 | Protein-altering is top | 22:46627780 | p.Ala268Val | T/C | TC | 0.123 | 0.018 | 7.40×10−12 | 0.50% | 0.01% | 0.01% | 0.23 |
| Asian | rs1800234 | Protein-altering is top | 22:46615880 | p.Val227Ala | C/T | TG | −0.094 | 0.018 | 3.17×10−7 | 4.21% | 0.07% | 0.15% | 0.12 | |
AAF, alternative allele frequency.
Position is reported in human genome build hg19.
Alleles are listed as alternative / reference allele on the forward strand of the reference genome.
Protein-altering is top: protein-altering variants are the most significant variants in the known loci.
Explaining index: Conditional on the coding variants, the adjusted P for index variants > 0.01.
Independent of index: Conditional on the index variants, the adjusted P for coding variants with exome-wide significance.
Table 4.
Loci where East Asian and GLGC samples identified the same putatively functional protein-altering variant
| Gene | rsID | Position | Variant | Alleles | Trait | Study | BETA | S.E. | P | AAF | Variance Explained | Note* |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GCKR | rs1260326 | 2:27730940 | p.Leu446Pro | C/T | TG | GLGC | −0.121 | 0.003 | 0.00 | 0.628 | 0.64% | Protein-altering is top |
| TG | Asian | −0.114 | 0.007 | 1.26×10−62 | 0.496 | 0.64% | Protein-altering is top | |||||
| MLXIPL | rs35332062 | 7:73012042 | p.Ala358Val | A/G | TG | GLGC | −0.124 | 0.004 | 5.22×10−205 | 0.117 | 0.30% | Protein-altering is top |
| TG | Asian | −0.109 | 0.011 | 2.03×10−23 | 0.109 | 0.23% | Explaining index | |||||
| LPL | rs328 | 8:19819724 | p.Ser474* | G/C | TG | GLGC | −0.184 | 0.004 | 0.00 | 0.098 | 0.58% | Explaining index |
| TG | Asian | −0.169 | 0.012 | 1.93×10−45 | 0.095 | 0.46% | Explaining index | |||||
| GPAM | rs2792751 | 10:113940329 | p.Ile43Val | C/T | TC | GLGC | −0.028 | 0.003 | 7.14×10−22 | 0.728 | 0.03% | Explaining index |
| TC | Asian | −0.043 | 0.007 | 5.67×10−9 | 0.706 | 0.07% | Protein-altering is top | |||||
| HNF1A | rs1169288 | 12:121416650 | p.Ile27Leu | C/A | TC | GLGC | 0.037 | 0.003 | 9.99×10−40 | 0.333 | 0.06% | Protein-altering is top |
| TC | Asian | 0.038 | 0.007 | 4.86×10−8 | 0.404 | 0.07% | Protein-altering is top | |||||
| TM6SF2 | rs58542926 | 19:19379549 | p.Glu167Lys | T/C | TC | GLGC | −0.129 | 0.005 | 7.03×10−155 | 0.074 | 0.22% | Protein-altering is top |
| TC | Asian | −0.066 | 0.013 | 4.25×10−7 | 0.070 | 0.06% | Protein-altering is top | |||||
| APOE | rs7412 | 19:45412079 | p.Arg176Cys | T/C | LDL-C | GLGC | −0.539 | 0.006 | 0.00 | 0.075 | 3.80% | Independent of index |
| LDL-C | Asian | −0.472 | 0.016 | 4.87×10−197 | 0.088 | 3.49% | Protein-altering is top |
AAF, alternative allele frequency.
Position is reported in human genome build hg19.
Alleles are listed as alternative / reference allele on the forward strand of the reference genome.
Protein-altering is top: protein-altering variants are the most significant variants in the known loci.
Explaining index: Conditional on the coding variants, the adjusted P for index variants >0.01.
Independent of index: Conditional on the index variants, the adjusted P for coding variants with exome-wide significance.
