

Abstract
Ocular hypertension is a causal risk-factor to developing glaucoma. This is associated with stiffening of the trabecular meshwork (TM), the primary site of resistance to aqueous-humor-outflow. The mechanisms underlying this stiffening or how pathologic extracellular matrix (ECM) affects cell function are poorly understood. It is recognized that mechanotransduction systems allow cells to sense and translate the intrinsic biophysical properties of ECM into intracellular signals to control gene transcription, protein expression, and cell behavior. Using an anterior segment perfusion model, we document that there are significantly more low flow regions that are much stiffer, and fewer high flow regions that are less stiff in glaucomatous TM (GTM) when compared to non-glaucomatous TMs (NTM). GTM tissue also has fewer cells overall when compared with NTM tissue. In order to study the role of pathologic ECM in glaucoma disease progression, we conducted studies using cell derived matrices (CDM). First, we characterized the mechanics, composition and organization of fibronectin in ECM deposited by GTM and NTM cells treated with glucocorticosteroids. Then, we determined that these GTM-derived ECM are able to induce stiffening of normal NTM cells, and alter their gene/protein expression to resemble that of a glaucomatous phenotype. Further, we demonstrate that GTM-derived ECM causes endoplasmic reticular stress in NTM. They also became resistant to being reorganized by these NTM cells. These phenomena were exacerbated by ECMs obtained from steroid treated glaucoma model groups. Collectively, our data demonstrates that CDMs represent a novel tool for the study of bidirectional interactions between TM cells and their immediate microenvironment.
Graphical Abstract
1. INTRODUCTION
Primary open angle glaucoma (POAG) is a disease of optic nerve degeneration where ocular hypertension due to increased resistance to aqueous humor outflow is a primary risk factor [1]. Most of the resistance to the outflow is thought to be provided by the extracellular matrix (ECM) of the trabecular meshwork (TM), specifically within the juxtacanalicular (JCT) or cribiform region and the basement membrane of the inner wall endothelium of the Schlemm’s canal (SC) [2, 3]. To date, the only rigorously validated treatment for glaucoma is the therapeutic lowering of intraocular pressure (IOP). However, none of the currently available drugs for glaucoma directly target the human TM [4–6], although recently FDA has approved Netarsudil and Latanoprostene Bunod. The long-term effects and efficacy of these new classes of drugs on TM cell or ECM function remains unclear. Optic nerve damage in glaucoma has been correlated with changes in the TM indicating the progression of visual loss is linked to alterations in this tissue [7].
ECM are dynamic three-dimensional structures whose composition, morphology, mechanics, and organization vary widely depending on the tissue. The interfaces between cells and ECM within these tissues are complex and the residing cells can sense, integrate, and respond to these intrinsic properties through modulation of gene/protein expression that ultimately govern cell fate and behavior. Matrix stiffness, a ubiquitous and potent biophysical cue that cells perceive and respond to, is dependent on the (i) type of structural fibrillar and non-fibrillar extracellular matrix (ECM) protein(s), (ii) presence of modifier ECM proteins, and (iii) morphology and organization of the ECM itself. The TM is a continuous structure comprised of fenestrated beams, matrix, matricellular proteins, glycosaminoglycans and proteoglycans. We have demonstrated that the apparent elastic modulus of the TM increases twenty fold with glaucoma in humans [8], and four fold with steroid treatment in rabbits [9]. More recently, it was demonstrated that matrix cross-linking profoundly altered ECM composition and outflow resistance in organ culture models [10]. However, flow across the TM is not uniform but is segmental with regions of high (HF) and low flow (LF). Recently, we reported that LF segments in non-glaucomatous organ cultures are stiffer than HF segments accompanied by a wide array of changes in protein composition and organization [11]. However, whether tissue mechanics across the segmental regions exhibiting differing flows in glaucomatous TM differ from non-glaucomatous donors is unknown. A significant attribute of glaucomatous TM is loss of cellularity accompanied by a dysfunctional ECM [12–14]. It remains unknown if cellularity differs segmentally, or whether ECM is related to loss of cells. Here, using an ex vivo organ perfusion model, we report the elastic modulus and cellularity of human glaucomatous TM in different segmental flow regions.
It is important to note that in vivo matrix biophysical properties are often accompanied by simultaneous changes in the biochemical constituents and/or organization. Indeed, in POAG or steroid induced ocular hypertension the ECM is altered significantly [15–20]. It is increasingly being accepted that, using mechanotransduction systems, cells can translate these intrinsic changes to intracellular signals that control gene transcription, protein expression, phosphorylation state and cell behavior. While a number of studies have been performed to characterize pathologic ECM, a limited number have investigated the impact that these ECM have on ‘normal’ cell behavior. This is in part due to the difficulties in obtaining glaucomatous TM for comprehensive phenotypic and mechanistic studies. Cell derived matrices (CDM) provide an alternative approach with the advantage that these matrices are cell type specific, i.e. normal or glaucomatous, and the effects of exogenous drugs/factors can be determined. Here, we characterize the biomechanics, composition and organization of glaucomatous TMs and CDMs, and report the effect that these cell derived matrices from multiple GTM or NTM cells have in modulating mechanics, gene and protein expression in cells derived from a single NTM donor when cultured upon them.
2. MATERIALS AND METHODS
2.1 Anterior segment perfusion culture
Anterior segment perfusion culture is an established technique to study outflow facility ex vivo [21]. Use of human donor eye tissue was approved by the Oregon Health and Science University Institutional Review Board and experiments were conducted in accordance with the tenets of the Declaration of Helsinki for the use of human tissue. Human eye tissue was obtained from cadavers (Lions VisionGift, Portland, OR, USA) and required prior informed consent of donors. This manuscript contains no information that could lead to identification of a study participant. Length of time from death to stationary culture was less than 48 hours. Anterior segments were placed into serum-free stationary organ culture for 5–7 days to facilitate recovery from post-mortem effects. The age range was 65–97 years and average age of the cadaver eyes for all experiments in this study was 76.3 ± 8.7 years (n = 16 normal eyes, and n=7 glaucomatous). Human anterior segments were perfused with serum-free Dulbecco’s Modified Eagle’s Medium (DMEM) containing 1% Penicillin/Streptomycin/Fungizone, at constant pressure of 8.8 mmHg for up to 72 hours. During the final stage of perfusion, fluorescently-labeled amine-modified 200 nm FluoSpheres or Cell Mask Orange (Thermo Fisher Scientific, Waltham, MA, USA) were diluted 1:1000 into PBS, vortexed vigorously, and 200 μl of that mixture was injected as a bolus directly in-line into the anterior segment organ culture and perfused for 1 hour. Specifically, for all mechanical characterization experiments, perfusion was done with Cell Mask Orange injected directly into the tube carrying the media from the reservoir to the anterior chamber in organ culture; it is worth noting that Cell Mask Orange is lipophilic and its lipophobic tail integrates into the plasma membranes of cells, thus labeling the cells where it passes through the outflow pathway. There is no rigid mechanical component that may present as an artifact during mechanical characterization. On the other hand, FluoSpheres are amine functionalized and thus bind to the negatively charged ECM proteins during perfusion.
Perfusion was stopped and anterior segments were imaged en face using a Leica DM500 microscope (Leica Microsystems, Buffalo Grove, IL, USA) prior to cutting into radial wedges of high flow (HF) or low flow (LF) regions. Details of this workflow in the form of a flowchart and how segmental regions were identified can be found in supplemental information (Supplementary methods, Fig. S1). In brief, these HF and LF regions were separated based on fluorescence intensity of the TM as was described previously using ImageJ software and “Plot Profile” to get graphical output of distance versus relative fluorescence intensity [22]. Images of the eye were merged in photoshop using the photomerge feature creating a composite collage image. Segments that did not automatically merge were done manually. The circumference of the TM (minus the gap created when the eye is cut to make it flat for imaging) was determined using a protractor overlay of the image. The same overlay application was used to determine the number of degrees of that circumference that were HF or LF with the remainder considered intermediate flow (IF). Normal HF regions were those measured to be in the top one third of the intensity, the LF regions were the lowest one third, and the IF regions were everything in between [11]. IF regions were not included in analyses herein. Most normal eyes have approximately a third HF, a third LF, and a third IF regions. In glaucomatous eyes, the relative distributions of HF/IF/LF/were quantified in this manuscript. The demographics of the donor tissues used for this study are listed in Table 1.
Table 1.
Demographical details of TM donors obtained from the eye bank for ex vivo experiments.
