

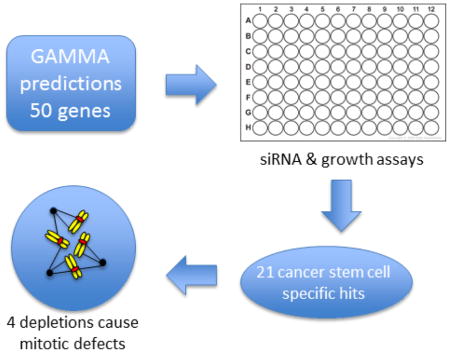
Abstract
The cancer stem cell model postulates that tumors are hierarchically organized with a minor population, the cancer stem cells, exhibiting unlimited proliferative potential. These cells give rise to the bulk of tumor cells, which retain a limited ability to divide. Without successful targeting of cancer stem cells, tumor reemergence after therapy is likely. However, identifying target pathways essential for cancer stem cell proliferation has been challenging. Here, using a transcriptional network analysis termed GAMMA, we identified 50 genes whose correlation patterns suggested involvement in cancer stem cell division. Using RNAi depletion, we found that 21 of these target genes showed preferential growth inhibition in a breast cancer stem cell model. More detailed initial analysis of 6 of these genes revealed 4 with clear roles in the fidelity of chromosome segregation. This study reveals the strong predictive potential of transcriptional network analysis in increasing the efficiency of successful identification of novel proliferation dependencies for cancer stem cells.
Keywords: cancer stem cells, breast cancer, mitosis, cell cycle, chromosome instability, cell division
Graphical Abstract
1. Introduction
The cancer stem cell (CSC) theory posits that, in at least some cancers, tumor cells are arranged in a hierarchical lineage with a minor population, the CSCs, capable of unlimited proliferation while the bulk of the tumor is comprised of partially differentiated cells with limited ability to divide [1]. A consequence is that only a subset of tumor cells, the CSCs, have the ability to generate tumors when transplanted [2–4]. A corollary of the CSC theory is that eradicating tumors and preventing recurrence requires elimination of CSCs. However, identifying specific pathways to target CSC’s has been a difficult challenge. Therefore, we used a transcriptional network algorithm called GAMMA to identify novel candidate targets, then tested the effects of depleting their expression in an established CSC model system.
Cell division is essential for tumor growth. The core pathways that mediate division are highly conserved from lower eukaryotes to mammals. However, mammals have evolved supplemental pathways. Proteins that participate in these supplemental pathways may be generally dispensable for the division in normal cells but may promote the fidelity of chromosome segregation. However, through mutation and epigenetic changes that accompany tumorigenesis, these pathways may become essential for cancer cell proliferation. This idea is supported by the fact that at least some cancers are highly vulnerable to inhibition of certain mitotic regulators [5, 6]. In brief, CSCs may become “addicted” to certain supplementary cell division pathways. Our goal was to test if our bioinformatic analysis could identify components of these pathways whose depletion would inhibit CSC growth.
2. Materials and Methods
2.1 Cell Culture
BPLE, BPLER, HMLE and HMLER cells (generously provided by Drs. Fabio Petrocca and Robert A. Weinberg, Massachusetts Institute of Technology) were maintained in WIT-T culture medium (Cellaria). All cell lines were maintained in 75 cm2 filter flasks in a humidified incubator at 37°C with 5% CO2. Cell lines were screened for mycoplasma by fixing cells on coverslips with 3:1 methanol:acetic acid and labeling with 1 μg/ml DAPI. Observation by fluorescence microscopy confirmed that all lines were free of mycoplasma contamination.
2.2 siRNA Screen
2.2.1 Cell culture
Cells were passaged by trypsinization (0.05% trypsin, 0.53 mM EDTA, 0.085% PBS). Optimal initial cell density was empirically determined as one that would be near confluency after a 7-day incubation, without overgrowth. Cells were plated with 100 μl of media in quadruplicate at 800, 600, 500, 400 and 300 cells/well for BPLE and BPLER and 600, 500, 400, 300 and 200 cells/well for HMLE and HMLER. 100 μl of media were added after 2 days to mimic experimental treatments. Cells were fixed, permeabilized, stained and read 7 days after initial plating. Optimal initial concentrations were 600 cells/well for BPLE and BPLER and 500 cells/well for HMLE and HMLER (Fig. S1).
