

Globular glial tauopathy (GGT) is a recently introduced 4-repeat (4R) tauopathy with frontotemporal lobar degeneration (FTLD) characterized by taupositive globular oligodendroglial (GOIs) and astrocytic (GAIs) inclusions [1]. To date, thirty-nine sporadic and ten hereditary GGT cases due to MAPT mutations (eight of a p.K317M, one of a p.K317N, and one of a p.P301L) have been reported [2].
Chorea is one of the abnormal involuntary and hyperkinetic movements which has rarely been reported in FTLD [3]. Here, we report a Korean GGT case presenting nonfluentagrammatic primary progressive aphasia (nfvPPA) who developed generalized chorea in the late stage of disease. To our knowledge, this is the first described GGT case exhibiting chorea as one of the clinical symptoms.
A 78-year-old right handed woman presented with a 4-year-history of progressive dysarthria, difficulty with expressive language and gait. The first symptom was slurred speech at age 74, followed by gait unsteadiness when walking on uneven or inclined surfaces. At age 77, she demonstrated hesitant speech and tended to express herself using one word (e.g. yes/no). Past medical history was significant for diabetes mellitus (DM). Family history was unremarkable. Neurological examination revealed severe dysarthria, generalized bradykinesia, mild clumsiness and rigidity on bilateral upper extremities, and reduced step length. In language evaluation, the patient showed severely impaired spontaneous speech with relatively preserved comprehension. Speech hesitancy, labored speech, varying degrees of phonological errors, sound distortion, and agrammatism were identified, but single-word comprehension and object knowledge were spared (Supplementary Table). Korean Mini-Mental Status Examination score was 25/30, Clinical Dementia Rating 0.5, Global Deteriorating Scale 4 and Barthel index 20. Brain MRIs revealed diffuse cortical atrophy, worse on the frontal than parietal with prominent left opercular atrophy and FDG -PET showed predominant left inferior frontal and opercular hypometabolism (Fig. 1A). Based on clinical and neuroimaging findings, her clinical diagnosis was nfvPPA [4]. At the age of 79, she developed generalized choreic movements involving the neck, trunk, and four extremities while walking or sitting. Genetic testing for Huntington’s disease (HD) was negative. At age 80, her choreiform movements became worse and had an exaggerated quality when she was agitated. At age 82, she reached a predominantly bedridden state and the chorea was detected only in the right lower extremity. She died 8 years after the disease onset.
Fig.1.
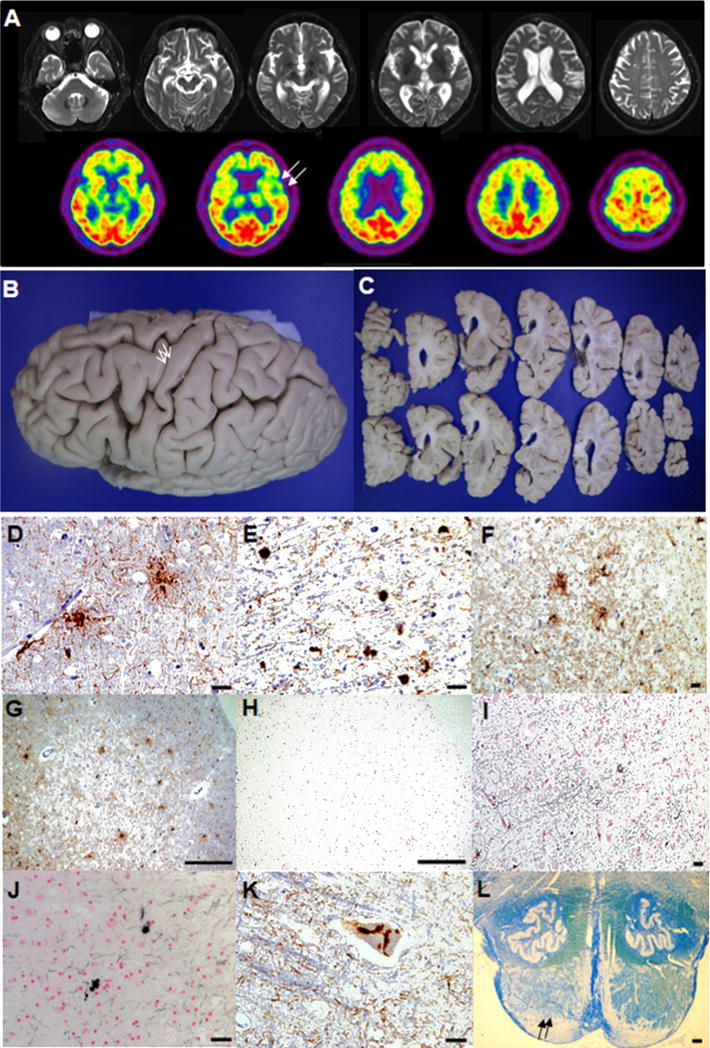
