

Abstract
Objective
The aim of this study was to preliminarily evaluate an oral small molecule p38α kinase inhibitor in patients with early Alzheimer's disease (AD) for the effects on brain amyloid plaque load and episodic memory function, and to establish pharmacokinetic–pharmacodynamics correlations if any effects identified on these parameters.
Methods
Sixteen patients with early AD received a highly selective p38α inhibitor (neflamapimod) for 84 days (12 weeks). To obtain a broad range of plasma drug exposures, subjects randomized to receive either 40 mg (n = 9) or 125 mg (n = 7) twice daily. Dynamic, 11C‐PiB positron emission scans were performed at baseline and at Day 84 and quantitatively analyzed by reference parametric mapping. Episodic memory assessed as Wechsler Memory Scale (WMS) immediate and delayed recall composites.
Result
In the 11C‐PiB analyses there were no main group level effects, though in the prespecified responder analysis (>7% reduction in 11C‐PiB signal) there were three responders in the 40 mg, and one in the 125 mg group. There were statistically significant increases from baseline in mean WMS immediate recall score and WMS delayed recall at both day 28 (P = 0.03 and P = 0.001) and day 84 (P = 0.001 and P < 0.001). Individual subject plasma drug concentration profiles were significantly positively correlated with the change in combined WMS immediate and delayed recall (P < 0.0001, r 2= 0.70). Within‐subject effect size was 0.59 for immediate recall and 0.67 for delayed recall.
Interpretation
Selective p38α inhibition in patients with early AD may improve episodic memory and potentially impact β‐amyloid production. These preliminary clinical findings support conduct of a longer duration placebo‐controlled study, particularly to confirm the effects on episodic memory function.
Introduction
A broad range of preclinical studies indicates that the alpha isoform of p38 mitogen‐activated protein kinase (p38α kinase) is a robust therapeutic target for Alzheimer's disease (AD).1, 2, 3, 4, 5 Traditionally p38α kinase is considered to be an inflammation‐related target as microglial p38α promotes production of proinflammatory cytokines6 and modulates microglial activation state,7, 8 and in the healthy state p38α expression within neurons is low.9 However, more recent findings indicate that neuronal p38α is increased in disease and under stress, and neuronal p38α expression has been implicated in amyloid‐beta and/or inflammation‐induced synaptic dysfunction,10, 11, 12, 13, 14 specifically impaired synaptic plasticity. Consistent with the biology of neuronal p38α, selective small molecule inhibitors of p38α rapidly (i.e., within 2–3 weeks) reverse spatial learning defects in the APP/PS1 mouse model,15 aged rats16 and in tauopathy (hTau) mouse model.17
Neflamapimod (previously code‐named VX‐745)18 is a highly selective oral small molecule inhibitor of p38α that after oral administration in animals achieves brain concentrations that are ~twofold higher than in blood.16, 19, 20 Following phase 1 studies in healthy volunteers and a phase 2a study in patients with rheumatoid arthritis, neflamapimod was repositioned as a CNS therapeutic.16 The primary preclinical animal pharmacology study was in aged rats, where the cognitive deficits are considered to result from interleukin‐1 beta (IL‐1β) induced impairment of synaptic plasticity. In that study, neflamapimod reversed spatial learning deficits in the Morris‐Water‐Maze test16 and at the highest dose level also reduced hippocamapal IL‐1β protein levels. In a pilot study in Tg2576 (Swedish mutation APP) transgenic mice that was conducted prior to the aged rats study #bib2 weeks neflamapimod treatment also showed strong trends toward reduced hippocampal amyloid plaque levels.16 Plasma drug concentrations were measured in the preclinical studies and correlated with prior animal and clinical results to determine the doses utilized in this study, with 40 mg twice daily projected to be the therapeutic dose level for effects cognition and amyloid plaque levels, whereas 125 mg twice daily projected to be the dose for effects on cytokine production.
Our objectives in this first clinical study of neflamapimod in patients with AD were to ascertain safety and to screen for potentially relevant pharmacological in the brain and/or clinical activity. As at the time of the study was initiated, the understanding of the role of p38α in brain was primarily around its activity in microglia, including potential effects on modulating microglia‐mediated amyloid clearance, we chose brain amyloid plaque levels by PET scan as the primary objective. However, there was already preliminary evidence suggesting a role of neuronal p38α in AD‐related synaptic dysfunction, and so we also decided to evaluate cognitive function, especially episodic memory.
Patients and Methods
Study design
This was a single center (VU University Medical Center, Amsterdam, Netherlands) double‐blind dose‐controlled clinical study. Sixteen subjects were planned for enrollment. No sample size calculations were performed as no comparisons between dose groups was planned. Subjects were randomized to receive either 40 mg or 125 mg twice daily to provide sufficient range of plasma drug levels to perform pharmacokinetic‐pharmacodynamics analyses if drug effects were demonstrated.