2.2.2 Transfections
Transfections were carried out using Lipofectamine RNAiMax (Invitrogen) 2 days after plating. siRNAs (Bioneer) were suspended in RNAse-free H2O at a concentration of 4 μM. 1–3 siRNAs were combined for each gene target (Table S1). Transfections were carried out in quadruplicate with 0.5 μl (10nM) siRNA mix used for each well and standard Lipofectamine RNAiMax protocol was followed. Transfection mix was made up in 100 μl of WIT-T media and added to each well bringing the total volume of the well to 200 μl of media.
To identify the optimal starting siRNA concentration BPLER cells were transfected in quadruplicate with 50 nM, 40 nM, 30 nM, 20 nM and 10 nM concentrations of siRNA targeting luciferase (negative control) and PLK1 (positive control). 10 nM luciferase siRNA transfection showed minimal growth inhibition and 10 nM PLK1 siRNA transfection had approximately the same level of inhibition as higher concentrations (Fig. S2A). This was repeated in all 4 cell lines with similar results (Fig. S2B).
2.2.3 Growth assays
Five days after transfection, the media was removed from each well, and cells were treated with 100 μl of 1% paraformaldehyde in PHEM buffer (60 mM PIPES, pH 6.9, 25 mM HEPES, 10 mM EGTA, 4 mM MgCl2) containing 0.05% Triton x-100 and 1:1000 dilution of commercial SYBRGold stock solution to fix and permeabilize the cells and label DNA. Plates were then incubated for 30 minutes and read on a Genios plate reader (Tecan) with the following settings: gain @ Optimal, 10 flashes/well, (FITC filter set), read from bottom, Lag 0, Integration 40. Growth was normalized by dividing the cell count of each well to the average of the control wells. 3 replicate experiments were performed for each cell line. After expression normalization, the Bioconductor package limma (http://www.bioconductor.org/packages/release/bioc/html/limma.html) was used to determine genes that showed significant differential expression under experimental conditions and cell types. A linear model was fitted to the expression data for each probe. Moderated t-statistics were computed by empirical Bayes shrinkage of the standard errors toward a common value. The P-values corresponding to the moderated t-statistics were adjusted for multiple testing by computing false discovery rates (fdr) using the method of Benjamini, Hochberg, and Yekutieli [7]. We used both fdr and fold change to select differentially expressed genes by requiring at least a twofold change (log2[fold]|≥1) and fdr ≤ 0.05.
Cell counts were determined using cell line specific equations generated by standard curves. Known cell counts ranging from 1250 to 35,000 cells/well were plated in quadruplicate and incubated at 37°C for 10 hours to allow cells to settle. The average fluorescent intensity was plotted against cell count and the resulting equation was used to extrapolate the cell count from the measured fluorescent intensity in the experimental groups (Fig. S3).
2.3 Cell Cycle Analysis
2.3.1 Immunolabeling
Cells were seeded on 25 mm coverslips in 6-well plates at approximately 12% confluency with 2 ml of media and transfected with 30 nM siRNA 24 h later. After another 48 h, cells were fixed in 1.5% % paraformaldehyde in PHEM buffer containing 0.05% Triton-X 100 solution for 15 minutes. Coverslips were blocked with 20% Boiled normal goat serum (BNGS) in MBST (10 mM MOPS, 150 mM NaCl, 0.05% Tween 20) for 20 minutes. Mouse anti-γTubulin antibody (Sigma: T5326) in MBST with 5% BNGS was applied overnight at 4°C. Coverslips were washed 3 times with PBST for 5 minutes and labeled with FITC-conjugated Goat anti-mouse secondary antibody (Jackson Immunoresearch: 115-096-062) at 5 μg/ml in MBST with 5% BNGS for 60 minutes at room temperature. Coverslips were then washed 3 times in PBST and labeled with 0.05 μg/ml DAPI in water for 3 min. Coverslips were washed with water and then mounted on slides with 9 μl vectashield (Vector laboratories) containing 1mM MgSO4 and the edges of the coverslip sealed with nail polish.
