

Abstract
A2B adenosine receptor (A2BAR) activation induces Gs-dependent cyclic AMP accumulation. However, A2BAR G protein-coupling to other signaling events, e.g. ERK1/2 and calcium, is not well documented. We explored Gi, Gq/11 and Gs coupling in 1321N1 astrocytoma, HEK293, and T24 bladder cancer cells endogenously expressing human A2BAR, using NECA or nonnucleoside BAY60-6583 as agonist, selective Gi, Gs and Gq/11 blockers, and CRISPR/Cas9-based Gq- and Gs-null HEK293 cells. In HEK293 cells, A2BAR-mediated ERK1/2 activity occurred via both Gi and Gs, but not Gq/11. However, HEK293 cell calcium mobilization was completely blocked by Gq/11 inhibitor UBO-QIC and by Gq/11 knockout. In T24 cells, Gi was solely responsible for A2BAR-mediated ERK1/2 stimulation, and Gs suppressed ERK1/2 activity. A2BAR-mediated intracellular calcium mobilization in T24 cells was mainly via Gi, although Gs may also play a role, but Gq/11 is not involved. In 1321N1 astrocytoma cells A2BAR activation suppressed rather than stimulated ERK1/2 activity. The ERK1/2 activity decrease was reversed by Gs downregulation using cholera toxin, but potentiated by Gi inhibitor pertussis toxin, and UBO-QIC had no effect. EPACs played an important role in A2BAR-mediated ERK1/2 signaling in all three cells. Thus, A2BAR may: couple to the same downstream pathway via different G proteins in different cell types; activate different downstream events via different G proteins in the same cell type; activate Gi and Gs, which have opposing or synergistic roles in different cell types/signaling pathways. The findings, relevant to drug discovery, address some reported controversial roles of A2BAR and could apply to signaling mechanisms in other GPCRs.
Keywords: G protein, GPCR, adenosine receptor, ERK1/2, calcium, A2B adenosine receptor
Graphical Abstract
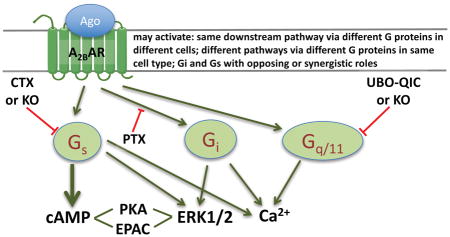
1. Introduction
The A2B adenosine receptor (A2BAR) is involved in many important physiological and pathophysiological functions, such as the hematopoietic stem cell emergence [1], inflammation [2–4], cell proliferation [5–7]; reactive oxygen species (ROS) production [8], erectile function [9], bone homeostasis [10], cardiac protection [11] and diabetes mellitus [12]. However, the signaling mechanisms related to those functions are not well understood. In particular, the mechanisms leading to ERK1/2 phosphorylation, one of those canonical signaling processes, have not been well explored and currently published data are inconsistent. For example, it has been suggested A2BAR-mediated ERK1/2 activity in HEK293 cells is via the Gq/11 protein [13]. However, Schulte and Fredholm [14] suggested that A2BAR-mediated ERK1/2 activity in CHO cells expressing the recombinant A2BAR is mediated via Gs rather than Gq/11. Epperson et al. [15] also found a lack of A2BAR-coupling to Gq protein in cardiac fibroblasts. PKC is a typical modulator of the A2BAR- and other GPCR-mediated ERK1/2 activity [16]. However, Fang and Olah [17] showed that EPAC1 but not PKC is involved in A2BAR-mediated ERK1/2 phosphorylation in human vascular endothelial cells (HUVEC). In rat skeletal muscle, A2BAR activation was found not to modulate ERK1/2, although it stimulated cAMP accumulation [18]. In trophoblast cells, it was found that A2BAR downregulates ERK1/2 activity [19]. ERK1/2 activity stimulated by adenosine-5′-N-ethyluronamide (NECA) in A2BAR-expressing CHO is via PI3K, but not PKA [14]. However, Sun et al. [20] showed that A2BAR activation stimulates ERK1/2 activity via PKA in sickle erythrocytes. Thus, it seems signal mechanisms diverge in various studies and in different cell types under different conditions. Indeed, A2BAR has been shown to cause both proliferation and antiproliferation in different cell types [5,21–27]. A2BAR has been reported to involve both pro- and anti-inflammatory actions [2,28–30], which might be related the different G protein-coupling in different types of immune cells. Thus, it is important to understand the types of G proteins or signaling pathways involved in A2BAR-mediated functions.
Recent studies show that various A2BAR agonists have different maximum effect (Emax) and signaling bias in different signaling pathways [31,32]. Nonnucleoside agonist BAY60-6583 stimulates ERK1/2 activity in HEK293 cells but not in T24 bladder cancer cells, both endogenously expressing the A2BAR, although nucleosides NECA and adenosine-5′-N-cyclopropyluronamide (CPCA) stimulated ERK1/2 activity in both cell types [31]. It was demonstrated that BAY60-6583 behaves as an antagonist and induces insulin secretion in a mouse pancreatic cell line, MIN-6 [31]. Some controversies concerning the A2BAR might also be due to different agonists used in those earlier studies.
In the present study, we explore the coupling of the native human A2BAR to Gi, Gq/11 and Gs in three different cell types, 1321N1 astrocytoma, HEK293, and T24 bladder cancer cells. We used NECA and BAY60-6583, selective blockers for Gi, Gs and Gq/11, as well as the clustered regularly interspaced short palindromic repeat-associated protein-9 nuclease (CRISPR/Cas9)-based Gq/11- and Gs-null HEK293 cells [33,34]. As ERK1/2 activity and calcium mobilization are involved in many A2B-mediated functions and are the convergence of multiple G protein-signaling pathways directed under the A2BAR, we explored the contributions of various G proteins and downstream signaling molecules to ERK1/2 activity and calcium mobilization. These pathways were compared in three different cell types endogenously expressing almost solely the A2BAR with other three AR subtypes being at very low levels. We found that the A2BAR may couple to different downstream signaling events via different G proteins in the same cell type, and the A2BAR may couple to the same downstream pathway via different G proteins in different cell types. Gi and Gs proteins activated by the A2BAR may play opposing or synergistic roles in different cell types and in different signaling pathways. Any one of the three G proteins, Gi, Gq/11, and Gs, may be solely responsible for a specific signaling event mediated via the A2BAR depending on cell type. The findings could be related to controversial roles of the A2BAR-mediated effects reported in literature, e.g. proliferation and antiproliferation, or proinflammatory vs. anti-inflammatory. These findings could be applicable to the study of signaling pathways of other GPCRs, and should be of considerable relevance to future drug discovery and development.
2. Materials and Methods
2.1. Materials
GO6983, CE3F4, ESI-09, HJC0350, H89, PSB603, NECA (adenosine-5′-N-ethyluronamide) and MRS2365 ([[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3-dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid monoester trisodium salt) were from Tocris (Ellisville, MO). CGS21680, Carbachol, CTX and PTX were from Sigma (St. Louis, MO). UBO-QIC was purchased from University of Bonn (Bonn, Germany). BAY60-6583 (LUF6210, termed hereafter ‘BAY’) was synthesized at Leiden/Amsterdam Center for Drug Research and was provided by Ad IJzerman (Leiden, The Netherlands). MRS3997 [35] and MRS5911 [36] were synthesized at NIDDK, National Institutes of Health (Bethesda, MD, USA), and were provided by H. Adachi and E. Kiselev (NIDDK, NIH), respectively. All compounds were dissolved in DMSO except that CTX and PTX were in water, and proper controls were included in all experiments. AlphaScreen cAMP kit, SureFire® p-ERK1/2 (Thr202/Tyr204) Assay Kit and AlphaScreen SureFire p-Akt 1/2/3 (p-Ser473) Assay Kit were purchased from PerkinElmer (Waltham, MA). Gs-null and Gq/11-null HEK293 cells were generated at Tohoku University, Sendai, Japan [33,34]. HEK293 human embryonic kidney, 1321N1 astrocytoma, and T24 bladder cancer cells were from ATCC (Mannasas, VA); all other reagents were from standard commercial sources and of analytical grade.
2.2. Detection of gene expression by quantitative real-time PCR (qRT-PCR)
Total mRNA was extracted as described in the protocol of the RNeasy® Mini Kit (Qiagen, Valencia, CA). Reverse transcription was performed using Superscript III First Strand Synthesis Supermix® kit (Invitrogen, Carlsbad, CA). The cDNA was amplified by PCR with gene-specific FAM-labeled MGB Taqman® probes (Applied Biosystems, Foster City, CA) in 96-well plates using a 7300 Real-Time PCR System (Applied Biosystems) and default thermocycler program. The gene expression levels were calculated by the 2−ΔΔCT method using GAPDH as the endogenous control.
