

Abstract
Objective
To identify rare causal variants underlying known loci that segregate with late‐onset Alzheimer's disease (LOAD) in multiplex families.
Methods
We analyzed whole genome sequences (WGS) from 351 members of 67 Caribbean Hispanic (CH) families from Dominican Republic and New York multiply affected by LOAD. Members of 67 CH and additional 47 Caucasian families underwent WGS as a part of the Alzheimer's Disease Sequencing Project (ADSP). All members of 67 CH families, an additional 48 CH families and an independent CH case‐control cohort were subsequently genotyped for validation. Patients met criteria for LOAD, and controls were determined to be dementia free. We investigated rare variants segregating within families and gene‐based associations with disease within LOAD GWAS loci.
Results
A variant in AKAP9, p.R434W, segregated significantly with LOAD in two large families (OR = 5.77, 95% CI: 1.07–30.9, P = 0.041). In addition, missense mutations in MYRF and ASRGL1 under previously reported linkage peaks at 7q14.3 and 11q12.3 segregated completely in one family and in follow‐up genotyping both were nominally significant (P < 0.05). We also identified rare variants in a number of genes associated with LOAD in prior genome wide association studies, including CR1 (P = 0.049), BIN1 (P = 0.0098) and SLC24A4 (P = 0.040).
Conclusions and Relevance
Rare variants in multiple genes influence the risk of LOAD disease in multiplex families. These results suggest that rare variants may underlie loci identified in genome wide association studies.
Introduction
Late‐onset Alzheimer's disease (LOAD) is the most common form of dementia in older adults, it lacks an effective treatment and represents an enormous societal burden. The disease is characterized by progressive deterioration of memory and cognitive functions, leading to loss of autonomy and ultimately requiring full‐time medical care. Pathologically, LOAD is defined by severe neuronal loss, aggregation of amyloid β (Aβ) in extracellular senile plaques, and formation of intra‐neuronal neurofibrillary tangles consisting of hyper‐phosphorylated tau protein.
Over the past decade, genetic research in LOAD has been dominated by genome‐wide association studies (GWAS), a hypothesis‐free scan of the genome using dense genotyping arrays based on common variants (single nucleotide polymorphisms, SNPs). Several genes within LOAD susceptibility loci cluster in specific pathways,1, 2, 3, 4, 5, 6 including amyloid processing, oxidative stress and immune or inflammatory pathways. Collectively, GWAS demonstrates that apart from the strongest risk factor, APOE‐ε4 a large number of loci with modest effect size also contribute to LOAD risk. Common variants identified through GWAS may not have functional consequences, simply reflecting linkage disequilibrium with the unobserved causal variants. It is also possible that these causal variants are rare and have large effects, such as TREM2, 7, 8, 9, 10, 11, 12, 13 and are not covered by commercially available GWAS platforms. In fact, putatively damaging variants have already been identified (for example TREM2, SORL1, and ABCA7) in some of these LOAD susceptibility loci, advancing our understanding of disease risk.14, 15, 16
Whole genome sequencing (WGS) provides a comprehensive and detailed investigation of human genetic variation allowing interrogation of coding and noncoding regions of the genome. Increasingly, WGS studies have provided the strongest evidence that rare genetic variants can have large cumulative effects on human diseases.17, 18, 19, 20 Family‐based studies represent an implicit enrichment strategy for identifying rare variants.21 Transmission of variants from parents to offspring maximizes the chance that multiple copies of rare variants exist in the pedigree. Moreover, compared with analyses using unrelated samples, sequencing in families provides data concerning identical‐by‐descent or sharing among relatives, greatly reducing false positives and permitting detection of sequencing errors, while facilitating the identification of alleles that cause genetic disorders.22, 23
In the current report, we focused on Caribbean Hispanic families multiply affected by LOAD. The frequency of LOAD among multiplex families from the Dominican Republic is known to be significantly higher (5‐fold) than expected for similarly aged individuals in a non‐Hispanic white population from the United States.24 Furthermore, we found that a moderate degree of inbreeding was present and a predictor of LOAD risk in this population.25 As part of the national Alzheimer's Disease Sequencing Project (ADSP), we identified several chromosomal regions with strong evidence for linkage in Caribbean Hispanic families with multiple LOAD cases.26 In the present study, we used WGS data (Data S1) from 67 families as discovery to detect rare variants in previously identified linkage regions and in previously reported LOAD candidate genes. Genotyping in additional WGS on additional 47 Caucasian families were used to replicate the findings from the CH families.
Material and Methods
Family characteristics
All participants (Table 1) were recruited after providing informed consent and with approval by the relevant institutional review boards both in the United States and in the Dominican Republic. Patients, unaffected family members and healthy unrelated controls were required to have had standard neuropsychological tests and neurological examinations to verify their clinical status and diagnosis. Most individuals have been evaluated on multiple occasions over the past 10 years. Families in which patients had known early‐onset disease mutations in APP, PSEN1, PSEN2, GRN, or MAPT were excluded from this analysis to increase power of discovery variants predisposing to LOAD. All selected probands came from families with three or more affected individuals recruited in the Dominican Republic and New York. Recruitment for this family study began in 1998, and was restricted to Caribbean Hispanics,27, 28 predominately from the Dominican Republic. As a part of the ADSP, a set of 67 CH families and 47 Caucasian families were selected for whole genome sequencing from approximately 1400 families reviewed. Selection was based on the number of affected individuals, the number of generations affected, age at onset of clinical symptoms, and presence of APOEε4 alleles.29
Table 1.