| EYE ID# | Use | Norm/Glau | AGE | GENDER | RACE |
|---|---|---|---|---|---|
| 12-0672OD | AFM | Glau | 86 | F | C |
| 12-1576OD | AFM | Glau | 88 | F | C |
| 12-1576OS | AFM | Glau | 88 | F | C |
| 11-1591OS | AFM | Norm | 68 | M | C |
| 11-1591OD | AFM | Norm | 68 | M | C |
| 12-0363OD | AFM | Norm | 96 | F | C |
| 12-0363OS | AFM | Norm | 96 | F | C |
| 16-1442OD | HF/LF% | Norm | 78 | M | C |
| 16-1442OS | HF/LF% | Norm | 78 | M | C |
| 16-1611OD | HF/LF% | Norm | 88 | M | C |
| 16-1747OD | HF/LF% | Norm | 55 | F | C |
| 16-1747OS | HF/LF% | Norm | 55 | F | C |
| 16-1748OD | HF/LF% | Norm | 81 | M | C |
| 16-1748OS | HF/LF% | Norm | 81 | M | C |
| 17-0015OS | HF/LF% | Norm | 82 | F | C |
| 17-0143OS | HF/LF% | Norm | 78 | F | C |
| 17-0147OS | HF/LF% | Norm | 82 | F | C |
| 17-0984OD | HF/LF% | Norm | 76 | M | C |
| 17-0987OD | HF/LF% | Norm | 75 | M | C |
| 17-0987OS | HF/LF% | Norm | 75 | M | C |
| 17-0992OD | HF/LF% | Norm | 77 | M | C |
| 16-1609OS | HF/LF% | Glau | 77 | M | C |
| 16-1159OS | HF/LF% | Glau | 84 | F | C |
| 17-0185OD | HF/LF% | Glau | 80 | F | O |
| 17-0185OS | HF/LF% | Glau | 80 | F | O |
| 17-0595OS | HF/LF% | Glau | 79 | F | C |
| 17-0600OD | HF/LF% | Glau | 82 | M | C |
| 17-0996OD | HF/LF% | Glau | 78 | M | C |
| 17-0996OS | HF/LF% | Glau | 78 | M | C |
| 11-0979OD | 1x/2x,Cell # | Glau | 92 | M | C |
| 11-0979OS | 1x/2x,Cell # | Glau | 92 | M | C |
| 12-0324OD | 1x/2x, Cell # | Glau | 85 | M | C |
| 12-0324OS | 1x/2x, Cell # | Glau | 85 | M | C |
| 12-0341OD | 1x/2x, Cell # | Norm | 86 | M | C |
| 12-0312OD | 1x/2x | Glau | 92 | M | C |
| 15-1310OS | 1x/2x | Glau | 83 | F | C |
| 16-1159OS | 1x/2x | Glau | 84 | F | C |
| 16-1159OD | 1x/2x | Glau | 84 | F | C |
| 10-0630OD | Cell # | Norm | 84 | F | C |
| 10-0630OS | Cell # | Norm | 84 | F | C |
| 10-0879OD | Cell # | Norm | 78 | M | C |
| 10-0879OS | Cell # | Norm | 78 | M | C |
| 11-0971OD | Cell # | Norm | 79 | F | C |
| 11-0024OD | Cell # | Norm | 83 | F | C |
| 11-0978OS | Cell # | Norm | 83 | F | C |
| 11-0981OD | Cell # | Norm | 80 | M | C |
| 11-1790OD | Cell # | Norm | 82 | M | C |
| 11-1790OS | Cell # | Norm | 82 | M | C |
| 12-0360OD | Cell # | Norm | 77 | F | C |
| 12-0360OS | Cell # | Norm | 77 | F | C |
| 12-0341OD | Cell # | Norm | 86 | M | C |
| 12-0341OS | Cell # | Norm | 86 | M | C |
| 10-0605OD | Cell # | Glau | 76 | M | C |
| 10-0605OS | Cell # | Glau | 76 | M | C |
| 10-0889OS | Cell # | Glau | 90 | M | C |
| 11-1860OD | Cell # | Glau | 87 | M | C |
“OD” = right eye; “OS” = left eye; “C” = Caucasian; “O” = Other; “AFM” = atomic force microscopy; “HF/LF%” = percentage of total of high flow and low flow regions; “1x/2x” = anterior segments perfused at physiological (1x) pressure for >24 hours, then challenged with elevated (2x) pressure for >72 hours.
2.2 In vitro cell culture
Primary human TM cells were isolated from donor (glaucomatous and non-glaucomatous) corneoscleral rims unsuitable for transplant SavingSight Eye Bank, St. Louis, MO, USA) as described previously [23]. Briefly, TM was dissected from corneo-scleral rims, cut into small pieces, and placed with 0.2% (w/v) collagen coated cytodex beads in growth medium [Dulbecco’s modified Eagle medium/Nutrient Mixture F-12 (50:50; DMEM/F-12) with 2.5 mM L-glutamine supplemented with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin/amphotericin (Life Technologies, Carlsbad, CA, USA)]. Cells that migrated out of the TM were maintained in growth media and used between passages two and seven. Since cells were obtained postmortem from de-identified donor tissue, these are not considered as Human Subjects Research. The demographics for the donor tissues used for this study are listed in Table 2.
Table 2.
Demographical details of TM donors obtained from the eye bank for in vitro experiments.
| Sample ID | Age (years) | Gender | Ocular history | Experiments |
|---|---|---|---|---|
| NTM62B | 62 | F | None |
|
| NTM70M | 70 | M | None | |
| NTM64A | 64 | M | None | |
| GTM73A | 73 | M | Glaucoma |
|
| GTM73B | 73 | F | Glaucoma | |
| GTM66 | 66 | F | Glaucoma | |
| GTM71A | 71 | M | Glaucoma |
|
| GTM63B | 63 | F | Glaucoma | |
| NTM50 | 50 | M | None |
|
2.2.1 Derivation and characterization of cell derived ECM
We used primary TM cells isolated from at least five donors. Culture of cells and derivation of ECM was performed as described previously [24]. Briefly, GTM and NTM were cultured in growth medium in the presence or absence of 100 nM dexamethasone (Dex; Sigma-Aldrich Corp., St. Louis, MO, USA) on 3-aminopropyl trimethoxysilane modified glass coverslips for 4 weeks. Cells were used between passages two and seven for all experiments, with media changes/treatment administered twice a week. Matrices were obtained by decellularization of cultures as described previously using 20 mM ammonium hydroxide and 0.05% Triton X-100 in deionized water [24, 25]. Freshly decellularized ECM was used for mechanical characterization, proteomics, and subsequent cell culture, while formaldehyde-fixed cell-derived matrices were used for immunocytochemistry. Results pertaining to ECM deposition and characterization are below in section 3.2.
2.2.2 Culture of cells on cell derived ECM
Decellularized ECM derived as above, was incubated in Hank’s balanced salt solution (HBSS) with DNAse I, protease/phosphatase inhibitor cocktail overnight. The samples were subsequently rinsed in HBSS five times, 5 min each wash and used immediately for cell culture. Freshly isolated primary TM cells from a non-glaucomatous 50 year-old male donor (NTM50) were plated on CDM or on glass coverslips and cultured in growth media (in absence of any EtOH or Dex) for 3 days, following which cell mechanical properties were determined by AFM. Separately, (NTM50 grown on CDM/glass) were fixed in 4% formaldehyde, or lysed in RIPA buffer for Western blotting and proteomics, or RNA was isolated for quantitative reverse transcriptase PCR (qRT-PCR). Results pertaining to the effect that CDM have on NTM50 cells are below in section 3.3.
2.3 Immunocytochemistry
All samples were fixed in 4% paraformaldehyde for 20 min, further permeabilized with 0.01% Triton X-100 in phosphate buffered saline (PBS, pH 7.4), blocked with 3% fish gelatin, and immunolabelled with anti-LAMP1 (catalog # 9091, Cell Signaling Technologies, Danvers, MA) or anti-Fibronectin (catalog # ab6584, Abcam, Cambridge, MA, USA) followed by species appropriate fluorophore-tagged secondary antibody. Where appropriate, samples were stained with F-actin (catalog # 00045, CF594 Phalloidin, Biotium, Fremont, CA) to confirm decellularization, and nuclei were counterstained with DAPI (catalog # D1306, FisherScientific, CA). All experiments were performed for all cell lineages, with at least 3 samples per cell lineage. For each immunostained sample, 5–10 random locations were imaged using a Leica DMi8 inverted fluorescence microscope. Due to the qualitative nature of imaging, no quantitative measurements were performed at this time. As appropriate, representative images from a single field of view for a single donor are illustrated in the results section(s).
2.4 RNA isolation and Quantitative Real-Time PCR
RNA was isolated using an RNA purification kit (GeneJET Plasmid Miniprep Kit; ThermoFisher Scientific, CA) following the manufacturer’s instructions and equal amounts were used for the qPCR reactions. The qPCR was performed using a reagent kit (SensiFAST™ Hi-ROX One-Step Kit; Bioline USA Inc, Taunton, MA) and commercially available and validated aptamers for 18S (Hs99999901_s1); CRYAB (Hs00157107_m1); and MYOC (Hs00165354_m1) in total volumes of 10 μL per reaction (ThermoFisher Scientific, CA). The reverse transcription reaction was performed in a qPCR machine (StepOne; ThermoFisher Scientific, CA) with the following parameters: 30 minutes at 50°C followed by 10 minutes at 95°C; 40 cycles of 60°C for 1 minute followed by 95°C for 15 seconds. Relative expression levels of mRNAs of interest were determined as described previously [26].
2.5 Atomic force microscopy and proteomic analysis
Elastic moduli of tissues, cells, and CDM were determined as described previously [24, 25, 27, 28]. For tissue mechanics measurements, TM from anterior segments wedges (where eyes were perfused with Cell Mask Orange) were dissected out and mounted with the SC/JCT side facing the cantilever using a glue-less method developed and validated by our group [28]. Briefly, force-distance curves were obtained in contact mode using PNP-TR cantilevers (nominal spring constant 0.32 N/m; Nano and More, USA) with a (i) pyramidal tip (for cells), (ii) modified with 5 μm diameter borosilicate beads (for CDM), and (iii) modified with 10 μm diameter borosilicate beads (for TM tissues). For all samples, force curves were obtained from at least 5–10 locations with 5 force curves at each location. Elastic modulus was determined using Sneddon model (for cells), and Hertzian model (for CDM and tissues); details of the equation for each are described elsewhere [29]. Thickness of the CDM were determined by a ‘scratch and indent’ method using pyramidal AFM cantilevers and measured between 7–13 μm in thickness. Composition of the ECMs deposited by both glaucomatous and non-glaucomatous cells was determined by shotgun proteomics as described previously [24, 25]. Briefly, CDM were solubilized in 4 M guanidine hydrochloride, precipitated, and subsequently trypsin digested prior to LC-MS/MS analysis. Further, NTM50 cells cultured on glass/CDM were solubilized in RIPA buffer, and processed for proteomic analysis as described previously [24, 25].
2.6 Western blotting
TM cell lysates were run on denaturing 4–12% gradient polyacrylamide ready-made gels (NuPAGE Bis-Tris gels, Life technologies, Grand Island, NY, USA) and transferred onto PVDF membranes electrophoretically. Blots were blocked with 10% non-fat dried milk for 1 hour then incubated overnight with specific primary antibodies at 4°C on a rotating shaker. The membranes were washed thrice with phosphate buffered saline with Tween-20 and incubated with corresponding HRP-conjugated secondary antibody for 90 minutes. The proteins were then visualized using ECL detection reagents (SuperSignal West Femto Maximum Sensitivity Substrate; Life technologies, Grand Island, NY, USA). Antibodies were purchased from the following sources: KDEL antibody (catalog # Ab12223, Abcam, Cambridge, MA, USA) was used to detect GRP78 and GRP94, ATF-4 (catalog # SC-200, Santa Cruz Biotechnology, Dallas, TX, USA)), CHOP (catalog # 13172, Novus Biologicals, Littleton, CO, USA), β-Actin (catalog # ab13822, Abcam, Cambridge, MA, USA), α-SMA (catalog # A5228, Sigma-Aldrich, MO, USA) and GAPDH (catalog # 3683, Cell signaling technology, Danvers, MA, USA).
