

Abstract
Key points
The mechanisms by which bacteria alter endothelial cell phenotypes and programme inflammatory angiogenesis remain unclear.
In lung endothelial cells, we demonstrate that toll‐like receptor 4 (TLR4) signalling induces activation of forkhead box protein C2 (FOXC2), a transcriptional factor implicated in lymphangiogenesis and endothelial specification, in an extracellular signal‐regulated kinase (ERK)‐dependent manner.
TLR4‐ERK‐FOXC2 signalling regulates expression of the Notch ligand DLL4 and signals inflammatory angiogenesis in vivo and in vitro.
Our work reveals a novel link between endothelial immune signalling (TLR pathway) and a vascular transcription factor, FOXC2, that regulates embryonic vascular development.
This mechanism is likely to be relevant to pathological angiogenesis complicating inflammatory diseases in humans.
Abstract
Endothelial cells (ECs) mediate a specific and robust immune response to bacteria in sepsis through the activation of toll‐like receptor (TLR) signalling. The mechanisms by which bacterial ligands released during sepsis programme EC specification and altered angiogenesis remain unclear. We postulated that the forkhead box protein C2 (FOXC2) transcriptional factor directs EC cell‐fate decisions and angiogenesis during TLR signalling. In human lung ECs, lipopolysaccharide (LPS) induced ERK phosphorylation, FOXC2, and delta‐like 4 (DLL4, the master regulator of sprouting angiogenesis expression) in a TLR4‐dependent manner. LPS‐mediated ERK phosphorylation resulted in FOXC2‐ERK protein ligation, ERK‐dependent FOXC2 serine and threonine phosphorylation, and subsequent activation of DLL4 gene expression. Chemical inhibition of ERK or ERK‐2 dominant negative transfection disrupted LPS‐mediated FOXC2 phosphorylation and transcriptional activation of FOXC2. FOXC2‐siRNA or ERK‐inhibition attenuated LPS‐induced DLL4 expression and angiogenic sprouting in vitro. In vivo, intraperitoneal LPS induced ERK and FOXC2 phosphorylation, FOXC2 binding to DLL4 promoter, and FOXC2/DLL4 expression in the lung. ERK‐inhibition suppressed LPS‐induced FOXC2 phosphorylation, FOXC2‐DLL4 promoter binding, and induction of FOXC2 and DLL4 in mouse lung ECs. LPS induced aberrant retinal angiogenesis and DLL4 expression in neonatal mice, which was attenuated with ERK inhibition. FOXC2+/− mice treated with LPS showed a mitigated increase in FOXC2 and DLL4 compared to FOXC2+/+ mice. These data reveal a new mechanism (TLR4‐ERK‐FOXC2‐DLL4) by which sepsis‐induced EC TLR signalling programmes EC specification and altered angiogenesis.
Keywords: FOXC2 and inflammatory angiogenesis, TLR4 regulates FOXC2, TLR and EC specification, TLR4‐ERK‐FOXC2‐DLL4 axis
Key points
The mechanisms by which bacteria alter endothelial cell phenotypes and programme inflammatory angiogenesis remain unclear.
In lung endothelial cells, we demonstrate that toll‐like receptor 4 (TLR4) signalling induces activation of forkhead box protein C2 (FOXC2), a transcriptional factor implicated in lymphangiogenesis and endothelial specification, in an extracellular signal‐regulated kinase (ERK)‐dependent manner.
TLR4‐ERK‐FOXC2 signalling regulates expression of the Notch ligand DLL4 and signals inflammatory angiogenesis in vivo and in vitro.
Our work reveals a novel link between endothelial immune signalling (TLR pathway) and a vascular transcription factor, FOXC2, that regulates embryonic vascular development.
This mechanism is likely to be relevant to pathological angiogenesis complicating inflammatory diseases in humans.
Introduction
Endothelial cells (ECs) that line the luminal surface of the vasculature regulate vascular tone, facilitate gas and solute exchange, have synthetic functions, and maintain vascular homeostasis by providing an anticoagulant interface (Ryan, 1986; Maniatis & Orfanos, 2008). ECs in the lung and other organs also play a pivotal role in the immune response to bacteria during sepsis, supporting the concept of ECs as conditional immune cells (Dauphinee & Karsan, 2006; Andonegui et al. 2009; Mai et al. 2013). Pulmonary ECs express multiple toll‐like receptors (TLRs), a sentinel receptor family, that mediates specific innate immune responses to conserved molecular motifs found in bacteria, viruses and fungi (Andonegui et al. 2003; Dauphinee & Karsan, 2006; Maniatis & Orfanos, 2008; Mai et al. 2013). The importance of EC TLR signalling in sepsis was reported by Andonegui et al. (2009) who showed that conditional knockout mice lacking immune cell TLR4 signalling, but with intact EC TLR4 signalling, were able to successfully clear gram‐negative bacteria. However, immune activation of lung ECs during sepsis also contributes to neutrophil influx, cytokine release, and tissue injury observed in acute respiratory distress syndrome (Andonegui et al. 2003; Maniatis & Orfanos, 2008). Lipopolysaccharide (LPS) released during sepsis is sensed by its cognate receptor TLR4, triggering canonical pathway signalling through myeloid differentiation factor 88 (MyD88), resulting in the activation of the NF‐kB‐dependent immune and inflammatory programme (Akira, 2006). While LPS‐induced lung EC immune activation contributes to bacterial clearance and acute lung injury, the consequences of EC TLR signalling on EC cell‐fate decisions central to angiogenesis and vasculogenesis remain unknown (Mattsby‐Baltzer et al. 1994; Menden et al. 2015). This is of translational significance as sepsis and inflammation programme deviant blood vessel formation observed in human diseases such as bronchopulmonary dysplasia (BPD) in premature babies, and rheumatoid arthritis and atherosclerosis in adults, the mechanistic origins of which remain unclear (Thebaud & Abman, 2007; Usman et al. 2015; Tas et al. 2016).
Angiogenesis, the process of new blood vessel formation from pre‐existing blood vessels, occurs either via sprouting or intussusceptive processes (Eilken & Adams, 2010). Sprouting angiogenesis is critical for vascular development as mice lacking delta‐like 4 (DLL4), the master regulator of sprouting angiogenesis, demonstrate embryonic lethality (Gale et al. 2004). ECs expressing DLL4 in a VEGF‐dependent manner specify a tip cell EC phenotype necessary for initiation of sprouting (Eilken & Adams, 2010). Tip cells specify a stalk cell EC phenotype in neighbouring ECs through Notch‐dependent lateral inhibition of DLL4, which is necessary for propagation of sprouts (Eilken & Adams, 2010). Regulation of EC cell‐fate decisions is central to sprouting angiogenesis and underlies both developmental and abnormal angiogenesis (Ridgway et al. 2006; Lobov et al. 2007). In this study, we investigated the mechanisms by which lung EC innate immune signalling alters EC‐fate decisions by regulating DLL4 expression. LPS stimulation activates multiple tyrosine kinase cascades in ECs, including ERK1/2, JNK and p38 kinase (Dauphinee & Karsan, 2006; Mai et al. 2013). We and others have shown that endothelial immune activation and pro‐inflammatory signalling is mediated through the MAPK p38 and JNK (Scholl et al. 2007; Cargnello & Roux, 2011). The extracellular signal‐regulated kinases (ERK1/2) are involved in multiple cellular processes such as proliferation and differentiation, gene regulation, and development (Scholl et al. 2007). ERK signalling promotes endothelial cell survival and sprouting during tumour angiogenesis, ERK inhibition causes abrogation of tube formation, and dominant‐negative ERK expression represses tumour growth (Mavria et al. 2006; Murphy et al. 2006). ERK is also considered to be a major determinant of lymphatic endothelial specification, and it also mediates VEGF‐dependent angiogenic signalling during embryonic development (Yu et al. 2014; Shin et al. 2016). The role of ERK signalling in mediating TLR4‐dependent angiogenic responses during endothelial innate immune signalling has not been characterized.