East Asian-specific association signals
We next attempted to identify variants that were associated with lipids in East Asian samples only. Within the known lipid loci, 363 independent variants were identified by sequential conditional analyses in GLGC exome-wide association studies (Supplementary Table 11). After conditioning on the independent variants in the corresponding loci, we identified 14 independent coding variant associations at 11 loci in East Asian samples with conditional P values < 4.5 × 10−7 (Table 5, Figure 1 and 3). Interestingly, all 14 East Asian-specific variants are included in the list of the putative functional variants we identified. Eight of these loci (EVI5, APOB, HMGCR, CD36, APOA1, CETP, LDLR, and PPARA) harbored at least one low-frequency or rare independent coding variant (MAF range from 4.21% to 0.03%). All of these variants are either monomorphic or have a frequency at least 1 order of magnitude lower in Europeans and, thus, showed only suggestive significance in ~ 300,000 GLGC individuals.
Table 5.
East Asian-specific variants associated with blood lipids (Conditional P < 4.5 ×10−7)
| Gene | Position | rsID | Alleles | Variant | Trait | East Asian | GLGC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||
| AAF | BETA | S.E. | P | P.adj | AAF | BETA | S.E. | P | ||||||
| EVI5 | 1:93159927 | rs117711462 | A/G | p.Arg354Cys | TC | 0.69% | 0.212 | 0.040 | 1.41×10−7 | 2.15×10−7 | 0.03% | 0.097 | 0.080 | 0.245 |
| APOB | 2:21228437 | rs376825639 | G/A | p.Ile3768Thr | TC | 0.15% | −0.659 | 0.097 | 8.44×10−12 | 9.96×10−12 | ||||
| LDL-C | 0.15% | −0.579 | 0.098 | 3.35×10−9 | 4.44×10−9 | |||||||||
| 2:21252807 | noRS | T/C | p.Cys478Tyr | TC | 0.09% | −0.876 | 0.138 | 2.08×10−10 | 1.65×10−10 | |||||
| LDL-C | 0.09% | −0.772 | 0.141 | 4.19×10−8 | 3.22×10−8 | |||||||||
| 2:21252534 | rs13306194 | A/G | p.Arg532Trp | TC | 12.39% | −0.114 | 0.010 | 1.45×10−29 | 2.01×10−17 | 0.19% | −0.084 | 0.031 | 6.74×10−3 | |
| LDL-C | 12.45% | −0.098 | 0.010 | 9.53×10−22 | 2.08×10−13 | 0.20% | −0.085 | 0.032 | 8.13×10−3 | |||||
| TG | 12.43% | −0.073 | 0.010 | 1.38×10−12 | 4.96×10−15 | 0.19% | −0.133 | 0.032 | 2.96×10−5 | |||||
| HMGCR | 5:74646765 | rs191835914 | C/A | p.Tyr311Ser | LDL-C | 1.73% | −0.190 | 0.026 | 2.20×10−13 | 2.68×10−9 | 0.04% | −0.117 | 0.067 | 0.079 |
| CD36 | 7:80302116 | rs148910227 | T/C | p.Arg386Trp | HDL-C | 0.31% | 0.342 | 0.058 | 3.17×10−9 | 3.60×10−9 | 0.02% | 0.215 | 0.084 | 0.010 |
| APOA1 | 11:116707736 | rs12718465 | T/C | p.Ala61Thr | HDL-C | 3.27% | −0.116 | 0.058 | 5.50×10−10 | 1.41×10−7 | 0.02% | 0.075 | 0.099 | 0.449 |
| ACACB | 12:109696838 | rs2075260 | A/G | p.Val2141Ile | TG | 74.34% | 0.043 | 0.008 | 3.95×10−8 | 7.64×10−8 | 80.23% | 0.011 | 0.003 | 5.32×10−4 |
| ALDH2 | 12:112241766 | rs671 | A/G | p.Glu457Lys | HDL-C | 20.43% | −0.048 | 0.008 | 1.16×10−8 | 1.85×10−8 | 0.08% | −0.005 | 0.052 | 0.928 |
| CETP | 16:56997025 | rs201790757 | G/T | p.Tyr74* | HDL-C | 0.03% | 1.117 | 0.182 | 8.97×10−10 | 4.33×10−10 | 0.001% | 0.719 | 0.352 | 0.041 |
| 16:57017292 | rs2303790 | G/A | p.Asp459Gly | HDL-C | 2.23% | 0.407 | 0.025 | 7.53×10−62 | 1.89×10−31 | 0.02% | 0.384 | 0.092 | 3.16×10−5 | |
| PKD1L3 | 16:71967927 | rs17358402 | T/C | p.Arg1572His | LDL-C | 5.40% | 0.085 | 0.015 | 2.11×10−8 | 1.86×10−9 | 24.44% | −0.013 | 0.003 | 8.47×10−5 |
| TC | 5.41% | 0.088 | 0.015 | 1.96×10−9 | 1.40×10−10 | 24.44% | −0.009 | 0.003 | 3.72×10−3 | |||||
| LDLR | 19:11217315 | rs200990725 | T/C | p.Arg257Trp | TC | 0.09% | 0.677 | 0.109 | 5.57×10−10 | 5.00×10−9 | 0.001% | 1.897 | 0.502 | 1.57×10−4 |
| LDL-C | 0.09% | 0.882 | 0.109 | 6.35×10−16 | 6.15×10−15 | 0.001% | 1.869 | 0.502 | 1.96×10−4 | |||||