2.3. cAMP accumulation assay
Cells were cultured in DMEM medium supplied with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin and 2 μmol/ml glutamine. For the assay of cAMP, cells were plated in 96-well plates in 100 μl medium overnight. After overnight culture, cells were first treated with adenosine deaminase (3 units/ml) and the phosphodiesterase inhibitor rolipram (10 μM) for 30 min, before the addition of agonists and incubate for another 20 min. For the test of antagonists or inhibitors, A2BAR antagonist PSB603 or Gq/11 inhibitor UBO-QIC was preincubated for 20 min and PTX (200 ng/ml) or CTX (500 ng/ml) was preincubated overnight before the addition of agonists. The reaction was terminated by removal of the supernatant, and cells were lysed upon the addition of 50 μl of lysis buffer (0.3% Tween-20). For determination of production of cAMP, an AlphaScreen® cAMP Kit was used following manufacturer’s instructions (PerkinElmer, Waltham, MA).
2.4. ERK1/2 Stimulation
The method used was as previously described [37]. HEK293 cells, 1321N1 astrocytoma cells or T24 bladder cancer cells endogenous expressing the A2BAR were seeded in a 96-well plate in complete growth medium. After cell attachment, medium was removed and cells were serum-starved overnight in 90 μl serum free medium. To examine the time-course, cells were stimulated with agonists at various time points. To obtain agonist concentration-response curves, HEK293 and T24 cells were stimulated for 5 min, and 1321N1 cells were stimulated for 10 min. Cells were pretreated with an antagonist or inhibitor 30 min (except that cells were pretreated with PTX (200 ng/ml) or CTX (500 ng/ml) overnight) before the addition of an agonist. After agonist treatment, the medium was removed and cells were lysed with 1× lysis buffer (20 μl) (PerkinElmer AlphaScreen SureFire® p-ERK1/2 (Thr202/Tyr204) Assay Kit). Lysate (4 μl/well) was transferred to a 384-well ProxiPlate Plus® (PerkinElmer, Waltham, MA). Acceptor beads were diluted 1:50 in a 1:5 mixture of activation buffer in the reaction mixture and added to the 384-well plate (5 μl/well). The plate was sealed and incubated for 2 h at room temperature. Donor beads (2 μl) diluted 1:20 in dilution buffer were added, and the plate was incubated for another 2 h at room temperature. The plate was measured using an EnVision® multilabel reader using standard AlphaScreen® settings as instructed by the manual.
2.5. Intracellular calcium mobilization
HEK293 cells, 1321N1 astrocytoma cells or T24 bladder cancer cells endogenously expressing the A2BAR were grown overnight in 100 μl of media in 96-well black plates at 37°C and 5% CO2. Cells were washed with Hank’s Buffer containing 20 mM HEPES (pH 7.4; without calcium and magnesium). The calcium 6 assay kit was used as instructed by mixing Hank’s Buffer containing 20 mM HEPES without calcium and magnesium. Probenecid (final concentration of 2.5 mM) was added to the loading dye to increase dye retention. Cells were incubated with 100 μl dye/probenecid for 60 min at room temperature. Cells were pretreated with various concentrations of antagonists for 30 min or PTX (200 ng/ml) overnight before the addition of agonists. All reagents used were prepared using Hank’s Buffer containing 20 mM HEPES. Samples were run in duplicate or triplicate using a FLIPR TETRA® (Molecular Devices, Sunnyvale, CA) at room temperature. Cell fluorescence (excitation at 485 nm; emission at 525 nm) was monitored following exposure to A2BAR agonists. Increases in intracellular calcium concentrations are reported as the maximum fluorescence value after exposure minus the basal fluorescence value.
2.6. Data analyses
Data were analyzed using Prism® 7.0 software (GraphPAD, San Diego, CA). Statistical significance of the differences was assessed using a Student’s t test (between two conditions) or a One-Way Analysis of Variance (ANOVA) followed by Bonferroni’s or Turkey’s multiple comparison tests where appropriate among multiple conditions. Differences yielding P<0.05 were considered as statistically different.
3. Results
3.1. Gene expression of four AR subtypes in three cell types
In order to study the coupling of the endogenous human A2BAR to different G proteins, we selected three cell types, HEK293, T24 and 1321N1, which endogenously express the A2BAR highly in comparison to other three AR subtypes. The reason for our use of AR agonist NECA is that it is still the only established full agonist for the A2BAR that is commercially available, although nonselective. The nonnucleoside agonist BAY60-6583 is a partial agonist, albeit selective. Previously, we have found that the A2BAR is highly expressed in HEK293, 1321N1 and T24 cells at a similar expression level [31]. However, the expression level of the A2BAR has not been strictly compared with that of the other three AR subtypes. Thus, in the present study we compared the expression patterns of the four ARs at these three cell lines. The gene expression of the A2BAR was found consistent with A2BAR agonist-induced cAMP accumulation in those three types of cells and several other types of cells [31]. Fig. 1 shows that A2BAR was the dominant AR in comparison to other ARs in all three cell types, which were used in subsequent experiments.
Figure 1.

Quantification using qRT-PCR of AR gene expression in HEK293 (A), T24 (B), and 1321N1 astrocytoma cells (C). Data were normalized based on endogenous GAPDH expression and were expressed as relative expression level. Results are expressed as mean ± SD and are from 3 independent experiments performed in triplicate or quadruplicate.
3.2. Coupling of the A2BAR to Gs, Gq and Gi in HEK293 cells
3.2.1. Cyclic AMP accumulation in HEK293 cells
Although Fig. 1a shows that the A2BAR is expressed at a much higher level compared with other three ARs, to assure that the effect of NECA is solely via the A2BAR, not the A2AAR, both of which couple to Gs, we examined the potential effect of the A2AAR-selective agonist CGS21680 and tested the effect a selective A2BAR antagonist PSB603 on NECA-stimulated cAMP accumulation in HEK293 cells. Fig. 2a shows that CGS21680 has negligible effect in comparison to NECA. The A2BAR-selective antagonist PSB603 at the concentration of 10 nM rightward shifts the NECA concentration-response curve to the right in a parallel manner, suggesting the effect of NECA is indeed solely via the A2BAR.
Figure 2.
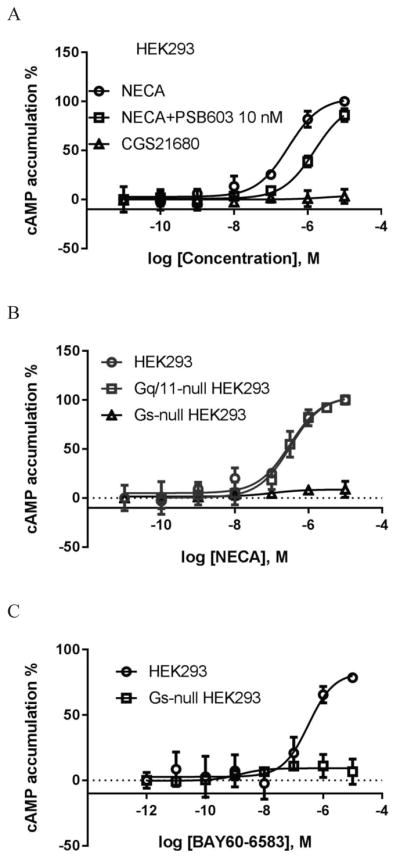
A2BAR-mediated cAMP accumulation in HEK293 cells. A. NECA induced cAMP accumulation in HEK293 cells. B. NECA-stimulated accumulation of cAMP in control, Gq/11-null and Gs-null HEK293 cells. C. cyclic AMP accumulation induced by non-nucleoside A2BAR agonist BAY60-6583. Cells were treated with agonists for 20 min. The A2BAR antagonist PSB603 was added 20 min before the addition of agonist NECA. Results are expressed as mean ± SD and are from at least three independent experiments performed in duplicate or triplicate. The reagent single concentrations used in all figures were carefully selected based on published and supplier information to maintain selectivity [31, 35–39, 41, 42].
We next examined NECA-induced cAMP accumulation in Gs-null HEK293 cells generated previously using CRISPR/Cas9 [34]. As a comparison, we also tested the effect of NECA in Gq-null HEK293 cells [37]. Fig. 2b shows that NECA-induced cAMP accumulation was completely eliminated by Gs knockout (KO), but unaffected by Gq/11 KO. The effect of BAY was also completely diminished by Gs KO (Fig. 2c), but not by Gq/11 KO, UBO-QIC or PTX (data not shown).
3.2.2. A2BAR-mediated ERK1/2 activity in HEK293 cells
In the following experiments (Fig. 3), we examined the G proteins possibly involved in A2BAR-mediated ERK1/2 activity in HEK293 cells. NECA (3 μM) induced stimulation of ERK1/2 activity in HEK293 typically 3–5-fold above basal value, reaching maximum at 5 min. The PKC inhibitor GO6983 completely eliminated the effect of NECA and diminished basal activity. The Gq/11 inhibitor UBO-QIC had a negligible effect, whereas both CTX and PTX significantly inhibited the effect of NECA at 5 min (Fig. 3a), suggesting that both Gs and Gi, but not Gq/11, are involved in NECA-induced ERK1/2 phosphorylation. The inhibition of ERK1/2 activity by PTX and GO6983 suggests that A2BAR-mediated ERK1/2 activity is at least partly via βγ release from Gi/o and subsequent activation of the PLC-PKC pathway, although crosstalk between intracellular second messenger systems and the activation of PKC by downstream pathways other than Gq/11 is also possible.