FOXC2 belongs to the forkhead family of transcription factors characterized by a DNA‐binding forkhead domain (Seo et al. 2006; Kume, 2009). FOXC2 plays an important role in vascular development, as targeted disruption of its locus causes branchial arch and vertebral anomalies resulting in death after embryonic day 13 to a few hours after birth (Iida et al. 1997; Kume, 2009). Loss of one copy of FOXC2 in humans causes hereditary lymphedema distichiasis (LD) syndrome and primary valve failure in veins of lower extremities (Fang et al. 2000). FOXC2 haploinsufficiency in mice phenocopies the human LD syndrome with lymphatic vascular defects and lymphedema (Fang et al. 2000). In mice, VEGF‐mediated PI3K signalling stimulates FOXC2‐mediated activation of DLL4 and HEY2 expression, demonstrating a role for FOXC2 in VEGF‐dependent regulation of DLL4 (Hayashi & Kume, 2008). In the same study, ERK inhibition enhanced FOXC2 signalling, indicating negative regulation of FOXC2 by ERK. In contrast, lymphatic endothelial cell‐FOXC‐KO mouse embryos show hyperactivation of the ERK pathway, suggesting that FOXC2 inhibits ERK activation (Fatima et al. 2016). While these data suggest that ERK and FOXC2 can regulate each other in the lymphatic endothelium in the context of vascular development, a non‐developmental role for ERK‐FOXC2 during innate immune signalling has not been elucidated. Specifically, whether endothelial TLR signalling directs EC specification and aberrant angiogenesis by activating FOXC2 signalling is unknown. In this study, we demonstrate that endothelial TLR4 signalling activates ERK, which regulates FOXC2‐dependent DLL4 expression in human and mouse lung ECs.
Methods
Ethical approval
Care of mice before and during experimental procedures was conducted in accordance with the policies at the University of Missouri‐Kansas City Lab Animal Resource Center and the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Protocols had prior approval from the University of Missouri‐Kansas City Institutional Animal Care and Use Committee. Investigators understand the ethical principles under which The Journal of Physiology operates and work described here complies with the animal ethics checklist published in The Journal (Grundy, 2015).
Animal model
Mice were housed at the University of Missouri‐Kansas City animal facility in a 12 h light/dark schedule as per the facility animal housing protocol. Experimental pups fed ad libitum with mothers and no specific fasting/feeding protocols were followed. Dams had unrestricted access to food and water. Our experiments did not involve anaesthetic or surgical procedures. FVB FOXC2 heterozygous (+/−) mice were obtained from Dr Tsutomu Kume, PhD (Northwestern University, Evanston, IL, USA) and were bred at the University of Missouri‐Kansas City animal facility with wild‐type FVB mice. Wild‐type mice, FVB and C57BL/6 strains, were obtained commercially from Charles River (Burlington, MA, USA), C57BL/6 TLR4−/− mice were obtained commercially from Jackson Labs (Ben Harbor, ME, USA). Mice were injected with lipopolysaccharide (LPS) injections (2 mg kg−1) or sterile saline (controls) intraperitoneally (i.p.) (Sigma, St Louis, MO, USA), with little to no mortality observed. The pups were left with the dam and closely monitored for signs of distress after treatments. Anaesthesia was not used with i.p. treatments as per our approved animal protocol. All animal experiments were performed on mouse pups between days of life 4–7. Chemical injections of a commercially available ERK inhibitor (U0126, ERK‐I) were i.p. injected at 20 mg kg−1, or sterile saline for controls, and given 1 h prior to LPS treatment. Mice were killed using a 100 mg kg−1 i.p. injection pentobarbital, exsanguinated after cessation of heartbeat, and the lungs were harvested and utilized as described below. Lung lobes were divided as specified: left lobe for histology; right lobes divided for RNA and protein.
Cell culture and reagents
Human primary pulmonary microvascular endothelial cells (HPMECs) from ScienCell (Carlsbad, CA, USA) were used between passages 3 and 4 for all experiments. These cells are derived from the lungs of newborn infants (ScienCell). HPMECs were grown in endothelial cell medium (ECM) supplemented with 5% fetal bovine serum (FBS), antibiotics, and endothelial cell growth serum (ECGS) as recommended by the manufacturer (ScienCell) in a humidified incubator containing 5% CO2 at 37°C. Ultrapure lipopolysaccharide (LPS, 100 ng mL−1) and the TLR4 blocking antibody (TLR4‐I, mab‐htlr4, 5 μm) were purchased commercially from Invivogen (San Deigo, CA, USA), and the ERK inhibitor (U0126, ERK‐l, 50 μm) was purchased commercially from SCBT (Duan et al. 2004; Chialda et al. 2005; Clark et al. 2010). For experiments with inhibitors, cells were pre‐treated with the chemicals for 1 h prior to the addition of LPS. Monkey retinal endothelial cells RF/6A (MRECs) were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and were grown in Eagles Minimum Essential Medium as recommended by ATCC.
Isolation of murine endothelial cells
For endothelial cell isolation, all lobes of the lung from 2 neonatal C57BL/6 pups (4–5 days old) were pooled per condition. For the FVB FOXC2+/+ and FOXC2+/− mice, the entire lung from one neonatal pup was used after their genotype was verified by PCR. The protocol for the isolation of mouse lung endothelial cells was described previously (Sobczak et al. 2010). Briefly, harvested lungs were minced with sterile scissors in ice‐cold DMEM and transferred to 15 mL of pre‐warmed 1 mg mL−1 collagenase solution in DMEM. The mixture was allowed to rotate for 45 min at 37°C. The digested tissue was then passed through a 14 g cannula attached to a 20 mL syringe several times, followed by passage through a 70 μm cell strainer, and washed with 20% FBS + DMEM. Cells were then centrifuged at 400 g for 5 min and the supernatant was aspirated off. The cell pellet was re‐suspended with 0.1% BSA in PBS. The suspension was incubated with anti‐PECAM‐1 antibody‐conjugated dynabeads from Life Technologies (Carlsbad, CA, USA) on a rocker for 15 min at room temperature as per the manufacturer's protocol. After precipitation, the cells were washed with PBS 3 times and protein or RNA was extracted following standard protocol as described below.