Demographics of the Caribbean‐Hispanic families and case–control cohorts used in WGS and validation genotyping
| Pedigrees Sequenced in ADSP discovery | |
| Number of pedigrees | 67 |
| Total number of subjects sampled | 860 |
| Average subjects sampled per pedigree | 12.84 ± 7.28 |
| Total number of subjects sequenced | 351 |
| Average samples sequenced in each pedigree | 5.24 ± 1.67 |
| Affected | 302 |
| Unaffected | 49 |
| Average affected per pedigree | 7.42 ± 3.61 |
| Age (sequenced individuals) | 73.02 ± 10.0 |
| Women (%) | 58.72 |
| APOE | 17.03% |
| Additional pedigrees used in validation genotyping | |
| Number of pedigrees | 48 |
| Total number of subjects sampled | 404 |
| Average subjects sampled per pedigree | 8.41 ± 4.8 |
| Average affected per pedigree | 5.08 ± 2.3 |
| Age | 70.76 ± 10.11 |
| Women (%) | 65 |
| APOE | 29.90% |
| Unrelated case–control set | |
| Total number of subjects sampled | 450 |
| No of affected individuals | 152 |
| Age (affecteds) | 85.5 ± 6.5 |
| Age | 79.3 ± 6.7 |
| Women (%) | 68.7 |
| APOE | 12.80% |
Whole Genome Sequence analysis of GWAS linkage peaks
Details about WGS methodology and variant calling pipelines can be found in Data S1. Three regions demonstrating linkage previously identified26 using GWAS data were prioritized for WGS data analysis. Results from genome‐wide linkage analysis revealed a strong linkage signal (HLOD = 3.6) on chromosomal region 3q29.26 We previously reported microsatellite linkage analyses in this region as strongly associated with LOAD in Caribbean Hispanics,30 including a single family that achieved a LOD score of 1.28. WGS data was generated for four of the family members with LOAD. Analysis of the WGS data was limited to an approximate 2 Mb region encompassing the 1‐LOD interval around linkage peak (3q29: 197,052,973 bp–199,212,658 bp). We also selected two chromosomal regions where we observed genome‐wide significant linkage and association signals across the families: 7p14.3 and 11q12.3.26 Annotation of the identified variants in these linkage regions was performed using the ANNOVAR software.31
Whole genome sequence variants prioritization
Consensus linkage regions
We followed a principled filtering strategy (Fig. 1, Table S4) to test the segregation of rare variants with LOAD status in the three linkage regions: 7p14.3 and 11q12.3 and 3q29 (Table S6). The first criterion used to prioritize the identified variants was based on familial segregation of cases. We required that at least 75% of the patients with LOAD and WGS data in the families were carriers. Additionally we also prioritized variants that were observed in at least five affected individuals in multiple Caribbean Hispanic families.
Figure 1.
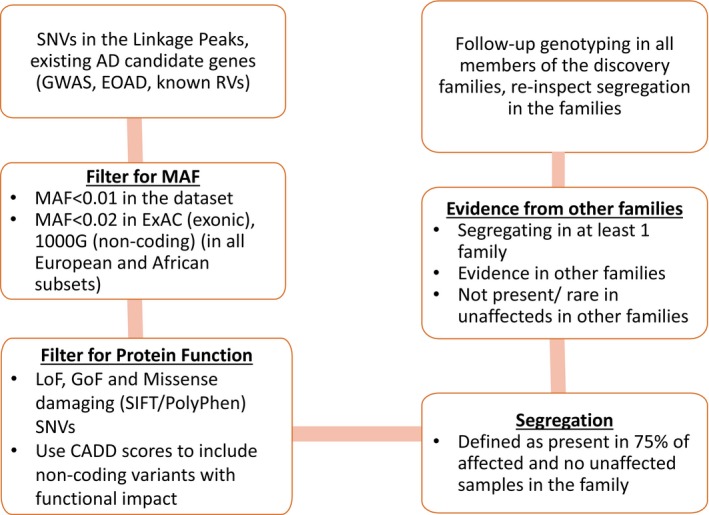
Filtering strategy to prioritize variants for follow‐up analyses in the validation sample.
We filtered variants with minor allele frequency (MAF) of less than 0.10 in the ADSP families and with an MAF of 0.02 in Caucasian and African American populations in ExAC database (for exonic variants)32 and 1000 genomes project (for intronic and intergenic variants)33 to restrict our analyses to rare variants in the population. Functional annotations were also used as filtering strategy, that is, variants were analyzed in order of increasing priority: exonic, inter‐genic, intronic and others (non‐coding RNA, 3′/5′ UTR, upstream/downstream gene). We also included any intronic or intergenic variants with a Combined Annotation Dependent Depletion (CADD)34 score of greater than 30 to include highly conserved and putative non‐exonic regulatory variants in the analyses. Further criteria for variant prioritization included, additional LOAD patients from different families carrying the variant and that the variant was either not present or present in very low frequency in the non‐demented family members.
Follow‐up genotyping validation
To confirm putative variants that segregated in 67 discovery families and candidate GWAS loci, we genotyped all of the family members in the discovery families, an additional 48 families and unrelated, unaffected controls of the same ancestry and age (Table 1). The validation cohort consisted of Caribbean Hispanic individuals from 115 multiplex families and 450 unrelated cases and controls from the same ethnic background (Table 1). Population allele frequencies for novel variants were estimated from unaffected persons in the Caribbean Hispanic population from samples/families that we have amassed over 15 years. Genotypes were generated using the KASPTM genotyping technology which uses allele‐specific polymerase chain reaction for accurate calling of single nucleotide variants (SNVs) and indels.35
Single variant association analyses
SNV genetic associations were restricted to individuals 60 years of age or older and were tested using General Linear Models in generalized estimating equations (GEE) to adjust for familial correlation in the data. The family ID was used as a repeat measure to account for correlation in the genotype data within family members. All analyses were adjusted for sex, age at last evaluation, education, APOE‐ε4 genotype and the first ten principal components.
LOAD GWAS and candidate loci
We tested genes associated with LOAD in large GWAS studies with multiple replication reports. We also included known early‐onset AD genes and genes implicated in earlier sequencing efforts in LOAD.1, 2, 3, 4, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15, 16 Candidate genes evaluated included: APP, PSEN1, PSEN2, GRN, MAPT, TREM2, PLD3, APOE, ABCA7, SORL1, CR1, BIN1, CD2AP, EPHA1, CLU, MS4A6A, PICALM, CD33, HLA‐DRB5, HLA‐DRB1, PTK2B, SLC24A4, RIN3, INPP5D, MEF2C, NME8, ZCWPW1, CELF1, FERMT2, CASS4, TREML2, and AKAP9.