2.7 Statistics
Statistical comparison for mechanics and gene expression between vehicle and Dex treated group/glaucoma vs non-glaucoma was done using Mann-Whitney U-test and results are indicated in the plots. One-way ANOVA with multiple comparisons was used to determine significance of AFM measurements on HF and LF tissues from perfused normal and glaucoma anterior segments, where p<0.05 was significant (N = 4 individual unpaired glaucoma eyes and 6 individual unpaired normal eyes used for AFM measurements). An unpaired two-tailed t-test was used to determine the statistical significance of comparisons of the percentage HF and LF regions from n = 12 individual normal donor eyes and 7 individual glaucomatous eyes (p<0.05 was significant), as well as to determine the statistical significance of the number of cells in glaucomatous TM (n = 8 individual glaucomatous eyes) compared to normal TM (n = 14 individual normal eyes).
2.7.1 Proteomics
Total spectral counts obtained by shotgun proteomics were analyzed as described previously [24, 25]. Data were analyzed using Scaffold Viewer (Proteome Software Inc, OR). Using the built-in features of the software, normalized total spectral counts from the various samples were compared for fold-change between the groups: (i) glaucoma vs non-glaucoma, (ii) EtOH vs Dex, (iii) TCP vs glaucoma vs non-glaucoma ECMs. The relative abundance between the two groups were compared using Fisher’s exact test [30]. For the dendrogram analyses of ‘ECM’ proteins, ANOVA was performed to compare the 4 groups: non-glaucoma EtOH vs non-glaucoma Dex, vs glaucoma EtOH vs Glaucoma Dex ECMs, and the resultant p-values were used. For the dendrogram analyses of ‘NTM50 on ECM’ proteins, ANOVA was performed to compare the 5 groups: TCP vs non-glaucoma EtOH vs non-glaucoma Dex, vs glaucoma EtOH vs Glaucoma Dex ECMs, and the resultant p-values were used. Functional annotation of the differentially expressed proteins were analyzed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) [31]. Only gene ontology (GO) clusters whose p-value was lesser than 0.05 were considered.
2.7.2 Principal component analysis (PCA)
Peptide MS1 spectral count at the protein level was used to determine the principal components using a covariance matrix in MATLAB. The scree plot is plotted as proportion of variance against the number of principal components (PCs). PCA is a data mining tool used to transform data to visualize covariance and ascertain the relation between the data populations (in this case, protein levels in NTM50 cells between TCP vs glaucoma vs non-glaucoma ECMs). Distribution along the X-axis demonstrates a strong correlation to PC1 while along the Y-axis is PC2. The differences observed between the groups (as appropriate; e.g.: TCP vs non-glaucoma vs glaucoma) are illustrated by ellipses. However, PCA was insufficient to document how the protein spectra clustered and as such we performed subsequent hierarchical cluster analysis.
2.7.3 Hierarchical cluster analysis
For each protein, an analysis of variance (ANOVA) model using the log2 normalized spectral counts were used to determine the significance of the abundance of the proteins across non-glaucoma and glaucoma patients. Fold-changes were calculated as the ratio of the average within sample spectral count difference between the treated and untreated conditions among glaucoma relative to non-glaucoma eyes. The subset of proteins with relative expression greater than a 2-fold change and whose p-value was less than 5% after implementation of False Discovery Rate (FDR) to control the Type I error rate due to multiple testing were identified. For this subset of proteins, cluster heatmaps were generated to display the protein abundance for each protein (row). Larger protein expression abundance is represented by blue squares and smaller by red squares. Hierarchical clustering using Ward’s [32] methods for agglomerations was used to order and structure the proteins and dendograms were displayed to show similarities among the proteins. Statistical software used for heatmap generation included SAS 9.2 (SAS Institute, Cary, NC) and R 3.1.1 using GPLOTS package heatmap.2 function.
3. RESULTS
3.1 Glaucomatous donors have altered segmental flow pattern distributions
First, we determined whether there was a relative shift in the amounts of HF and LF regions of glaucomatous compared to non-glaucomatous TM tissue. Using a fluorescent tracer, we determined that glaucomatous donors, had significantly less HF and more LF regions in comparison with their non-glaucomatous counterpart (Fig. 1A). The decrease in the amount of HF regions in glaucoma as compared with HF regions in normal was not statistically significant, nor was the increase amount of LF regions in glaucoma as compared with LF regions in normal. When these regions were examined further by AFM, a statistically significant 8–10 fold increase in elastic moduli of LF regions of the glaucomatous TM was observed compared to LF regions of non-glaucomatous TM (Fig. 1B). On average, a 50% decrease was observed in the moduli of the HF region of glaucomatous TMs, when compared with HF regions of non-glaucomatous TM, although it did not reach statistical significance due to relatively small number of biological replicates. We also noted a significant reduction in total cell numbers across the entire TM in glaucomatous donors compared with non-glaucomatous donors (Fig. 1C). Next, we determined if glaucomatous TM were capable of producing a typical homeostatic response to elevated pressure using perfused anterior segment organ culture (Fig. 1D). Under normal 1x pressure, the normalized flow rate observed was 1.8 μl/min. When the perfusion pressure was elevated to 2x pressure (17.6 mm Hg perfusion pressure), the normalized flow rate immediately doubled to approximately 3.3 μl/min. Over the next 72 hours, the flow rate varied but did not significantly change from the initial 2x response. Anterior segments that successfully mount a homeostatic response typically reduce the outflow resistance slowly over this time frame and the flow rate steadily increases [33]. For reference, we have included a typical line for a normal response from earlier studies [34]. Here, the glaucomatous TMs were unable to trigger this homeostatic resistance adjustment. These observations led us to next determine if there were significant differences in ECM deposited by non-glaucomatous and glaucomatous TM cells using an in vitro cell derived extracellular matrix platform.
Figure 1.
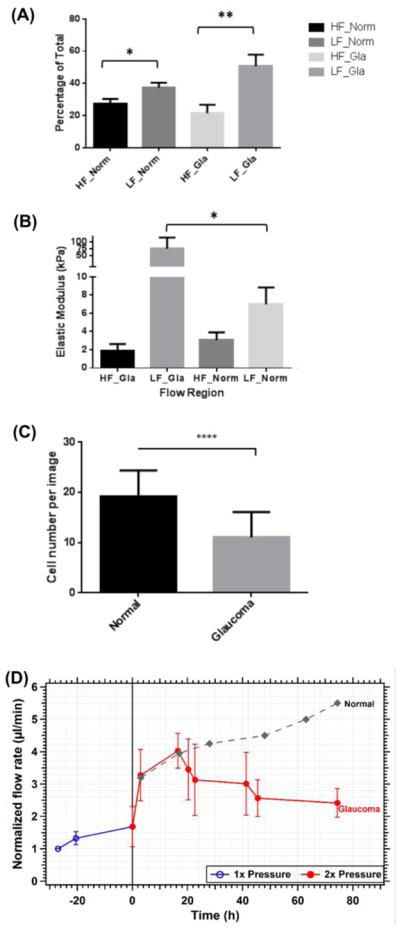
(A) Relative percentage of high flow (HF) and low flow (LF) segmental regions in normal (norm) and glaucomatous (Gla) TM, *p=0.029 and **p=0.006; mean values are the percentage of total relative fluorescence units around the circumference of the eye; n = 12 individual normal eyes and n = 7 individual glaucomatous eyes; (B) elastic moduli of segmental regions of the TM; * p=0.04; n = 6 individual normal eyes and n = 6 individual glaucomatous eyes, (C) number of cells observed in the TM from glaucomatous and non-glaucomatous donor tissues, **** p=0.0001; mean cell numbers were from n = 14 individual normal eyes and n = 8 individual glaucomatous eyes. (D) Graphical representation of normalized flow rate in glaucomatous anterior segment perfusion culture calculated at normal (1x or 8.8 mm Hg) and elevated (2x or 17.6 mm Hg) perfusion pressures. In contrast, flow rates in normal anterior segment perfusion culture have previously been shown to steadily increase over time in response to elevated pressure [33, 34].
3.2 ECM deposited by glaucomatous TM cells differ from that deposited by non-glaucomatous donors
3.2.1 Matrix mechanics
Force vs. indentation curve analysis was performed on ECM deposited by five glaucomatous donor TM cell lines. Data revealed significant donor-to-donor variability in moduli values. However, for all 5 donors, elastic moduli of ECMs deposited by all cells with dex treatment was significantly greater than vehicle control treated cells (Fig. 2A). ECM deposited by glaucomatous cells were stiffer than those deposited by non-glaucomatous cells for both vehicle control (EtOH) and Dex treated groups (Fig. 2B). Regardless of whether the ECM was derived from NTM or GTM cells, ECM after Dex treatment was 2–3 fold stiffer.
Figure 2.

(A) Elastic modulus of extracellular matrix derived from five different glaucomatous TM donors cultured in the absence (EtOH vehicle control) presence of 100 nM dexamethasone (Dex). At least 7–10 random locations were probed with 5 force curves obtained at each location. Results are mean ± standard deviation.*p<0.05, **p<0.01, ***p<0.001, t-test (with Bonferroni correction) compared with vehicle control (EtOH). (B) Comparison of elastic moduli of cell derived matrices obtained from glaucomatous (5 donors) and non-glaucomatous (3 donors) TM cell cultures. Data are the mean ± standard deviation of all force curves obtained for all donors at all locations. ***p<0.001, t-test compared with normal (non-glaucomatous, and ##p<0.01, t-test compared with vehicle control (EtOH). (C) Representative image of fibronectin organization in the matrix deposited by non-glaucomatous and glaucomatous cells. (D) Matrix proteins from glaucomatous and non-glaucomatous donors cluster differently. Heat map and dendrogram based on mean differences in protein expression between each group. Data are from 3 donors for each group.