Quantification of mRNA expression using real‐time PCR
Total RNA was extracted from HPMECs and mouse lung EC or tissue using the PureLink RNA Mini Kit (Life Technologies) following the manufacturer's instructions and cDNA was synthesized from 1 μg of RNA using an iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA, USA) according to the manufacturer's instructions. Real‐time PCR was run on a Bio‐Rad IQ5 with SYBR green mastermix. The primers for mouse and human DLL4, FOXC2, and 18S were purchased commercially from Sigma. 18S was used as the housekeeping gene. The relative gene expression was calculated using the Pflaffl method (Pfaffl, 2001).
Immunoblotting for quantifying changes in protein expression
HPMECs and mouse lung tissue were homogenized in RIPA lysis buffer containing commercially available protease and phosphatase inhibitors (Sigma) after LPS treatment, with the clarified lysates used for Western blotting. Immunoblotting was done following standard protocol. The primary antibodies used were: goat anti‐FOXC2, goat anti‐DLL4, and rabbit anti‐p65 (Santa Cruz Biotechnology (SCBT), Santa Cruz, CA, USA), rabbit anti‐phospho‐P44/42 MAPK (ERK(Thr202/Tyr204)), rabbit anti‐P44/42 MAPK (ERK), and rabbit anti‐phospho‐p65 (Cell Signaling, Danvers, MA, USA), and mouse anti‐β‐Actin (Sigma). Densitometry was performed using ImageJ Software (NIH, Bethesda, MD, USA) and changes were normalized to β‐actin or the corresponding non‐phosphorylated antibody. To quantify apoptosis after LPS and ERK‐I treatments, cell lysates were immunoblotted using a rabbit anti‐cleaved caspase 3 (Cell Signaling) antibody. To quantify the HPMEC survival with and without ERK‐I and LPS, Trypan Blue (Life Technologies) staining was used with a cell counter following the manufacturer protocol, with the average of 2 reads done per condition.
Immunoprecipitation for phosphorylation studies
HPMECs grown to 90% confluence in 60‐mm dishes had various treatments, and lysates were used for immunoprecipitation studies. Whole lung homogenates obtained after various timed i.p. treatments were used for studies. The immunoprecipitation protocol was followed as described previously (Menden et al. 2013, 2015). Briefly, 500 μg of protein was incubated with the primary antibody overnight at 4°C and for 2 h with protein A sepharose beads. Upon completion, the beads were washed twice with ice‐cold TBS, after which 100 μL of TBS and 2× Laemmli buffer was added to each sample and the beads were boiled for 10 min. Proteins were separated SDS‐PAGE gel and blotted for phosphorylated proteins. After the blots were developed using enhanced chemiluminescence (ECL) they were stripped with Restore Plus stripping buffer (Thermo‐Fisher, Rockford, IL, USA) and probed with the primary antibody specific to the protein immunoprecipitated. The primary antibody used for immunoprecipitation was goat anti‐FOXC2 (SCBT). For experiments with FOXC2 phosphorylation the following antibodies were used: mouse anti‐phospho‐serine [(p)Ser], mouse anti‐phospho‐threonine [(p)Thr] and mouse anti‐phospho‐tyrosine [(p)Tyr] (SCBT). For experiments with FOXC2 binding proteins, rabbit anti P44/42 MAPK (ERK) (Cell Signaling) was used. Densitometry was performed using ImageJ Software (NIH) and changes in phosphorylation were normalized to FOXC2.
siRNA‐mediated FOXC2 gene silencing
siRNA sequences targeting human FOXC2 (siFOXC2) were purchased from SCBT and gene silencing was performed as per the manufacturers recommendations as reported before (Menden et al. 2013, 2015). For the non‐silenced cells, control siRNA (consisting of scrambled sequence that does not interfere with cellular function) was used (SCBT). Briefly, HPMECs were cultured with antibiotic‐free ECM until 80% confluent. The media was then aspirated and cells washed twice with the siRNA transfection medium. The plates were then incubated with either the control siRNA or siFOXC2 strand (8 μg) in transfection medium and incubated for 16 h. Subsequently, the reagents were aspirated off and normal ECM was gently put on the plates. After silencing, the cells were grown for another 48 h and then treated with LPS. Silencing efficiency was determined by Western blotting using a goat anti‐FOXC2 (SCBT) antibody.
Chromatin immunoprecipitation (ChIP)
For HPMECs, the ChIP procedure followed Pierce Magnetic ChIP Kit's protocol for human samples and the mouse samples followed the MAGnify Chromatin Immunoprecipitation System's protocol (Thermo‐Fisher). A FOXC2 antibody (Abcam, Cambridge, MA, USA) was used to pull‐down FOXC2 binding DNA, and the immunoprecipitated DNA was analysed by PCR using specific primers corresponding to the DLL4 promoter that were designed based on FOXC2 binding sites to DLL4 promoter (Hayashi & Kume, 2008). The primers for human samples were sense 5ʹ‐ACCACCAAGTCATCAC CTTCCTCAC‐3ʹ and antisense 5ʹ‐TCGCACCTGCCGGTCAATAAATC‐3ʹ. The primers for mouse samples were sense 5ʹ‐AGTCATCATCTTCCTCTCTCCTCCCTCAGC‐3ʹ and antisense 5ʹ‐TCGCACCTGCC GGTCAATAAATC‐3ʹ. Several primer pairs were designed to cover the 2‐kb human FOXC2 promoter region to examine if FOXC2 can bind to its own promoter region. One band can be amplified from FOXC2 antibody pulled‐down DNA with sense primer 5ʹ‐GTGATTGGCT CAAAGTTCCG‐3ʹ and antisense primer 5ʹ–TGAGAGCGAGAGAGCGCGAGAGA‐3ʹ.
Design of wild‐type/variant constructs and transfection
Total RNA was extracted from HPMECs using the PureLink RNA Mini Kit (Life Technologies) and the cDNA library was generated with SuperScript III Reverse Transcriptase (Thermo‐Fisher) following the manufacturer's protocol. Human ERK1 and ERK2 cDNA was amplified and cloned to pIRES‐EGFP‐puro (Addgene, Cambridge, MA, USA). ERK1 K71R and ERK2 K54R was constructed with PCR strategy and cloned into pIRES‐EGFP‐puro. Constitutively active FOXC2‐CA, which was fused with FOXC2 DNA binding domain and VP16 activation domain (Gerin et al. 2009), was amplified with 5ʹ‐ACTACTAGTCCACCATGGCGGCGCCTAAGGACCTGGT‐3ʹ and 5ʹ‐AGTCTCGAGCTACCCACCGTACTCGTCAATTCC‐3ʹ by PCR, and then cloned into pIRES‐EGFP‐puro. (FOXC2‐CA was a gift from Ormond A. MacDougald.) HPMECs grown in 6‐well tissue culture plates were transfected overnight with 2 μg of the indicated plasmids or empty plasmids (vector) with Lipofectamine 3000 (Thermo‐Fisher) as per the manufacturer's protocol. Cells were allowed to recover for 24 h, and then treated with LPS for indicated time‐points.