2.3.2 Cell Cycle Profiling
100 fields on each coverslip were imaged with a 15-plane z-series at 0.25 μm steps spanning the chromatin. Each z-series was summed, and individual nuclei were counted and analyzed with Metamorph software (Molecular Devices) using the cell cycle plugin. Intensity gates for scoring cells in G1, S, G2/M were established in the control and applied to each of the experimental sets. Mitotic indices were determined by manually counting the proportion of cells containing condensed chromosomes.
3. Results and Discussion
3.1 A Bioinformatics Approach to Candidate Identification
Stem-cell mitotic regulators remain poorly characterized, and their identification is complicated by several factors. First, stem cells are a minor population of the dividing cells within a tissue or tumor. Second, accessory mitotic regulators may be difficult to discern because their depletion only decreases the very high fidelity of chromosome segregation but are not indispensable for the division process itself. Third, proteins may have evolved functions in multiple areas of cell cycle control or cell physiology, making the characterization of their roles more difficult. To address these challenges, we used a predictive algorithm called Global Microarray Meta-Analysis (GAMMA) to identify candidate stem cell mitotic regulators. GAMMA is a bioinformatics approach that uses public microarray and RNAseq datasets from NCBI’s Gene Expression Omnibus (GEO) repository to identify transcripts that are correlated across many experimental conditions [8, 9]. Using a “guilt by association” principle, groups of transcripts that are highly correlated with each other are likely to share similar biological associations, such as playing a role in the same disease or phenotype, and being involved in the same pathway. Using a k-Nearest Neighbors approach, GAMMA identifies the 40 most correlated transcripts for each gene, then uses literature mining to identify what the correlated genes have in common in MEDLINE, in terms of their being co-mentioned with diseases, phenotypes, chemicals, and other genes [10]. GAMMA has been successfully validated in several studies [11–15]. Selecting for high GAMMA scores in genes related to cancer, stem cells, and mitosis, we evaluated 50 candidate genes with minimal previous characterization (Table 1).
Table 1.
GAMMA scores for candidate genes arranged alphabetically. GAMMA scores are determined by the overall literature correlation between search query and the gene of interest’s correlated genes. Higher scores are better fit. GAMMA scores from the search queries Cancer, Stem Cell, and Mitosis were used to choose genes for growth assay testing
| Gene Symbol | Entrez ID | Cancer Score | Stem Cell Score | Mitosis Score |
|---|---|---|---|---|
| ARHGAP11A | 9824 | 362 | 16 | 129 |
| BCAS2 | 10286 | 456 | 33 | 54 |
| BCAT1 | 586 | 590 | 140 | 41 |
| C12orf24 | 29902 | 171 | 13 | 66 |
| C16orf59 | 80178 | 239 | 13 | 63 |
| C20orf20 | 55257 | 349 | 18 | 30 |
| C5orf13 | 9315 | 405 | 23 | 35 |
| C9orf40 | 55071 | 461 | 12 | 92 |
| CACYBP | 27101 | 376 | 32 | 33 |
| CEP72 | 55722 | 246 | 18 | 37 |
| CKAP2L | 150468 | 38 | 13 | 47 |
| COPS3 | 8533 | 401 | 42 | 77 |
| CSAG1 | 158511 | 145 | 13 | 36 |
| DDX5 | 1655 | 384 | 24 | 63 |
| DEK | 7913 | 172 | 16 | 31 |
| DONSON | 29980 | 297 | 31 | 80 |
| FAM102B | 284611 | 350 | 21 | 78 |
| GNL2 | 29889 | 611 | 24 | 27 |
| GTSE1 | 51512 | 334 | 18 | 84 |
| IGF2BP1 | 10642 | 317 | 94 | 28 |
| IGF2BP3 | 10643 | 491 | 18 | 48 |
| ITGB3BP | 23421 | 339 | 29 | 67 |
| LYPD1 | 116372 | 463 | 75 | 37 |