3.2.2 Matrix morphology
Cells were removed by NH4OH treatment and well preserved ECMs were obtained. Complete decellularization was confirmed by immunocytochemistry by staining for fibronectin, nuclear material and filamentous actin. All ECM samples stained positively for fibronectin, which is a typical ECM component made by these cells. A qualitative representative image from one donor ECM for each condition is illustrated (Fig. 2C). At this time, we were unable to perform any quantitative evaluation of ECM organization, which is quite complex. Significant differences in staining patterns for fibronectin was observed between both control and Dex treated groups and between glaucomatous and non-glaucomatous groups. In ECM from dex treated non-glaucomatous cells, fibronectin was observed to be deposited as a fibrillar sheet in comparison with a less fibrillary morphology and minimal organization observed in the ECM of EtOH treated cells. In contrast, fibronectin in ECMs deposited by glaucomatous cells was more fibrillar in both Dex treated and untreated groups. With Dex treatment, fibronectin was observed to be denser than untreated samples (Fig. 2C, inset).
3.2.3 Matrix composition
To ascertain quantitative differences in biochemical composition of ECMs derived from non-glaucomatous and glaucomatous cells treated with vehicle control (EtOH) or Dex, nano-scale liquid chromatography tandem-mass spectrometry was performed. In the spectral data, analysis revealed significant differences in protein expression between the groups (Fig. 2D). Hierarchical clustering documented composition of ECMs to be strikingly different between the non-glaucomatous and glaucomatous donors. Interestingly, minimal differences were observed between EtOH vs Dex treatment in ECM deposited by glaucomatous cells. However, consistent with our previous observations, the composition of ECMs differed significantly between the treatment groups in non-glaucomatous cells.
3.3 ECMs deposited by primary TM cells (+/− Dex) from non-glaucomatous and glaucomatous donors differentially modulate non-glaucomatous cell behavior
In order to investigate the effect that substratum have on cell behavior, primary NTM cells from a 50 year old male (NTM50) were plated on glass surfaces, or on decellularized ECMs derived from either non-glaucomatous or glaucomatous donors in the presence or absence of Dex, for 3 days.
3.3.1 Gene expression
mRNA expression of two genes, myocilin (MYOC) and αB-crystallin (CRYAB), were found to be differentially altered in NTM50 cells cultured on the various substrates (Fig. 3). Greater than 4-fold changes in expression were observed when NTM50 cells were cultured on ECMs derived from three different glaucomatous donors in comparison with cultures on non-glaucomatous ECM or glass/TCP substrates. On ECMs derived from two of the three Dex treated glaucomatous cells NTM50 cells overexpressed MYOC and CRYAB at greater levels than their EtOH counterparts. Expression of MYOC, but not CRYAB, were found to be elevated in NTM50 cells cultured on ECMs derived from non-glaucomatous cells in comparison with TCP substrates.
Figure 3.

Expression of (A) myocilin (MYOC) and (B) αB-Crystallin in NTM50 cells cultured on matrix derived from either glaucomatous (GTM) or non-glaucomatous (NTM) donor cells. ECM from both donors were obtained when cells were treated with 100 nM dex or vehicle control (EtOH). Data are mean ± standard deviation from 3 reactions for each sample. *p<0.05, **p<0.01, ***p<0.001, t-test compared with vehicle control (EtOH), and ### p<0.001, t-test (with Bonferroni correction) compared with NTM50 cells cultured on tissue culture plastic (TCP) or ECM from non-glaucomatous donors.
3.3.2 Cell mechanics
Elastic moduli of NTM50 cells were determined using a pyramidal AFM cantilever by obtaining force vs. indentation curves over the nucleus (Fig. 4A). The modulus of cells on glass substrates was 2.83 ± 0.15 kPa (mean ± standard error). NTM50 cells, on ECM derived from Dex treated non-glaucomatous cells, demonstrated a ~2 fold elevation in elastic modulus for two of three donor ECMs in comparison with that observed on glass surfaces. No statistically significant differences were observed when they were cultured on ECM from EtOH treated non-glaucomatous cells, although they trended to possess slightly lower elastic moduli. In contrast, cell stiffness was significantly greater when NTM50 cells were cultured on both ECMs derived from glaucomatous donors with a greater response observed in the Dex treated group (~3 fold on GTM EtOH and ~5 fold on GTM Dex) in comparison with glass surfaces.
Figure 4.

(A) Elastic modulus of NTM50 cells cultured on either glass of ECM derived from either glaucomatous (GTM) or non-glaucomatous (NTM) donor cells. At least 7–10 random individual cells were probed with 5 force curves obtained at each location. Results are mean ± standard deviation. ***p<0.001, t-test (with Bonferroni correction) compared with vehicle control (EtOH), and ### p<0.001, t-test compared with NTM50 cells cultured on ECM from non-glaucomatous donors. (B) Western blots demonstrating αSMA levels in NTM50 cells cultured on cell derived matrices. Graph is the quantification of relative optical density of αSMA protein band normalized to β-actin. *p<0.05, t-test compared with vehicle control (EtOH).
3.3.3 αSMA expression
Concurrent with changes in cell mechanics, we documented that NTM50 cells were more responsive to modulating αSMA expression (Fig. 4B) when cultured on non-glaucomatous ECM (EtOH vs Dex) than on glaucomatous donors (EtOH vs Dex). For both donor groups, αSMA expression was always greater in NTM50 cells cultured on ECM derived after Dex treatment.
3.3.4 Protein expression
In addition to specific changes documented in MYOC, CRYAB or αSMA expression, we determined global changes in protein expression in NTM50 cells cultured on the various substrates. Spectral data were analyzed to ascertain differential protein expression between the various groups: i.e. NTM50 cells cultured on (i) TCP, (ii) NTM EtOH ECM, (iii) NTM Dex ECM, (iv) GTM EtOH ECM, and (v) GTM Dex ECM. Principal component analysis (Fig. 5A) revealed the data to group in three distinct groups: TCP vs NTM ECM vs GTM ECM. Specifically, proteins expressed in cells cultured on TCP surfaces grouped on the right hand side along principal component 1 (PC1; gray ellipse); proteins in NTM50 cells cultured on non-glaucomatous CDM grouped in the positive quadrant along PC2 (blue ellipse) while those in cell cultured on glaucomatous CDM grouped in the negative quadrant along PC2 (pink ellipse). There were no specific groupings (along PC1 or PC2) in proteins of NTM50 cells cultured on CDMs derived from either NTM or GTM cells treated with EtOH or Dex. These together suggest that protein expression profiles in NTM50 cells negatively correlate with each other comparing culture conditions of NTM CDM or GTM CDM. A closer look by hierarchical clustering identified the correlation between 18 proteins between these 5 groups (Fig. 5B). From these two analyses it was apparent that NTM50 cells behaved differently when cultured on the non-physiological plastic substrates. We then further analyzed for differences in protein expression in NTM50 cells by two groups: i.e. when combined on glaucomatous vs non-glaucomatous ECM (Fig. 6). Proteins of particular interest that were over expressed in cells cultured on glaucomatous ECM are: Pentraxin 3 (PTX3), growth differentiation factor 15 (GDF15), connective tissue growth factor (CTGF), fibulin 1 (FBLN1), and inter-alpha trypsin inhibitor (ITIH). A number of proteins involved in metabolic processes were down regulated in these cells. Using the Database for Annotation, Visualization, and Integrated Discovery (DAVID)[31] tool for gene ontology analysis, we present the functional classification of the proteins that are altered (Table 3).
Figure 5.

(A) Principal component analysis (PC1 vs PC2) demonstrating different clusterings for protein levels in NTM50 cells cultured on TCP or cell derived matrices. (B) Heat map and dendrogram based on mean differences in protein expression NTM50 cells between each group.
Figure 6.

Histogram illustrating log to the base 2-fold changes of protein expression in NTM50 cells when cultured on ECM derived from glaucomatous vs non-glaucomatous cells. Proteins that were upregulated are in red and those downregulated are in blue comparing NTM50 on GTM ECM with NTM ECM.
Table 3.
Functional classification of proteins, as defined by GO classification, that are altered in NTM50 cells cultured on either GTM or NTM ECM by at least 2-fold.
| Proteins downregulated in NTM50 cells on GTM vs NTM ECM | |
|---|---|
| Category | Gene |
| Cell-cell adhesion | LIMA1, HDLBP, KIF5B, PDLIM5, IQGAP1, EIF4G1, RPS26, ATIC, RARS, SND1, LRRC59, USO1, FASN, SPTBN1, CNN2, DBN1, EMD, AHNAK, SPTAN1 |
| Translational initiation | EIF4G1, EIF6, RPS26, RPL32, RPS17, RPS9, RPLP2, RPL4, RPS24 |
| ER to Golgi vesicle-mediated transport | SEC23A, SEC31A, COPG1, USO1, SEC22B, SPTBN1, DYNC1H1, SPTAN1 |
| Translation | WARS, RPS26, RPL32, RPS17, RRBP1, RPS9, RPLP2, RPL4, RPS24 |
| Regulation of cellular amino acid metabolic process | PSMA2, PSMD14, PSME2, PSMD1, NQO1 |
| Regulation of mRNA stability | PSMA2, EIF4G1, PSMD14, PSME2, PSMD1, HSPB1 |
| Wnt signaling pathway, planar cell polarity pathway | PSMA2, AP2B1, PSMD14, PSME2, PSMD1 |
| Negative regulation of canonical Wnt signaling pathway | PSMA2, DAB2, PSMD14, PSME2, PSMD1 |
| MAPK cascade | PSMA2, PSMD14, PSME2, PSMD1, SPTBN1, SPTAN1 |
| Negative regulation of apoptotic process | DAB2, SQSTM1, DDB1, HSPB1, GLO1, NQO1, NCL |
| Positive regulation of phagocytosis | C3, PTX3, AHSG |
| Positive regulation of ERK1 and ERK2 cascade | HMGB1, CD44, C3, CTGF |
| Cell adhesion | CD44, CTGF, IGFBP7, MFGE8, MYH10 |
| Hyaluronan metabolic process | ITIH2, ITIH3 |
| Positive regulation of collagen biosynthetic process | CTGF, F2 |
| Positive regulation of cysteine-type endopeptidase activity involved in apoptotic process | HMGB1, CTGF |
| SMAD protein signal transduction | AFP, GDF15 |
| Positive regulation of JNK cascade | HMGB1, CTGF |
| Inflammatory response | HMGB1, C3, PTX3 |
3.3.5 Stress response
Next, we determined whether ECM derived from cells are capable of modulating the stress response in NTM50 cells. First, we immunostained for lysosomal marker, LAMP1, imaged 5 random field of views from each sample at 40X for 3 CDM samples per condition, and quantified the pixel intensity sum per unit area. We observed that the relative intensity and number of cells that have a greater amount of LAMP1 staining was higher when cultured upon glaucomatous ECM than non-glaucomatous ECM. Further, cells plated on ECM derived after Dex treatment tended to overexpress LAMP1. Representative images and quantitative measurements from NTM50 cells cultured on one donor ECM for each condition is represented in Fig. 7A. Next, we determined if endoplasmic reticulum (ER) stress was present in these cells. Indeed, on glaucomatous ECM derived after Dex treatment, a prominent increase in expression of ATF4, GRP78, and CHOP was observed (Fig. 7B).