Figure 3.


Stimulation of ERK1/2 activity in HEK293 cells. A. Effect of Gq/11 inhibitor UBO-QIC, Gi inhibitor PTX, Gs down-regulator CTX and PKC inhibitor GO (GO6983) on NECA-stimulated ERK1/2 activity. Cells were pretreated with UBO-QIC and GO6983 for 30 min or PTX and CTX overnight before the addition of agonist. #Significantly different from control (NECA) (P<0.05, One-Way ANOVA followed by post hoc test (Bonferroni)). B. NECA-stimulated ERK1/2 activity in Gs- and Gq/11-null HEK293 cells. C. ERK1/2 activity stimulated by the agonist MRS2365 of the Gq/11-coupled P2Y1 receptor. D. ERK1/2 activity in Gq/11-null HEK293 cells. E. Comparison of ERK1/2 activity induced by the adenylyl cyclase activator forskolin and A2BAR (nonselective) agonist NECA (expressed as 100%), and the inhibition of PKA inhibitor H89 and nonselective EPAC inhibitor ESI-09 on the effect of NECA. F. Comparison of the effects of forskolin and NECA in HEK293 cells overexpressing the A2BAR (HEK293-A2B), and the effects Gi, Gs, and Gq/11 inhibitors on NECA-stimulated ERK1/2 activity. #Significantly different from control (NECA) (P<0.05, One-Way ANOVA followed by post hoc test (Bonferroni)). Data are mean ± SD from three independent experiments performed in triplicate.
Fig. 3b shows that NECA induced increases of ERK1/2 activity in control HEK293 cells and Gq/11-null HEK293 cells, but not in Gs-null HEK293 cells, further confirming that Gs but not Gq/11 plays a major role in ERK1/2 activity mediated via the endogenous A2BAR. As a control, both UBO and Gq/11 KO diminished ERK1/2 activity stimulated by the P2Y1 receptor-selective agonist MRS2365 (Fig. 3c). To confirm the involvement of Gi proteins in NECA-induced ERK1/2 activity, PTX was applied to Gq/11-null HEK293 cells. Fig. 3d shows that similar to the control HEK293 cells, PTX but not UBO significantly diminished the effect of NECA. The ERK1/2 activity stimulated by NECA at 5 min in the presence of PTX was significantly different from the control (p<0.05). UBO did not affect NECA-stimulated ERK1/2 activity in Gq/11-null HEK293 cells (p>0.05 compared with control).
As both PKA and EPAC are possibly involved in Gs-mediated events, we next examined the steps downstream of Gs mediating ERK1/2 activity. Fig. 4e shows that the adenylyl cyclase activator forskolin and the A2BAR agonist NECA stimulate ERK1/2 activity to a similar extent, and the effect of NECA was diminished 48±6% by the PKA inhibitor H89, and 92±5% by the nonselective EPAC inhibitor ESI-09 [39]. This suggested that both PKA and EPACs are downstream of Gs-mediated ERK1/2 activity in HEK293 cells, and EPACs probably play a major role in this event. It remains to be examined whether EPAC1 or EPAC2 plays a more significant role in A2BAR-mediated ERK1/2 activity.
Figure 4.

Intracellular calcium mobilization in HEK293 cells. A. Agonist NECA-mediated response. B. P2Y1 receptor agonist MRS2365-induced response and the effect of the Gq/11 inhibitor UBO (UBO-QIC). C. NECA-induced calcium increase in HEK293 cells overexpressing the A2BAR (HEK293-A2B). Cells were pretreated with UBO-QIC for 30 min before the addition of agonists. Results were expressed as mean ± SD from 3 independent experiments performed in triplicate.
As described above, Gi and Gs but not Gq/11 are involved in endogenous A2BAR-stimulated ERK1/2 activity. We were curious if the expression level of the A2BAR may or may not affect G protein-coupling, as cells overexpressing the recombinant A2BAR were used in many previous studies [14,40]. Fig. 3f shows that overexpression of the A2BAR in HEK293 cells led to a dramatic increase in NECA-induced ERK1/2 activity. In HEK293 cells expressing the recombinant A2BAR, forskolin only induced a 4-fold increase of ERK1/2 activity, whereas NECA induced a >40-fold increase of ERK1/2. Interestingly, UBO, which did not affect NECA-induced ERK1/2 activity in control HEK293 cells, induced a modest but significant decrease with A2BAR overexpression (28±5%; P<0.05 compared with NECA group). On the other hand, both CTX and PTX induced a >70% inhibition of ERK1/2 activity in control HEK293 cells, but only about 50% inhibition in HEK293 cells overexpressing the recombinant A2BAR. Thus, G protein-coupling patterns of the A2BAR in the overexpressed cells may not accurately represent the coupling of the native A2BAR.
3.2.3. A2B-mediated intracellular calcium mobilization in HEK293 cells
Fig. 4a shows that NECA only induced a slight increase of intracellular calcium (about 10–15% compared with that induced by the selective P2Y1 receptor agonist MRS2365 (Fig. 4b)). However, neither NECA nor MRS2365 induced calcium transients in Gq/11-null HEK293 cells (Fig. 4a, 4b).
As NECA only induced a small increase in calcium transients in control HEK293 cells, we further tested this in HEK293 cells overexpressing the recombinant A2BAR. Fig. 3c shows that NECA induced a robust calcium response in HEK293 cell overexpressing the A2BAR but not in Gq/11-null HEK293 cells overexpressing the A2BAR. Also, the Gq/11 inhibitor UBO completely inhibited the effect of NECA.
Comparing Fig. 4a and Fig. 4c, it was noted that the A2BAR-overexpression increased the NECA-induced calcium response, and the signal-to-noise ratio increased ~5-fold. However, the potencies of NECA in control HEK293 and in HEK293-A2BAR cells were found not significantly different (385±86 vs. 496±138 nM, P>0.05, unpaired t-test). Similarly, potencies of NECA to induce ERK1/2 activity in control HEK293 cells and in HEK293-A2BAR cells (475±66 vs. 561±122 nM) were also not significantly different (P>0.05, unpaired t-test). Fig. 5a shows the raw ERK1/2 activity data; Fig. 5b shows the normalized data. However, A2BAR overexpression significantly increased the potency (over 100-fold) rather than the Emax of NECA in stimulation of cyclic AMP accumulation (Fig. 5c). Thus, A2BAR overexpression changed coupling efficiency differently at different signaling pathways, i.e. it may change agonist Emax or agonist potency depending on the signaling pathway measured.
Figure 5.

Comparison of the effects of overexpression of the A2BAR on ERK1/2 activity and cAMP accumulation in HEK293 cells. A. Overexpression of the A2BAR increased agonist response without affect agonist potency. B. Data from Figure 5A were normalized in order to make the two curves discernible and comparable. C. Overexpression of the A2BAR increased agonist potency. NECA was incubated with cells for 5 min and 20 min, respectively. Each data point represents the mean ± SD from three experiments in duplicate.
3.3. Coupling of the A2BAR to Gs, Gi and Gq/11 in T24 bladder cancer cells
We demonstrated previously that NECA-induced cAMP accumulation in T24 cells is exclusively via the A2BAR [31]. NECA also induced a robust response in calcium mobilization and ERK1/2 phosphorylation. However, the G proteins or signaling molecules involved in those events have not been explored. In the following experiments, we examined the G proteins that potentially mediate those signaling events.
3.3.1. A2B-mediated cAMP accumulation in T24 bladder cancer cells
Fig. 6 shows that both NECA and BAY induced robust cAMP accumulation, which was not affected by PTX and/or UBO-QIC, suggesting neither Gi or Gq/11 proteins have an impact on A2BAR-mediated cAMP accumulation in T24 cells under normal conditions.
Figure 6.

A2BAR agonist-induced cAMP accumulation in T24 bladder cancer cells. A. NECA. B. BAY (BAY60-6583). Cells were pretreated with UBO-QIC for 30 min or PTX overnight before the addition of agonists and incubated for another 20 min. Data are expressed as mean ± SD from three separate experiments in duplicate or triplicate.
3.3.2. A2B-mediated ERK1/2 activity in T24 bladder cancer cells
Fig. 7a shows that NECA induced a maximum stimulation of ERK1/2 activity of 2- to 3-fold above basal value at about 5 min. Similar to that in HEK293 cells, the PKC inhibitor GO6983 eliminated NECA-induced ERK1/2 activity and diminished basal ERK1/2 activity in T24 cells. Interestingly, unlike that in HEK293 cells, NECA-stimulated ERK1/2 activity was completely blocked by PTX, but was significantly enhanced by CTX, suggesting Gs and Gi play opposing roles in A2BAR-mediated ERK1/2 phosphorylation in T24 cells. The effect of UBO was negligible. To confirm this, we ran a concentration response curve of NECA in the absence or presence of UBO. Fig. 7b shows that UBO mainly affected the basal ERK1/2 activity, without affecting the potency of NECA. Fig. 7c shows that the concentration-response of NECA was enhanced and diminished by CTX and PTX, respectively. The results suggest that Gi proteins solely mediate a strong A2BAR-stimualted ERK1/2 activity, however, under normal conditions, a large part of the stimulatory effect of A2BAR is masked by Gs.