Gene‐based association analyses
Gene‐based F‐SKAT36 analyses were conducted for genes in the three consensus linkage regions. The F‐SKAT test was restricted to variants defined as exonic and rare (<2% in Europeans, African Americans and overall in the ExAC database). To define a testable‐set, we used two sets of variants in the gene‐based test based on functional annotation: (1) Stop‐loss, stop gain and missense damaging variants (as defined by SIFT37 or PolyPhen38) and (2) including all non‐synonymous variants. We also tested gene‐sets that included both loss of function variants and variants that had a high CADD score (>30). The CADD score filter captures putatively non‐functional variants in the non‐coding regions. For most genes (Table S5), we did not observe variants in non‐coding regions with a high CADD score. In addition functional characterization of GWAS loci have indicated that cis‐regulation is a common mechanism underlying these associations.39 The most frequent elements affected are transcriptional enhancers and silencers that regulate transcription through long‐range interactions, typically located more than 1Kb from their target genes. Therefore, to examine the top GWAS signals,4 we have considered a chromosomal region of 1 Mb upstream and 1 Mb downstream the reported GWAS loci. We conducted F‐SKAT analyses of genes within ±1 Mb encompassing the reported LOAD‐GWAS SNPs. Each gene was independently assessed for association with LOAD using F‐SKAT with the modified R code to adjust for family structure. We tested models of association also adjusting for (1) age, sex, population substructure, ancestry proportions for African‐American and native American ancestry (Data S1) and (2) including APOE‐ε4 dosage with other covariates. Analyses were subsequently repeated adjusting for the possible effect of the reported GWAS SNP to assess the additional contribution of rare variants in conferring LOAD risk. We also conducted region‐based FSKAT test combining variants from all the genes in the 7p14.3 and 11q12.3 regions to assess the joint burden of all variants together in the region.
Results
Families
As detailed in Table 1, 67 Caribbean‐Hispanic families underwent WGS. The families consisted of 351 individuals (302 individuals with LOAD and 49 unaffected family members) with average age of 73.02 ± 10 years, 59% were women. Seventeen percent were carriers of APOE‐ε4 allele. On average, approximately five individuals were sequenced within each of the families. For validation genotyping, we genotyped all the members of the 67 families, an additional 48 Hispanic families and an independent elderly case control cohort of the same ethnic background. The additional 48 Hispanic families had similar characteristics to the discovery families (Table 1). These families also had, on average, five affected individuals, but had a higher frequency for the APOE ε4 allele (29.9%).
Linkage region in a single Caribbean‐Hispanic family at 3q29
After applying quality control metrics, we identified a total of 6551 sequence variants under the 3q29 linkage peak. Applying the filters in the prioritization pipeline, we identified 11 rare SNVs: six intronic and five inter‐genic variants. The 11 identified SNVs were genotyped in 1,720 Caribbean Hispanic unrelated individuals. The strongest association with LOAD (β = 0.83, SE = 0.45, P = 0.064) was found for an intronic variant rs186972238 in the LRCH3 gene. A complex pattern of inheritance with incomplete penetrance emerged when the variant segregation pattern was evaluated within each of these families.
Locus 7q14.3
A total of 35,376 high quality SNVs were observed in 67 families from the WGS experiment and subsequent QC, of which 26,654 SNVs were observed a frequency of <0.10 in the ADSP families. Applying the filtering criteria described above (Fig. 1), we observed 12 non‐synonymous and one synonymous mutation that were observed in at least two affected individuals and absent in unaffected members (Table S1).
Locus 11q12.3
A total of 19,106 SNVs were observed in the 11q12.3 linkage regions in the 67 ADSP families of which 149 SNVs were putatively damaging (stop‐loss, stop‐gain or damaging classified by SIFT or PolyPhen) and novel or rare (Fig. 1). Forty variants were observed to be perfectly or imperfectly segregating in at least one family. Remarkably, p. V707I in TMEM132A was observed in two families with complete segregation in one family. (Table S2)
Analyses of loci under linkage peaks
We selected 14 variants under the linkage peaks (10 from 11q12.3 and 4 from 7q14.3) for follow‐up genotyping in additional family members and unrelated controls. We chose variants that fulfilled at least one of the following criteria: (1) variants observed in two or more Caribbean Hispanic families in at least five affected LOAD individuals, and further prioritized if observed in affected individuals in Caucasian families or showing segregation with LOAD and (2) variants observed in at least four affected LOAD individuals and no unaffected individuals from one Caribbean Hispanic family with CADD score >15. We tested SNV association with LOAD using a GEE model to adjust for familial correlation. We found nominally significant association (P < 0.05) for two SNVs each in MYRF and ASRGL1, respectively, (Table 2).
Table 2.
Results from follow‐up genotyping
| NO APOE‐ε4a | APOE‐ε4b | ||||||
|---|---|---|---|---|---|---|---|
| SNV | BETA | SE | P | BETA | SE | P | GENE |
| 1‐207680070‐C‐T | 0.13 | 0.21 | 5.2E‐01 | 0.15 | 0.21 | 4.6E‐01 | CR1 |
| 1‐207741237‐C‐T | 1.61 | 0.99 | 1.0E‐01 | 1.60 | 0.96 | 9.6E‐02 | CR1 |
| 7‐29035428‐C‐T | 0.82 | 0.44 | 6.3E‐02 | 0.80 | 0.43 | 6.2E‐02 | CPVL |
| 7‐30876316‐C‐T | −0.58 | 0.42 | 1.7E‐01 | −0.50 | 0.41 | 2.3E‐01 | FAM188B |
| 7‐31682771‐C‐T | 0.87 | 1.25 | 4.9E‐01 | 0.86 | 1.26 | 4.9E‐01 | CCDC129 |
| 7‐91630531‐C‐T | 1.75 | 0.86 | 4.1E‐02 | 1.84 | 0.88 | 3.8E‐02 | AKAP9 |
| 7‐91667736‐A‐G | 0.35 | 0.35 | 3.2E‐01 | 0.35 | 0.38 | 3.5E‐01 | AKAP9 |
| 7‐143091417‐C‐T | 0.18 | 0.68 | 8.0E‐01 | 0.19 | 0.67 | 7.7E‐01 | EPHA1 |
| 11‐60703423‐G‐A | 0.06 | 0.45 | 8.9E‐01 | 0.00 | 0.43 | 1.0E+00 | TMEM132A |
| 11‐61015721‐G‐A | 0.93 | 0.54 | 8.5E‐02 | 0.81 | 0.54 | 1.3E‐01 | PGA5 |
| 11‐61120560‐G‐A | ‐0.08 | 0.58 | 8.9E‐01 | ‐0.07 | 0.57 | 9.0E‐01 | CYB561A3 |
| 11‐61250246‐G‐A | 0.10 | 0.50 | 8.4E‐01 | ‐0.02 | 0.47 | 9.6E‐01 | PPP1R32 |
| 11‐61546888‐G‐A | ‐0.55 | 0.26 | 3.8E‐02 | ‐0.49 | 0.26 | 5.9E‐02 | MYRF |
| 11‐62286666‐C‐T | 0.72 | 0.73 | 3.2E‐01 | 0.75 | 0.75 | 3.2E‐01 | AHNAK |
| 11‐62343562‐G‐C | 1.09 | 0.50 | 2.8E‐02 | 0.96 | 0.51 | 5.9E‐02 | ASRGL1 |
| 11‐62344743‐C‐T | 0.72 | 0.73 | 3.2E‐01 | 0.75 | 0.75 | 3.2E‐01 | MIR3654 |
| 11‐62400108‐G‐A | ‐0.31 | 0.36 | 4.0E‐01 | ‐0.26 | 0.36 | 4.7E‐01 | GANAB |
| 11‐62886345‐G‐A | 0.21 | 0.54 | 6.9E‐01 | 0.24 | 0.54 | 6.6E‐01 | SLC3A2 |
| 14‐93142861‐T‐C | 0.29 | 0.42 | 5.0E‐01 | 0.31 | 0.42 | 4.6E‐01 | RIN3 |
| 19‐1057919‐C‐T | 0.62 | 0.70 | 3.8E‐01 | 0.58 | 0.67 | 3.9E‐01 | ABCA7 |
Model‐ LOAD ~ Gene + Age + Sex + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components.