Figure 7.

(A) LAMP1 expression (red) was observed to be greater, and (B) endoplasmic reticular stress markers (ATF4, GRP78, and CHOP) were overexpressed in NTM50 cultured on glaucomatous ECM. Graph is the quantification of relative optical density of αSMA protein band normalized to β-actin. *p<0.05, t-test compared with vehicle control (EtOH), and *p<0.05, t-test compared with non-glaucomatous group.
3.3.6 Remodeling of the ECM
A critical observation with glaucoma, is the presence of a dysfunctional ECM in the TM. We therefore sought to explore the effect that freshly plated cells have on remodeling the fibronectin deposited by glaucomatous and/or non-glaucomatous donors. When plated on ECM from non-glaucomatous donors (Fig. 2), NTM50 cells were capable of re-organizing fibronectin. Notably NTM50 cells cultured on these CDM were able to re-organize the underlying FN selectively. While a number of ‘fibronectin free’ spots were observed right beneath the NTM50 cells in both NTM EtOH and NTM Dex groups (Fig. 8; Fig. S2), there were no regions devoid of fibronectin observed when cells were cultured on GTM Dex ECM, although partial absence of fibronectin was observed in the GTM EtOH group.
Figure 8.

Representative images demonstrating changes in fibronectin (green) organization when NTM50 cells (nuclei in blue) are cultured on ECM derived from glaucomatous and non-glaucomatous cells.
4. DISCUSSION
4.1 Loss of homeostasis in glaucoma
Ocular hypertension has long been associated with changes in the ECM, but few have identified a causal relationship between hypertension and ECM. Using a laser induced model of experimental glaucoma in non-human primates (NHPs), we recently demonstrated that normal animals subjected to prolonged elevated pressure are capable of compensation by remodeling their ECM, even when only one clock hour of the TM was left unlasered [35]. The use of NHPs for mechanistic studies are expensive and practically limiting to dissect the dynamic interactions between cells and their ECM. More commonly, ex vivo anterior segment organ perfusion models are currently exploited to study segmental flow, and mechanistic changes in TM/Schlemm’s canal with altered pressure/flow [36–38]. The segmental nature of the TM has been known for decades, and has been demonstrated in a number of species. Classical studies from de Kater et al [39], using cationic ferritin, showed that aqueous outflow is distinct in regions and hypothesized that glaucoma may be a disease of segmentation. In the same year, Johnson observed segmental pigmentation in the TM and posed a question whether this contributes as a factor in pigment dispersion glaucoma [40]. To the best of our knowledge, no one has documented with great certainty what percentage of a hypertensive eye can be expected to be HF/LF/IF, or how such segmental distribution differs with POAG, and when it does what might its functional correlate be. Our group and others, over the past several years have hypothesized that ocular hypertension may present manifestations in segmental outflow regions but had not documented these until now.
In the present study, glaucomatous eyes at elevated pressure, resulted in a decreased ability to adjust to the pressure increase. We also observed a significant increase in the amount of stiffer LF regions, and a concurrent decrease in the amount of softer HF regions in glaucomatous TM as compared with normal TM. Non-glaucomatous eyes when perfused at constant 2x pressure to stimulate the IOP homeostatic outflow resistance response, demonstrate a slow and sustained adjustment to normalized outflow facility over several days [33] that likely involves dynamic alteration in TM ECM. This is supported by recent data from our labs where HF regions in normal donor eyes compensate by becoming softer via ECM remodeling when challenged with 2X pressure over 72 h [11]. The lack of adjustment to elevated pressure, in the present study, demonstrates that glaucomatous TM are incapable of maintaining homeostasis. Simultaneously, we observed that LF regions challenged with 2X pressure were significantly stiffer, and on par with glaucomatous TM stiffness as previously reported [41], compared to LF regions challenged at 1X pressure. Also, softening of the TM in HF regions in glaucomatous eyes suggests that the cells in these regions retain some ability, albeit subdued, to remodel the ECM to facilitate aqueous outflow in an attempt to compensate for IOP fluctuations. These together informed us that a dynamic reciprocity exists between cells and their immediate microenvironment that allows for ECM remodeling to accommodate for outflow facility.
4.2 Comparing ECM deposited by glaucomatous and non-glaucomatous TM cells
Over the past decade, cell derived matrices from a variety of cell types have been utilized to induce differentiation of stem cells into a chondrogenic or osteogenic phenotype [42–45]. However, to the authors’ knowledge, very few studies have applied cell derived matrices to study disease mechanisms, and there are currently none that describe glaucoma or ocular hypertension. Limited studies have compared ECM derived from normal vs diseased cells or their ability to regulate cellular phenotype. Our data demonstrates that the ECM deposited by TM cells from glaucomatous donors differs significantly in mechanics and organization accompanied by relative differences in matrix and matricellular proteins. Importantly, we observed that ECM deposited by vehicle control (EtOH) treated glaucomatous cells were as stiff as those deposited by dex treated non-glaucomatous donors [as reported previously in [24]]. Dex treatment of glaucomatous cells only made the ECM stiffer. Using scratch and indentation on an AFM we measured that the CDMs were at least 7–10 μm thick; Hertzian curve fit analyses were performed over indentation depths no greater than 650 nm (where the elastic modulus and indentation depth are linear in relation); since this indentation depth is <10% of the thickness sampled we rule out any substratum effects that could dictate matrix stiffness [46]. That ECM deposited by GTM cells in vitro is stiffer than that deposited by NTM cells is itself interesting considering that GTM tissue is stiffer than NTM tissue [41]. That being said, the mechanical integrity and make-up of cell derived matrices are vastly different from native tissues.
In vivo, numerous studies have documented fibronectin to be overexpressed in glaucomatous TM [47, 48] but few have revealed differences in how this scaffolding protein may be organized to contribute to outflow resistance. Organ culture or animal based models have yet to address the causal nature of the increased resistance: whether this is because of abundance of certain matrix proteins, their organization, or their crosslinking, or a cell mediated response, or some mix of all of the above. In this study, interestingly, no significant changes in the expression of large structural ECM proteins were observed between the groups, although a number of smaller matricellular/modifier proteins were changed. Among the major ECM proteins and of particular relevance to glaucoma, fibronectin is a critical protein that is of specific interest to us. An excellent review on the relevance of fibronectin in the trabecular meshwork can be found in Faralli et al [49]. While the expression levels of fibronectin weren’t different between the various groups in vitro, as observed by proteomics, the morphology of fibronectin and its distribution were. Fibronectin was observed to be dense, fibrillary, and appeared to be more crosslinked when deposited by glaucomatous cells. Although quantitative differences in fibronectin organization weren’t evaluated in this study, prior investigations document that increased cell-generated traction forces and surrounding ECM stiffness facilitate the assembly and organization of fibronectin fibrils [50, 51]. Further, fibronectin plays a direct role and also serves as a scaffolding protein for the deposition and assembly of a number of ECM proteins such as collagen, fibrillin, tenascin-C, latent TGFβ binding protein etc, and thus contributes significantly to matrix stability, biomechanics and signaling [52–56]. Importantly, fibronectin controls the availability of active TGFβ in the extracellular mileu, thus limiting severity of resulting fibrosis [57]. We therefore infer that the cohesive arrangement of fibronectin from glaucomatous and dex treated TM cells are due to a combination of cell-generated and matrix derived factors, and further posit that such changes result in dramatic cellular consequences.
4.3 Cellular responses to cell derived matrices
Consequent to changes in ECM we demonstrate their potent ability to modulate gene/protein expression, cellular mechanics, and resultant cellular function. One of the most significant effects was the marked stiffening of non-glaucomatous cells cultured on glaucomatous ECM. We postulate that this increase in stiffness was mediated by (i) a stiffer ECM deposited by glaucomatous cells, (ii) a more fibrillary, dense, crosslinked matrix, (iii) secreted factors in the matrix, or (iv) a combination of all of the above. That substratum mechanical properties can modulate cell stiffness has been demonstrated by us and others in various cell types [58–61]. To our knowledge, this is the first demonstration that cell derived matrices from diseased or normal tissues can differentially modulate cell mechanics. We observed significant differences in αSMA expression, a critical cytoskeletal protein implicated in fibrotic diseases [62–64]. The clinical implications for such a response, or how αSMA upregulation translates to tissue contractility and contributes to ocular hypertension remains to be investigated. Overexpression of αSMA has been observed in TM and other cells under a range of stimuli, e.g. uniaxial strain [65], TGFβ [66–68], glucocorticosteroid stimulus [24], increased substratum stiffness [69], and ER stress [70], all of which have been postulated to play a role in glaucoma pathogenesis. Accompanying these, we observed significant overexpression of myocilin and αB-crystallin in NTM cells cultured on glaucomatous ECM; both genes have been associated with glaucoma and are inducible by TGFβ and dexamethasone [71, 72]. αB-Crystallin is a small heat shock protein (HSPB5) that stabilizes polymerized actin contributing to formation of stress fibers and influences cell mechanics [73, 74], and its expression is increased in glaucomatous eyes [75]. The role of myocilin in TM cells is unclear, although its intracellular accumulation leads to ER stress [76], apoptosis [77], can result in modulation of Wnt pathway [78, 79], and mutations in the gene can cause glaucoma [80–83]. Whether overexpression of these genes is an adaptive response or a shift towards a pathologic phenotype remains to be elucidated.