Diagnosis and main criteria for inclusion: Male or female subjects age 60–85 years with either mild cognitive impairment due to AD or mild AD, with demonstrated elevated brain amyloid plaque load, and a Mini‐Mental State Examination (MMSE) score between 20 and 28.
The study was conducted under the Competent Authority in the Netherlands, the CCMO. The protocol and informed consent form was reviewed by, and approval granted by the VU Medical Center Medical Review Ethics Committee (Medisch Ethische Toetsings Commissie, METc). All patients signed the METc approved informed consents form prior to participation in the study. The trial was registered at clinicaltrials.gov prior to study start (NCT024231220), and with EudraCT as protocol 2014‐002855‐25.
Assessments
The dynamic amyloid PET scanning method of van Berckel and colleagues at the VU Medical Center was followed.21 In this method, dynamic emission scanning consists of 23 frames with progressive increases in frame duration (1 × 15 #bib3 × 5, 3 × 10, 2 × 30, 3 × 60, 2 × 150, 2 × 300, and 7 × 600 sec) for a total scan duration of 90 min followed by a full quantitative data analysis of the images. Image analysis was performed utilizing a parametric method based on the simplified reference tissue method (SRTM2, also known as RPM2)22 and the PET signal is reported as BPND (binding potential, nondisplacable), according to standard nomenclature.23 To provide a reference to other clinical trials, where generally the more simply to acquire, but semiquantitative Standard Uptake Value ratio (SUVr) is utilized, the images were also analyzed using SUVr relative to cerebellar gray matter from 60 to 90 min.
Resting state fMRI and MEG were conducted to gain performance data in advance of potential inclusion in future studies (see Data S1).
The Wechsler Memory Scale (WMS) included evaluations of both immediate recall and delayed recall utilizing WMS‐IV® by Pearson Clinical. The WMS was administered in Dutch at baseline (Day 1, first dose administration) and at Days 28 and 84 of study drug administration. Standardized MMSE (Version 2.0) was also applied in Dutch, and administered at screening and Day 1 predosing, and on Days 28 #bib56, and 84 during study drug administration.
Plasma drug concentrations were determined utilizing a LC‐MS/MS assay at Charles River Laboratories, Edinburgh.
Statistical methods
All analyses were defined in a Statistical Analysis Plan that was developed and implemented during the first half of patient enrollment. The primary pharmacodynamics variable in the study cortical‐specific11C‐PiB binding (i.e., BPND) by 90 min dynamic PET scanning, evaluated as percentage change from Baseline to Day 84 in cortical specific 11C‐PiB binding (i.e. BPND) by 90 min dynamic PET scanning and number and proportion of responders (>7% reduction in BPND). Due to the nonsymmetrical nature of the change from baseline values, a nonparametric approach, Wilcoxon signed rank test was used in the percentage change analysis. The response criterion was based on the published within subject variability in test–retest conditions of 2–3% with reference parametric mapping of dynamic22‐PiB PET scanning. For MMSE and WMS, as the evaluation was to screen for improvement, the null hypothesis was no improvement (rather than no change), and therefore within‐subject change in these measures were assessed with a one‐sided Wilcoxon sign rank test. The episodic memory components of the WMS were analyzed as composite measures of immediate and delayed recall. WMS immediate recall composite score at each testing sessions consisted of the sum of the scores on Logical Memory (Logical Memory) I, Verbal Paired Associates (VPA) I, and Visual Reproduction (VR) I. WMS delayed recall composite score consisted of the sum of LM II, VPA II, and VR II.
The area under plasma drug time (0–12 h)–concentration curve (AUC0–12) was derived for each subject from a population pharmacokinetic analysis that incorporated sparse sampling in this study and intensive sampling from another neflamapimod study in early AD (NCT02423200). Linear regression (y = bx) was performed to evaluate the impact of plasma drug concentration on WMS outcome.
Results
Baseline subject characteristics
Sixteen patients were enrolled (9 randomized to 40 mg; 7–125 mg). For efficacy analyses, data on 15 patients were available as one patient developed MRI‐induced panic attack on the last study visit and could not complete either PET scan or cognitive testing. Seven men and two women were enrolled in the 40 mg group and three men and four women in 125 mg dose group. The median age at baseline was 66.5 years (range: 60–76); and the median baseline MMSE score was 24 (range: 20–28; median 23 and 24 in 40 mg and 125 mg groups, respectively). None of the patients received concomitant symptomatic AD therapy.