| PPARA | 22:46615880 | rs1800234 | C/T | p.Val227Ala | TG | 4.21% | −0.094 | 0.018 | 3.17×10−7 | 3.36×10−7 | 0.15% | −0.058 | 0.037 | 0.118 |
AAF, alternative allele frequency.
Position is reported in human genome build hg19.
Alleles are listed as alternative / reference allele on the forward strand of the reference genome.
P.adj, conditioning on the independent variants in the corresponding loci identified by GLGC exome-wide association studies (Supplementary Table 11).
Discussion
This study represents the largest discovery effort for coding variation that influences lipid levels in the East Asian population, enabling us to systemically evaluate protein-altering variants that identify candidate functional genes. Meta-analyses in East Asian and multi-ancestry samples using an exome-chip genotyping array identified twelve novel loci, five of which harbored non-synonymous variants. In the 38 known loci that were replicated, we identified 31 protein-altering variants that likely point to 25 functional lipid genes. Moreover, the same 16 putative functional genes were identified by significant association with protein-altering variants in both European and East Asian samples--at 9 of those genes by identifying independent protein-altering variants in the two ancestries.
Among the novel genetic loci identified, several have been implicated in cardiovascular and metabolic phenotypes, which may provide mechanistic insight into the regulation of lipid levels and potential targets for treatments. The significant novel variant associated with both lipids and CAD is located in intron of MCU. MCU encodes mitochondrial inner membrane calcium uniporter that mediates calcium uptake into mitochondria. It has been found that mitochondrial calcium plays an important role in the regulation of metabolism in the heart20. CD163 encodes a macrophage specific receptor involved in the clearance and endocytosis of hemoglobin-haptoglobin complexes by macrophages. Soluble CD163 was recently proposed as a biomarker of the well-known variables metabolic syndrome, including HDL-cholesterol21. ACVR1C encodes activin receptor-like kinase 7 (ALK7), one of the type I transforming growth factor-β receptors. ALK7 has recently been demonstrated to play an important role in the maintenance of metabolic homeostasis22. ALK7 is highly expressed in adipose tissue of humans and is correlated with body fat and lipids. The ALK7 dysfunction could cause increased lipolysis in adipocytes and leads to decreased fat accumulation. MGAT1 encodes Mannosyl (Alpha-1,3-)-Glycoprotein Beta-1,2-N-Acetylglucosaminyltransferase, which is involved in the synthesis of protein-bound and lipid-bound oligosaccharides. It has been found that the variant in MGAT1 was associated with body weight and obesity23. Of note, CD163 and PDGFC were also found to be associated with lipid levels by the East Asian lipids GWAS meta-analysis since our manuscript was submitted24. To further clarify the possible transcriptional mechanisms underlying the identified loci in associations with lipids, we investigated the relationships of the novel variants and proxies with expression quantitative trait loci (eQTLs) using the GTEx eQTL browser. Significant cis-eQTLs effects in human tissues were found at five loci at a significance of P < 4.5 × 10−7 (Supplementary Table 12). We further predicted putatively regulatory variants in seven novel noncoding regions in 81 cell type lines using deltaSVM scores25, and found that the variants in PDGFC, LOC100996634, and MCU had high regulatory potential with extreme deltaSVM scores greater than 10 in absolute value (Supplementary Figure 4).