We observed that on glaucomatous ECM, proteins involved in cell-cell adhesion, mRNA stability, or ER to golgi vesicle mediated transport were downregulated while those that regulate the ERK cascade, collagen and hyaluronan biosynthetic process, and SMAD signal transduction were all upregulated in NTM50 cells. mRNA instability has been associated with a number of diseases including cancer, inflammatory disease, and Alzheimer’s disease [84]. Activation of Smad proteins is a critical part of the TGFβ pathway that is implicated in TM dysfunction with aberrant expression of ECM proteins [85, 86]. These together suggested that NTM50 cells were indeed shifting towards a ‘stressed’ phenotype when cultured upon glaucomatous matrices. ER stress and the unfolded protein response have long been implicated in fibrosis [87–89] and in glaucoma, where alleviation of ER stress was found to rescue steroid induced glaucoma phenotype in a murine model [90]. Indeed, in this study, we observed upregulation of ER stress markers in TM cells cultured on glaucomatous ECM and on ECM derived from dex treated cells, demonstrating that pathologic ECM can promote a pathologic phenotype in normal cells. It is likely that the resultant ER stress was a direct result of accumulation of myocilin, or other matricellular modulators, as recently suggested [91]. Other unknown mechanisms may also play a role. Cellular response to ER stress is strongly modulated by their interaction with ECM proteins [92–94]. Subsequently, while low levels of ER stress may lead to an adaptive response towards survival, greater amounts can result in accumulation of abnormal/misfolded proteins (including those of the ECM) thus resulting in an aberrant feedback loop. Accumulation of such proteins may impair a cell’s ability to effect adequate ECM turnover. Here, we observed a significant number of regions where fibronectin was unaltered when NTM50 cells were cultured on GTM ECM. In contrast, fibronectin free regions were observed when NTM50 cells were cultured on NTM ECM. This suggests that glaucomatous ECM may be resilient to being remodeled by non-glaucomatous cells in vitro. Further studies are necessary to determine if this is true and if so what the mechanisms underlying such resistance would be.
5. SUMMARY
We summarize the key findings in this manuscript below:
There is a loss of homeostasis to accommodate for outflow facility under elevated pressure in glaucomatous eyes.
This is accompanied by an increase in the number of stiffer LF regions and a simultaneous decrease in softer HF regions under 2X pressure when compared with 1X pressure in glaucomatous eyes.
Glaucomatous TM cells deposit a stiffer matrix when compared with non-glaucomatous TM cells in vitro and this stiffness is increased under dex stimulus.
Fibronectin organization in ECM derived from GTM cells are different from those derived from NTM cells.
Expression of protein levels in normal TM cells (NTM50) differ significantly depending on type of substrate: i.e. TCP vs glaucomatous ECM vs non-glaucomatous ECM.
Normal cells (NTM50) cultured on ECM derived from GTM cells and dex treated cells develop ER stress, are stiffer, and express greater markers of cell contractility thus pushing it towards a more patho-mimetic phenotype.
GTM ECM appears to be more resilient to re-organization by NTM50 cells.
Correlating these to our observations in the ex vivo anterior segment perfusion culture model of glaucomatous eyes, these data collectively suggest that, cells in the diseased TM over time lose their ability to reorganize the tissue, via multiple yet unknown mechanisms, impairing tissue homeostasis. This results in a gradual increase in more crosslinked and rigid LF regions that contribute to sustained increases in IOP. Data point towards the contribution of ER stress perhaps due to erroneous ECM degradation/accumulation in this process, although further studies are required to establish causality. We recognize that CDMs do not replace the native tissue present in vivo, however our data suggests that CDMs are capable of mimicking the diseased phenotypes observed in vivo and are thus a potent model for studying cell-ECM interactions. In addition, these results raise concerns when developing stem cell based therapeutic modalities to renew the trabecular meshwork and treat ocular hypertension/glaucoma. Collectively, we demonstrate that in the absence of adequate animal models, CDM will be greatly beneficial to study disease mechanisms and test therapeutics and contribute towards reducing the use of animals during early research stages.
Supplementary Material
STATEMENT OF SIGNIFICANCE.
Extracellular matrix (ECM) changes are prevalent in a number of diseases. The precise mechanisms by which changes in the ECM contribute to disease progression is unclear, primarily due to absence of appropriate models. Here, using glaucoma as a disease model, we document changes in cell derived matrix (CDM) and tissue mechanics that contribute to the pathology. Subsequently, we determine the effect that ECMs from diseased and healthy individuals have on healthy cell behaviors. Data emanating from this study demonstrate that CDMs are a potent tool for the study of cell-ECM interactions.
Acknowledgments
This study was supported by Bright Focus National Glaucoma Research Awards (VKR, JAV), startup funding at UHCO (VKR), and National Institute of Health grants EY003279, EY008247, EY025721 (TSA) and P30 EY010572, and by an unrestricted grant to the Casey Eye Institute from Research to Prevent Blindness, New York, NY. The authors would like to thank Dr. Paul Russell for his valuable insight and discussions through the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Quigley HA. Glaucoma. Lancet. 2011;377(9774):1367–77. doi: 10.1016/S0140-6736(10)61423-7. [DOI] [PubMed] [Google Scholar]
- 2.Acott TS, Kelley MJ. Extracellular matrix in the trabecular meshwork. Exp Eye Res. 2008;86(4):543–61. doi: 10.1016/j.exer.2008.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stamer WD, Acott TS. Current Understanding of Conventional Outflow Dysfunction in Glaucoma. Curr Opin Ophthalmol. 2012;23(2):135–143. doi: 10.1097/ICU.0b013e32834ff23e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The AI. The Advanced Glaucoma Intervention Study (AGIS): 7. the relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol. 2000:429–440. doi: 10.1016/s0002-9394(00)00538-9. [DOI] [PubMed] [Google Scholar]
- 5.G. Collaborative Normal-Tension Glaucoma Study. Comparison of glaucomatous progression between untreated patients with normal-tension glaucomaa and patients with therapeutically reduced intraocular pressures. Am J Ophthalmol. 1998;126:487–497. doi: 10.1016/s0002-9394(98)00223-2. [DOI] [PubMed] [Google Scholar]
- 6.Heijl A, Leske MC, Bengtsson B, Hyman L, Hussein M. For the Early Manifest Glaucoma Trial Group: reduction of intraocular pressure and glaucoma progression. Arch Ophthalmol. 2002:1268–1279. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- 7.Gottanka J, Johnson DH, Martus P, Lutjen-Drecoll E. Severity of optic nerve damage in eyes with POAG is correlated with changes in the trabecular meshwork. J Glaucoma. 1997;6(2):123–132. [PubMed] [Google Scholar]
- 8.Last JA, Pan T, Ding Y, Reilly CM, Keller K, Acott TS, Fautsch MP, Murphy CJ, Russell P. Elastic Modulus Determination of Normal and Glaucomatous Human Trabecular Meshwork. Invest Ophthalmol Vis Sci. 2011;52:2147–2152. doi: 10.1167/iovs.10-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghunathan VK, Morgan JT, Park SA, Weber D, Phinney BS, Murphy CJ, Russell P. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Investigative Ophthalmology & Visual Science. 2015;56(8):4447–4459. doi: 10.1167/iovs.15-16739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang YF, Sun YY, Acott TS, Keller KE. Effects of induction and inhibition of matrix cross-linking on remodeling of the aqueous outflow resistance by ocular trabecular meshwork cells. Sci Rep. 2016;6:30505. doi: 10.1038/srep30505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vranka JA, Staverosky JA, Reddy AP, Wilmarth PA, David LL, Acott TS, Russell P, Raghunathan VK. Biomechanical rigidity and quantitative proteomics analyses of segmental regions of the ocular trabecular meshwork at physiologic and elevated pressures. Invest Ophthalmol Vis Sci. 2017 doi: 10.1167/iovs.17-22759. Accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alvarado J, Murphy C, Juster R. Trabecular meshwork cellularity in primary open-angle glaucoma and nonglaucomatous normals. Ophthalmology. 1984;91(6):564–79. doi: 10.1016/s0161-6420(84)34248-8. [DOI] [PubMed] [Google Scholar]
- 13.Alvarado J, Murphy C, Polansky J, Juster R. Age-related changes in trabecular meshwork cellularity. Invest Ophthalmol Vis Sci. 1981;21(5):714–27. [PubMed] [Google Scholar]
- 14.Grierson I, Howes RC. Age-related depletion of the cell population in the human trabecular meshwork. Eye. 1987;1(2):204–210. doi: 10.1038/eye.1987.38. [DOI] [PubMed] [Google Scholar]
- 15.Overby DR, Bertrand J, Tektas O-Y, Boussommier-Calleja A, Schicht M, Ethier CR, Woodward DF, Stamer WD, Lütjen-Drecoll E. Ultrastructural changes associated with dexamethasone-induced ocular hypertension in mice. Invest Ophthalmol Vis Sci. 2014;55(8):4922–33. doi: 10.1167/iovs.14-14429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lütjen-Drecoll E, Tektas OY. Structural Changes in the Trabecular Meshwork with Primary Open Angle Glaucoma*. In: Dartt DA, editor. Encyclopedia of the Eye. Academic Press; Oxford: 2010. pp. 224–228. [Google Scholar]