11C‐PiB PET scan results
There were minor trends in percentage change on 11C‐PiB binding with the median (mean) percentage change in global cortical BPND values being −4.57% (−5.36%) in the 40 mg group and +4.57% (+5.39%) in the 125 mg group (Fig. 1A). The responder analysis of global cortical amyloid plaque load (Fig. 1B) demonstrated four of 15 subjects (26.7%) meeting the predefined definition of response with >7% reduction in BPND, including three of eight subjects in the 40 mg group and one of seven subjects in the 125 mg group. Percentage reduction in global cortical BPND in the three responders in the 40 mg dose group was −11.6%, −11.9%, and −40.5%, respectively, and in the one responder in the 125 mg dose group was −7.7%. Interestingly, the responder in the 125 mg dose group was the subject with the lowest plasma drug levels in that group. Additional exploratory analysis (Fig. 2A) showed that responses (≥7% reduction) were limited to subjects who had baseline BPND less than 0.9. To provide a comparison to other studies, we analyzed the 11C‐PiB results utilizing the Standardized Uptake Value ratio (SUVr; Fig. 2B). The results were similar although not identical to those utilizing reference parametric mapping, with confirmation that the three responders by the prior analysis in the 40 mg dose group also showing reductions in brain amyloid plaque levels of −15%, −16.3% and −21.8% by the SUVr analysis; however, the BPND responder in the 125 mg dose group had a only −1% change in the SUVr analysis.
Figure 1.
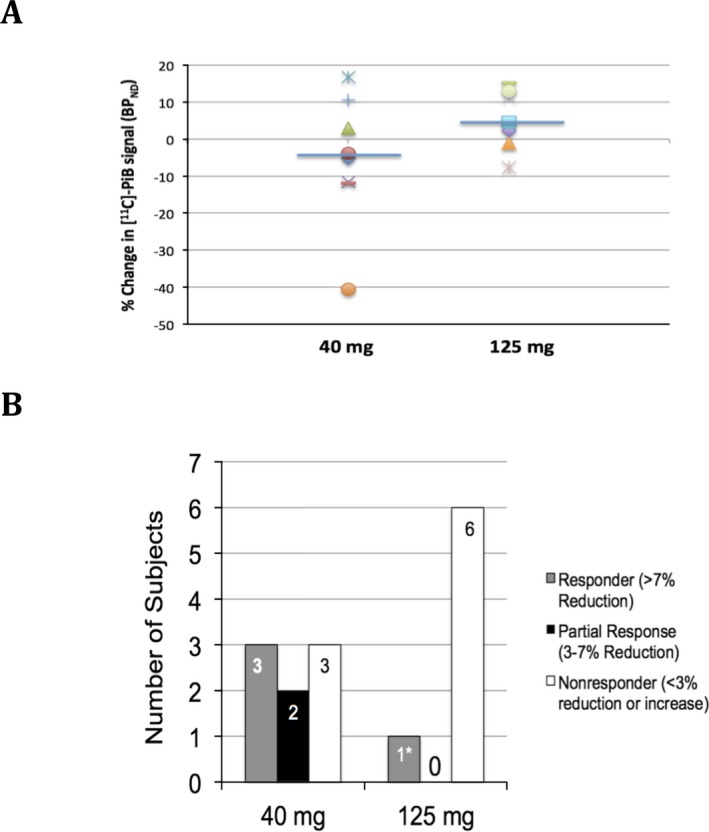
11C‐PiB Amyloid PET scan results. (A) Percentage change in global cortical BPND parameter from reference parametric mapping analysis. Median (horizontal blue line) with individual results shown as different marker for each subject. (B) Responder analysis, with response defined as >7% reduction from baseline in BPND parameter at Day 84 (week 12). * – this patient had the lowest plasma drug concentration levels in the 125 mg dose group, nearly approximating the subject with the highest plasma drug concentration in the 40 mg dose group.
Figure 2.