Figure 7.


A2BAR agonist-stimulated ERK1/2 activity in T24 bladder cancer cells. A. Effect of Gq/11 inhibitor UBO-QIC, Gi inhibitor PTX, Gs down-regulator CTX and PKC inhibitor GO (GO6983) on NECA-stimulated ERK1/2 activity. Cells were pretreated with UBO-QIC and GO6983 for 30 min or PTX and CTX overnight before the addition of agonist. B. concentration-response curves of NECA in the presence and absence of UBO (UBO-QIC). C. Concentration-response curves of NECA in the presence and absence of PTX and CTX (the Emax of NECA in the absence of CTX or PTX was expressed as 100%). D. Comparison of forskolin- and NECA-stimulated ERK1/2 activity, and the effects of PKC inhibitor GO6983 (10 μM), PKA inhibitor H89 (10 μM) and EPAC1 inhibitor CE3F4 (10 μM) and EPAC2 inhibitor HJC0530 (3 μM). Control and NECA+GO groups were expressed as 100% and 0%, respectively. *Significantly different from control (P<0.05). # significantly different from NECA group (P<0.05). E. Effect of downregulation of Gs by CTX on the ERK1/2 activity stimulated by the nonnucleoside agonist BAY60-6583. Each data point represents the mean ± SD from three independent experiments in triplicate.
We further explored the pathways downstream of Gs that mediates the inhibitory effect on ERK1/2 activity. Fig. 7d shows that forskolin significantly diminished whereas NECA stimulated ERK/12 activity in T24 cells, suggesting that the role of Gs is probably opposite to that in HEK293 cells (Fig. 4e). The effect of NECA was significantly enhanced by both the PKA inhibitor H89 and the EPAC2 inhibitor HJC0350 [41], but not by the EPAC1 inhibitor CE3F4 [42].
As shown earlier [31] in T24 cells, only NECA stimulated ERK1/2 activity (about 2-fold above basal value), whereas the nonnucleoside agonist BAY did not increase ERK1/2 activity, which was thought to be presumably due to the partial agonist activity of BAY. In the present study, we further examined the role of Gs in this lack of ERK1/2 stimulation by BAY. Fig. 7e shows that indeed BAY does not stimulate ERK1/2 activity. However, in the presence of CTX, BAY induced a robust increase (3-fold) of ERK1/2 activity. Thus, the previous observation that BAY did not stimulate ERK1/2 activity in T24 cells is due to the fact that Gs masks the stimulatory effect of the A2BAR.
3.3.3. A2B-mediated intracellular calcium mobilization in T24 bladder cancer cells
Unlike in HEK293 cells, activation of the native A2BAR induced a robust increase of intracellular calcium. Fig. 8 shows that NECA-induced calcium increase is almost completely diminished by PTX, and also significantly affected by CTX, but not influenced by UBO-QIC, suggesting that calcium mobilization in T24 cells is mostly mediated via Gi proteins, and to a lesser extent by Gs, but not via the Gq/11 proteins.
Figure 8.
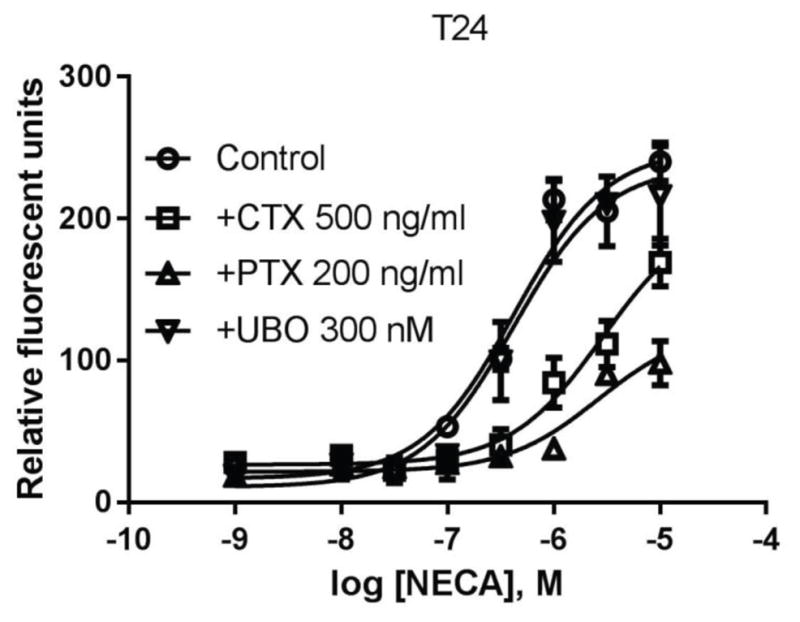
A2BAR-mediated calcium mobilization in T24 bladder cancer cells. Cells were pretreated with UBO-QIC 30 min or PTX and CTX overnight before the addition of agonist NECA. Data are expressed as mean ± SD from 3–4 separate experiments performed in duplicate.
3.4. Coupling of the A2BAR to Gs, Gi and Gq in 1321N1 astrocytoma cells
3.4.1. A2BAR stimulated cAMP accumulation in 1321N1 astrocytoma cells endogenously expressing the A2BAR
NECA concentration-dependently induced cAMP accumulation in 1321N1 astrocytoma cells endogenously expressing the A2BAR, which was not significantly affected by the Gi inhibitor PTX or the Gq/11 inhibitor UBO (Fig 9).
Figure 9.

A2BAR-stimulated cAMP accumulation in1321N1 astrocytoma cells. Cells were pretreated with UBO for 30 min or PTX overnight before the addition of agonist and incubated for an additional 20 min. Results are expressed as mean ± SD from three independent experiments performed in duplicate.
3.4.2. A2BAR suppressed ERK1/2 activity in 1321N1 astrocytoma cells
Unlike the role of the native A2BAR in HEK293 and T24 cells which increases ERK1/2 activity, the activation of the A2BAR in 1321N1 astrocytoma cells diminished rather than stimulated ERK1/2 activity. The suppressing effect of NECA reached a maximum at about 5–10 min, and the suppression lasted for at least 60 min. Fig. 10 shows the time course of NECA-induced and the A2BAR-mediated suppression of ERK1/2 activity.
Figure 10.


A2BAR-mediated ERK1/2 activity in 1321N1 astrocytoma cells. A. Time-course of NECA-suppressed ERK1/2 activity. B. Effect of Gq/11 inhibitor UBO-QIC, Gi inhibitor PTX and Gs downregulator CTX on NECA-suppressed ERK1/2 activity. Cells were pretreated with UBO-QIC for 30 min or PTX and CTX overnight before the addition of agonist and incubated for another 10 min. *Significantly different from control (P<0.05). #Significantly different from NECA group (P<0.05). C. Comparison of the suppression of ERK1/2 activity by adenylyl cyclase stimulator forskolin and agonist NECA, and the effects of PKA and EPAC inhibitors on NECA-induced suppression of ERK1/2 activity. Control and NECA+GO groups were expressed as 100% and 0%, respectively. *Significantly different from control (P<0.05, unpaired t test). #Significantly different from NECA group (P<0.05, One-Way ANOVA followed by post hoc test (Bonferroni)). D. Concentration-response of ERK1/2 activity suppressed by four A2BAR agonists. Results are expressed as mean ± SD from at least three independent experiments performed in duplicate or triplicate.
As described above, the suppression of ERK1/2 activity mediated via the A2BAR in 1321N1 astrocytoma cells reached a maximum at 5–10 min, thus in the following experiment, the stimulation time was selected as 10 min. The decrease by NECA was partially reversed by CTX, further diminished by PTX, but not affected by UBO (Fig. 10b), suggesting a Gs-mediated mechanism. CTX and PTX significantly increased and decreased the effect of NECA, respectively (P<0.05, compared with NECA group; One-Way Analysis of Variance (ANOVA) followed by post-test (Bonferroni)).
Fig. 10c shows that both forskolin and NECA diminish ERK1/2 activity in 1321N1 astrocytoma cells, and forskolin actually produced an effect larger than that of NECA (P<0.05, unpaired t-test). The effect of NECA was partially reversed by H89 and further decreased by EPAC1 inhibitor CE3F4, but it was not affected by EPAC2 inhibitor HJC0350. This suggested that PKA is at least partially responsible for NECA-induced decrease of ERK1/2 activity in 1321N1 astrocytoma cells, and EPAC1 plays a role that is the opposite of PKA. The decreased ERK1/2 activity by NECA was further decreased by GO6983, but completely reversed by the selective A2BAR antagonist PSB603.
We have shown previously [31] that NECA is a full agonist, whereas BAY, MRS5911 and MRS3997 are partial agonists in stimulation of ERK1/2 activity in HEK293 cells. Here, Fig. 10d shows that all agonists concentration-dependently decreased ERK1/2 activity in 1321N1 astrocytoma cells, with NECA being more efficacious than BAY and MRS3997 in suppressing the ERK1/2 activity.
3.4.3. A2BAR activation fails to stimulate intracellular calcium mobilization in 1321N1 astrocytoma cells
Unlike in T24 cells, activation of the A2BAR in 1321N1 astrocytoma cells did produce any intracellular calcium mobilization (Fig. 11). As a control, carbachol produced a robust response via the activation of native muscarinic receptors.