Model‐ LOAD ~ Gene + Age + Sex + number of APOE ε4 alleles + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components. (nominally significant variants are highlighted in bold).
Segregation of variants in established GWAS loci
We evaluated the segregation with LOAD of the 147 observed rare functional variants in 30 candidate genes implicated in LOAD by GWAS,4, 5 next‐generation sequencing or associated with early‐onset Alzheimer's disease. Sixteen of these variants were segregating in at least one family (Table S3). Remarkably, five missense variants in AKAP9 segregated with LOAD status in one or more families, p.R434W (Figs. 2 and 3, Tables S9 and S10) and p.I1448V (Fig. S1) were subsequently genotyped in the all members of these families, and in an additional 48 families and an independent case–control set to determine complete segregation and test association with LOAD. We also genotyped two missense variants in CR1 and one variant each, from EPHA1, RIN3, and ABCA7 (Table 2) and used GEE adjusting for familial correlation. p.R434W was nominally associated with LOAD adjusting for age and sex (OR = 5.77; 95% confidence interval [CI] 1.07–30.9), P = 0.041) and APOE‐ε4 (OR = 6.3; CI: 1.11–35.54, P = 0.038). p.R434W also segregated with LOAD status in two large families (Fig. 2). p.R434W was predicted to be damaging by SIFT and Polyphen and had a highly deleterious CADD score of 32. Previously implicated variants in AKAP9 40 (rs144662445 and rs149979685) were each observed in one LOAD patient.
Figure 2.
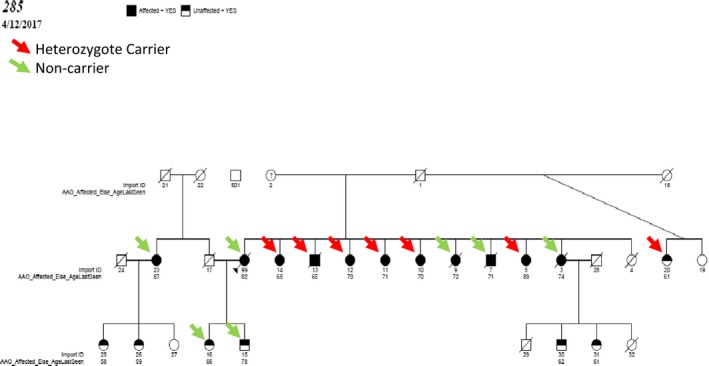
Segregation pattern of AKAP9 p.R434W in family 285 (Refer to Table S8 for conversion for EFIGA to ADSP ids).
Figure 3.
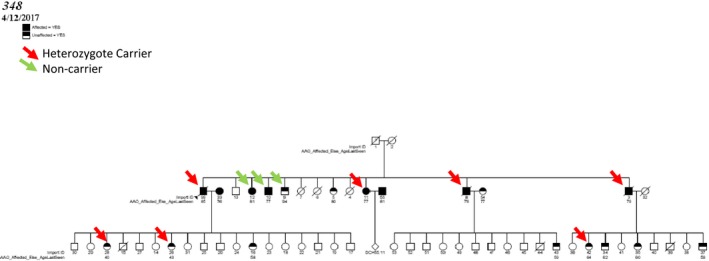
Segregation pattern of AKAP9 p.R434W in family 348 (Refer to Table S9 for conversion for EFIGA to ADSP ids).
F‐SKAT gene based tests
Nominal association was found for CR1 (P = 0.049), SLC24A4 (P = 0.040) and BIN1 (P = 0.0098). CR1 and SLC24A4 were also significant in replication study of the Caucasian families (P = 0.040 and 0.002, respectively). (Table 3). However, only rare variants in BIN1 (P = 0.026) remained significant after adjusting for the significant GWAS‐SNP that was previously associated with LOAD (Table 4).
Table 3.
F‐SKAT analyses of AD candidate GWAS loci
| GENE | N_LOF | No APOEa | APOEb |
|---|---|---|---|
| ABCA7 | 12 | 0.6655 | 0.7486 |
| AKAP9 | 9 | 0.6659 | 0.5895 |
| BIN1 | 3 | 0.0098 | 0.0139 |
| CASS4 | 2 | 0.3814 | 0.3871 |
| CIT | 6 | 0.4767 | 0.5128 |
| CR1 | 9 | 0.0490 | 0.0830 |
| DSG2 | 5 | 0.4623 | 0.3587 |
| EPHA1 | 3 | 0.8159 | 0.8659 |
| FRMD4A | 2 | 0.1807 | 0.1418 |
| INPP5D | 4 | 0.5328 | 0.6067 |
| MAPT | 5 | 0.7163 | 0.7605 |
| PSEN1 | 2 | 0.5914 | 0.7302 |
| RIN3 | 5 | 0.8367 | 0.8526 |
| SLC24A4 | 3 | 0.0433 | 0.0505 |
| SORL1 | 3 | 0.4285 | 0.4552 |
| TREM2 | 2 | 0.9846 | 0.9619 |
Model‐ LOAD ~ Gene + Age + Sex + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components.