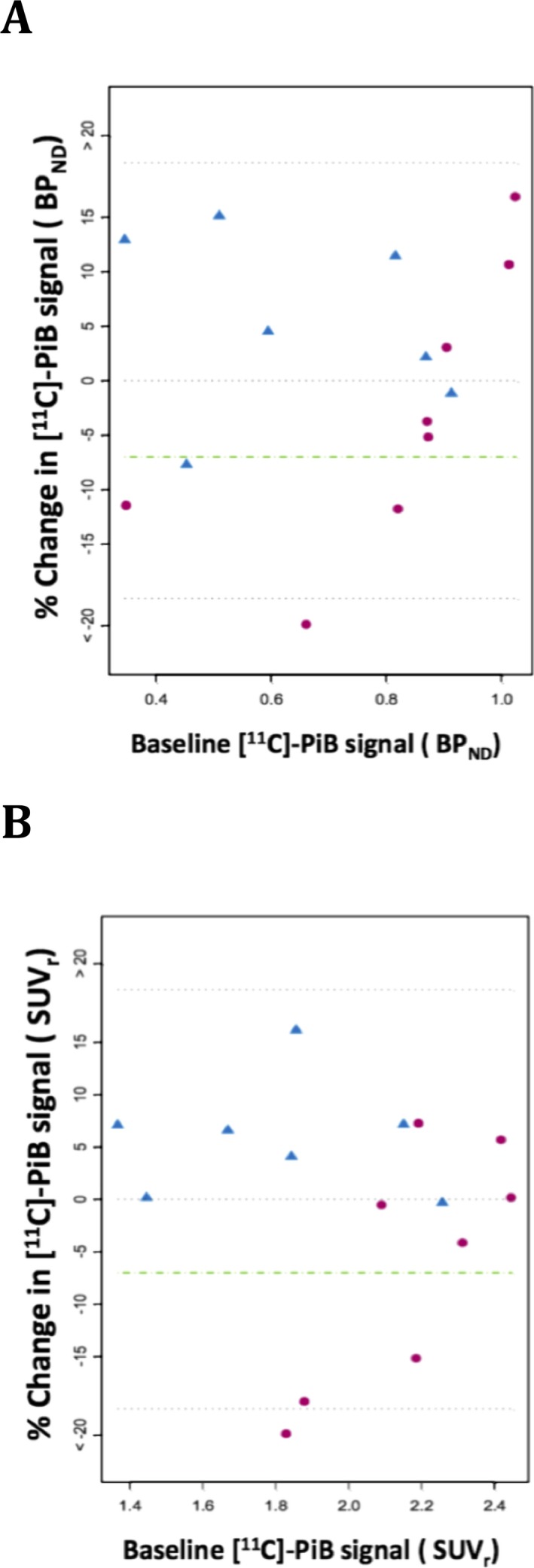
Percent change in amyloid load by 11C‐PiB Amyloid PET as a function of baseline amyloid levels. (A) Percentage change in global cortical BPND parameter as a function of baseline BPND parameter. (B) Percentage change in global cortical SUVr parameter as a function of baseline SUVr parameter. Red circles represent 40 mg dose group, blue triangles represent 125 mg dose group, dashed green line represents the responder definition of a 7% reduction in BPND or SUVr.
Episodic memory
Episodic memory is considered to be a direct measure of synaptic dysfunction in the hippocampus and the most clinically relevant measure of disease progression in early AD.24, 25 In this study episodic memory was assessed through immediate and delayed recall composites of components of the WMS. Mean WMS immediate recall composite scores (range: 0–136) increased from 48.4 ± 3.8 (SEM) at baseline to 58.4 ± 4.3 at Day 84 (Fig. 3A, P = 0.005 by one‐sided Wilcoxon sign rank test for improvement). Mean WMS delayed recall composite scores increased from 13.2 ± 2.3 at baseline to 22.1 ± 4.1 at Day 84 (Fig. 3B, P < 0.001). All subcomponents of WMS episodic memory composites showed improvement (P < 0.05) at Day 84 (Table 1). No changes were observed in the Symbol Span component of WMS, the component that does not depend on episodic memory.
Figure 3.
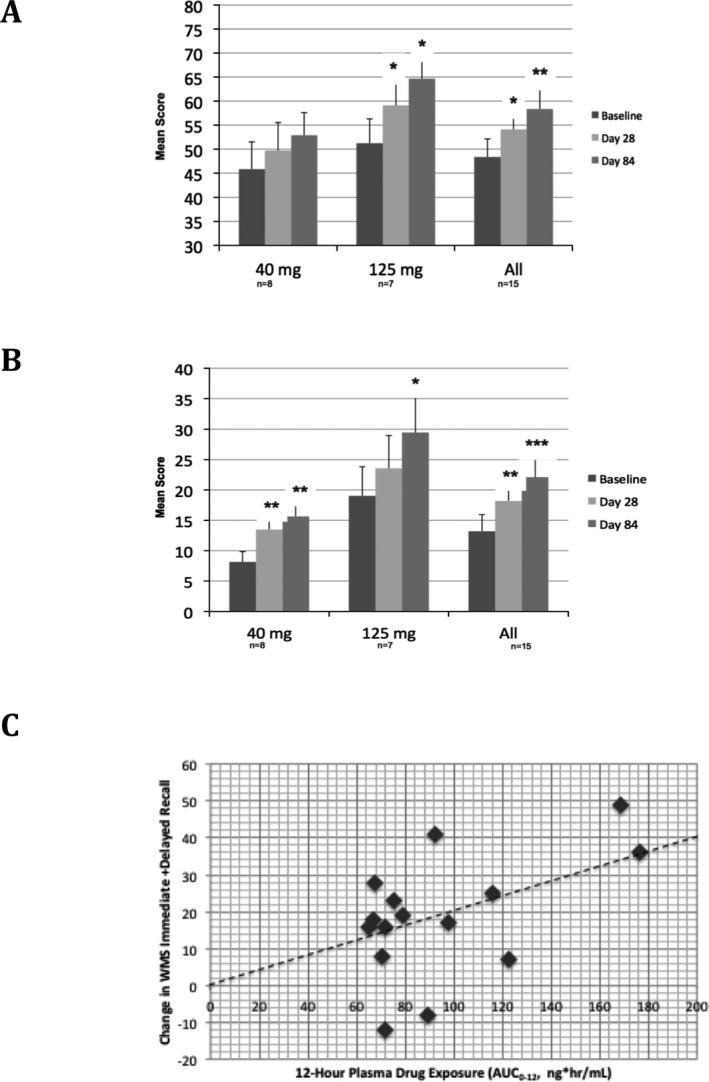
Wechsler Memory Scale immediate and delayed recall results. (A) Immediate recall composite results. (B) Delayed recall composite results. For A and B, mean and SEM are shown; * ‐P < 0.05, ** ‐P < 0.01, *** ‐P < 0.001, by one‐sided Wilcoxon sign rank test. (C) PK‐PD relationship of individual subject 12‐h area‐under‐curve of plasma drug concentrations (AUC 0–12) with individual subject change in combined immediate and delayed recall (P < 0.0001, r 2 = 0.7 for the correlation). The dashed line represents the output from the best‐fit model using linear regression, y = bx.