Angiogenic tube and network formation assay on Matrigel
The in vitro angiogenesis network formation assay on Matrigel using lung endothelial cells was performed as described previously (Akhtar et al. 2002; Menden et al. 2015). Briefly, HPMECs grown to ∼80% confluence in 6‐well culture plates were silenced either with siFOXC2 or control siRNA. After silencing, the medium was replaced with serum‐free medium, followed by treatment with LPS for 10 h. For experiments with ERK plasmids after transfection (see above), the medium was changed and the cells were allowed to grow for another 24 h. Subsequently, the medium was replaced with serum‐free medium, treated with LPS for 10 h, and underwent the in vitro angiogenesis assay on Matrigel. Cells were then detached with Tryple Express (Life Technologies), re‐suspended in basal ECM and 6 × 104 cells in 300 μL of media were plated on to a 24‐well Matrigel matrix coated plate (Corning, Corning, MA, USA). Angiogenesis was assessed 12 h after cells were seeded on Matrigel. Calcein AM fluorescent dye (Corning) was used to enhance the visibility of tube and network formation on Matrigel. For experiments with ERK‐I, cells were treated with the inhibitor for 1 h prior to LPS, followed by the above experimental steps. Angiogenesis was evaluated by counting the number of tube and network formations in one quadrant (the same one for each condition) and multiplying by four. Only tubular structures connecting two cell clusters were considered for measurements, whereas cell clusters with at least three tubular structures emanating out were considered to be a network. Representative images were taken using an Olympus 1 × 71 microscope fluorescence microscope with attached camera at 4× zoom.
Quantification of lung DLL4 immunofluorescent staining
The left lung of the mouse pups was fixed in formalin, blocked in paraffin, and 4 μm sections were cut onto slides. The slides were used for immunofluorescent staining using a validated mouse anti‐DLL4 IgG2a antibody and mouse anti‐PECAM IgG1 antibodies (SCBT) with the corresponding Alexafluor secondary antibodies (Thermo‐Fisher). The slides mounted in ProLong Gold with DAPI (Thermo‐Fisher), which stains the nucleus. Images were taken at 63× magnification using a Zeiss LSM510 confocal microscope with an attached camera. Staining is indicated with a channel of colour: DLL4 is red, PECAM is green, and DAPI is blue. At least three mice were in each experimental group and quantification of the percentage of DLL4/PECAM positive cells per PECAM positive cells per high power field (HPF) are reported as the average of ten images taken per mouse.
Retinal angiogenesis assay and DLL4 quantification
The eyes from 4 day old pups were fixed in formalin at 4°C overnight and the retinas were excised following a protocol from Tual‐Chalot et al. (2013). After the retinas were isolated, they were fixed in methanol and stored at −80°C until use. The retinas underwent a staining protocol consisting of washing with PBS, blocking overnight with BSA and serum, primary antibody incubation overnight, washes with PBS prior to incubation with the corresponding Alexafluor secondary antibody (Thermo‐Fisher) and isolectin B4‐Alexa 488 for 3 h, nuclear staining for 1 h with Hoechst stain (Thermo‐Fisher), further washes with PBS, and mounting the retinas on a slide with Prolong Gold mount (Thermo‐Fisher). DLL4 was used as the primary antibody (SCBT). Images were taken at 25× magnification using a Zeiss LSM510 confocal microscope with an attached camera. At least three mice were in each experimental group and quantification of the percentage of DLL4/Hoescht/Isolectin B4 positive cells per Hoescht/Isolectin B4 positive cells per high power field (HPF) are reported as the average of five images taken per mouse, per condition. The Isolectin B4 staining was quantified for branching points using a high power field (HPF) image taken at 25× and ImageJ's add‐on for angiogenesis analysis (ImageJ). The number of capillaries, and the number of capillary junctions (branching points) are reported as the average of at least 5 images taken per mouse, with at least 3 mice used per condition.
Data analysis
Data are presented as means ± SD. P < 0.05 was considered significant. A minimum of three animals were used for each experimental group. Fold changes in protein levels were determined relative to expression in control wild‐type mice, and were compared between groups using ANOVA. For changes in phosphorylation, the phosphorylated to total protein ratio was calculated for each condition and compared between groups. For mRNA studies, fold change was calculated relative to expression in control wild‐type mice and compared between experimental groups using ANOVA. The Bonferroni test was used in conjunction with ANOVA to correct for multiple comparisons. For DLL4/PECAM quantification, the average number of cells with positive staining per HPF was compared between groups. For Matrigel‐based angiogenesis assays the average of tubes and networks from one quadrant (see Methods above) from ≥3 experiments was compared between groups using ANOVA with a post hoc Bonferroni correction. For the retinal angiogenesis assay, the number of capillaries and capillary junctions from 5 high power fields per retina were quantified (see Methods above) from ≥3 experiments and compared between groups using ANOVA.
Results
TLR4 activation stimulates DLL4 expression in human pulmonary microvascular endothelial cells (HPMECs) and in mouse lung EC (Fig. 1)
Figure 1. LPS stimulates DLL4 expression in HPMECs and mouse lung.
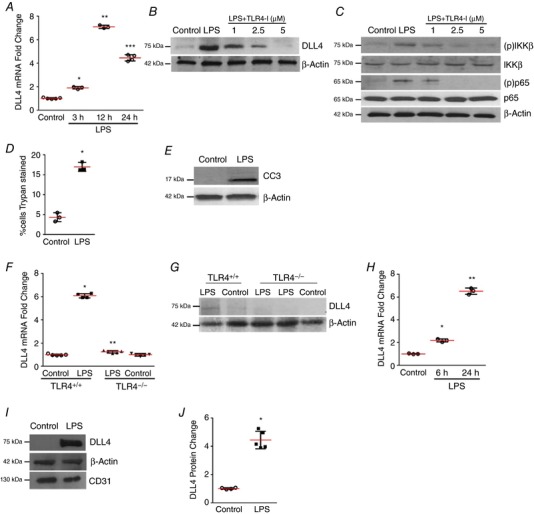
A, HPMEC RNA obtained at 3, 12 and 24 h after LPS was used to quantify DLL4 expression by qRT‐PCR, P < 0.01 (Control vs. LPS at 3 h*, 12 h** and 24 h***), n = 3. B, DLL4 protein in HPMECs was quantified by immunoblotting 24 h after LPS treatment, with or without TLR4‐blocking antibody (TLR4‐I), n ≥ 3. C, DLL4, IKK‐β phosphorylation [(p)IKKβ], and phosphorylated NF‐κB [(p)p65] in HPMECs were quantified 30 min after treatment with LPS, and with or without TLR4‐I, n ≥ 3. D, HPMEC cell death (%) was quantified by Trypan Blue staining 24 h after LPS treatment, P < 0.01 (*Control vs. LPS), n = 3. E, cleaved caspase 3 protein level was quantified by immunoblotting HPMEC lysates 24 h after LPS treatment. F, total RNA obtained from whole lung of 7‐day old TLR4+/+ and TLR4−/− mice 24 h after i.p. saline or i.p. LPS treatment was used to probe DLL4 mRNA expression by qRT‐PCR. Fold change in expression is relative to TLR4+/+ pups with saline injection, P < 0.05 (*TLR4+/+ vs. TLR4+/+ LPS; **TLR4+/+ LPS vs. TLR4−/− LPS), n ≥ 4 mice per group. G, DLL4 protein expression in whole lung lysates of 7‐day old TLR4+/+ and TLR4−/− mice, n ≥ 3. H, DLL4 mRNA was quantified in mouse lung EC isolated at 6 and 24 h after i.p. LPS treatment by qRT‐PCR, P < 0.01 (Control vs. LPS at 6 h* and 24 h**), n ≥ 3 mice per group. I, DLL4 protein expression was quantified in mouse lung EC isolated 24 h after i.p. LPS, with densitometric quantification shown graphically (J), * P < 0.05 (Control vs. LPS), n ≥ 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
To examine whether LPS alters endothelial cell‐fate decisions we examined DLL4 expression in HPMECs. LPS strongly induced DLL4 RNA expression at 3, 12 and 24 h and protein expression at 24 h in HPMECs (Fig. 1 A and B). LPS‐induced DLL4 expression and TLR4 signalling (evidenced by IKKβ and p65 phosphorylation) was mitigated in a dose‐dependent manner by pre‐treating cells with a TLR4‐blocking antibody (Fig. 1 C). Stimulation with LPS in HPMECs was associated with increased cell death as shown by increased Trypan Blue staining and cleaved caspase 3 (CC3) levels at 24 h (Fig. 1 D and E). To confirm our in vitro data, we injected mice on day of life 6 with LPS intraperitoneally (i.p.), and examined whole lung expression of DLL4 at 24 h. LPS robustly induced DLL4 RNA and protein expression in the lung of wild‐type mice, but not in the lungs of TLR4−/− mice (Fig. 1 F and G). To characterize cell‐type specific expression, we isolated lung ECs from 5‐day old mouse pups. LPS robustly induced DLL4 RNA at 6 and 24 h, and protein in lung ECs at 24 h (Fig. 1 H–J). These data show that LPS induces expression of the EC tip cell specification marker DLL4 in a TLR4‐dependent fashion in human and mouse lung EC.