(A)Brain MRIs at age 78 demonstrated diffuse cortical atrophy, worse on the frontal than parietal with dilated ventricle (upper row). 18[F]-fluorodeoxyglucose (FDG)-PET showed glucose hypometabolism in bilateral frontotemporal area including severe glucose hypometabolism in the left frontal opercular area (lower row, arrows). (B)Widening of the superior and middle frontal sulci, and grossly preserved precentral cortex (arrows) were detected in the formalin-fixed left hemisphere. (C)The coronal sections of the left hemisphere displayed moderate atrophy in dorsolateral frontal and inferior frontal cortices with mildly dilated frontal horn and the lateral ventricle. (D)GAIs in the middle frontal cortex (CP-13, scale bar = 25 μm). (E)GOIs and coiled bodies in the subcortical white matter of superior frontal sulcus (CP-13, scale bar = 25 μm). (F)4R tau positive GAIs and threads in the precentral cortex (RD 4, scale bar = 250 μm). (G)Tau immunoreactive GAIs in the prefrontal cortex (CP-13, scale bar = 250 μm). (H, I)No evidence of Gallyas positive GAIs was detected in the same prefrontal cortex as (G) (Gallyas, scale bar = 250 μm) and the superior frontal sulcus (Gallyas, scale bar = 250 μm). (J)Gallyas positive GOIs and coiled bodies in the subcortical white matter of superior frontal sulcus (Gallyas, scale bar = 25 μm). (K)Tau immunoreactive neuronal cytoplasmic inclusions in Betz cell of precentral cortex (CP-13, scale bar = 25 μm). (L)Myelin pallor (arrows) was seen in the pyramid of the pontomedullary junction (Luxol Fast Blue stain, scale bar = 500 μm).
Gross observation revealed moderate frontal atrophy with the brain weight of 1012g (Fig. 1B, C). Microscopic features are illustrated in Fig. 1D-L, including numerous tau immunoreactive GAIs in the affected cortices and profound subcortical white matter glial tau pathology (GOIs and coiled bodies). Both GAIs and GOIs were immunore-active for 4R tau. However, while GOIs and coiled bodies were positive with Gallyas silver stain, most GAIs were negative with Gallyas, which is consistent with GGT. Genetic screening for the MAPT gene did not reveal any known pathogenic variants. Tau haplotype was H1/H1.
The three pathological subtypes of GGT has been proposed based on the location of tau inclusions [1]. Our case might be classified as Type III, because the main 4R-pathologies were mostly observed in both gray and white matter of the frontotemporal cortex including the precentral gyrus with demyelination of the corticospinal tract. However, one recent case series suggested that subtyping GGT cases seemed to be a debatable issue [5].
In terms of clinical features, GGT is known to present with a varying combination of frontotemporal dementia (FTD), motor neuron disease (MND) and/or extrapyramidal symptoms [1]. Out of these diverse phenotypes, bvFTD and nfvPPA are the most frequently described [5]. The clinical presentation of the current case was nfvPPA without any clinical evidence of MND at the initial assessment. Nevertheless, we could assume that MND might have been developed in advanced stages based on postmortem examinations of the involvement of motor cortex and the degeneration of the corticospinal tract.