Our data provided a more comprehensive understanding of the genetic architecture of lipid susceptibility by discovering novel lipid genes and revealing allelic heterogeneity across different ancestry populations. We detected multiple independent association signals or new lead variants in known lipid-associated loci that frequently displayed no or moderate linkage disequilibrium (LD) with the corresponding GWAS index variants in European populations. Specially, we identified 14 East Asian-specific variants that could not be explained by all the independent variants in the corresponding loci identified by GLGC samples. Our study demonstrated the benefits of distinct LD patterns between ancestry groups in dissecting validated loci. We also found substantial inter-ancestry differences in the identification of rare coding variants across populations, which may have been subjected to natural selection during human evolution or genetic drift. All the low-frequency or rare functional coding variants identified in East Asians (MAF range from 0.03% to 4.21%) appeared to be population-specific, and were monomorphic or not present in 1000 Genomes European individuals, this allelic heterogeneity across different ancestry populations have been partly reported6,11. However, we observed that these rare variants were not monomorphic in over 300,000 GLGC individuals, but had 15 to 160-fold lower frequencies (MAF range from 0.001% to 0.15%) in Europeans than East-Asians (Supplementary Table 13 and Supplementary Figure 5), with little power to detect association in Europeans. Similarly, the low-frequency and rare coding variants identified in GLGC samples were extremely rare or monomorphic in East Asian samples (Supplementary Figure 6 and Supplementary Table 11). Overall, our finding demonstrated that rare and low frequency coding variants are more likely to be population-specific, which underscores the value of discovering ancestry-specific rare variants in diverse populations, particularly for low frequency variation.
Since most GWAS index variants are located in non-coding regions, the identification of associated protein-coding variants may allow us to prioritize functional genes and variation. Among the 38 known loci that reached chip-wide significance in our data, coding variants at 16 loci (42.1%) were found to completely account for the original association signal. At an additional 9 loci, an independent protein-altering variant indicated a likely functional gene. The coding variants are more likely to have consistent effect sizes across ethnic groups compared to non-coding variants. For the GWAS index variants that could not be replicated in East Asian samples, the effect sizes were poorly correlated with those observed in Europeans. In contrast, the effect sizes of the putatively-functional coding variants in the same loci are strongly related across ethnic groups (Supplementary Figure 7). Trans-ancestry comparison provided additional credible evidence to support the same 16 genes as putative functional genes. The functional genes pointed to by coding variants are either well-known genes or genes with previously unknown roles in lipid metabolism (such as GPAM and PMFBP1), which may be good candidates for functional assessment. More importantly, we found the effects of these putative functional coding variants on LDL cholesterol, triglyceride, and total cholesterol were highly correlated with the effect on CAD, but the effect on HDL cholesterol levels were not correlated with CAD. Our findings are consistent with the recent genetic studies that both LDL cholesterol and triglyceride levels but not HDL cholesterol levels are causally related to CAD risk26–29.
This large-scale exome wide association study allowed us to detect a larger number of low-frequency and rare variants, 30% of which were not polymorphic in the previous exome-wide study involving 12,685 Chinese individuals11. Nonetheless, the exome array offered moderate coverage for rare variants observed in ExAC East Asian samples. Power calculations indicated that the available sample size provided 80% power to detect variants with effect size of 0.27 s.d. and MAF as low as 0.5% at P < 4.5 × 10−7. However, we had considerably less power to evaluate extremely rare variants (MAF < 0.1%). Studies in larger sample sizes and of sequenced samples are therefore needed to fully investigate associations of rare variants with lipid levels.