Model‐ LOAD ~ Gene + Age + Sex + number of APOE ε4 alleles + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components.
Table 4.
Results from FSKAT gene‐based analyses of all the genes within 1 MB of the AD loci implicated in the Lambert et al. report
| GENE | Chr | NO ADJUSTMENT FOR IGAP SNPa | ADJUSTING FOR IGAP SNPb | Top IGAP SNP | Nearest Gene | ||||
|---|---|---|---|---|---|---|---|---|---|
| N_NS | NO APOE | APOE | N_NS | NO APOE | APOE | ||||
| MSH5 | 6 | 5 | 1.77E‐03 | 0.00246474 | 5 | 1.81E‐03 | 2.61E‐03 | rs9271192 | HLA‐DRB5 |
| HLA‐DQA2 | 6 | 3 | 7.37E‐03 | 6.50E‐03 | 3 | 6.64E‐03 | 6.01E‐03 | rs9271192 | HLA‐DRB5 |
| CYP3A43 | 7 | 6 | 5.69E‐03 | 4.09E‐03 | 6 | 6.92E‐03 | 5.09E‐05 | rs1476679 | ZCWPW1 |
| TAF6 | 7 | 4 | 3.97E‐02 | 3.11E‐02 | 4 | 1.44E‐02 | 1.67E‐04 | rs1476679 | ZCWPW1 |
| MAP3K2 | 2 | 3 | 1.56E‐02 | 1.48E‐02 | 3 | 1.46E‐02 | 1.28E‐02 | rs6733839 | BIN1 |
| ZSCAN21 | 7 | 2 | 8.40E‐02 | 1.06E‐01 | 2 | 1.80E‐02 | 4.10E‐03 | rs1476679 | ZCWPW1 |
| BIN1 | 2 | 4 | 2.77E‐02 | 3.36E‐02 | 4 | 2.59E‐02 | 2.88E‐02 | rs6733839 | BIN1 |
| CHGA | 14 | 4 | 3.39E‐02 | 3.31E‐02 | 4 | 3.46E‐02 | 3.56E‐02 | rs10498633 | SLC24A4 |
| NAT16 | 7 | 2 | 7.30E‐02 | 8.31E‐02 | 2 | 4.55E‐02 | 1.05E‐01 | rs1476679 | ZCWPW1 |
| LGMN | 14 | 4 | 5.24E‐02 | 2.74E‐02 | 4 | 4.70E‐02 | 2.29E‐02 | rs10498633 | SLC24A4 |
| TNXB | 6 | 25 | 4.79E‐02 | 5.19E‐02 | 25 | 4.91E‐02 | 5.38E‐02 | rs9271192 | HLA‐DRB5 |
| TFR2 | 7 | 6 | 1.04E‐01 | 8.82E‐02 | 6 | 1.12E‐01 | 1.14E‐02 | rs1476679 | ZCWPW1 |
Model‐ LOAD ~ Gene + Age + Sex + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components (without and without APOE ε4 adjustment).
Model‐ LOAD ~ Gene + Age + Sex + number of APOE ε4 alleles + proportion of African ancestry + proportion of Native American Ancestry + First ten principal components + Corresponding IGAP SNP (without and without APOE ε4 adjustment).
As described in Table 4, the strongest LOAD associations after adjusting for the effect of the corresponding common variants were found in genes located in three loci: HLA‐DRB5, ZCWPW1 and BIN1. MSH5 (P = 0.0026) and HLA‐DQA2 (P = 0.006) located in HLA‐DRB5 region, and CYP3A43 gene (5.09E‐05) located in ZCWPW1 region were significantly associated with LOAD. LGMN in the SLC24A4 region was modestly associated with LOAD in a model adjusted for APOE dosage (P = 0.033).
Discussion
A variant in AKAP9, (p.R434W), a gene previously associated with LOAD in a case–control study among African Americans,40 segregated in two large families and was nominally associated with LOAD, with fivefold increased risk adjusted for age, sex, and APOE‐ε4. The two different variants in AKAP9 were previously identified, were considered rare in populations African‐descent, and were not present in European‐descent or East Asian‐descent individuals in the 1000 Genomes database. AKAP9 is located on chromosome 7q21–22, and it encodes a member of the A kinase anchoring protein (AKAP) family. The A‐kinase anchor proteins (AKAPs) are structurally diverse proteins that bind to the regulatory subunits of protein kinase A (PKA), confining the holoenzyme to discrete locations within the cell. Alternate splicing of this gene results in at least two isoforms that localize to the centrosome and the Golgi apparatus, and interact with numerous signaling proteins from multiple signal transduction pathways.41 AKAP9 is also expressed in the cerebral cortex, hippocampus, and cerebellum, and is involved in the cytoskeletal attachment of NMDA receptors, important for controlling synaptic plasticity and memory function.42
In addition, missense mutations in Myelin Gene Regulatory Factor (MYRF) and Asparaginase‐Like Protein 1 (ASRGL1) genes under linkage peak on chromosome 11q12.3 segregated completely in one LOAD family and tested nominally significant (P < 0.05) in association with LOAD in follow‐up genotyping. The MYRF gene encodes a transcription factor that is required for central nervous system myelination and may regulate oligodendrocyte differentiation. It increases expression of genes that affect myelin production. Mutant huntingtin mice show progressive neurological symptoms and early death, as well as age‐dependent demyelination and reduced expression of myelin genes that are downstream of MYRF.43A missense mutation in ASRGL1 has been associated with autosomal recessive retinal degeneration,44 but it has not been previously related to Alzheimer's disease or neurodegenerative disorders. In patients with retinal degeneration, variants in ASRGL1 are thought to be the cause of protein misfolding, intracellular mis‐routing and aggregation of misfolded proteins. It is expressed in brain, specifically in astrocytes and oligodendrocytes.45
Examination of known LOAD genes (e.g., from large GWAS‐based meta‐analyses) confirmed the role of rare functional variation in CR1 (P = 0.049), BIN1 (P = 0.0098), and SLC24A4 (P = 0.040). CR1 encodes a transmembrane glycoprotein in the innate immune system. It is a receptor for the complement components C3b and C4b. It has been consistently identified as a risk factor in Alzheimer's disease.3, 4, 46 BIN1 encodes the bridging integrator 1 gene, which has also been consistently associated with Alzheimer's disease in GWAS studies.4, 47 BIN1 is present in neurons, oligodendrocytes and microglia, and its primary role is thought to be in endocytosis and trafficking in neurons and immune response in glia cells.48 SLC24A4 is a solute carrier that has been associated with pigmentation traits in European populations. However, more relevant to LOAD is its association with blood pressure in African Americans, as LOAD may be influenced by vascular disease.49
Of the genes identified within the 2 Mb region surrounding the previously identified GWAS loci from the IGAP study,1, 2, 3, 4, 5, 6 several were nominally significant. CYP3A43 is a member of the cytochrome P450 superfamily of enzymes. The cytochrome P450 proteins catalyze many reactions involved in drug metabolism and synthesis of cholesterol, steroids and other lipids, and are part of a cluster of cytochrome P450 genes on chromosome 7q21.1. MSH5 encodes a member of the mutS family of proteins that are involved in DNA mismatch repair or meiotic recombination processes. Women with premature ovarian failure were found to carry a mutation in MSH5 suggesting a role in meiotic recombination. Genetic variants in a gene within the major histocompatibility complex, HLA‐DQA2, were associated with LOAD in this study. However, this gene has low expression in brain and has been associated with multiple inflammatory disorders.