FOXC2 regulates LPS‐mediated DLL4 expression in HPMECs (Fig. 2)
Figure 2. FOXC2 regulates LPS‐mediated DLL4 expression in HPMECs.
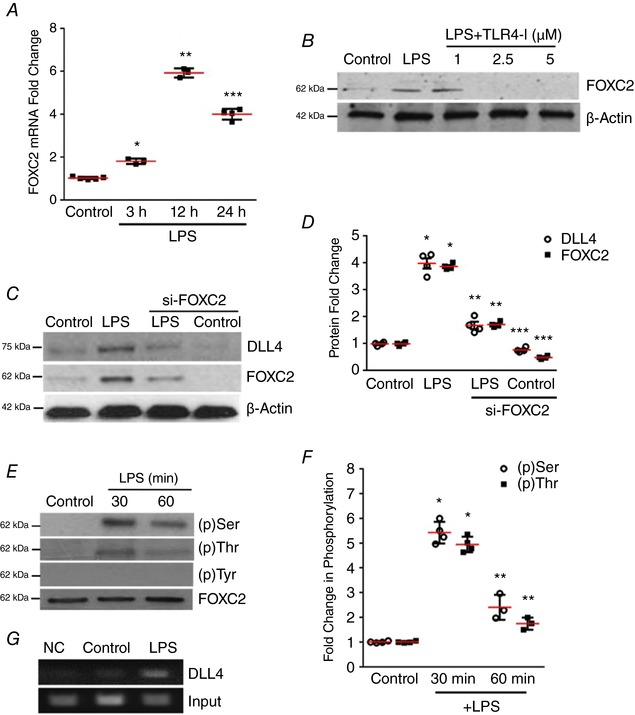
A, FOXC2 mRNA expression in HPMECs was quantified by qRT‐PCR after 3, 12 and 24 h LPS treatment, P < 0.01 (Control vs. LPS at 3 h*, 12 h** and 24 h***), n = 4. B, FOXC2 protein in HPMECs was probed by immunoblotting 24 h after treatment with LPS, and with or without TLR4‐I, n ≥ 3. C, DLL4 and FOXC2 protein were quantified by immunoblotting after 24 h LPS treatment in control siRNA or FOXC2 siRNA transfected HPMECs, with densitometry shown graphically (D), P < 0.05 (*Control vs. LPS; **LPS vs. si‐FOXC2 LPS; ***Control vs. siFOXC2 Control), n ≥ 3. E and F, FOXC2 protein was immunoprecipitated from lysates at 30 and 60 min after LPS, and serine, threonine and tyrosine phosphorylation was assessed by immunoblotting (E), and quantified by densitometry (F), P < 0.01 (Control vs. LPS at 30 min* and 60 min**), n = 3. G, HPMEC nuclear lysates obtained 30 min after LPS were used to quantify FOXC2 binding to the DLL4 promotor by the chromatin immunoprecipitation assay (ChIP), n ≥ 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
To determine whether LPS‐induced DLL4 expression is transcriptionally regulated by FOXC2, we examined whether TLR4 signalling activates FOXC2. We noted that LPS induced FOXC2 RNA and protein expression in HPMECs (Fig. 2 A and B) in a TLR4‐dependent manner. Attenuating FOXC2 with siRNA decreased basal and LPS‐stimulated FOXC2 protein expression (Fig. 2 C and D). LPS‐induced DLL4 expression was suppressed in HPMECs treated with siFOXC2 but not scrambled siRNA (Fig. 2 C and D). To determine whether FOXC2 regulates DLL4 in HPMECs we initially examined whether LPS activated FOXC2. Phosphorylation of FOXC2 at several of its serine and threonine residues results in activation and nuclear translocation of FOXC2 (Ivanov et al. 2013). Co‐immunoprecipitation studies revealed that LPS‐induced serine and threonine, but not tyrosine phosphorylation of FOXC2 at 30 and 60 min (Fig. 2 E and F). To examine whether FOXC2 directly binds to the DLL4 promoter after LPS stimulation we performed chromatin immunoprecipitation (ChIP) studies. We found that FOXC2 binds to the DLL4 promoter under basal conditions but there was increased binding of FOXC2 to the DLL4 promoter 30 min after LPS treatment (Fig. 2 G). These data reveal that LPS‐mediated DLL4 expression in HPMECs is mediated through FOXC2.
ERK2 regulates FOXC2‐dependent DLL4 expression in LPS‐treated lung ECs (Fig. 3 and Fig. 4)
Figure 3. ERK regulates FOXC2‐dependent DLL4 expression in LPS‐treated HPMECs.