| MTA1 | 9112 | 256 | 21 | 30 |
| NASP | 4678 | 215 | 36 | 57 |
| NME1 | 4830 | 570 | 17 | 53 |
| NUP205 | 23165 | 265 | 41 | 47 |
| PAICS | 10606 | 305 | 13 | 66 |
| PBK | 55872 | 555 | 37 | 142 |
| PLS1 | 5357 | 422 | 14 | 24 |
| POLA2 | 23649 | 473 | 30 | 67 |
| POLD3 | 10714 | 452 | 32 | 64 |
| PUS7 | 54517 | 283 | 30 | 35 |
| QSER1 | 79832 | 230 | 15 | 59 |
| RAD51AP1 | 10635 | 269 | 13 | 69 |
| RBBP8 | 5932 | 346 | 14 | 52 |
| SFXN1 | 94081 | 227 | 44 | 69 |
| SLMO2 | 51012 | 228 | 45 | 22 |
| SMS | 6611 | 400 | 24 | 26 |
| SNRPB | 6628 | 328 | 13 | 29 |
| SPAG5 | 10615 | 468 | 24 | 162 |
| SRM | 6723 | 547 | 18 | 113 |
| TADA1L | 117143 | 428 | 51 | 52 |
| TBCB | 1155 | 642 | 18 | 35 |
| TPRKB | 51002 | 206 | 12 | 32 |
| TRIP13 | 9319 | 317 | 14 | 115 |
| TROAP | 10024 | 247 | 29 | 226 |
| UHRF1 | 29128 | 352 | 13 | 94 |
| XRCC6 | 2547 | 312 | 17 | 58 |
| YWHAQ | 10971 | 473 | 16 | 21 |
3.2 siRNA Screen of BPLER and BPLE
A significant challenge in identifying gene dependence in CSCs is the lack of reliable experimental comparisons in growth assays. When tumor cells are placed in culture, growth rates of various subpopulations of cancer cells may vary wildly as cultures are often heterogeneous. Furthermore, although CSC populations have been successfully isolated by fluorescence-activated cell sorting (FACS), these populations quickly lose their stem cell characteristics and differentiate in culture [16]. To address these problems, we focused on BPLER cells, a tumorigenic cell line derived from normal breast epithelium sequentially transformed with SV40LT, hTERT and hRAS(V12) [17]. These were compared with non-tumorigenic BPLE cells, which lack transformation with hRAS. BPLER cells share several characteristics associated with cancer stem cells, expressing high levels of CSC markers CD166 [18, 19], EpCAM [3, 19, 20], BMI1 [19, 21], ALDH1A [17, 22], p63 [19, 23] and a high molecular weight variant of CD44 (CD44v) [3, 17, 19, 20, 24]. They have a tumor-initiating cell frequency greater than 1 in 100 cells, form heterogeneous tumors and metastasize [17]. The expression profile of BPEC (the parental line of BPLE and BPLER) varies from that of differentiated cells in either the luminal or myoepithelial layers, but the expression profiles of BPLER-derived tumors do cluster with those of basal-like breast cancer, thought to originate from luminal progenitor cells [17, 20, 25]. Significant for our purposes, BPLER cells do not differentiate in culture, allowing us to carry out growth assays in a majority CSC-like population [19].
The sequential transformation origin of BPLER cells also offers another advantage over tumor-derived CSC cell lines, direct comparison with the parental line, BPLE. It can be difficult to identify the cell type of origin for a particular cancer. However, a direct comparison to the parental line is essential in identifying gene addiction. By using BPLER cells to test gene addiction in CSCs, we performed side-by-side growth assay comparisons with BPLE cells, which have a similar proliferation rate.
siRNA screens in BPLER and BPLE were carried out in 96-well plates for 5 days. Data were normalized to an internal negative control targeting luciferase in order to minimize the effects of experimental variations between plates (i.e. slight differences in starting cell numbers, time between plating and transfection). We found that growth of BPLER cells, in comparison with BPLE cells, showed significantly greater inhibition for 21 of the 50 genes (42%) upon depletion with siRNA (Figure 1a and S4).
Figure 1.