In conclusion, we identified 12 new loci associated with lipid levels. We also identified coding variants that highlight 25 likely functional genes at previously known loci, including several with previously undiscovered roles in lipids. We also found an abundance of population specific coding variant associations that underlie lipid traits, highlighting the importance of including individuals of diverse ancestry background. At the same time, our data demonstrate that integrating genomic data across diverse ancestry groups may enable us to determine functional variants and genes for further functional study.
Methods
Study cohorts
Twenty three studies including both population-based studies and case-control studies of coronary artery disease (CAD) and type 2 diabetes (T2D) were genotyped with the Illumina HumanExome array resulting in a total of 47,532 participants, all of whom were of East Asian ancestry (Supplementary Table 14). All participants provided written informed consent, and ethics approval for their data generation and analyses was individually obtained for each contributing study. The relevant human genetic data was also approved by Ministry of Science and Technology of China. For GLGC exome study, the studies contributed association results for exome chip genotypes and plasma lipid levels (Supplementary notes and Supplementary Table 15).
Phenotypes
For most East-Asian subjects (83%), TC, HDL-C, and TG were measured at > 8 hours of fasting. LDL-C levels were directly measured in 18 studies (88% of total study individuals) and were estimated using the Friedewald formula in the remaining studies, with missing values assigned to individuals with triglycerides >400 mg/dl. We adjusted the TC values for individuals on lipid-lowering medication by replacing their TC values by TC/0.8 with lipid medication status available. If measured LDL-C was available in a study, the treated LDL-C value was divided by 0.7. No adjustment for individuals using medication was made for HDL-C and TG.
Exome array genotyping and quality control
All study participants were genotyped on the HumanExome Bead-Chip (Illumina), and most cohorts (83%) also included the custom Asian Vanderbilt content. This custom content was added to the standard Illumina HumanExome BeadChip to improve the coverage of low frequency variants in Asian populations. The variants were selected from 1077 (581 Chinese women and 496 Singapore Chinese) whole exome sequenced East Asian samples generously provided by Wei Zheng and Jianjun Liu30. Additional approximately 29K common variants were added to the array including previously identified GWAS variants selected from the GWAS catalogue. Genotype calling was performed with GenTrain version 2.0 in GenomeStudio V2011.1 (Illumina) in combination with zCall version 2.231. Within each study, individuals with low genotype completion rates, individuals expressing gender mismatches or a high level of heterozygosity, and related individuals, and PCA outliers were excluded from further analysis (Supplementary Table 16). In addition, variants that did not meet the 95% or 98% genotyping threshold or showed deviation from the Hardy-Weinberg equilibrium were removed.
Statistical analyses
Within each cohort, HDL-C, LDL-C, TG and TC measurements were transformed using the inverse normal distribution after adjustment of each trait for age, age2, and study-specific covariates, including principal components in order to account for population structure. In studies ascertained on diabetes or cardiovascular disease status, cases and controls were analyzed separately.
We performed both single variant and gene-level association tests. Single variant analyses in each cohort were carried out using either RAREMETALWORKER or RVTESTS32, both of which generate single variant score statistics and their covariance matrix between single marker statistics. The test statistics, as visualized in a quantile–quantile plot, appeared well-calibrated (Supplementary Figure 8). Gene-based tests were restricted to variants that were predicted to alter the coding sequence of the gene product (defined as missense, stop-gain, stop-loss, or splice-site variants) in order to enhance the likelihood of identifying causal variants and to reduce the multiple testing burden. For each trait, we ran four gene-based tests: a variable threshold burden test with a MAF cutoff of <5% or <1% and a sequence kernel association test (SKAT) with a MAF cutoff of <5% or <1%. Next, the meta-analyses of single variant and gene-level association tests were performed using RAREMETALS33 for HDL-C, LDL-C, TC and TG. For single variants, we applied a significance threshold of P < 4.5 × 10−7, corresponding to a Bonferroni correction for 110,986 polymorphic variants that had at least 20 minor alleles. For gene-level tests, we used a significance threshold of P < 2.8 × 10−6, corresponding to a Bonferroni correction for 17,614 gene-level tests.