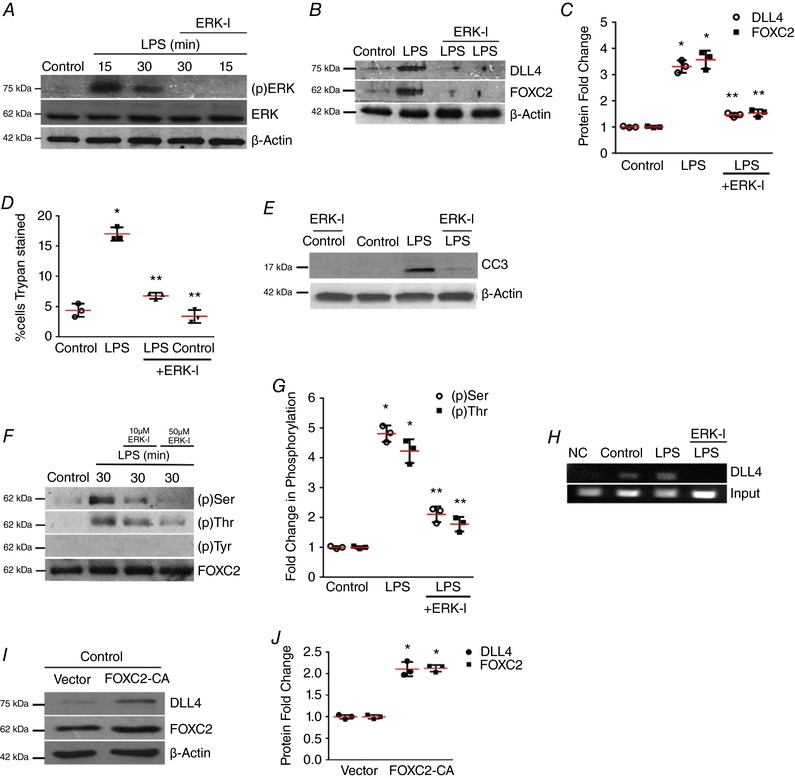
A, ERK phosphorylation [(p)ERK] was quantified at 15 and 30 min after LPS with or without ERK‐I pre‐treatment, n ≥ 3. B and C, DLL4 and FOXC2 protein were quantified at 24 h after LPS with or without ERK‐I, with densitometric quantification shown graphically (C), P < 0.01 (*Control vs. LPS; **LPS vs. ERK‐I + LPS), n = 3. D, HPMEC cell death (%) was quantified by Trypan Blue staining 24 h after LPS treatment with or without ERK‐I (50 μm), P < 0.01 (*Control vs. LPS: **LPS vs. ERK‐I + LPS), n = 3. E, CC3 protein level was assessed by immunoblotting after LPS treatment with or without ERK‐I. F, FOXC2 protein was immunoprecipitated from HPMECs at 30 min after LPS treatment with or without ERK‐I (10 and 50 μm), and serine, threonine and tyrosine phosphorylation quantified by densitometry (G), P < 0.001 (*Control vs. LPS: **LPS vs. ERK‐I + LPS), n = 3. H, HPMEC nuclear lysates obtained 30 min after LPS with or without ERK‐I treatments were used to quantify FOXC2 binding to the DLL4 promoter by ChIP, n ≥ 3. I, DLL4 and FOXC2 protein were assessed after transfection with FOXC2‐CA (CA, constitutively active) or empty plasmid, and quantified by densitometry (J), * P < 0.01 (Control vs. FOXC2‐CA), n = 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 4. ERK2 regulates FOXC2‐dependent DLL4 expression in LPS‐treated HPMECs.
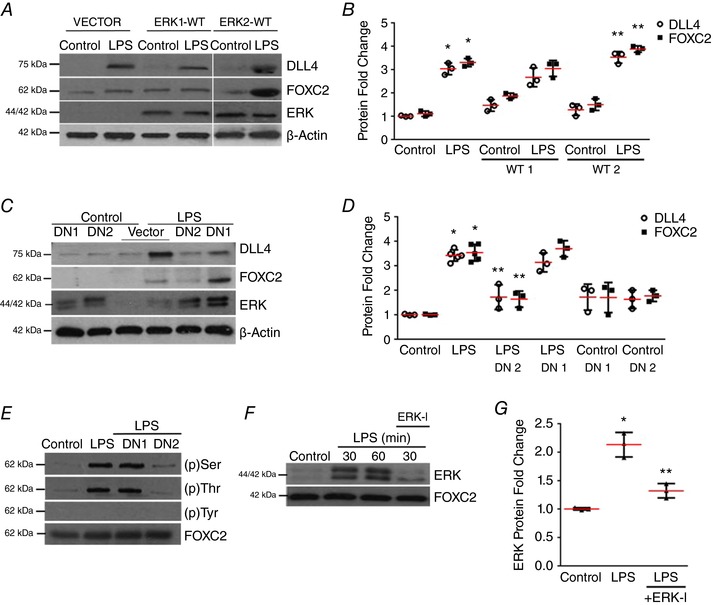
A and B, DLL4, FOXC2 and ERK protein were assessed by immunoblotting 24 h after LPS treatment in vector, wild‐type ERK1 or ERK2 plasmid (ERK1‐WT, ERK2‐WT) transfected HPMECs, and quantified by densitometry (B), P < 0.01 (*Control vs. LPS, **LPS vs. ERK2‐WT + LPS), n ≥ 3. C, DLL4, FOXC2, and ERK protein were assessed by immunoblotting 24 h after LPS treatment in vector, ERK1‐DN (DN1) or ERK2‐DN (DN2) plasmid transfected HPMECs, and quantified by densitometry (D), P < 0.001 (*Control vs. LPS; **LPS vs. ERK2‐DN + LPS), n = 3. E, FOXC2 was immunoprecipitated 30 min after LPS treatment in cells transfected with control, ERK1‐DN, ERK2‐DN plasmid with serine, threonine and tyrosine FOXC2 phosphorylation quantified, n = 3. F and G, ERK binding to FOXC2 was examined in FOXC2 immunoprecipitates obtained at 30 and 60 min after ERK‐I and LPS treatment, and quantified by densitometry (G), P < 0.01 (*Control vs. LPS; ** LPS vs. ERK‐I + LPS), n = 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
To define the mechanisms by which LPS activates FOXC2 we targeted the extracellular signal‐related kinase (ERK). LPS induced ERK phosphorylation (Thr202/Tyr204), required for its activation, in HPMECs at 15 and 30 min (Fig. 3 A). Inhibition of ERK with a specific ERK‐inhibitor (ERK‐I, U0126, 50 μm) suppressed LPS‐mediated DLL4 and FOXC2 expression (Fig. 3 A–C) in HPMECs (Duan et al. 2004; Chialda et al. 2005; Clark et al. 2010). To ascertain whether ERK inhibition did not suppress DLL4 expression by inducing cell death, we examined the effect of ERK‐I on HPMEC apoptosis. ERK‐I (50 μm) inhibited the LPS‐mediated increase in Trypan Blue staining and CC3 expression in HPMECs (Fig. 3 D and E). Co‐immunoprecipitation studies showed that ERK inhibition suppressed LPS‐induced FOXC2 serine and threonine phosphorylation, indicating that ERK mediates FOXC2 activation in HPMECs (Fig. 3 F and G). To determine whether ERK mediates FOXC2 binding to the DLL4 promoter we performed ChIP studies. ERK inhibition suppressed FOXC2 binding to the DLL4 promoter after LPS (Fig. 3 H). As we noted that LPS induced FOXC2 transcriptional activity and FOXC2 expression in HPMECs, we investigated whether FOXC2 regulates its own expression. We expressed FOXC2‐CA (FOXC2 DNA binding domain fused with a synthetic peptide) to evaluate FOXC2 auto‐regulation (Gerin et al. 2009). FOXC2‐CA transfected cells had increased expression of FOXC2 and DLL4 protein verifying that FOXC2 induces its own expression (Fig. 3 I and J). These data reveal that ERK‐inhibition suppresses LPS‐mediated FOXC2 phosphorylation, FOXC2 transcriptional activation, and DLL4 expression in lung EC.