Most published GGT cases are sporadic. To date, three pathogenic variants (p.K317M, p.K317N, p.P301L) in the MAPT gene have been associated with GGT [2]. While the most common subtype of sporadic GGT is Type I, Type III is frequently reported in familial cases. However, mutation screening of the MAPT gene in our case (Type III) revealed a negative result.
Of great interest was that the patient developed chorea in the late stage of disease. Chorea is one of the key clinical features of HD associated with striatal atrophy. Even though the association between HD phenocopy syndrome and C9ORF72 expansions has recently been described, chorea rarely occurs in patients with underlying FTLD, especially FTLD-tau [3]. Likewise, chorea has never been reported in GGT as a combined extrapyramidal symptom. Although Fu et al. reported three sporadic Japanese GGT cases showing “involuntary movements”, detailed description was not given [6]. Because diabetes was well controlled when chorea developed, it is not as likely that diabetes was the cause of the patient’s choreic movement. Chorea can be caused by diffuse or focal damage of the cerebral cortex, basal ganglia, substantia nigra, subthalamic nucleus, or cerebellum [3] which can be affected in GGT or other 4R tauopathies (e.g. Progressive supranuclear palsy). Further clinicopathological comparison studies between GGT cases with and without chorea are needed to improve our understanding of the pathophysiological mechanism of chorea in GGT.
Supplementary Material
Acknowledgments
We thank our patient and his family for donating brain to the Pusan National University Hospital Brain Bank to contribute to dementia research. We are grateful to Peter Davies, Feinstein Institute for Medical Research, North Shore-Long Island Jewish Health Care System who provided CP-13 antibody. This study was supported by the Original Technology Research Program for Brain Science through the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (No. 2014M3C7A1064752).
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.parkreldis.2017.09.006.
Footnotes
Financial disclosure
None reported.
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standard
The study has been approved by the institutional review boards and has therefore been performed in the accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Contributor Information
Eun-Joo Kim, Department of Neurology, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Myung Jun Lee, Department of Neurology, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Jae-Hyeok Lee, Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, South Korea.
Young Min Lee, Department of Psychiatry, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Jin-Hong Shin, Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, South Korea.
Myung-Jun Shin, Department of Rehabilitation Medicine, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Kyung-Un Choi, Department of Pathology, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Na-Yeon Jung, Department of Neurology, Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, South Korea.
Kyoungjune Pak, Department of Nuclear Medicine, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Chungsu Hwang, Department of Pathology, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Jae Woo Ahn, Department of Pathology, Pusan National University Hospital, Pusan National University School of Medicine and Medical Research Institute, Busan, South Korea.
Suk Sung, Department of Anatomy, Pusan National University School of Medicine and Medical Research Institute, Yangsan, South Korea.
Salvatore Spina, Department of Neurology, Memory and Aging Center, University of California, San Francisco, CA, USA.
Lea T. Grinberg, Department of Neurology, Memory and Aging Center, University of California, San Francisco, CA, USA
William W. Seeley, Department of Neurology, Memory and Aging Center, University of California, San Francisco, CA, USA
Gi Yeong Huh, Department of Forensic Medicine, Pusan National University School of Medicine and Medical Research Institute, Yangsan, South Korea.
References
- 1.Ahmed Z, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol. 2013;126:537–544. doi: 10.1007/s00401-013-1171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tacik P, et al. Clinicopathologic heterogeneity in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) due to microtubuleas sociated protein tau (MAPT) p.P301L mutation, including a patient with globular glial tauopathy, Neuropathol. Appl Neurobiol. 2017;43:200–214. doi: 10.1111/nan.12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawakami I, et al. Chorea as a clinical feature of the basophilic inclusion body disease subtype of fused-in-sarcoma-associated frontotemporal lobar degeneration. Acta Neuropathol Commun. 2016;4:36. doi: 10.1186/s40478-016-0304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorno-Tempini ML, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014. doi: 10.1212/WNL.0b013e31821103e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burrell JR, et al. Expanding the phenotypic associations of globular glial tau subtypes. Alzheimers Dement (Amst) 2016;4:6–13. doi: 10.1016/j.dadm.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu YJ, et al. Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, Parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol. 2010;120:21–32. doi: 10.1007/s00401-010-0649-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.