Table 1.
Wechsler memory scale components
| Mean (SEM) | P‐valuea | ||||
|---|---|---|---|---|---|
| Baseline | Day 28 | Day 84 | Day 28 | Day 84 | |
| Immediate recall | |||||
| LM I (0–53) | 17.8 (1.5) | 19.4 (2.1) | 21.7 (2.3) | 0.089 | 0.008 |
| VPA I (0–40) | 10.9 (2.0) | 13.5 (2.0) | 14.1 (2.1) | 0.013 | 0.023 |
| VR I (0–43) | 19.7 (1.9) | 21.2 (1.8) | 22.5 (2.3) | 0.229 | 0.026 |
| Delayed recall | |||||
| LM II (0–39) | 4.1 (1.1) | 7.1 (1.5) | 7.1 (1.7) | 0.002 | 0.005 |
| VPA II (0–10) | 3.1 (0.6) | 3.6 (0.7) | 3.9 (0.7) | 0.138 | 0.006 |
| VR II (0–43) | 6.0 (6.9) | 7.5 (6.2)) | 11.1 (8.4) | 0.062 | 0.005 |
| Recognition | |||||
| LM II (Recognition, 0–23) | 15.4 (0.8) | 15.6 (0.6) | 16.0 (0.5) | 0.489 | 0.312 |
| VPA II (Recognition, 0–30) | 22.8 (1.1) | 22.7 (1.3) | 22.7 (1.2) | 0.523 | 0.627 |
| VR II (Recognition, 0–7) | 2.3 (0.4) | 3.1 (0.4) | 3.0 (0.4) | 0.062 | 0.029 |
| Symbol span (0–50) | 10.7 (1.7) | 11.9 (1.5) | 11.2 (1.6) | 0.116 | 0.546 |
LM, logical memory; VPA, verbal paired associates; VR, visual reproduction.
One‐sided Wilcoxon sign rank test for change from baseline for all subjects (N = 15).
PK‐PD modeling of the WMS data utilizing a simple linear regression model (y = bx) indicated that plasma drug concentration area‐under‐curve over the 12‐h dosing interval (AUC0–12) of neflamapimod were strongly correlated with change in combined WMS immediate and delayed recall score from baseline to Day 84 (Fig. 3c, r 2 = 0.70, P < 0.0001 for AUC0–12 being predictor of change in combined WMS immediate/delayed recall), supporting a true drug effect of neflamapimod on episodic memory. To further evaluate the effect on episodic memory, we calculated the within‐subject effect size (ES; i.e., cohen's d) for improvement from baseline to Day 84 for the episodic memory composites. For immediate recall the ES was 0.59 and for delayed recall the ES was 0.67.
Additional outcomes
MMSE scores were generally stable, and for the study as a whole marginally improved (Table S1). Plasma biomarkers (tau, neurofilament light chain, brain‐derived nerve growth factor), MRI atrophy measures, fMRI, and MEG‐based functional connectivity measures were assessed for purposes of planning for future clinical studies. These measures showed either no change or minor changes consistent with disease progression (Tables S2–S6). For example, median change in normalized brain volume by MRI was −0.56% in the 40 mg dose group and −0.4% in the 125 mg dose group, both consistent with expectations of approximately 3% annual decline in brain volume in an early AD patient population.26, 27
Safety
No clinically relevant safety signals were identified in this study, with no severe, serious, or adverse events resulting in discontinuation of study drug reported during the study. The most common adverse event was diarrhea reported in three subjects, all within the 40 mg dose group; two of these events were mild in severity, with the 3rd event, that was moderate in severity, considered as not related as the event did not recur during an additional 56 days treatment after a brief treatment interruption. Abnormal laboratory values were infrequent and no consistent trend on any laboratory parameter was evident.