Growth screen for 50 stem cell cancer candidate gene produces by siRNA screening. (a) The 21 gene targets that resulted in significant growth inhibition in BPLER compared to BPLE. (b) heat map showing growth inhibition of all 4 cell lines. Growth inhibition is measured relative that of non-targeting control siRNA wells for each cell line. 100% (bright red) is complete growth inhibition and 0% (bright blue) is no growth inhibition. (c) VENN diagram showing numbers and overlap of genes that induced growth inhibition by more than 50% in each cell line.
3.3 siRNA Screens of HMLE and HMLER
To determine if the observed over-reliance by BPLER cells was related to their stem cell-like properties, we repeated the siRNA screen in HMLER and HMLE cell lines. As with BPLER cells the original source for HMLE and HMLER was normal breast tissue [17]. Unlike BPLER cells, HMLER cells form homogenous tumors in mice, are poorly vascularized and usually benign, and the expression profile of their parental line (HMEC) suggests a more differentiated state along the myoepithelial lineage [17]. HMLER cells have lower expression of stem cell markers, the standard form of CD44 (CD44s) rather than the high molecular weight variant associated with stem cells [19, 24], and lower levels of stem cell markers, CD166, EpCAM, BMI1, ALDH1A and p63 compared with BPLER cells [17, 19]. They also have a much lower frequency of tumor initiating cells (<1 in 105), and they do not form metastases in mice [17, 20]. The shared origin, generation method, and propagation rates of HMLER cells and BPLER cells make them suitable for comparison.
Screens were carried out as described previously. Though growth inhibition was detected in many of the depletions, no significant differences in proliferation between HMLER and HMLE cells occurred for any of the targets (Fig. S5). This observation suggests that differential growth inhibition for the 21 genes for BPLER cells compared with BPLE cells is related to their putative stem cell origin. Overall results comparing all 4 cell lines are summarized in figures 1b and 1c.
3.4 Cell Cycle Analysis
To gain insight into the mechanisms underlying growth inhibition for a subset of the 21 genes, we investigated changes to the cell cycle profiles following depletion of 6 hits, CSAG1, ITGB3BP, NME1, SLMO2, SMS and YWHAQ, that specifically inhibited BPLER cells. Fixed-cell fluorescent microscopy measuring DNA content was conducted on BPLER cells following siRNA. No significant differences were found in the proportions of cells in G1, S and G2/M (Fig. 2a and 2b). The mitotic indices were determined manually with over 20,000 cells analyzed for each target. We found that depletion of ITGB3BP, NME1 and SLMO2 each resulted in a significantly decreased mitotic index (Fig. 2c).
Figure 2.

Cell Cycle alteration testing by siRNA in BPLER cells for 6 selected genes. (a) Cell cycle profile of control BPLER cells. (b) Percentages of cells in each phase of the cell cycle for indicated siRNA depletions. Intensity gates for scoring cells in G1, S, G2/M were established in control BPLER cells and applied to each of the experimental sets. No significant changes were observed in the proportion of cells in any cell cycle phase. (c) Mitotic indices. ITGB3BP (p = 0.0001), NME1 (p = 0.0019) and SLMO2 (p = 0.0002) depletions resulted in significant decreases of mitotic index. (d) At least 200 mitotic cells from each of 4 experiments were counted (n>800) and classified into 4 categories: prophase, cells with an intact nuclear envelope and condensed chromosomes; prometaphase, cells after nuclear envelope breakdown and before formation of the metaphase plate, metaphase, cells having all chromosomes aligned at the metaphase plate, and anaphase, cells with separated chromatids before nuclear reformation. (e) Proportion of cells at each phase of mitosis. Numbers represent the percentage of total mitotic cells (n>800). Prometaphase cells were increased (p = 0.0012) and metaphase cells decreased (p = 0.0001) after depletion of SMS. Significance was determined using one-way ANOVA and adjusted for multiple testing using the Dunnett method of statistical hypothesis testing [39]. Note: asterisks correspond to alpha level (*p < 0.05. **p < 0.01. ***p < 0.001. **** p < 0.0001) Error bars represent standard deviation from 4 separate experiments.