Family‐based WGS on previously localized linkage regions remains a particularly powerful strategy for causal variant identification. Several new disease susceptibility genes have been successfully identified using linkage analysis coupled with WGS, in complex phenotype disorders such as hearing impairment,50 familial goiters,51 and familial hypertension.52
Taken together the results here imply that rare variants in multiple genes are likely to increase the risk of LOAD. Large families multiply affected by LOAD are extremely helpful in identifying novel rare variants even in genes previously investigated by other means. For example, loci identified by genome wide array studies have pointed to a large number of genes many of which have rare variants suggesting that these loci were in linkage disequilibrium with one or more of the mutations. How variants in these multiple genes lead to a common phenotype of LOAD needs to be investigated. However, investigations of gene functions may point to potential targets for new treatments or preventive measures.
Conflict of Interest
The authors have no conflicts of interest to disclose.
Supporting information
Data S1. Methods.
Figure S1. Details of carriers of pV707I variant in TMEM132A in Caribbean Hispanic family 860.
Table S1. WGS rare exonic variants segrgating with LOAD in chromosome 7q14.3 linkage region.
Table S2. Interesting variants segregatating with affection status in Chr 11 Linkage region.
Table S3. Rare missense variants in candidate LOAD genes segregating with AD in Caribbean Hispanic families.
Table S4. Variant count across the chromosomes and linkage regions at each filtering step (corresponding to Fig. 1).
Table S5. Gene‐based test for variants sets that including variants with CADD>30.
Table S6. Caribbean Hispanic families contributing to the linkage signals on chromosomes 7 and 11.
Table S7. Details of carriers of pv707I variant in TMEM132A.
Table S8. For family 284, individual and family IDs are displayed as EFIGSA IDs.
Table S9. For familiy 348, individual and family IDs are displayed as EFIGA IDs.
Acknowledgments
EFIGA and WHICAP cohorts: Data collection for this project was supported by the Genetic Studies of Alzheimer's disease in Caribbean Hispanics (Estudio familiar de la genética de la enfermedad de Alzheimer, also known as EFIGA) and the Washington Heights and Inwood Community Aging Project (WHICAP) funded by the National Institute on Aging (NIA) and by the National Institutes of Health (NIH) (5R37AG015473, RF1AG015473, R56AG051876, 1RF1AG054023). We acknowledge the EFIGA and WHICAP study participants and the research and support staff for their contributions to this study. ADSP: The Alzheimer's Disease Sequencing Project (ADSP) is comprised of two Alzheimer's Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer's Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non‐governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak‐Vance, Mayeux, and Haines); U01AG049505 to Dr. Seshadri; U01AG049506 to Dr. Boerwinkle; U01AG049507 to Dr. Wijsman; and U01AG049508 to Dr. Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr. Goate and U01AG052410 to Dr. Pericak‐Vance. Data generation and harmonization in the Follow‐up Phases is supported by U54AG052427 (to Drs. Schellenberg and Wang).Cohorts sequenced in ADSP: The ADGC cohorts include: Adult Changes in Thought (ACT), the Alzheimer's Disease Centers (ADC), the Chicago Health and Aging Project (CHAP), the Memory and Aging Project (MAP), Mayo Clinic (MAYO), Mayo Parkinson's Disease controls, University of Miami, the Multi‐Institutional Research in Alzheimer's Genetic Epidemiology Study (MIRAGE), the National Cell Repository for Alzheimer's Disease (NCRAD), the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐LOAD), the Religious Orders Study (ROS), the Texas Alzheimer's Research and Care Consortium (TARC), Vanderbilt University/Case Western Reserve University (VAN/CWRU), the Washington Heights‐Inwood Columbia Aging Project (WHICAP) and the Washington University Sequencing Project (WUSP), the Columbia University Hispanic‐ Estudio Familiar de Influencia Genetica de Alzheimer (EFIGA), the University of Toronto (UT), and Genetic Differences (GD). The CHARGE cohorts, with funding provided by 5RC2HL102419 and HL105756, include the following: Atherosclerosis Risk in Communities (ARIC) Study which is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), Austrian Stroke Prevention Study (ASPS), Cardiovascular Health Study (CHS), Erasmus Rucphen Family Study (ERF), Framingham Heart Study (FHS), and Rotterdam Study (RS). CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the National Heart, Lung, and Blood Institute (NHLBI), with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the National Institute on Aging (NIA). A full list of principal CHS investigators and institutions can be found at CHS‐NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The three LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), and the Washington University Genome Institute (U54HG003079). NCRAD and NIAGADS: Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer's Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer's Disease Centers (ADCs), and the National Alzheimer's Coordinating Center (NACC, U01AG016976) and the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA, and at the Database for Genotypes and Phenotypes (dbGaP) funded by NIH. This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations.