To identify which isoform of ERK, i.e. ERK1 or ERK2, mediates LPS‐dependent DLL4 expression in HPMECs, and to confirm the specificity of the results obtained with ERK‐I, we cloned the wild‐type (WT) ERK1 and ERK2 (ERK1‐WT, ERK2‐WT) and dominant‐negative (ERK1‐DN, ERK2‐DN) in a pIRES‐EGFP‐puro plasmid. Cells transfected with ERK2‐WT plasmid, but not cells transfected with ERK1‐WT plasmid, showed an additional increase in LPS‐mediated DLL4 and FOXC2 expression (Fig. 4 A and B). LPS‐induced DLL4 and FOXC2 expression at 24 h was attenuated with ERK2‐DN, but not ERK1‐DN (Fig. 4 C and D) in HPMECs. Consistent with the above results, ERK2‐DN, but not ERK1‐DN, suppressed LPS‐induced FOXC2 serine/threonine phosphorylation (Fig. 4 E). We next probed whether ERK regulation of FOXC2 phosphorylation involves binding of ERK to FOXC2. Co‐immunoprecipitation revealed that ERK bound to FOXC2 after LPS, suggesting a direct role for ERK in regulating FOXC2 phosphorylation (Fig. 4 F and G). Treatment with ERK‐I, which inhibits ERK phosphorylation, suppressed ERK binding to FOXC2 after LPS (Fig. 4 F and G). These studies identify ERK2 as the isoform that mediates LPS‐induced FOXC2 phosphorylation, DLL4 and FOXC2 expression.
ERK regulates LPS‐induced in vitro angiogenesis in HPMECs (Fig. 5)
Figure 5. ERK regulates LPS‐induced in vivo angiogenesis in HPMECs.
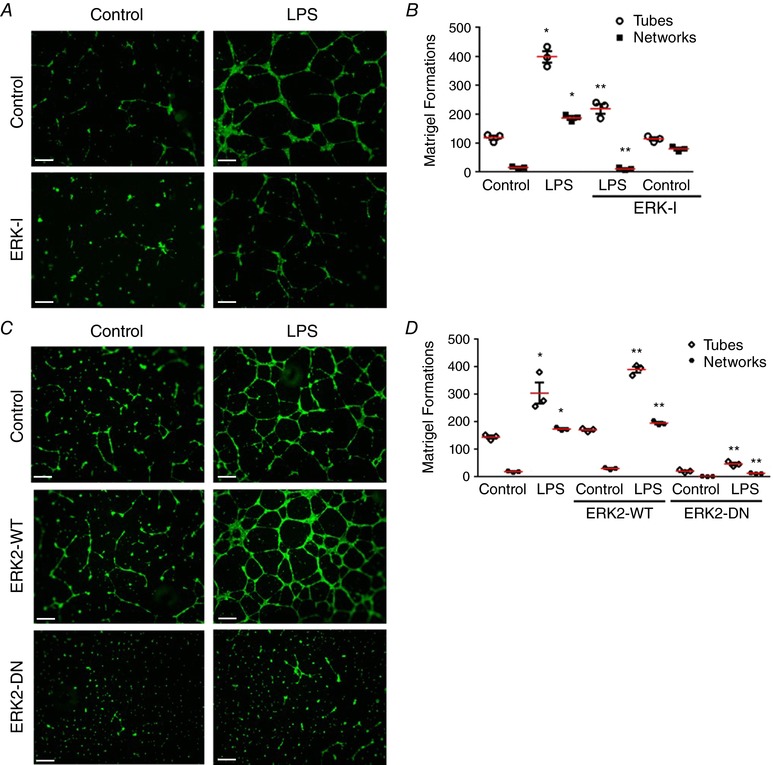
A–D, in vitro angiogenesis was assessed after treatment with LPS, ERK‐I, ERK2‐WT and ERK2‐DN in HPMECs using a Matrigel‐based assay. A and C, fluorescent microscope images depicting angiogenic tube and network formation were captured at 4×. B, graphical representation summarizing data from Fig. 5 A for tube and network formations, P < 0.01 (*Control vs. LPS; **LPS vs. ERK‐I + LPS), n = 3. D, graphical representation summarizing data from Fig. 5 B, P < 0.001 (*Control vs. LPS; **LPS vs. ERK2‐WT+LPS and LPS vs. ERK2‐DN+LPS), n = 3. Scale bar represents 500 μm.
To evaluate the role of ERK‐mediated DLL4 and FOXC2 expression in LPS‐mediated angiogenesis we used a Matrigel‐based in vitro angiogenesis assay (Akhtar et al. 2002). LPS strongly induced formation of angiogenic tubes (number of formations) and networks (tube interconnections) in HPMECs (Fig. 5 A and B). LPS‐induced tube and network formation was suppressed in cells pre‐treated with the ERK inhibitor ERK‐I (Fig. 5 A and B). As our prior studies indicated that ERK2 mediated LPS‐induced FOXC2 and DLL4 expression, we performed angiogenesis experiments with ERK2‐WT and ERK2‐DN. We noted that HPMECs transfected with ERK2‐WT showed a modest increase in tube and network formation at baseline (Fig. 5 C and D). ERK2‐DN suppressed both baseline and LPS‐induced angiogenic tube and network formation (Fig. 5 C and D). These data suggest that ERK2 mediates LPS‐induced in vitro angiogenesis in fetal lung ECs.
ERK regulates FOXC2 activation and DLL4 expression in the mouse whole lung (Fig. 6) and mouse lung ECs in vivo (Fig. 7)
Figure 6. ERK regulates FOXC2 activation and DLL4 expression in the developing lung.