Discussion
The means by which p38α kinase have been theorized to impact AD disease progression broadly fall into three categories1, 2, 3, 4, 5: (1) as an anti‐inflammatory through reducing proinflammatory cytokine production from immune cells, (2) promoting amyloid plaque clearance through modulating microglial phenotype, and (3) reversing inflammation and/or amyloid‐beta induced synaptic dysfunction, particularly impaired synaptic plasticity in the hippocampus. The first mechanism (i.e., inhibition of proinflammatory cytokine production) is the classical drug effect of p38α kinase inhibitors that formed the basis for evaluating this class of inhibitors for rheumatoid arthritis and other peripheral inflammatory disorders. With neflamapimod, assessment of its anti‐inflammatory activity in the CNS was conducted in a separate phase 2a study (NCT02423200), in which CSF inflammatory cytokines were measured. The results of that study, which were limited by levels of the primary cytokine targets of p38α being below the limit of detection and/or quantitation, will be reported separately. In the current study, we assessed the other two mechanisms, that is, effects on amyloid plaque clearance and/or synaptic dysfunction.
The results of this study did not support our primary postulated effect of p38α kinase inhibition on amyloid plaque clearance, as there were no consistent effects on brain amyloid plaque load in the main group level analysis. However, there were apparent reductions in amyloid plaque load in a subset of patients with lower baseline amyloid plaque load. In particular, as within subject variability in BPND is less than 3% between two PET scans,21, 22 the three responder subjects in the 40 mg dose group are highly likely to have had significant reductions in brain amyloid plaque load during the study. We believe these reduction in brain amyloid plaque load are consistent with the scientific reports published during and after the conduct of the study that indicates neuronal p38α through modulating autophagy‐lysosome protein degradation appears to play a “critical role” in amyloid beta generation and plaque production.28, 29 Moreover, a very recent publication indicates that genetic knockout of neuronal p38α in AD‐transgenic mice prevents both the development of amyloid plaque pathothology and synaptic dysfunction and memory loss.30 Accordingly, we hypothesize that any effects on amyloid plaque load evident would be due to inhibition of p38α within the neuron leading to a decrease in amyloid plaque production, rather than an effect on p38α within microglia. The heterogeneous response within the active 40 mg dose group could be explained with a model in which there is consistent reduction in amyloid plaque production due to a direct pharmacological effect on amyloid‐beta generation, but only patients with efficient endogenous clearance would demonstrate reduction in brain amyloid plaque load. Consistent with this hypothesis the responders were all patients with lower levels of baseline brain amyloid plaque load less than the 0.9 for the study, as these would be the patients that would be expected to have higher endogenous amyloid plaque clearance levels.
The reductions in brain amyloid plaque load were largely confined to the 40 mg dose group. The lesser of an amyloid plaque load at the higher dose level of 125 mg could be explained by the expected additional effect of expected anti‐inflammatory activity of neflamapimod at the higher dose range that could, through reducing amyloid plaque clearance, offset the effect on amyloid plaque production at the lower dose. The 125 mg twice daily dose is equivalent in humans to the dose in aged rats that reduced IL‐1β levels in the hippocampus. In addition, neflamapimod at 250 mg twice daily in patients with rheumatoid arthritis demonstrated an anti‐inflammatory effect with reductions in serum C‐reactive‐protein and IL‐6 levels (Clinical Study Report VX00‐745‐102, EIP Pharma on file).
The effect we found on episodic memory is in line with a broad scientific literature that indicates that p38α in involved in oligomeric amyloid‐beta and inflammation‐induced synaptic dysfunction and to stress‐ and age‐related synaptic dysfunction in the hippocampus.4, 5, 10, 11, 12, 13, 14, 31 In our clinical study, the ES that we saw for immediate and delay recall compares favorably to ES of ≤0.2 for WMS immediate or delayed recall at week 12 in the placebo‐treated subjects in two trials of Souvenaid in a similar patient population (mild AD, baseline MMSE = 24).32, 33 Although with the absence of a placebo group our results could reflect practice effect, we think this is unlikely since the learning effects on episodic memory tests that are well recognized in cognitively intact older individuals are less prominent, or absent, in biomarker‐positive early AD.34, 35 The highly correlated relationship between increasing plasma drug concentration of neflamapimod with increasing improvement in immediate/delayed also suggests that there is a true drug treatment effect.
Whether the effects of p38α kinase inhibition on synaptic dysfunction are related to the effects on amyloid beta generation remains unknown. When neuronal p38α is knocked out in the APP/PS1 mouse the effects on amyloid beta generation and synaptic function occur concurrently, suggesting the prevention of synaptic dysfunction in that model is due to reducing amyloid beta generation.30 However, small molecule p38α also reverse spatial learning and spatial working memory deficits in aged rats16 and in an inflammation‐induced tauopathy model,17 respectively; neither of which demonstrate amyloid pathology. In this study, there were no correlations between amyloid plaque reduction and improvement in episodic memory function. However, brain amyloid plaque levels by PET scanning is not a measure of amyloid beta generation, rather it is an integrated measure of both amyloid plaque production and clearance. As a result, there might be a consistent underlying effect on amyloid beta generation with neflamapimod treatment that was not detected by PET scanning.