We then analyzed the 6 genes to determine effects on progression through the stages of mitosis. Accumulation of cells at different mitotic stages can indicate defects in various mitotic processes and give insight into the pathways affected by gene depletion. Mitotic cells were classified into prophase, prometaphase, metaphase and anaphase stages (Fig. 2d). Depletion of SMS resulted in significantly increased percentages of cells in prometaphase with unaligned chromosomes suggesting that SMS depletion inhibited or delayed chromosome alignment at the metaphase plate, (Fig. 2e). A corresponding decrease in metaphase cells without change in the proportion of anaphase cells suggests that SMS depletion induced cells to progress to anaphase without full metaphase alignment.
We also quantified the percentage of mitotic errors associated with depletion of the 6 selected targets. Mitotic errors resulting in missegregation of chromosomes, and spindle defects are indicators of chromosome instability that may promote tumorigenesis at low levels but inhibit tumor cell growth at high levels [26, 27]. The two major classes of mitotic error observed in our study were multipolar spindle formation and chromosome missegregation during anaphase. The anaphase errors included chromosomes lagging in the spindle midzone and chromosome bridges (Fig. 3a). Multipolar cells were identified by immunofluorescent labeling of the spindle pole protein, γ-Tubulin (Fig. 3b). Both anaphase errors and multipolarity drive genetic instability that can lead to catastrophic division and unbalanced chromosome segregation and cell death. Depletion of NME1, SMS, and YWHAQ significantly increased the occurrence of anaphase errors (Fig. 3c). Depletion of CSAG1 resulted in significantly increased multipolarity (Fig. 3d).
Figure 3.

Mitotic errors observed following siRNA inhibition in 4 of the 6 selected genes. (a) examples of anaphase errors. The two types of anaphase errors observed were lagging chromosomes (arrow, top panels) and chromosome bridges (arrow, bottom panels). (b) Percent of anaphase cells exhibiting errors. Numbers are represented as percentage of total anaphase cells. NME1 (P=0.0059), SMS (P=0.0384) and YWHAQ (P=0.0388) inhibition were significant. (c) example of a multipolar cell. Multipolarity was determined using γ-Tubulin staining. (d) Percent of mitotic cells exhibiting multipolarity. CSAG1 (P=0.0041) inhibition was significant. Note: asterisks correspond to alpha level (*p < 0.05. **p < 0.01. ***p < 0.001. **** p < 0.0001). Error bars represent the standard deviation of 4 separate experiments.
3.5 Screening for CSC Targets
Whole genome screens with RNAi or CRISPR-Cas9 have proven powerful [28, 29], but they can overlook important contributors to cell physiology. In many such studies, only targets with the strongest effects stand out and are considered hits, since these are typically measured against the background inhibitory activity of all targets in the screen [30]. Predictive bioinformatics can focus the search by narrowing investigation on a contingent of genes likely to be important. The downside of many of these algorithms is that they base their predictions on gene expression levels solely within the cell type of interest. GAMMA expands the search in a different way. While it uses expression patterns to predict function, it is not contingent on the expression of the gene itself in a given cell type. By comparing the expression patterns of genes across thousands of data sets, we were able to predict putative cancer stem cell genes with a high success rate without relying on differential expression. Only 4 of the 21 genes whose depletion selectively inhibited growth in BPLER cells showed differential expression in BPLEs [17]. Thus, algorithmic analysis of transcriptional correlation networks can identify relevant genes that would not be revealed by focusing on those that are differentially expressed.
Anti-mitotic drugs were historically thought to act by arresting cells in mitosis through activation of the mitotic spindle checkpoint, though this view has been challenged [31]. Tumors typically contain both numerical and structural chromosome abnormalities proposed to be, at least in part, consequences of errors in mitosis. The generation of these errors is termed chromosome instability. There remains debate about whether chromosome instability is a driver or consequence of tumorigenesis [32, 33]. But increasing chromosome instability in tumor cells to levels incompatible with cell survival may provide a means of tumor therapy [34–38]. Therefore, identifying essential genes and pathways specific for CSC for proliferation is an important goal. We purposely focused on protein targets with little previous characterization in order to find novel targets for CSC inhibition. Further study will be necessary to determine the biochemical pathways in which these proteins participate and whether these pathways are amenable to therapeutic manipulation with small molecule inhibitors or other methods.