- 17.Tektas OY, Lütjen-Drecoll E. Structural changes of the trabecular meshwork in different kinds of glaucoma. Exp Eye Res. 2009;88(4):769–775. doi: 10.1016/j.exer.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 18.Gottanka J, Johnson DH, Grehn F, Lutjen-Drecoll E. Histologic findings in pigment dispersion syndrome and pigmentary glaucoma. J Glaucoma. 2006;15(2):142–51. doi: 10.1097/00061198-200604000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Johnson D, Flügel Johannes C, Hoffmann F, Futa R, Lütjen-Drecoll E. ULtrastructural changes in the trabecular meshwork of human eyes treated with corticosteroids. Arch Ophthalmol. 1997;115(3):375–383. doi: 10.1001/archopht.1997.01100150377011. [DOI] [PubMed] [Google Scholar]
- 20.Rohen JW, Lütjen-Drecoll E, Flügel C, Meyer M, Grierson I. Ultrastructure of the Trabecular Meshwork 10in Untreated Cases of Primary Open-Angle Glaucoma (POAG) Exp Eye Res. 1993;56(6):683–692. doi: 10.1006/exer.1993.1085. [DOI] [PubMed] [Google Scholar]
- 21.Johnson DH, Tschumper RC. Human trabecular meshwork organ culture. A new method. Investigative ophthalmology & visual science. 1987;28(6):945–53. [PubMed] [Google Scholar]
- 22.Vranka JA, Bradley JM, Yang Y-F, Keller KE, Acott TS. Mapping Molecular Differences and Extracellular Matrix Gene Expression in Segmental Outflow Pathways of the Human Ocular Trabecular Meshwork. PLoS One. 2015;10(3):e0122483. doi: 10.1371/journal.pone.0122483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morgan JT, Wood JA, Walker NJ, Raghunathan VK, Borjesson DL, Murphy CJ, Russell P. Human trabecular meshwork cells exhibit several characteristics of, but are distinct from, adipose-derived mesenchymal stem cells. J Ocul Pharmacol Ther. 2014;30(2–3):254–66. doi: 10.1089/jop.2013.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raghunathan VK, Morgan JT, Park SA, Weber D, Phinney BS, Murphy CJ, Russell P. Dexamethasone Stiffens Trabecular Meshwork, Trabecular Meshwork Cells, and Matrix. Invest Ophthalmol Vis Sci. 2015;56(8):4447–59. doi: 10.1167/iovs.15-16739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghunathan VK, Morgan JT, Chang YR, Weber D, Phinney B, Murphy CJ, Russell P. Transforming Growth Factor Beta 3 Modifies Mechanics and Composition of Extracellular Matrix Deposited by Human Trabecular Meshwork Cells. ACS Biomaterials Science & Engineering. 2015;1(2):110–118. doi: 10.1021/ab500060r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghunathan VK, Morgan JT, Dreier B, Reilly CM, Thomasy SM, Wood JA, Ly I, Tuyen BC, Hughbanks M, Murphy CJ, Russell P. Role of substratum stiffness in modulating genes associated with extracellular matrix and mechanotransducers YAP and TAZ. Invest Ophthalmol Vis Sci. 2013;54(1):378–86. doi: 10.1167/iovs.12-11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang YR, Raghunathan VK, Garland SP, Morgan JT, Russell P, Murphy CJ. Automated AFM force curve analysis for determining elastic modulus of biomaterials and biological samples. J Mech Behav Biomed Mater. 2014;37(0):209–18. doi: 10.1016/j.jmbbm.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgan JT, Raghunathan VK, Thomasy SM, Murphy CJ, Russell P. Robust and artifact-free mounting of tissue samples for atomic force microscopy. BioTechniques. 2014;56(1):40–2. doi: 10.2144/000114126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKee CT, Last JA, Russell P, Murphy CJ. Indentation versus tensile measurements of Young’s modulus for soft biological tissues. Tissue engineering Part B, Reviews. 2011;17(3):155–64. doi: 10.1089/ten.teb.2010.0520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteome Res. 2006;5(11):2909–18. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
- 31.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocols. 2008;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 32.Ward JH. Hierarchical Grouping to Optimize an Objective Function. Journal of the American Statistical Association. 1963;58(301):236–244. [Google Scholar]
- 33.Acott TS, Kelley MJ, Keller KE, Vranka JA, Abu-Hassan DW, Li X, Aga M, Bradley JM. Intraocular Pressure Homeostasis: Maintaining Balance in a High-Pressure Environment. J Ocul Pharmacol Ther. 2014;30(2–3):94–101. doi: 10.1089/jop.2013.0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abu-Hassan DW, Li X, Ryan EI, Acott TS, Kelley MJ. Induced pluripotent stem cells restore function in a human cell loss model of open-angle glaucoma. Stem Cells. 2014 doi: 10.1002/stem.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raghunathan V, Eaton JS, Christian BJ, Morgan JT, Ver Hoeve JN, Yang CC, Gong H, Rasmussen CA, Miller PE, Russell P, Nork TM, Murphy CJ. Biomechanical, ultrastructural, and electrophysiological characterization of the non-human primate experimental glaucoma model. Sci Rep. 2017;7(1):14329. doi: 10.1038/s41598-017-14720-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao W, Tovar-Vidales T, Yorio T, Wordinger RJ, Clark AF. Perfusion-cultured bovine anterior segments as an ex vivo model for studying glucocorticoid-induced ocular hypertension and glaucoma. Invest Ophthalmol Vis Sci. 2011;52(11):8068–75. doi: 10.1167/iovs.11-8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Erickson-Lamy K, Rohen JW, Grant WM. Outflow facility studies in the perfused human ocular anterior segment. Exp Eye Res. 1991;52(6):723–731. doi: 10.1016/0014-4835(91)90024-9. [DOI] [PubMed] [Google Scholar]
- 38.Fautsch MP, Bahler CK, Jewison DJ, Johnson DH. Recombinant TIGR/MYOC increases outflow resistance in the human anterior segment. Invest Ophthalmol Vis Sci. 2000;41(13):4163–8. [PubMed] [Google Scholar]
- 39.de Kater AW, Melamed S, Epstein DL. Patterns of aqueous humor outflow in glaucomatous and nonglaucomatous human eyes. A tracer study using cationized ferritin. Arch Ophthalmol. 1989;107(4):572–6. doi: 10.1001/archopht.1989.01070010586035. [DOI] [PubMed] [Google Scholar]
- 40.Johnson DH. Does pigmentation affect the trabecular meshwork? Arch Ophthalmol. 1989;107(2):250–4. doi: 10.1001/archopht.1989.01070010256032. [DOI] [PubMed] [Google Scholar]
- 41.Last JA, Pan T, Ding Y, Reilly CM, Keller K, Acott TS, Fautsch MP, Murphy CJ, Russell P. Elastic modulus determination of normal and glaucomatous human trabecular meshwork. Invest Ophthalmol Vis Sci. 2011;52(5):2147–52. doi: 10.1167/iovs.10-6342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dzobo K, Turnley T, Wishart A, Rowe A, Kallmeyer K, van Vollenstee F, Thomford N, Dandara C, Chopera D, Pepper M, Parker M. Fibroblast-Derived Extracellular Matrix Induces Chondrogenic Differentiation in Human Adipose-Derived Mesenchymal Stromal/Stem Cells in Vitro. International Journal of Molecular Sciences. 2016;17(8):1259. doi: 10.3390/ijms17081259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yong-Beom P, Sinji S, Jin AK, Jin-Chul H, Young-Cheol L, Chul-Won H. Effect of chondrocyte-derived early extracellular matrix on chondrogenesis of placenta-derived mesenchymal stem cells. Biomedical Materials. 2015;10(3):035014. doi: 10.1088/1748-6041/10/3/035014. [DOI] [PubMed] [Google Scholar]
- 44.Shraddha T, Corina AG, Maqsood A, Cindy K, Clemens AvB, Daniel S, Hugo AMF, Lorenzo M. Mesenchymal stromal cell-derived extracellular matrix influences gene expression of chondrocytes. Biofabrication. 2013;5(2):025003. doi: 10.1088/1758-5082/5/2/025003. [DOI] [PubMed] [Google Scholar]
- 45.Zeitouni S, Krause U, Clough BH, Halderman H, Falster A, Blalock DT, Chaput CD, Sampson HW, Gregory CA. Human mesenchymal stem cell-derived matrices for enhanced osteoregeneration. Sci Transl Med. 2012;4(132):132ra55. doi: 10.1126/scitranslmed.3003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas G, Burnham NA, Camesano TA, Wen Q. Measuring the Mechanical Properties of Living Cells Using Atomic Force Microscopy. Journal of visualized experiments: JoVE. 2013;(76):50497. doi: 10.3791/50497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Babizhayev MA, Brodskaya MW. Fibronectin detection in drainage outflow system of human eyes in ageing and progression of open-angle glaucoma. Mech Ageing Dev. 1989;47(2):145–157. doi: 10.1016/0047-6374(89)90017-1. [DOI] [PubMed] [Google Scholar]
- 48.Hann CR, Springett MJ, Wang X, Johnson DH. Ultrastructural localization of collagen IV, fibronectin, and laminin in the trabecular meshwork of normal and glaucomatous eyes. Ophthalmic Res. 2001;33(6):314–24. doi: 10.1159/000055687. [DOI] [PubMed] [Google Scholar]
- 49.Faralli JA, Schwinn MK, Gonzalez JM, Jr, Filla MS, Peters DM. Functional properties of fibronectin in the trabecular meshwork. Exp Eye Res. 2009;88(4):689–693. doi: 10.1016/j.exer.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lemmon CA, Chen CS, Romer LH. Cell Traction Forces Direct Fibronectin Matrix Assembly. Biophys J. 2009;96(2):729–738. doi: 10.1016/j.bpj.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Halliday NL, Tomasek JJ. Mechanical Properties of the Extracellular Matrix Influence Fibronectin Fibril Assembly in Vitro. Exp Cell Res. 1995;217(1):109–117. doi: 10.1006/excr.1995.1069. [DOI] [PubMed] [Google Scholar]
- 52.Chung CY, Erickson HP. Glycosaminoglycans modulate fibronectin matrix assembly and are essential for matrix incorporation of tenascin-C. J Cell Sci. 1997;110(Pt 12):1413–9. doi: 10.1242/jcs.110.12.1413. [DOI] [PubMed] [Google Scholar]