To identify putative functional coding variants accounting for the effects at known lipid loci, we performed reciprocal conditional analyses to control for the effects of known lipid GWAS or coding variants. Loci where the initial lead variant had conditional P > 0.01 were considered to be explained by the variants used in the conditional analyses. To dissect East Asian-specific association signals in the reported loci, we also performed conditional association analysis for variants within 1MB of each locus using covariance matrices between single variant association statistics. Details of the methods can be found in Liu et al32. To evaluate whether two or more independent association signals, we performed sequential conditional association analyses using the lead variant at each locus as a covariate until results after conditional analysis were no longer significant (P > 4.5× 10−7). We estimated the linkage disequilibrium (LD) metric r2 using the cohort-combined variants and LD matrices. LD for variants not included on the exome array was estimated from the 1000 Genomes Project East Asian individuals.
To further assess whether the identified functional coding variants also relate to coronary artery disease (CAD), we tested their associations with CAD in PUUMA-MI11, HKU-TRS, HuCAD34, and two GWAS samples35 (the Beijing Atherosclerosis Study (BAS) and the China Atherosclerosis Study (CAS)) involving 9,661 CAD cases and 18,558 controls. The effect estimates and s.e. were meta-analysed using METAL by the fixed-effect inverse-variance method36. We also looked up the CAD association in the largest publicly available CAD GWAS analyses (CARDIoGRAMplusC4D) of ~185,000 CAD cases and controls17.
In silico Replication Samples
The in silico replication study was conducted using additional independent individuals of European ancestry from the HUNT study18 and GLGC GWAS2, and Chinese subjects from Chinese lipids GWAS19. HUNT is a population-based cohort of 62,168 individuals with genome-wide genotypes (Illumina Human CoreExome), imputation from the Haplotype Reference Consortium panel, and non-fasting lipid phenotypes. The Chinese lipids GWAS was a meta-analysis consisting of over 13,000 Han Chinese who underwent standardized collection of blood lipid measurements in five independent genome wide association studies. These studies included the China Atherosclerosis Study (CAS), the Beijing Atherosclerosis Study (BAS), Genetic Epidemiology Network of Salt-Sensitivity (GenSalt) study37, and the China Atherosclerosis Study phase II (CAS-II).
Heritability and proportion of variance explained estimates
We estimate the proportion of variance explained by the set of independently associated variants. Joint effects of variants in a locus were approximated by , where U⃗NETA is single variant score statistics and is the covariance matrix between them. Covariance between single variant genetic effects were approximated by the inverse of the variance-covariance matrix of score statistics, i.e. . The explained phenotype variance by independently associated variants in a locus is given by .
Annotation
Variants were annotated as missense, splice, stop-gain/loss, synonymous or noncoding using ANNOVAR (version 2012-05-25)38. Variant identifiers and chromosomal positions are listed with respect to the hg19 genome build.
DeltaSVM analysis
DeltaSVM uses a gapped k-mer support vector machine to estimate the effect of a variant in a cell-type-specific manner25. Precomputed weights were available from a total of 222 ENCODE DHS samples—99 from the Duke University (Duke) set and 123 from the University of Washington (UW) set39. For the current study, genetic variants were scored for deltaSVM in 81 cell lines from four tissues (blood, blood vessel, heart and liver). For each of the seven novel noncoding regions, all proxies (r2 > 0.8) were identified using data from 1000 genome.
Data Availability
Summary statistics have been made available for download from http://csg.sph.umich.edu/abecasis/public/lipids2017EastAsian. Additional supporting data are provided in the supplementary material.
Supplementary Material
Acknowledgments
We thank all the participants of this study for their contributions. X. Lu is supported by the National Science Foundation of China (81422043, 91439202, 81370002, and 81773537) and CAMS Innovation Fund for Medical Sciences (2016-I2M-1-009, 2016-I2M-1-011). C.J.W. is supported by HL135824 and S.K. and C.J.W. are supported by HL127564. Additional acknowledgments of funding sources for the primary studies are provided in the Supplementary Note.