When possible, we used siRNA’s that were validated by the manufacturer. However, because we focused on novel, uncharacterized targets and because we targeted 50 genes, we did not specifically validate depletion of message or protein. Thus, we cannot exclude the possibility that negative results may be attributable to failure of the siRNA’s to deplete their target. In all cases where we lacked validated siRNA’s we used pools of three siRNA’s predicted to provide strong depletion to minimize the false negatives. However, both for technical reasons (failure of the siRNA’s to be effective) and for biological reasons (protein targets that are not expressed in the cell lines we used, proteins with long half-lives, or proteins that could function sufficiently at depleted concentrations) negative results cannot be fully guaranteed accurate. Additionally, we cannot rule out all possibility that positive hits may be affected by potential off-target effects of our siRNA depletions. This must be tested by using multiple single siRNA’s, using complimentary targeting strategies such as CRISPR-Cas9 or expression of degron regulatable proteins, and by rescue experiments using siRNA-resistant expression constructs. These experiments are ongoing for several of our most promising targets.
In summary, using the transcriptional network analysis tool GAMMA, we tested 50 genes whose products were potentially involved in CSC proliferation. We assessed these in a breast cancer CSC model system and found 21 genes whose repression led to significant inhibition of proliferation. Further analysis of 6 of these genes implicated 4 in specific mitotic phenotypes. Further research will be needed to test the precise pathways and cell cycle stages in which these gene products execute their important functions and to test whether they are amenable to inhibition for targeting CSC proliferation in tumors.
Supplementary Material
Highlights.
Bioinformatics used to predict novel therapeutic targets for cancer stem cells
Fifty candidate genes tested by siRNA depletion in a breast cancer stem cell model
Specific growth inhibition of cancer stem cells detected in twenty-one depletions
Chromosome segregation defects identified in four of the hits
Acknowledgments
We thank Dr. Fabio Petrocca and Dr. Robert A. Weinberg for generously providing cell lines. The work in JDW’s laboratory was supported by the National Institutes of Health [grant number 5P20GM103636]. The work in GJG’s laboratory was supported by the National Institutes of Health [grant number RO1GM111731], by the Oklahoma Center for Adult Stem Cell Research, and by the McCasland Foundation.
Abbreviation
- CSC
cancer stem cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822–829. doi: 10.1016/j.cell.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 2.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 5.Mita AC, Mita MM, Nawrocki ST, Giles FJ. Survivin: key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin Cancer Res. 2008;14:5000–5005. doi: 10.1158/1078-0432.CCR-08-0746. [DOI] [PubMed] [Google Scholar]
- 6.Perez de Castro I, de Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 7.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Annals of Statistics. 2001;29:1165–1188. [Google Scholar]
- 8.Wren JD. A global meta-analysis of microarray expression data to predict unknown gene functions and estimate the literature-data divide. Bioinformatics. 2009;25:1694–1701. doi: 10.1093/bioinformatics/btp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dozmorov MG, Giles CB, Wren JD. Predicting gene ontology from a global meta-analysis of 1-color microarray experiments. BMC Bioinformatics. 2011;12(Suppl 10):S14. doi: 10.1186/1471-2105-12-S10-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wren JD, Bekeredjian R, Stewart JA, Shohet RV, Garner HR. Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics. 2004;20:389–398. doi: 10.1093/bioinformatics/btg421. [DOI] [PubMed] [Google Scholar]