Figure 11.
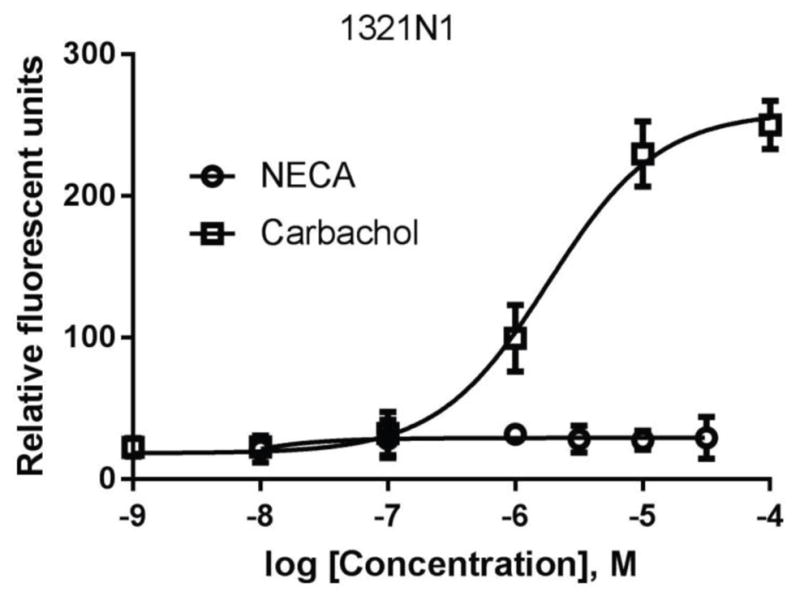
Intracellular calcium mobilization in 1321N1 astrocytoma cells. Cells were plated in 96-well black plates with clear bottom and were grown overnight. Calcium 6 dye was loaded as instructed by kit manual and incubated for 60 at room temperature before the addition of agonists carbachol or NECA. Each data point represents the mean ± SD from three experiments performed in triplicate.
4. Discussion
By using three cell types, HEK293 (including wild-type, Gs- and Gq/11-null HEK293), 1321N1 astrocytoma and T24 bladder cancer cells, it was demonstrated in the present study that the endogenous A2BAR may couple to the same downstream signaling pathway via different G proteins in different cell types, and it may couple to different signaling pathways via different G proteins in the same type of cells (Table 1). Furthermore, Gs and Gi may have both opposing effects and synergistic effects in mediating downstream events, depending on cell type and signaling pathway. This could be a reason for the A2BAR displaying both proliferative and antiproliferative effects in different cell types [5,21–24]. It could also be related to the proinflammatory and anti-inflammatory roles of the A2BAR in different types of inflammatory cells or different functions of the same cell types [2, 28–30]. It remains to be explored how the ubiquitous A2BAR couples to G proteins and downstream signaling events in other cell types and interacts with adenosine signaling via other ARs in the same cell type, considering the important adenosinergic functions throughout the body. Also, differential signaling of the A2BAR in other peripheral tissues (e.g. cardiac and vascular tissue) versus CNS regions, and signaling changes in disease states deserve further exploration. The potential Gi coupling could also explain the cardioprotective effects of the A2BAR [43,44]. The differential signaling observed here for the A2BAR raises concerns regarding the reliability of in vitro data as it relates to drug discovery in general.
Table 1.
Summary of A2BAR-induced signaling.
| Activity | Agonista | Evidencea | Figure |
|---|---|---|---|
| HEK293 kidney cells | |||
| Gs-mediated cAMP production | NECA, Bay | GsKO | 2A–C |
| Gs-mediated ERK1/2 stimulation | NECA | GsKO, CTX | 3A,B |
| Gs-mediated ERK1/2 via PKA, EPAC | NECA | H89, ESI | 3E |
| ERK1/2 activity via PKC | NECA | GO | 3A |
| Gi-mediated ERK1/2 stimulation | NECA | PTX | 3A,D,F |
| Gq-mediated Ca2+ mobilization | NECA | UBO, Gq/11KO | 4A,C |
| A2B-overexpressing HEK293 kidney cells | |||
| Gs-mediated cAMP production left-shifted | NECA | C/R curve | 5C |
| T24 bladder cancer cells | |||
| Gs-mediated cAMP production | NECA, Bay | PTX/UBO (−) | 6 |
| ERK1/2 activity via PKC | NECA | GO | 7A |
| Gi-mediated ERK1/2 stimulation | NECA | PTX | 7C |
| Gs/PKA-suppressed ERK1/2 activity | NECA, Bay | CTX, H89 | 7A,C–E |
| ERK1/2 activity suppressed by EPAC2 | NECA | HJC0350 | 7D |
| ERK1/2 stimulation requires PKC | NECA | GO | 7D |
| Gi-mediated Ca2+ mobilization | NECA | PTX | 8 |
| 1321N1 astrocytoma cells | |||
| Gs-mediated cAMP production | NECA | UBO/PTX (−) | 9 |
| Gs-suppressed ERK1/2 activity | NECA | forsk, CTX | 10A,B |
| Gs-suppression of ERK1/2 is PKC-depend. | NECA | GO | 10C |
| ERK1/2 inhibition via EPAC1 | NECA | CE3F4 | 10C |
| Gi-stimulated ERK1/2 activity | NECA | PTX | 10B |
| ERK1/2 inhibition (other agonists) | Bay, MRS5911, MRS3997 | 10D | |
| Lack of A2BAR-induced Ca2+ mobilization | NECA | Ca2+ dye | 11 |
Bay, BAY60-6583; C/R, concentration-response; ESI, ESI-09; forsk, forskolin; GO, GO6983; UBO, UBO-QIC.
The possibility of a GPCR coupling to multiple G proteins has been proposed previously, including for β2 and β3 adrenergic [45,46]; luteinizing hormone [47], follicle stimulating hormone [48], δ-opioid [49], and cannabinoid CB1 [50] receptors. It has been suggested that FMRFamide related mixed action in vivo might be related to dual Gi/Gs coupling, based on studies using PTX and CTX [51]. The coupling to multiple G proteins has been the best characterized for the β2 receptor, including the demonstration that both Gi and Gs pathways under the β2 receptor are involved in heart function [52–54]. However, the nature of dual coupling has not been explored previously by strictly comparing the native receptors in different cell types. The mechanisms behind this Gi-Gs dual coupling phenomenon have been controversial. For example, a possibility of ligand-specific coupling of β2 receptor to G proteins was suggested [54]. PKA phosphorylation of receptor might switch the β2 receptor coupling from Gs to Gi [55]. Hill and Baker [56] proposed that expression levels and locations of G proteins and receptors in the cell membrane, rather than phosphorylation by PKA are responsible for the Gi coupling. Crespo et al [57] suggested that β2-mediated MAPK activation in COS-7 cells is via G protein βγ subunits. It has also been indicated that β2 receptor coupling to Gi is indirect and dependent on A1AR [58].
However, the mechanisms inferred from many earlier studies were from the use of transfected cell lines, and may not represent the mechanisms in native cells. Furthermore, the coupling to different G proteins has not been strictly compared in different cell types, which could have different expression levels of receptors and G proteins, as well as different locations of G proteins in different membrane microdomains. With the recent availability of the selective Gq/11 inhibitor UBO-QIC [37,59] and Gs- and Gq/11-null HEK293 cells generated using CRISPR/Cas9 [34,37,60], we were able to compare the G protein-coupling unambiguously. For example, intracellular calcium mobilization in HEK293 cells was completely eliminated by either Gq/11 KO or by UBO suggesting Gq/11-mediated mechanism; In T24 cells, the calcium increase was significantly diminished by PTX but not by UBO, suggesting a Gi-mediated mechanism. Also, by using both HEK293 cells and the Gq/11-null HEK293 cells, we were able to observe the diminishing effect of PTX on NECA-stimulated ERK1/2 activity, clearly suggesting a Gi- but not Gq/11-mediated mechanism.
It has been well documented that in addition to PKA, EPACs, also downstream mediators of Gs, are critical in many signaling cascades [61–63]. However, the role of the EPACs in A2BAR signaling has not been well documented. In the present study, we found that EPACs have different roles in different cell types and in different signaling pathways. For example, in HEK293 cells, A2BAR-mediated ERK1/2 activity is via Gs followed by both PKA and EPACs; in T24 cells, both PKA and EPAC2, but not EPAC1, inhibit Gs-mediated suppression of ERK1/2 activity. In 1321N1 cells, EPAC1 and PKA have opposing role in A2BAR-mediated ERK1/2 activity, whereas EPAC2 plays a negligible role. These pathways remain to be examined in other cell types.
As A2BAR activation may only lead to a Gi-dependent signaling event that is relevant to a specific condition (e.g. ERK1/2 activity in T24 cells) or to Gs-dependent signaling (ERK1/2 activity in1321N1 cells), it is important to understand the signaling mechanisms including the G proteins involved in a specific disease. Receptor-selective drugs may not have sufficient specificity for certain specific conditions, as they may produce both Gi and Gs signaling events.