ADSP consortium members
Baylor College of Medicine: Michelle Bellair, Huyen Dinh, Harsha Doddapeneni, Shannon Dugan‐Perez, Adam English, Richard A. Gibbs, Yi Han, Jianhong Hu, Joy Jayaseelan, Divya Kalra, Ziad Khan, Viktoriya Korchina, Sandra Lee, Yue Liu, Xiuping Liu, Donna Muzny, Waleed Nasser, William Salerno, Jireh Santibanez, Evette Skinner, Simon White, Kim Worley, Yiming Zhu. Boston University: Alexa Beiser, Yuning Chen, Jaeyoon Chung, L. Adrienne Cupples, Anita DeStefano, Josee Dupuis, John Farrell, Lindsay Farrer, Daniel Lancour, Honghuang Lin, Ching Ti Liu, Kathy Lunetta, Yiyi Ma, Devanshi Patel, Chloe Sarnowski, Claudia Satizabal, Sudha Seshadri, Fangui Jenny Sun, Xiaoling Zhang. Broad Institute : Seung Hoan Choi, Eric Banks, Stacey Gabriel, Namrata Gupta. Case Western Reserve University : William Bush, Mariusz Butkiewicz, Jonathan Haines, Sandra Smieszek, Yeunjoo Song. Columbia University : Sandra Barral, Phillip L De Jager, Richard Mayeux, Christiane Reitz, Dolly Reyes, Giuseppe Tosto, Badri Vardarajan. Erasmus Medical University : Shahzad Amad, Najaf Amin, M Afran Ikram, Sven van der Lee, Cornelia van Duijn, Ashley Vanderspek. Medical University Graz : Helena Schmidt, Reinhold Schmidt; Mount Sinai School of Medicine : Alison Goate, Manav Kapoor, Edoardo Marcora, Alan Renton; Indiana University : Kelley Faber, Tatiana Foroud; National Center Biotechnology Information : Michael Feolo,Adam Stine; National Institute on Aging : Lenore J. Launer; Rush University : David A. Bennett; Stanford University : Li Charlie Xia; University of Miami : Gary Beecham, Kara Hamilton‐Nelson, James Jaworski, Brian Kunkle, Eden Martin, Margaret Pericak‐Vance, Farid Rajabli, Michael Schmidt; University of Mississippi : Thomas H. Mosley; University of Pennsylvania : Laura Cantwell, Micah Childress, Yi‐Fan Chou, Rebecca Cweibel, Prabhakaran Gangadharan, Amanda Kuzma, Yuk Yee Leung, Han‐Jen Lin, John Malamon, Elisabeth Mlynarski, Adam Naj, Liming Qu, Gerard Schellenberg, Otto Valladares, Li‐San Wang, Weixin Wang, Nancy Zhang; University of Texas Houston : Jennifer E. Below, Eric Boerwinkle, Jan Bressler, Myriam Fornage, Xueqiu Jian, Xiaoming Liu; University of Washington : Joshua C. Bis, Elizabeth Blue, Lisa Brown, Tyler Day, Michael Dorschner, Andrea R. Horimoto, Rafael Nafikov, Alejandro Q. Nato Jr., Pat Navas, Hiep Nguyen, Bruce Psaty, Kenneth Rice, Mohamad Saad, Harkirat Sohi, Timothy Thornton, Debby Tsuang, Bowen Wang, Ellen Wijsman, Daniela Witten; Washington University : Lucinda Antonacci‐Fulton, Elizabeth Appelbaum, Carlos Cruchaga, Robert S. Fulton, Daniel C. Koboldt, David E. Larson, Jason Waligorski, Richard K. Wilson.
Consortium authors are listed in Acknowledgements.
Funding Statement
This work was funded by National Institute on Aging grants 5R37AG015473, R01 AG033193, R56AG051876, RF1AG015473, U01 AG032984, U01AG049505, U01AG049506, U01AG049507, U01AG049508, U01AG052410, U01AG052411, U54AG052427, and UF1AG047133.
Contributor Information
Richard Mayeux, Email: rpm2@cumc.columbia.edu.
The Alzheimer's Disease Sequencing Project:
Mosley, Laura Cantwell, Micah Childress, Yi‐Fan Chou, Rebecca Cweibel, Prabhakaran Gangadharan, Amanda Kuzma, Han‐Jen Lin, John Malamon, Elisabeth Mlynarski, Adam Naj, Liming Qu, Gerard Schellenberg, Otto Valladares, Weixin Wang, Nancy Zhang, Below, Eric Boerwinkle, Jan Bressler, Myriam Fornage, Xueqiu Jian, Xiaoming Liu, Bis, Elizabeth Blue, Lisa Brown, Tyler Day, Michael Dorschner, Nafikov, Pat Navas, Hiep Nguyen, Bruce Psaty, Kenneth Rice, Mohamad Saad, Harkirat Sohi, Timothy Thornton, Debby Tsuang, Bowen Wang, Ellen Wijsman, Elizabeth Appelbaum, Carlos Cruchaga, Daniel C Koboldt, and Jason Waligorski
References
- 1. Harold D, Abraham R, Hollingworth P, et al. Genome‐wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099. [DOI] [PubMed] [Google Scholar]
- 4. Lambert JC, Ibrahim‐Verbaas CA, Harold D, et al. Meta‐analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 2013;45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late‐onset Alzheimer's disease. Nat Genet 2011;43:436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome‐wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging. 2013;34:1711e1715–1711e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bertram L, Parrado AR, Tanzi RE. TREM2 and neurodegenerative disease. N Engl J Med. 2013;369:1565. [DOI] [PubMed] [Google Scholar]
- 9. Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reitz C, Mayeux R. Alzheimer's Disease Genetics C. TREM2 and neurodegenerative disease. N Engl J Med 2013;369:1564–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruiz A, Dols‐Icardo O, Bullido MJ, et al. Assessing the role of the TREM2 p.R47H variant as a risk factor for Alzheimer's disease and frontotemporal dementia. Neurobiol Aging 2014;35:444e441–444e444. [DOI] [PubMed] [Google Scholar]
- 13. Slattery CF, Beck JA, Harper L, et al. R47H TREM2 variant increases risk of typical early‐onset Alzheimer's disease but not of prion or frontotemporal dementia. Alzheimer's Dement 2014;10:602–608e604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Guennec K, Nicolas G, Quenez O, et al. ABCA7 rare variants and Alzheimer disease risk. Neurology 2016;86:2134–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vardarajan BN, Zhang Y, Lee JH, et al. Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 2015;77:215–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vardarajan BN, Ghani M, Kahn A, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome‐wide association studies loci. Ann Neurol 2015;78:487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fritsche LG, Igl W, Bailey JN, et al. A large genome‐wide association study of age‐related macular degeneration highlights contributions of rare and common variants. Nat Genet 2016;48:134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao J, Akinsanmi I, Arafat D, et al. A burden of rare variants associated with extremes of gene expression in human peripheral blood. Am J Hum Genet 2016;98:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet 2016;48:634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet 2015;97:199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Preston MD, Dudbridge F. Utilising family‐based designs for detecting rare variant disease associations. Ann Hum Genet 2014;78:129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naj AC, Lin H, Vardarajan BN, et al. Quality Control (QC) and Multi‐Pipeline Genotype Consensus Calling Strategies for 578 whole genomes and 10,692 whole exomes in the Alzheimer's Disease Sequencing Project (ADSP). Presented at the 63rd Annual Meeting of The American Society of Human Genetics, 9th October 2015, Baltimore. 2015.