In summary, though the size of this study and the lack of a placebo‐controlled group limits the ability to draw conclusions, our results suggest that selective p38α inhibition with neflamapimod may improve episodic memory and in parallel may have the potential to impact amyloid plaque production in AD. The results are consistent with the scientific literature indicating increased neuronal p38α activity leads to both synaptic dysfunction and increased amyloid‐beta generation through impairing autophagy‐mediated protein turnover. A 6‐month placebo‐controlled study has recently been initiated to confirm these preliminary clinical findings, particularly with respect to the effects on episodic memory function.
Author Contributions
PS, ND, and JA designed study, conducted/managed study, evaluated results and were primary authors of the manuscript. ND was the primary treating physician. AL, MY, and BVB managed and analyzed PET studies. AG conducted MEG analyses, and AMW conducted fMRI and MRI analyses. HMC was responsible for all statistical analyses.
Conflict of Interest
JA is founder and CEO of EIP Pharma, the company that is developing neflamapimod as a treatment for AD. The other authors have no conflict to declare.
Supporting information
Table S1. Summary statistics of mini‐mental state examination overall score.
Table S2. Change in mini‐mental state examination overall score from baseline.
Table S3. Plasma biomarker results.
Table S4. Summary statistics of resting fMRI metrics by visit by treatment.
Table S5. Structural MRI analyses.
Table S6. Magnetoencephalogram (MEG) summary statistics.
Data S1. Supplementary materials.
Acknowledgments
Marieke Reimert, Kim Verheul, and Marceline Hos for their work in conducting the study and managing patients at the Brain Research Center (http://www.brainresearchcenter.nl). Elisabeth Garrison at Voisin Consulting was responsible for all regulatory filings and project management. Biomarker evaluations conducted in laboratory of Charlotte Teunissen. The study was supported by funding from EIP Pharma LLC, Cambridge, MA, USA.
Funding Statement
This work was funded by EIP Pharma LLC, Cambridge, MA, USA grant .
References
- 1. Munoz L, Ammit AJ. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology 2010;58:561–568. [DOI] [PubMed] [Google Scholar]
- 2. Yasuda S, Sugiura H, Tanaka H, et al. p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent Nerv Syst Agents Med Chem 2011;11:45–59. [DOI] [PubMed] [Google Scholar]
- 3. Correa SA, Eales KL. The Role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct 2012;2012:649079 https://doi.org/10.1155/2012/649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sanderson TM, Hogg EL, Collingridge GL, Correa SA. Hippocampal metabotropic glutamate receptor long‐term depression in health and disease: focus on mitogen‐activated protein kinase pathways. J Neurochem 2016;139:200–214. [DOI] [PubMed] [Google Scholar]
- 5. Lee JK, Kim NJ. Recent advances in the inhibition of p38 MAPK as a potential strategy for the treatment of Alzheimer's Disease. Molecules 2017;22:E1287 https://doi.org/10.3390/molecules22081287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bachstetter AD, Van Eldik LJ. The p38 map kinase family as regulators of proinflammatory cytokine production in degenerative diseases of the CNS. Aging Dis. 2010;1:199–211. [PMC free article] [PubMed] [Google Scholar]
- 7. Bachstetter AD, Rowe RK, Kaneko M, et al. The p38alpha MAPK regulates microglial responsiveness to diffuse traumatic brain injury. J Neurosci 2013;33:6143–6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Adolfsson O, Pihlgren M, Toni N, et al. An effector‐reduced anti‐beta‐amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci 2012;32:9677–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lawson SK, Dobrikova EY, Shveygert M, Gromeier M. p38alpha mitogen‐activated protein kinase depletion and repression of signal transduction to translation machinery by miR‐124 and ‐128 in neurons. Mol Cell Biol 2013;33:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watterson DM, Grum‐Tokars VL, Roy SM, et al. Development of novel in vivo chemical probes to address CNS protein kinase involvement in synaptic dysfunction. PLoS ONE 2013;8:e66226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li S, Jin M, Koeglsperger T, et al. Soluble Abeta oligomers inhibit long‐term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B‐containing NMDA receptors. J Neurosci 2011;31:6627–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tong L, Prieto GA, Kramar EA, et al. Brain‐derived neurotrophic factor‐dependent synaptic plasticity is suppressed by interleukin‐1beta via p38 mitogen‐activated protein kinase. J Neurosci 2012;32:17714–17724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prieto GA, Snigdha S, Baglietto‐Vargas D, et al. Synapse‐specific IL‐1 receptor subunit reconfiguration augments vulnerability to IL‐1beta in the aged hippocampus. Proc Natl Acad Sci USA 2015;112:E5078–E5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koppensteiner P, Trinchese F, Fa M, et