Cell type-specific or G protein-coupling specific agonists for the A2BAR in theory could produce fewer side effects, considering the ubiquitous presence and the important functions of the A2BAR. Selectively enhancing a specific A2BAR signaling pathway may be a useful strategy for the treatment of heart failure [64]. On the other hand, it is possible that the balance between Gi and Gs coupling might be required for a particular physiological response, considering the ubiquitous presence of adenosine throughout the body. Adenosine is released under stress conditions, the imbalanced response of the A2BAR in different cells or tissues and the balanced Gi-Gs coupling within the same type of cells may represent fine-tuned AR systems.
Considering the impact of ERK and calcium in many important cell functions such as cancer drug addiction, cell proliferation and insulin secretion [24,65,66], it is important to consider both activation or inhibition of the A2BAR based on the cell types and the relevant pathophysiological conditions. For example, A2BAR activation may have both proliferative and antiproliferative effects depending on cell types [5,21–24]. A2BAR signaling has also been suggested to induce different types of cardioprotection in a tissue-dependent way [67].
In summary, A2BAR may couple to the same downstream signaling pathway via different G proteins in different cell types, and it may couple to different signaling pathways via different G proteins in the same type of cells. Any one of the three G proteins, Gi, Gq/11, or Gs, may be solely responsible for a specific signaling event mediated via the A2BAR depending on cell type. Furthermore, Gs and Gi may have both opposing effects and synergistic effect in mediating downstream events depending on cell type and signaling pathway. The findings could be related to controversial roles of the A2BAR reported in literature, e.g. proliferation and antiproliferation. This could also be a reason for the A2BAR to induce both proinflammatory and anti-inflammatory effects in different types of inflammatory cells or different functions of the same cell types. It remains to be explored how the A2BAR couples to G proteins and downstream signaling events in other cell types, considering the important functions of the A2BAR is involved and the ubiquitous presence of the A2BAR throughout the body. The balance of Gi-Gs coupling in one cell type may be of important physiological relevance, and the imbalance of Gi-Gq-Gs coupling among various cell types, tissues and organs should be considered for disease treatment. It remains to be examined whether other GPCRs behave in a similar way or not, as strict comparisons among cell types and signaling pathways, and multiple agonists with diverse chemical structures in native cell systems are rare in the literature.
Acknowledgments
Supported the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases (ZIADK031117), National Institutes of Health, Bethesda, MD, USA. AI was funded by the PRIME from Japan Agency for Medical Research and Development (AMED) and Japan Society for the Promotion of Science KAKENHI Grant Number 17K08264. The authors thank Jurgen Wess (NIDDK, NIH) for advice, Ad IJzerman (Leiden, The Netherlands) for providing BAY60-6583 (LUF6210), E. Kiselev (NIDDK, NIH) for providing MRS5911, and H. Adachi (NIDDK, NIH) for MRS3997.
Abbreviations
- BAY60-6583
2-[[6-amino-3,5-dicyano-4-[4-(cyclopropylmethoxy)phenyl]-2-pyridinyl]thio]-acetamide
- cAMP
3′,5′-cyclic adenosine monophosphate
- CE3F4
5,7-dibromo-6-fluoro-3,4-dihydro-2-methyl-1(2H)-quinolinecarboxaldehyde
- CGS21680
2-[p-(2-carboxyethyl)phenyl-ethylamino]-5′-N-ethylcarboxamidoadenosine
- CRISPR/Cas9
clustered regularly interspaced short palindromic repeat-associated protein-9 nuclease
- CTX
cholera toxin
- EPAC
exchange protein directly activated by cAMP
- ERK
extracellular-signal-regulated kinase
- ESI-09
α-[(2-(3-chlorophenyl)hydrazinylidene]-5-(1,1-dimethylethyl)-β-oxo-3- isoxazolepropanenitrile
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase;
- GO6983
3-[1-[3-(dimethylamino)propyl]-5-methoxy-1H-indol-3-yl]-4-(1H-indol-3-yl)-1H- pyrrole-2,5-dione
- GPCR
G protein-coupled receptor
- H89
N-[2-[[3-(4-bromophenyl)-2-propenyl]amino]ethyl]-5-isoquinolinesulfonamide dihydrochloride
- HEK
human embryonic kidney
- HJC0350
2,4-dimethyl-1-[(2,4,6-trimethylphenyl)sulfonyl]-1H-pyrrole
- MRS2365
[[(1R,2R,3S,4R,5S)-4-[6-amino-2-(methylthio)-9H-purin-9-yl]-2,3- dihydroxybicyclo[3.1.0]hex-1-yl]methyl] diphosphoric acid monoester
- MRS3997
2-(2-(6-bromo-indol-3-yl)ethyloxy)adenosine
- MRS5911
N6-(4-iodophenyl)-adenosine
- NECA
adenosine-5′-N-ethyluronamide
- PKA
protein kinase A
- PTX
pertussis toxin
- UBO-QIC
L-threonine, (3R)-N-acetyl-3-hydroxy-L-leucyl-(αR)-α-hydroxybenzenepropanoyl-2, 3-didehydro-N-methylalanyl-L-alanyl-N-methyl-L-alanyl-(3R)-3-[[(2S,3R)-3-hydroxy-4-methyl-1-oxo-2-[(1-oxopropyl)amino]pentyl]oxy]-L-leucyl-N,O-dimethyl-, (7 →1)-lactone.
Footnotes
Conflict of interest: The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jing L, Tamplin OJ, Chen MJ, Deng Q, Patterson S, Kim PG, Durand EM, McNeil A, Green JM, Matsuura S, Ablain J, Brandt MK, Schlaeger TM, Huttenlocher A, Daley GQ, Ravid K, Zon LI. Adenosine signaling promotes hematopoietic stem and progenitor cell emergence. J Exp Med. 2015;212(5):649–63. doi: 10.1084/jem.20141528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feoktistov I, Biaggioni I. Role of adenosine A2B receptors in inflammation. Adv. Pharmacol. 2011;61:115–44. doi: 10.1016/B978-0-12-385526-8.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu H, Zhang Y, Wu H, D’Alessandro A, Yegutkin GG, Song A, Sun K, Li J, Cheng NY, Huang A, Edward Wen Y, Weng TT, Luo F, Nemkov T, Sun H, Kellems RE, Karmouty-Quintana H, Hansen KC, Zhao B, Subudhi AW, Jameson-Van Houten S, Julian CG, Lovering AT, Eltzschig HK, Blackburn MR, Roach RC, Xia Y. Beneficial role of erythrocyte adenosine A2B receptor-mediated AMP-activated protein kinase activation in high-altitude hypoxia. Circulation. 2016;134(5):405–21. doi: 10.1161/CIRCULATIONAHA.116.021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ohta A, Sitkovsky M. Extracellular adenosine-mediated modulation of regulatory T cells. Front Immunol. 2014;5:304. doi: 10.3389/fimmu.2014.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei Q, Costanzi S, Balasubramanian R, Gao ZG, Jacobson KA. A2B adenosine receptor blockade inhibits growth of prostate cancer cells. Purinergic Signal. 2013;9:271–80. doi: 10.1007/s11302-012-9350-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mittal D, Sinha D, Barkauskas D, Young A, Kalimutho M, Stannard K, Caramia F, Haibe-Kains B, Stagg J, Khanna KK, Loi S, Smyth MJ. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. 2016;76(15):4372–82. doi: 10.1158/0008-5472.CAN-16-0544. [DOI] [PubMed] [Google Scholar]
- 7.Ntantie E, Gonyo P, Lorimer EL, Hauser AD, Schuld N, McAllister D, Kalyanaraman B, Dwinell MB, Auchampach JA, Williams CL. An adenosine-mediated signaling pathway suppresses prenylation of the GTPase Rap1B and promotes cell scattering. Sci Signal. 2013;6(277):ra39. doi: 10.1126/scisignal.2003374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang X, Xin W, Yang XM, Kuno A, Rich TC, Cohen MV, Downey JM. A2B adenosine receptors inhibit superoxide production from mitochondrial complex I in rabbit cardiomyocytes via a mechanism sensitive to Pertussis toxin. Br J Pharmacol. 2011;163:995–1006. doi: 10.1111/j.1476-5381.2011.01288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ning C, Wen J, Zhang Y, Dai Y, Wang W, Zhang W, Qi L, Grenz A, Eltzschig HK, Blackburn MR, Kellems RE, Xia Y. Excess adenosine A2B receptor signaling contributes to priapism through HIF-1α mediated reduction of PDE5 gene expression. FASEB J. 2014;28(6):2725–35. doi: 10.1096/fj.13-247833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Corciulo C, Wilder T, Cronstein BN. Adenosine A2B receptors play an important role in bone homeostasis. Purinergic Signal. 2016;12(3):537–47. doi: 10.1007/s11302-016-9519-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eltzschig HK, Bonney SK, Eckle T. Attenuating myocardial ischemia by targeting A2B adenosine receptors. Trends Mol Med. 2013;19(6):345–54. doi: 10.1016/j.molmed.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonioli L, Blandizzi C, Csóka B, Pacher P, Haskó G. Adenosine signalling in diabetes mellitus--pathophysiology and therapeutic considerations. Nat Rev Endocrinol. 2015;11(4):228–41. doi: 10.1038/nrendo.2015.10. [DOI] [PubMed] [Google Scholar]