- 23. Roach JC, Glusman G, Smit AF, et al. Analysis of genetic inheritance in a family quartet by whole‐genome sequencing. Science 2010;328:636–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vardarajan BN, Faber KM, Bird TD, et al. Age‐specific incidence rates for dementia and Alzheimer disease in NIA‐LOAD/NCRAD and EFIGA families: National Institute on Aging Genetics Initiative for Late‐Onset Alzheimer Disease/National Cell Repository for Alzheimer Disease (NIA‐LOAD/NCRAD) and Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA). JAMA Neurol 2014;71:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vardarajan BN, Schaid DJ, Reitz C, et al. Inbreeding among Caribbean Hispanics from the Dominican Republic and its effects on risk of Alzheimer disease. Genet Med 2015;17:639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barral S, Cheng R, Reitz C, et al. Linkage analyses in Caribbean Hispanic families identify novel loci associated with familial late‐onset Alzheimer's disease. Alzheimer's Dement 2015;11:1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee JH, Cheng R, Barral S, et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Arch Neurol 2011;68:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Romas SN, Santana V, Williamson J, et al. Familial Alzheimer disease among Caribbean Hispanics: a reexamination of its association with APOE. Arch Neurol 2002;59:87–91. [DOI] [PubMed] [Google Scholar]
- 29. Beecham GW, Bis JC, Martin ER, et al. The Alzheimer's disease sequencing project: study design and sample selection. Neurol Genet 2017;3:e194; Under Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee JH, Cheng R, Santana V, et al. Expanded genomewide scan implicates a novel locus at 3q28 among Caribbean Hispanics with familial Alzheimer disease. Arch Neurol 2006;63:1591–1598. [DOI] [PubMed] [Google Scholar]
- 31. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Genomes Project C , Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Abrams KR, Gillies CL, Lambert PC. Meta‐analysis of heterogeneously reported trials assessing change from baseline. Stat Med 2005;24:3823–3844. [DOI] [PubMed] [Google Scholar]
- 36. Yan Q, Tiwari HK, Yi N, et al. A sequence kernel association test for dichotomous traits in family samples under a generalized linear mixed model. Hum Hered 2015;79:60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 38. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 2013;93:779–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Logue MW, Schu M, Vardarajan BN, et al. Two rare AKAP9 variants are associated with Alzheimer's disease in African Americans. Alzheimer's Dement 2014;10:609–618e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carnegie GK, Means CK, Scott JD. A‐kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life 2009;61:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin JW, Wyszynski M, Madhavan R, et al. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci 1998;18:2017–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huang B, Wei W, Wang G, et al. Mutant huntingtin downregulates myelin regulatory factor‐mediated myelin gene expression and affects mature oligodendrocytes. Neuron 2015;85:1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Biswas P, Chavali VR, Agnello G, et al. A missense mutation in ASRGL1 is involved in causing autosomal recessive retinal degeneration. Hum Mol Genet 2016;25:2483–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Konopka G, Friedrich T, Davis‐Turak J, et al. Human‐specific transcriptional networks in the brain. Neuron 2012;75:601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fonseca MI, Chu S, Pierce AL, et al. Analysis of the putative role of CR1 in Alzheimer's Disease: genetic association, expression and function. PLoS ONE 2016;11:e0149792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kamboh MI, Demirci FY, Wang X, et al. Genome‐wide association study of Alzheimer's disease. Transl Psychiat 2012;2:e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tan MS, Yu JT, Tan L. Bridging integrator 1 (BIN1): form, function, and Alzheimer's disease. Trends Mol Med 2013;19:594–603. [DOI] [PubMed] [Google Scholar]
- 49. Rosenthal SL, Kamboh MI. Late‐onset Alzheimer's disease genes and the potentially implicated pathways. Curr Genet Med Rep 2014;2:85–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Santos‐Cortez RL, Faridi R, Rehman AU, et al. Autosomal‐recessive hearing impairment due to rare missense variants within S1PR2. Am J Hum Genet 2016;98:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yan J, Takahashi T, Ohura T, et al. Combined linkage analysis and exome sequencing identifies novel genes for familial goiter. J Hum Genet 2013;58:366–377. [DOI] [PubMed] [Google Scholar]
- 52. Louis‐Dit‐Picard H, Barc J, Trujillano D, et al. KLHL3 mutations cause familial hyperkalemic hypertension by impairing ion transport in the distal nephron. Nat Genet 2012;44:456–460; S451‐453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Methods.
Figure S1. Details of carriers of pV707I variant in TMEM132A in Caribbean Hispanic family 860.
Table S1. WGS rare exonic variants segrgating with LOAD in chromosome 7q14.3 linkage region.
Table S2. Interesting variants segregatating with affection status in Chr 11 Linkage region.
Table S3. Rare missense variants in candidate LOAD genes segregating with AD in Caribbean Hispanic families.
Table S4. Variant count across the chromosomes and linkage regions at each filtering step (corresponding to Fig. 1).
Table S5. Gene‐based test for variants sets that including variants with CADD>30.
Table S6. Caribbean Hispanic families contributing to the linkage signals on chromosomes 7 and 11.
Table S7. Details of carriers of pv707I variant in TMEM132A.
Table S8. For family 284, individual and family IDs are displayed as EFIGSA IDs.
Table S9. For familiy 348, individual and family IDs are displayed as EFIGA IDs.