Footnotes
URLs.
Genotype-Tissue Expression (GTEx) Portal, http://www.gtexportal.org/home
Genezoom, http://genome.sph.umich.edu/wiki/Genezoom
ExAC, http://exac.broadinstitute.org
RareMETALS, http://genome.sph.umich.edu/wiki/RareMETALS
RVTESTS, http://genome.sph.umich.edu/wiki/RvTests
RAREMETALWORKER, http://genome.sph.umich.edu/wiki/RAREMETALWORKER
Author Contributions
Drafting of the manuscript: X.Lu, C.J.W, G.M.P., D.J.L., D.G., K.L.M. Project coordination: C.J.W., D.G., X.Lu, P.C.S., S.K., K.L.M., Y.E.C. Central meta-analysis group: X.Lu, D.J.L., G.M.P., H.Z. eQTL analysis: X.Lu, J.B.N. DeltaSVM analysis X.Lu, W.Zhou. Cohort data analyst: X.Lu, G.M.P., D.J.L., Y.Wu, H.Z., J.Li, C.S.T., R.D., J.Long, X.G., C.N.S., Y.C., Y.Wang, C.Y.Y.C, Q.F., J.S., X.Y., W.Zhao, M.H., J.B.N. Cohort genotyping: W.Zhou, H.L., C.C.K., J.Liu, L.W., F.W., J.S., W.H. Cohort phenotyping: H.L., M.X., X.Liu, Y.Z., L.S., Y.G., Y.Hu, K.Y., J.H., Q.C., S.C., A.B.F., L.S.A., P.G., S.D., K.H., L.F. Cohort Principal investigators: X.Lu, W.H.S., S.S.C., A.B.F., L.S.A., P.G., S.D., R.V., Y.I.C., X.O.S., K.S.L.L, T.Y.W., S.K.G., Z.M., K.H., L.F., H.T., Y.Huo, C.C., Y.E.C., W.Zheng, E.S.T., W.G., X.Lin, W.H., G.A., S.K., K.L.M., T.W., P.C.S., D.G., C.J.W.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Teslovich TM, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–13. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willer CJ, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–83. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Surakka I, et al. The impact of low-frequency and rare variants on lipid levels. Nat Genet. 2015;47:589–97. doi: 10.1038/ng.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asselbergs FW, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet. 2012;91:823–38. doi: 10.1016/j.ajhg.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim YJ, et al. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat Genet. 2011;43:990–995. doi: 10.1038/ng.939. [DOI] [PubMed] [Google Scholar]
- 6.Wu Y, et al. Trans-ethnic fine-mapping of lipid loci identifies population-specific signals and allelic heterogeneity that increases the trait variance explained. PLoS Genet. 2013;9:e1003379. doi: 10.1371/journal.pgen.1003379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert NG, et al. Risk factors and biomarkers of age-related macular degeneration. Prog Retin Eye Res. 2016 doi: 10.1016/j.preteyeres.2016.04.003. S1350–9462(16)30012-X. [DOI] [PMC free article] [PubMed]
- 8.Herder C, Kowall B, Tabak AG, Rathmann W. The potential of novel biomarkers to improve risk prediction of type 2 diabetes. Diabetologia. 2014;57:16–29. doi: 10.1007/s00125-013-3061-3. [DOI] [PubMed] [Google Scholar]
- 9.Wierzbicki AS, Oben J. Nonalcoholic fatty liver disease and lipids. Curr Opin Lipidol. 2012;23:345–52. doi: 10.1097/MOL.0b013e3283541cfc. [DOI] [PubMed] [Google Scholar]
- 10.Kathiresan S. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med. 2008;358:2299–300. doi: 10.1056/NEJMc0707445. [DOI] [PubMed] [Google Scholar]
- 11.Tang CS, et al. Exome-wide association analysis reveals novel coding sequence variants associated with lipid traits in Chinese. Nat Commun. 2015;6:10206. doi: 10.1038/ncomms10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peloso GM, et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet. 2014;94:223–32. doi: 10.1016/j.ajhg.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lange LA, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014;94:233–45. doi: 10.1016/j.ajhg.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmen OL, et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat Genet. 2014;46:345–51. doi: 10.1038/ng.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanna S, et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikpay M, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krokstad S, et al. Cohort Profile: the HUNT Study, Norway. Int J Epidemiol. 2013;42:968–77. doi: 10.1093/ije/dys095. [DOI] [PubMed] [Google Scholar]