- 13.Gao Z, Chen T, Weber MJ, Linden J. A2B adenosine and P2Y2 receptors stimulate mitogen- activated protein kinase in human embryonic kidney-293 cells. cross-talk between cyclic AMP and protein kinase c pathways. J. Biol. Chem. 1999;274(9):5972–80. doi: 10.1074/jbc.274.9.5972. [DOI] [PubMed] [Google Scholar]
- 14.Schulte G, Fredholm BB. The Gs-coupled adenosine A2B receptor recruits divergent pathways to regulate ERK1/2 and p38. Exp. Cell. Res. 2003;290:168–76. doi: 10.1016/s0014-4827(03)00324-0. [DOI] [PubMed] [Google Scholar]
- 15.Epperson SA, Brunton LL, Ramirez-Sanchez I, Villarreal F. Adenosine receptors and second messenger signaling pathways in rat cardiac fibroblasts. Am J Physiol Cell Physiol. 2009;296(5):C1171–7. doi: 10.1152/ajpcell.00290.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei H, Ahn S, Shenoy SK, Karnik SS, Hunyady L, Luttrell LM, Lefkowitz RJ. Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Nat Acad Sci USA. 2003;100(19):10782–7. doi: 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang Y, Olah ME. Cyclic AMP-dependent, protein kinase A-independent activation of extracellular signal-regulated kinase 1/2 following adenosine receptor stimulation in human umbilical vein endothelial cells: role of exchange protein activated by cAMP 1 (Epac1) J Pharmacol Exp Ther. 2007;322(3):1189–200. doi: 10.1124/jpet.107.119933. [DOI] [PubMed] [Google Scholar]
- 18.Lynge J, Schulte G, Nordsborg N, Fredholm BB, Hellsten Y. Adenosine A 2B receptors modulate cAMP levels and induce CREB but not ERK1/2 and p38 phosphorylation in rat skeletal muscle cells. Biochem Biophys Res Commun. 2003;307(1):180–7. doi: 10.1016/s0006-291x(03)01125-2. [DOI] [PubMed] [Google Scholar]
- 19.Darashchonak N, Sarisin A, Kleppa MJ, Powers RW, von Versen-Höynck F. Activation of adenosine A2B receptor impairs properties of trophoblast cells and involves mitogen-activated protein (MAP) kinase signaling. Placenta. 2014;35(9):763–71. doi: 10.1016/j.placenta.2014.06.369. [DOI] [PubMed] [Google Scholar]
- 20.Sun K, Zhang Y, Bogdanov MV, Wu H, Song A, Li J, Dowhan W, Idowu M, Juneja HS, Molina JG, Blackburn MR, Kellems RE, Xia Y. Elevated adenosine signaling via adenosine A2B receptor induces normal and sickle erythrocyte sphingosine kinase 1 activity. Blood. 2015;125(10):1643–52. doi: 10.1182/blood-2014-08-595751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubey RK, Fingerle J, Gillespie DG, Mi Z, Rosselli M, Imthurn B, Jackson EK. Adenosine attenuates human coronary artery smooth muscle cell proliferation by inhibiting multiple signaling pathways that converge on cyclin D. Hypertension. 2015;66(6):1207–19. doi: 10.1161/HYPERTENSIONAHA.115.05912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bot I, de Vries H, Korporaal SJ, Foks AC, Bot M, van Veldhoven J, Ter Borg MN, van Santbrink PJ, van Berkel TJ, Kuiper J, IJzerman AP. Adenosine A2B receptor agonism inhibits neointimal lesion development after arterial injury in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2012;32(9):2197–205. doi: 10.1161/ATVBAHA.112.252924. [DOI] [PubMed] [Google Scholar]
- 23.Jackson EK, Ren J, Gillespie DG. 2′,3′-cAMP, 3′-AMP, and 2′-AMP inhibit human aortic and coronary vascular smooth muscle cell proliferation via A2B receptors. Am. J. Physiol. Heart Circ. Physiol. 2011;301(2):H391–401. doi: 10.1152/ajpheart.00336.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Y, Chu X, Deng F, Tong L, Tong G, Yi Y, Liu J, Tang J, Tang Y, Xia Y, Dai Y. The adenosine A2b receptor promotes tumor progression of bladder urothelial carcinoma by enhancing MAPK signaling pathway. Oncotarget. 2017;8(30):48755–68. doi: 10.18632/oncotarget.17835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merighi S, Bencivenni S, Vincenzi F, Varani K, Borea PA, Gessi S. A2B adenosine receptors stimulate IL-6 production in primary murine microglia through p38 MAPK kinase pathway. Pharm. Res. 2017;117:9–19. doi: 10.1016/j.phrs.2016.11.024. [DOI] [PubMed] [Google Scholar]
- 26.Costa MA, Barbosa A, Neto E, Sá-e-Sousa A, Freitas R, Neves JM, Magalhães-Cardoso T, Ferreirinha F, Correia-de-Sá P. On the role of subtype selective adenosine receptor agonists during proliferation and osteogenic differentiation of human primary bone marrow stromal cells. J Cell Physiol. 2011;226:1353–66. doi: 10.1002/jcp.22458. [DOI] [PubMed] [Google Scholar]
- 27.Faria M, Magalhães-Cardoso T, Lafuente-de-Carvalho JM, Correia-de-Sá P. J Pharmacol Exp Ther. 2006;319:405–13. doi: 10.1124/jpet.106.107821. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Dai Y, Wen J, Zhang W, Grenz A, Sun H, Tao L, Lu G, Alexander DC, Milburn MV, Carter-Dawson L, Lewis DE, Zhang W, Eltzschig HK, Kellems RE, Blackburn MR, Juneja HS, Xia Y. Detrimental effects of adenosine signaling in sickle cell disease. Nat Med. 2011;17(1):79–86. doi: 10.1038/nm.2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Dikov MM, Blackburn MR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene ablation on proinflammatory adenosine signaling in mast cells. J Immunol. 2008;180(11):7212–20. doi: 10.4049/jimmunol.180.11.7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Auchampach JA, Jin X, Wan TC, Caughey GH, Linden J. Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol. Pharmacol. 1997;52(5):846–60. doi: 10.1124/mol.52.5.846. [DOI] [PubMed] [Google Scholar]
- 31.Gao ZG, Balasubramanian R, Kiselev E, Wei Q, Jacobson KA. Probing biased/partial agonism at the G protein-coupled A2B adenosine receptor. Biochem. Pharmacol. 2014;90(3):297–306. doi: 10.1016/j.bcp.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinz S, Lacher SK, Seibt B, Müller CE. BAY60-6583 acts as a partial agonist at adenosine A2B receptors. J. Pharmacol. Exp. Ther. 2014;349:427–36. doi: 10.1124/jpet.113.210849. [DOI] [PubMed] [Google Scholar]
- 33.Hu J, Stern M, Gimenez LE, Wanka L, Zhu L, Rossi M, Meister J, Inoue A, Beck-Sickinger AG, Gurevich VV, Wess J. A G protein-biased designer G protein-coupled receptor useful for studying the physiological relevance of Gq/11-dependent signaling pathways. J Biol Chem. 2016;291:7809–20. doi: 10.1074/jbc.M115.702282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stallaert W, van der Westhuizen ET, Schönegge AM, Plouffe B, Hogue M, Lukashova V, Inoue A, Ishida S, Aoki J, Le Gouill C, Bouvier M. Purinergic receptor transactivation by the β2-adrenergic receptor increases intracellular Ca2+ in nonexcitable cells. Mol. Pharmacol. 2017;91(5):533–44. doi: 10.1124/mol.116.106419. [DOI] [PubMed] [Google Scholar]
- 35.Adachi H, Palaniappan KK, Ivanov AA, Bergman N, Gao ZG, Jacobson KA. Structure-activity relationships of 2,N6,5′-substituted adenosine derivatives with potent activity at the A2B adenosine receptor. J. Med. Chem. 2007;50:1810–27. doi: 10.1021/jm061278q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Zwart M, de Groote M, van der Klein PAM, van Dun S, von Frijtag Drabbe Künzel JK, IJzerman AP. Phenyl-substituted N6-phenyladenosines and N6-phenyl-5′-N- ethylcarboxamidoadenosines with high activity at human adenosine A2B receptors. Drug Devel. Res. 2000;49:85–93. [Google Scholar]