- 53.Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005;280(19):18871–80. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- 54.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20(5–24):495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sabatier L, Chen D, Fagotto-Kaufmann C, Hubmacher D, McKee MD, Annis DS, Mosher DF, Reinhardt DP. Fibrillin assembly requires fibronectin. Mol Biol Cell. 2009;20(3):846–58. doi: 10.1091/mbc.E08-08-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sottile J, Hocking DC. Fibronectin polymerization regulates the composition and stability of extracellular matrix fibrils and cell-matrix adhesions. Mol Biol Cell. 2002;13(10):3546–59. doi: 10.1091/mbc.E02-01-0048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kawelke N, Vasel M, Sens C, Au A, Dooley S, Nakchbandi IA. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-beta. PLoS One. 2011;6(11):e28181. doi: 10.1371/journal.pone.0028181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Raghunathan VK, Thomasy SM, Strom P, Yanez-Soto B, Garland SP, Sermeno J, Reilly CM, Murphy CJ. Tissue and cellular biomechanics during corneal wound injury and repair. Acta Biomater. 2017 doi: 10.1016/j.actbio.2017.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Byfield FJ, Reen RK, Shentu T-P, Levitan I, Gooch KJ. Endothelial actin and cell stiffness is modulated by substrate stiffness in 2D and 3D. J Biomech. 2009;42(8):1114–1119. doi: 10.1016/j.jbiomech.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 61.Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93(12):4453–61. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kalluri R. EMT: when epithelial cells decide to become mesenchymal-like cells. J Clin Invest. 2009;119(6):1417–9. doi: 10.1172/JCI39675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112(12):1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2005;46(8):2857–2868. doi: 10.1167/iovs.05-0075. [DOI] [PubMed] [Google Scholar]
- 66.Torrejon KY, Papke EL, Halman JR, Bergkvist M, Danias J, Sharfstein ST, Xie Y. TGFβ2-induced outflow alterations in a bioengineered trabecular meshwork are offset by a rho-associated kinase inhibitor. 2016;6:38319. doi: 10.1038/srep38319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torrejon KY, Pu D, Bergkvist M, Danias J, Sharfstein ST, Xie Y. Recreating a human trabecular meshwork outflow system on microfabricated porous structures. Biotechnol Bioeng. 2013;110(12):3205–3218. doi: 10.1002/bit.24977. [DOI] [PubMed] [Google Scholar]
- 68.Tamm ER, Siegner A, Baur A, Lutjen-Drecoll E. Transforming growth factor-beta 1 induces alpha-smooth muscle-actin expression in cultured human and monkey trabecular meshwork. Exp Eye Res. 1996;62(4):389–97. doi: 10.1006/exer.1996.0044. [DOI] [PubMed] [Google Scholar]
- 69.Quinlan AMT, Billiar KL. Investigating the role of substrate stiffness in the persistence of valvular interstitial cell activation. Journal of Biomedical Materials Research Part A. 2012;100A(9):2474–2482. doi: 10.1002/jbm.a.34162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heindryckx F, Binet F, Ponticos M, Rombouts K, Lau J, Kreuger J, Gerwins P. Endoplasmic reticulum stress enhances fibrosis through IRE1α-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol Med. 2016;8(7):729–744. doi: 10.15252/emmm.201505925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Welge-Lussen U, May CA, Eichhorn M, Bloemendal H, Lutjen-Drecoll E. AlphaB-crystallin in the trabecular meshwork is inducible by transforming growth factor-beta. Invest Ophthalmol Vis Sci. 1999;40(10):2235–41. [PubMed] [Google Scholar]
- 72.Zode GS, Kuehn MH, Nishimura DY, Searby CC, Mohan K, Grozdanic SD, Bugge K, Anderson MG, Clark AF, Stone EM, Sheffield VC. Reduction of ER stress via a chemical chaperone prevents disease phenotypes in a mouse model of primary open angle glaucoma. The Journal of Clinical Investigation. 2011;121(9):3542–3553. doi: 10.1172/JCI58183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao Ch M. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J Mol Biol. 2007;366(3):756–67. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 74.Wang K, Spector A. alpha-crystallin stabilizes actin filaments and prevents cytochalasin-induced depolymerization in a phosphorylation-dependent manner. Eur J Biochem. 1996;242(1):56–66. doi: 10.1111/j.1432-1033.1996.0056r.x. [DOI] [PubMed] [Google Scholar]
- 75.Lütjen-Drecoll E, May CA, Polansky JR, Johnson DH, Bloemendal H, Nguyen TD. Localization of the stress proteins alpha B-crystallin and trabecular meshwork inducible glucocorticoid response protein in normal and glaucomatous trabecular meshwork. Invest Ophthalmol Vis Sci. 1998;39(3):517–25. [PubMed] [Google Scholar]
- 76.Joe MK, Sohn S, Hur W, Moon Y, Choi YR, Kee C. Accumulation of mutant myocilins in ER leads to ER stress and potential cytotoxicity in human trabecular meshwork cells. Biochem Biophys Res Commun. 2003;312(3):592–600. doi: 10.1016/j.bbrc.2003.10.162. [DOI] [PubMed] [Google Scholar]
- 77.Yam GH-F, Gaplovska-Kysela K, Zuber C, Roth J. Aggregated Myocilin Induces Russell Bodies and Causes Apoptosis: Implications for the Pathogenesis of Myocilin-Caused Primary Open-Angle Glaucoma. The American Journal of Pathology. 2007;170(1):100–109. doi: 10.2353/ajpath.2007.060806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kwon H-S, Lee H-S, Ji Y, Rubin JS, Tomarev SI. Myocilin Is a Modulator of Wnt Signaling. Mol Cell Biol. 2009;29(8):2139–2154. doi: 10.1128/MCB.01274-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shen X, Ying H, Yue BYJT. Wnt Activation by Wild Type and Mutant Myocilin in Cultured Human Trabecular Meshwork Cells. PLoS One. 2012;7(9):e44902. doi: 10.1371/journal.pone.0044902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fingert JH, Héon E, Liebmann JM, Yamamoto T, Craig JE, Rait J, Kawase K, Hoh ST, Buys YM, Dickinson J. Analysis of myocilin mutations in 1703 glaucoma patients from five different populations. Hum Mol Genet. 1999;8(5):899–905. doi: 10.1093/hmg/8.5.899. [DOI] [PubMed] [Google Scholar]
- 81.Gobeil S, Letartre L, Raymond V. Functional analysis of the glaucoma-causing TIGR/myocilin protein: Integrity of amino-terminal coiled-coil regions and olfactomedin homology domain is essential for extracellular adhesion and secretion. Exp Eye Res. 2006;82(6):1017–1029. doi: 10.1016/j.exer.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 82.McDowell CM, Luan T, Zhang Z, Putliwala T, Wordinger RJ, Millar JC, John SWM, Pang I-H, Clark AF. Mutant human myocilin induces strain specific differences in ocular hypertension and optic nerve damage in mice. Exp Eye Res. 2012;100(0):65–72. doi: 10.1016/j.exer.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Senatorov V, Malyukova I, Fariss R, Wawrousek EF, Swaminathan S, Sharan SK, Tomarev S. Expression of Mutated Mouse Myocilin Induces Open-Angle Glaucoma in Transgenic Mice. The Journal of Neuroscience. 2006;26(46):11903–11914. doi: 10.1523/JNEUROSCI.3020-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hollams EM, Giles KM, Thomson AM, Leedman PJ. mRNA Stability and the Control of Gene Expression: Implications for Human Disease. Neurochem Res. 2002;27(10):957–980. doi: 10.1023/a:1020992418511. [DOI] [PubMed] [Google Scholar]
- 85.Sethi A, Jain A, Zode GS, Wordinger RJ, Clark AF. Role of TGFbeta/Smad signaling in gremlin induction of human trabecular meshwork extracellular matrix proteins. Invest Ophthalmol Vis Sci. 2011;52(8):5251–9. doi: 10.1167/iovs.11-7587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zode GS, Sethi A, Brun-Zinkernagel A-M, Chang IF, Clark AF, Wordinger RJ. Transforming growth factor-β2 increases extracellular matrix proteins in optic nerve head cells via activation of the Smad signaling pathway. Mol Vis. 2011;17:1745–1758. [PMC free article] [PubMed] [Google Scholar]
- 87.Inagi R, Shoji K, Nangaku M. Oxidative and Endoplasmic Reticulum (ER) Stress in Tissue Fibrosis. Current Pathobiology Reports. 2013;1(4):283–289. [Google Scholar]
- 88.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle R-M, Seeger W, Weaver TE, Guenther A. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proceedings of the National Academy of Sciences. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zode GS, Sharma AB, Lin X, Searby CC, Bugge K, Kim GH, Clark AF, Sheffield VC. Ocular-specific ER stress reduction rescues glaucoma in murine glucocorticoid-induced glaucoma. J Clin Invest. 2014;124(5):1956–65. doi: 10.1172/JCI69774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kasetti RB, Maddineni P, Millar JC, Clark AF, Zode GS. Increased synthesis and deposition of extracellular matrix proteins leads to endoplasmic reticulum stress in the trabecular meshwork. Sci Rep. 2017;7(1):14951. doi: 10.1038/s41598-017-14938-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lynch Jeffrey M, Maillet M, Vanhoutte D, Schloemer A, Sargent Michelle A, Blair NS, Lynch Kaari A, Okada T, Aronow Bruce J, Osinska H, Prywes R, Lorenz John N, Mori K, Lawler J, Robbins J, Molkentin Jeffery D. A Thrombospondin-Dependent Pathway for a Protective ER Stress Response. Cell. 2012;149(6):1257–1268. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nugent AE, McBurney DL, Horton WE. The presence of extracellular matrix alters the chondrocyte response to endoplasmic reticulum stress. J Cell Biochem. 2011;112(4):1118–1129. doi: 10.1002/jcb.23025. [DOI] [PubMed] [Google Scholar]
- 94.Kasetti RB, Phan TN, Millar JC, Zode GS. Expression of Mutant Myocilin Induces Abnormal Intracellular Accumulation of Selected Extracellular Matrix Proteins in the Trabecular Meshwork. Invest Ophthalmol Vis Sci. 2016;57(14):6058–6069. doi: 10.1167/iovs.16-19610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.