- 19.Lu X, et al. Genetic Susceptibility to Lipid Levels and Lipid Change Over Time and Risk of Incident Hyperlipidemia in Chinese Populations. Circ Cardiovasc Genet. 2016;9:37–44. doi: 10.1161/CIRCGENETICS.115.001096. [DOI] [PubMed] [Google Scholar]
- 20.Williams GS, Boyman L, Lederer WJ. Mitochondrial calcium and the regulation of metabolism in the heart. J Mol Cell Cardiol. 2015;78:35–45. doi: 10.1016/j.yjmcc.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parkner T, Sørensen LP, Nielsen AR, Fischer CP, Bibby BM, Nielsen S, Pedersen BK, Møller HJ. Soluble CD163: a biomarker linking macrophages and insulin resistance. Diabetologia. 2012;55:1856–62. doi: 10.1007/s00125-012-2533-1. [DOI] [PubMed] [Google Scholar]
- 22.Carlsson LM, et al. ALK7 expression is specific for adipose tissue, reduced in obesity and correlates to factors implicated in metabolic disease. Biochem Biophys Res Commun. 2009;382:309–14. doi: 10.1016/j.bbrc.2009.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johansson A, et al. Linkage and genome-wide association analysis of obesity-related phenotypes: association of weight with the MGAT1 gene. Obesity (Silver Spring) 2010;18:803–8. doi: 10.1038/oby.2009.359. [DOI] [PubMed] [Google Scholar]
- 24.Spracklen CN, et al. Association analyses of East Asian individuals and trans-ancestry analyses with European individuals reveal new loci associated with cholesterol and triglyceride levels. Hum Mol Genet. 2017;26:1770–1784. doi: 10.1093/hmg/ddx062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee D, et al. A method to predict the impact of regulatory variants from DNA sequence. Nat Genet. 2015;47:955–61. doi: 10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Do R, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–52. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenson RS, Davidson MH, Hirsh BJ, Kathiresan S, Gaudet D. Genetics and causality of triglyceride-rich lipoproteins in atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2014;64:2525–40. doi: 10.1016/j.jacc.2014.09.042. [DOI] [PubMed] [Google Scholar]
- 28.Helgadottir A, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48:634–9. doi: 10.1038/ng.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voight BF, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, et al. Rare coding variants and breast cancer risk: evaluation of susceptibility Loci identified in genome-wide association studies. Cancer Epidemiol Biomarkers Prev. 2014;23:622–8. doi: 10.1158/1055-9965.EPI-13-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldstein JI, et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28:2543–5. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu DJ, et al. Meta-analysis of gene-level tests for rare variant association. Nat Genet. 2014;46:200–4. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng S, Liu D, Zhan X, Wing MK, Abecasis GR. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics. 2014;30:2828–9. doi: 10.1093/bioinformatics/btu367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu X, et al. Coding-sequence variants are associated with blood lipid levels in 14,473 Chinese. Hum Mol Genet. 2016;25:4107–4116. doi: 10.1093/hmg/ddw261. [DOI] [PubMed] [Google Scholar]
- 35.Lu X, et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat Genet. 2012;44:890–4. doi: 10.1038/ng.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genome wide association scans. Bioinformatics. 2010;26:2190–1. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.GenSalt Collaborative Research G. GenSalt: rationale, design, methods and baseline characteristics of study participants. J Hum Hypertens. 2007;21:639–46. doi: 10.1038/sj.jhh.1002207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Summary statistics have been made available for download from http://csg.sph.umich.edu/abecasis/public/lipids2017EastAsian. Additional supporting data are provided in the supplementary material.