- 37.Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, Büllesbach KM, Bald T, Inoue A, Shinjo Y, Galandrin S, Shridhar N, Hesse M, Grundmann M, Merten N, Charpentier TH, Martz M, Butcher AJ, Slodczyk T, Armando S, Effern M, Namkung Y, Jenkins L, Horn V, Stößel A, Dargatz H, Tietze D, Imhof D, Galés C, Drewke C, Müller CE, Hölzel M, Milligan G, Tobin AB, Gomeza J, Dohlman HG, Sondek J, Harden TK, Bouvier M, Laporte SA, Aoki J, Fleischmann BK, Mohr K, König GM, Tüting T, Kostenis E. The experimental power of FR900359 to study Gq-regulated biological processes. Nat Commun. 2015;6:10156. doi: 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gao ZG, Verzijl D, Zweemer A, Ye K, Göblyös A, IJzerman AP, Jacobson KA. Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem. Pharmacol. 2011;82:658–68. doi: 10.1016/j.bcp.2011.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu Y, Chen H, Boulton S, Mei F, Ye N, Melacini G, Zhou J, Cheng X. Biochemical and pharmacological characterizations of ESI-09 based EPAC inhibitors: defining the ESI-09 “therapeutic window”. Sci Rep. 2015;5:9344. doi: 10.1038/srep09344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Linden J, Thai T, Figler H, Jin X, Robeva AS. Characterization of human A2B adenosine receptors: radioligand binding, western blotting, and coupling to Gq in human embryonic kidney 293 cells and HMC-1 mast cells. Mol. Pharmacol. 1999;56(4):705–13. [PubMed] [Google Scholar]
- 41.Chen H, Tsalkova T, Chepurny OG, Mei FC, Holz GG, Cheng X, Zhou J. Identification and characterization of small molecules as potent and specific EPAC2 antagonists. J Med Chem. 2013;56(3):952–62. doi: 10.1021/jm3014162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Courilleau D, Bisserier M, Jullian JC, Lucas A, Bouyssou P, Fischmeister R, Blondeau JP, Lezoualc’h F. Identification of a tetrahydroquinoline analog as a pharmacological inhibitor of the cAMP-binding protein Epac. J Biol Chem. 2012;287(53):44192–202. doi: 10.1074/jbc.M112.422956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohen MV, Downey JM. Adenosine: trigger and mediator of cardioprotection. Basic Res Cardiol. 2008;103(3):203–15. doi: 10.1007/s00395-007-0687-7. [DOI] [PubMed] [Google Scholar]
- 44.Boros D, Thompson J, Larson DF. Adenosine regulation of the immune response initiated by ischemia reperfusion injury. Perfusion. 2016;31(2):103–10. doi: 10.1177/0267659115586579. [DOI] [PubMed] [Google Scholar]
- 45.Xiao RP. Cell logic for dual coupling of a single class of receptors to Gs and Gi proteins. Circ. Res. 2000;87(8):635–7. doi: 10.1161/01.res.87.8.635. [DOI] [PubMed] [Google Scholar]
- 46.Soeder KJ, Snedden SK, Cao W, Della Rocca GJ, Daniel KW, Luttrell LM, Collins S. The beta3-adrenergic receptor activates mitogen-activated protein kinase in adipocytes through a Gi-dependent mechanism. J Biol Chem. 1999;274(17):12017–22. doi: 10.1074/jbc.274.17.12017. [DOI] [PubMed] [Google Scholar]
- 47.Herrlich A, Kühn B, Grosse R, Schmid A, Schultz G, Gudermann T. Involvement of Gs and Gi proteins in dual coupling of the luteinizing hormone receptor to adenylyl cyclase and phospholipase C. J Biol Chem. 1996;271(28):16764–72. doi: 10.1074/jbc.271.28.16764. [DOI] [PubMed] [Google Scholar]
- 48.Crépieux P, Marion S, Martinat N, Fafeur V, Vern YL, Kerboeuf D, Guillou F, Reiter E. The ERK-dependent signalling is stage-specifically modulated by FSH, during primary Sertoli cell maturation. Oncogene. 2001;20(34):4696–709. doi: 10.1038/sj.onc.1204632. [DOI] [PubMed] [Google Scholar]
- 49.Wu G, Lu ZH, Ledeen RW. Interaction of the delta-opioid receptor with GM1 ganglioside: conversion from inhibitory to excitatory mode. Brain Res Mol Brain Res. 1997;44(2):341–6. doi: 10.1016/s0169-328x(96)00281-1. [DOI] [PubMed] [Google Scholar]
- 50.Chen XP, Yang W, Fan Y, Luo JS, Hong K, Wang Z, Yan JF, Chen X, Lu JX, Benovic JL, Zhou NM. Structural determinants in the second intracellular loop of the human cannabinoid CB1 receptor mediate selective coupling to Gs and Gi. Br J Pharmacol. 2010;161(8):1817–34. doi: 10.1111/j.1476-5381.2010.01006.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Raffa RB, Stone DJ., Jr Could dual G-protein coupling explain [D-Met2]FMRFamide’s mixed action in vivo? Peptides. 1996;17(8):1261–5. doi: 10.1016/s0196-9781(96)00196-9. [DOI] [PubMed] [Google Scholar]
- 52.Zhu W, Petrashevskaya N, Ren S, Zhao A, Chakir K, Gao E, Chuprun JK, Wang Y, Talan M, Dorn GW, 2nd, Lakatta EG, Koch WJ, Feldman AM, Xiao RP. Gi-biased β2AR signaling links GRK2 upregulation to heart failure. Circ Res. 2012 Jan 20;110(2):265–74. doi: 10.1161/CIRCRESAHA.111.253260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to Gs and Gi proteins. Sci STKE. 2001;2001(104):re15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 54.Wenzel-Seifert K, Seifert R. Molecular analysis of beta2-adrenoceptor coupling to Gs-, Gi-, and Gq-proteins. Mol. Pharmacol. 2000;58(5):954–66. doi: 10.1124/mol.58.5.954. [DOI] [PubMed] [Google Scholar]
- 55.Zamah AM, Delahunty M, Luttrell LM, Lefkowitz RJ. Protein kinase A-mediated phosphorylation of the beta2-adrenergic receptor regulates its coupling to Gs and Gi. Demonstration in a reconstituted system. J. Biol. Chem. 2002;277(34):31249–56. doi: 10.1074/jbc.M202753200. [DOI] [PubMed] [Google Scholar]
- 56.Hill SJ, Baker JG. The ups and downs of Gs- to Gi-protein switching. Br J Pharmacol. 2003;138(7):1188–9. doi: 10.1038/sj.bjp.0705192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crespo P, Cachero TG, Xu N, Gutkind JS. Dual effect of beta-adrenergic receptors on mitogen-activated protein kinase. Evidence for a beta gamma-dependent activation and a G alpha s-cAMP-mediated inhibition. J Biol Chem. 1995;270(42):25259–65. doi: 10.1074/jbc.270.42.25259. [DOI] [PubMed] [Google Scholar]
- 58.Duarte T, Menezes-Rodrigues FS, Godinho RO. Contribution of the extracellular cAMP- adenosine pathway to dual coupling of β2-adrenoceptors to Gs and Gi proteins in mouse skeletal muscle. J. Pharmacol. Exp. Ther. 2012;341(3):820–8. doi: 10.1124/jpet.112.192997. [DOI] [PubMed] [Google Scholar]
- 59.Gao ZG, Jacobson KA. On the selectivity of the Gαq inhibitor UBO-QIC: A comparison with the Gαi inhibitor pertussis toxin. Biochem Pharmacol. 2016;107:59–66. doi: 10.1016/j.bcp.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Furness SGB, Liang YL, Nowell CJ, Halls ML, Wookey PJ, Dal Maso E, Inoue A, Christopoulos A, Wootten D, Sexton PM. Ligand-Dependent Modulation of G Protein Conformation Alters Drug Efficacy. Cell. 167(3):739–49.e11. doi: 10.1016/j.cell.2016.09.021. 201. [DOI] [PubMed] [Google Scholar]
- 61.de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–7. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 62.Rehmann H, Arias-Palomo E, Hadders MA, Schwede F, Llorca O, Bos JL. Structure of Epac2 in complex with a cyclic AMP analogue and RAP1B. Nature. 2008;455(7209):124–27. doi: 10.1038/nature07187. [DOI] [PubMed] [Google Scholar]
- 63.Dekkers BG, Racké K, Schmidt M. Distinct PKA and Epac compartmentalization in airway function and plasticity. Pharmacol Ther. 2013;137(2):248–65. doi: 10.1016/j.pharmthera.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Chakir K, Depry C, Dimaano VL, Zhu WZ, Vanderheyden M, Bartunek J, Abraham TP, Tomaselli GF, Liu SB, Xiang YK, Zhang M, Takimoto E, Dulin N, Xiao RP, Zhang J, Kass DA. Galphas-biased beta2-adrenergic receptor signaling from restoring synchronous contraction in the failing heart. Sci. Transl. Med. 2011;3(100):100ra88. doi: 10.1126/scitranslmed.3001909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song JY, Niessen HWM, Rozeman EA, Geukes Foppen MH, Blank CU, Peeper DS. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature. 2017;550(7675):270–4. doi: 10.1038/nature24037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Petersen OH. Calcium signalling and secretory epithelia. Cell Calcium. 2014;55(6):282–9. doi: 10.1016/j.ceca.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 67.Seo SW, Koeppen M, Bonney S, Gobel M, Thayer M, Harter PN, Ravid K, Eltzschig HK, Mittelbronn M, Walker L, Eckle T. Differential tissue-specific function of Adora2b in cardioprotection. J Immunol. 2015;195(4):1732–43. doi: 10.4049/jimmunol.1402288. [DOI] [PMC free article] [PubMed] [Google Scholar]