- 11.Towner RA, Jensen RL, Vaillant B, Colman H, Saunders D, Giles CB, Wren JD. Experimental validation of 5 in-silico predicted glioma biomarkers. Neuro Oncol. 2013;15:1625–1634. doi: 10.1093/neuonc/not124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Towner RA, Jensen RL, Colman H, Vaillant B, Smith N, Casteel R, Saunders D, Gillespie DL, Silasi-Mansat R, Lupu F, Giles CB, Wren JD. ELTD1, a potential new biomarker for gliomas. Neurosurgery. 2013;72:77–90. doi: 10.1227/NEU.0b013e318276b29d. discussion 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clemmensen SN, Bohr CT, Rorvig S, Glenthoj A, Mora-Jensen H, Cramer EP, Jacobsen LC, Larsen MT, Cowland JB, Tanassi JT, Heegaard NH, Wren JD, Silahtaroglu AN, Borregaard N. Olfactomedin 4 defines a subset of human neutrophils. J Leukoc Biol. 2012;91:495–500. doi: 10.1189/jlb.0811417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lupu C, Zhu H, Popescu NI, Wren JD, Lupu F. Novel protein ADTRP regulates TFPI expression and function in human endothelial cells in normal conditions and in response to androgen. Blood. 2011;118:4463–4471. doi: 10.1182/blood-2011-05-355370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ. Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr Biol. 2009;19:1467–1472. doi: 10.1016/j.cub.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10:R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ince TA, Richardson AL, Bell GW, Saitoh M, Godar S, Karnoub AE, Iglehart JD, Weinberg RA. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell. 2007;12:160–170. doi: 10.1016/j.ccr.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 18.Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW, Hoey T, Gurney A, Huang EH, Simeone DM, Shelton AA, Parmiani G, Castelli C, Clarke MF. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158–10163. doi: 10.1073/pnas.0703478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Witt AE, Lee CW, Lee TI, Azzam DJ, Wang B, Caslini C, Petrocca F, Grosso J, Jones M, Cohick EB, Gropper AB, Wahlestedt C, Richardson AL, Shiekhattar R, Young RA, Ince TA. Identification of a cancer stem cell-specific function for the histone deacetylases, HDAC1 and HDAC7, in breast and ovarian cancer. Oncogene. 2017;36:1707–1720. doi: 10.1038/onc.2016.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrocca F, Altschuler G, Tan SM, Mendillo ML, Yan H, Jerry DJ, Kung AL, Hide W, Ince TA, Lieberman J. A genome-wide siRNA screen identifies proteasome addiction as a vulnerability of basal-like triple-negative breast cancer cells. Cancer Cell. 2013;24:182–196. doi: 10.1016/j.ccr.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 22.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sauder CA, Koziel JE, Choi M, Fox MJ, Grimes BR, Badve S, Blosser RJ, Radovich M, Lam CC, Vaughan MB, Herbert BS, Clare SE. Phenotypic plasticity in normal breast derived epithelial cells. BMC Cell Biol. 2014;15:20. doi: 10.1186/1471-2121-15-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013;238:324–338. doi: 10.1177/1535370213480714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Visvader JE, Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci U S A. 2013;110:E4134–4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, Tan IB, Richardson AL, Szallasi Z, Swanton C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011;71:3447–3452. doi: 10.1158/0008-5472.CAN-10-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9:554–566. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- 30.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 31.Weaver BA. How Taxol/paclitaxel kills cancer cells. Mol Biol Cell. 2014;25:2677–2681. doi: 10.1091/mbc.E14-04-0916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duesberg P, Rasnick D, Li R, Winters L, Rausch C, Hehlmann R. How aneuploidy may cause cancer and genetic instability. Anticancer Res. 1999;19:4887–4906. [PubMed] [Google Scholar]
- 33.Zimonjic D, Brooks MW, Popescu N, Weinberg RA, Hahn WC. Derivation of human tumor cells in vitro without widespread genomic instability. Cancer Res. 2001;61:8838–8844. [PubMed] [Google Scholar]
- 34.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 35.Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci U S A. 2009;106:19108–19113. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci U S A. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci U S A. 2004;101:4459–4464. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Colombo R, Caldarelli M, Mennecozzi M, Giorgini ML, Sola F, Cappella P, Perrera C, Depaolini SR, Rusconi L, Cucchi U, Avanzi N, Bertrand JA, Bossi RT, Pesenti E, Galvani A, Isacchi A, Colotta F, Donati D, Moll J. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010;70:10255–10264. doi: 10.1158/0008-5472.CAN-10-2101. [DOI] [PubMed] [Google Scholar]
- 39.Dunnett CW. A Multiple Comparison Procedure for Comparing Several Treatments with a Control. J Am Stat Assoc. 1955;50:1096–1121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.