al. Time‐dependent reversal of synaptic plasticity induced by physiological concentrations of oligomeric Abeta42: an early index of Alzheimer's disease. Sci Rep 2016;6:32553 https://doi.org/10.1038/srep32553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roy SM, Grum‐Tokars VL, Schavocky JP, et al. Targeting human central nervous system protein kinases: an isoform selective p38alphaMAPK inhibitor that attenuates disease progression in Alzheimer's disease mouse models. ACS Chem Neurosci 2015;6:666–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alam JJ. Selective brain‐targeted antagonism of p38 MAPKalpha reduces hippocampal IL‐1beta levels and improves morris water maze performance in aged rats. J Alzheimers Dis 2015;48:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maphis N, Jiang S, Xu G, et al. Selective suppression of the alpha isoform of p38 MAPK rescues late‐stage tau pathology. Alzheimers Res Ther 2016;8:54 https://doi.org/10.1186/s13195-016-0221-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duffy JP, Harrington EM, Salituro FG, et al. The Discovery of VX‐745: a novel and selective p38alpha kinase inhibitor. ACS Med Chem Lett 2011;2:758–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis MI, Hunt JP, Herrgard S, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol 2011;29:1046–1051. [DOI] [PubMed] [Google Scholar]
- 20. Uitdehaag JC, Verkaar F, Alwan H, et al. A guide to picking the most selective kinase inhibitor tool compounds for pharmacological validation of drug targets. Br J Pharmacol 2012;166:858–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Berckel BN, Ossenkoppele R, Tolboom N, et al. Longitudinal amyloid imaging using 11C‐PiB: methodologic considerations. J Nucl Med 2013;54:1570–1576. [DOI] [PubMed] [Google Scholar]
- 22. Tolboom N, Yaqub M, Boellaard R, et al. Test‐retest variability of quantitative [11C]PIB studies in Alzheimer's disease. Eur J Nucl Med Mol Imaging 2009;36:1629–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Innis RB, Cunningham VJ, Delforge J, et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 2007;27:1533–1539. [DOI] [PubMed] [Google Scholar]
- 24. Gold CA, Budson AE. Memory loss in Alzheimer's disease: implications for development of therapeutics. Expert Rev Neurother 2008;8:1879–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tromp D, Dufour A, Lithfous S, et al. Episodic memory in normal aging and Alzheimer disease: insights from imaging and behavioral studies. Ageing Res Rev 2015;24(Pt B):232–262. [DOI] [PubMed] [Google Scholar]
- 26. Schuff N, Woerner N, Boreta L, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain 2009;132:1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Risacher SL, Shen L, West JD, et al. Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging 2010;31:1401–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schnoder L, Hao W, Qin Y, et al. Deficiency of Neuronal p38alpha MAPK attenuates amyloid pathology in Alzheimer disease mouse and cell models through facilitating lysosomal degradation of BACE1. J Biol Chem 2016;291:2067–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alam J, Scheper W. Targeting neuronal MAPK14/p38alpha activity to modulate autophagy in the Alzheimer disease brain. Autophagy 2016;12:2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colie S, Sarroca S, Palenzuela R, et al. Neuronal p38alpha mediates synaptic and cognitive dysfunction in an Alzheimer's mouse model by controlling beta‐amyloid production. Sci Rep 2017;7:45306 https://doi.org/10.1038/srep45306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cortez I, Bulavin DV, Wu P, et al. Aged dominant negative p38alpha MAPK mice are resistant to age‐dependent decline in adult‐neurogenesis and context discrimination fear conditioning. Behav Brain Res 2017;322:212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scheltens P, Kamphuis PJ, Verhey FR, et al. Efficacy of a medical food in mild Alzheimer's disease: a randomized, controlled trial. Alzheimers Dement 2010;6:1–10e11. [DOI] [PubMed] [Google Scholar]
- 33. Scheltens P, Twisk JW, Blesa R, et al. Efficacy of souvenaid in mild Alzheimer's disease: results from a randomized, controlled trial. J Alzheimers Dis 2012;31:225–236. [DOI] [PubMed] [Google Scholar]
- 34. Hassenstab J, Ruvolo D, Jasielec M, et al. Absence of practice effects in preclinical Alzheimer's disease. Neuropsychology 2015;29:940–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Goldberg TE, Harvey PD, Wesnes KA, et al. Practice effects due to serial cognitive assessment: implications for preclinical Alzheimer's disease randomized controlled trials. Alzheimers Dement (Amst). 2015;1:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary statistics of mini‐mental state examination overall score.
Table S2. Change in mini‐mental state examination overall score from baseline.
Table S3. Plasma biomarker results.
Table S4. Summary statistics of resting fMRI metrics by visit by treatment.
Table S5. Structural MRI analyses.
Table S6. Magnetoencephalogram (MEG) summary statistics.
Data S1. Supplementary materials.