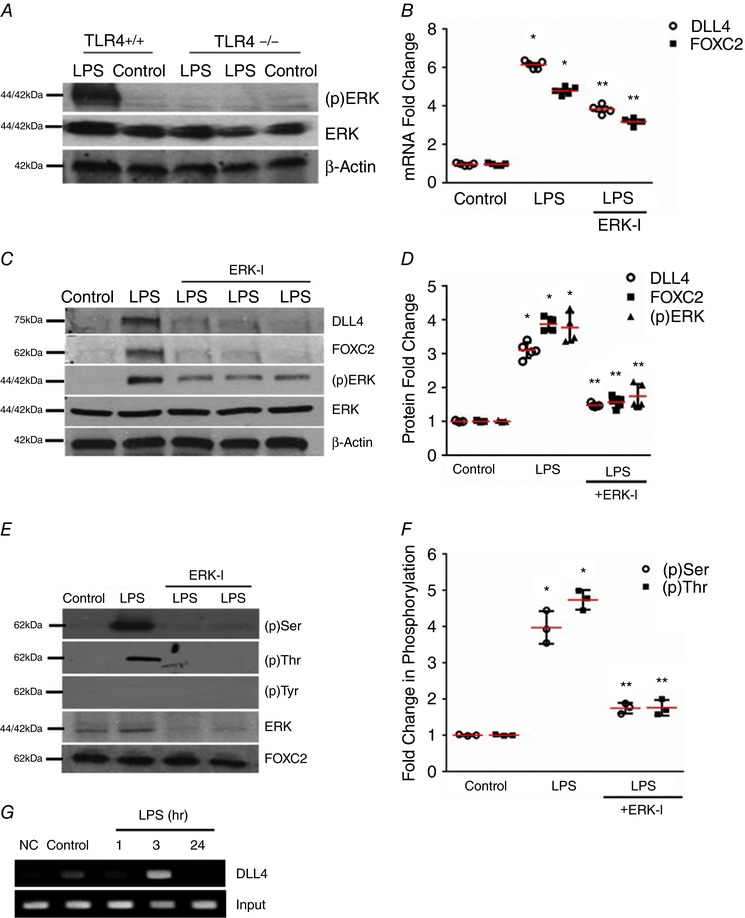
A, ERK phosphorylation was quantified by immunoblotting using an anti‐phospho ERK antibody in lung homogenates obtained from TLR4+/+ and TLR4−/− mice 24 h after i.p. LPS, n ≥ 4 mice in each group. B, lung DLL4 and FOXC2 mRNA were quantified by qRT‐PCR 24 h after i.p. LPS with or without i.p. ERK‐I (20 mg kg−1), P < 0.01 (*Control vs. LPS; **LPS vs. ERK‐I + LPS), n ≥ 3 mice in each group. C and D, DLL4, FOXC2, and (p)ERK were quantified by Western blotting 24 h after i.p. LPS with or without i.p. ERK‐I, with densitometry analysis shown graphically (D), P < 0.05 (*Control vs. LPS; **LPS vs. ERK‐I + LPS), n ≥ 4 mice. E and F, lung FOXC2 phosphorylation was examined by immunoprecipitation 3 h after i.p. LPS with or without ERK‐I, and quantified by densitometry (F), P < 0.001 (*Control vs. LPS: **LPS vs. ERK‐I + LPS), n ≥ 3. G, whole lung nuclear homogenates obtained at 1, 3, and 24 h after i.p. LPS were used for the ChIP assays to quantify binding of FOXC2 to the DLL4 promotor, n ≥ 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 7. ERK regulates FOXC2 activity and DLL4 expression in mouse lung EC in vivo .
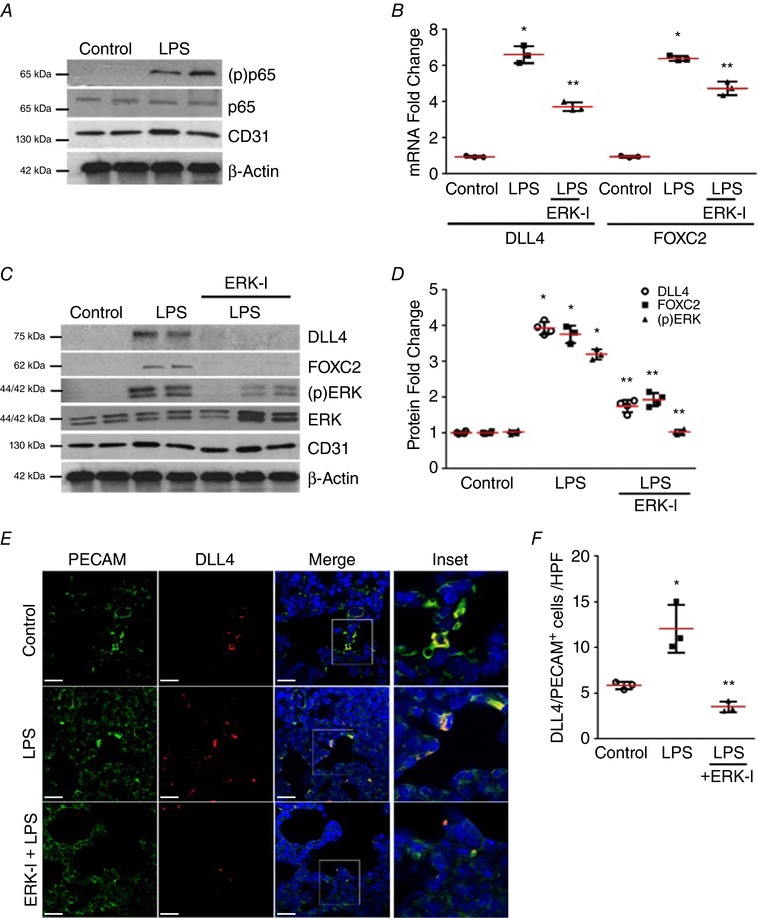
A–D, mouse lung EC isolated from 4‐day old mice 24 h after LPS treatment with or without i.p. 20 mg kg−1 ERK‐I were used. A, phosphorylation of the NF‐κB sub‐unit p65 was assessed in lung EC by Western blotting using an anti‐(p) p65 antibody, n ≥ 4 mice in each group. B, lung EC RNA was used to quantify DLL4 and FOXC2 mRNA by qRT‐PCR, P < 0.01 (*Control vs. LPS: ** LPS vs. ERK‐I + LPS), n = 3. C, DLL4, FOXC2, and (p)ERK were examined in lung EC by Western blotting, and quantified by densitometry (D), P < 0.01 (*Control vs. LPS; **LPS vs. ERK‐I + LPS), n ≥ 3. E and F, mouse lung paraffin sections obtained after LPS and ERK‐I treatments were used for studies. E, confocal microscopy depicting PECAM (green), DLL4 (red), and DAPI (nucleus‐blue) immunofluorescent images. Images were captured at 63×, with the inset being the magnification of the boxed area in the Merge image. F, graph summarizing percentage of PECAM+/DLL4+ cells per high power field (HPF), P < 0.01 (*Control vs. LPS; ** LPS vs. ERK‐I + LPS), n ≥ 3 mice. Scale bar represents 20 μm.
We next evaluated whether activation of innate immune signalling alters EC specification in the developing lung. LPS induced ERK phosphorylation (Thr202/Tyr204) in the whole lung of wild‐type mice, but not in TLR4−/− mice, at 24 h (Fig. 6 A). DLL4 and FOXC2 RNA and protein were strongly induced by LPS in the lung at 24 h (Fig. 6 B and C). ERK‐inhibition with ERK‐I (i.p. 20 mg kg−1) inhibited LPS‐induced ERK phosphorylation, DLL4 and FOXC2 expression in the mouse lung (Fig. 6 B–D). Immunoprecipitation showed that FOXC2 serine and threonine phosphorylation induced by LPS in the lung was suppressed with ERK inhibition (Fig. 6 E and F). Further, ERK binding to FOXC2 after i.p. LPS was attenuated with ERK inhibition (Fig. 6 E). We next examined whether FOXC2 binds to the DLL4 promoter in the lung after systemic LPS challenge. ChIP studies showed binding of FOXC2 to the DLL4 promoter 3 h after LPS (Fig. 6 G). These data suggest that ERK inhibition attenuates LPS‐induced FOXC2 activation, and DLL4/FOXC2 expression in the saccular lung.
To determine the lung EC‐specific effect of ERK treatment on FOXC2 activation and DLL4 expression we performed experiments in mouse lung ECs isolated after i.p. LPS. Activation of canonical TLR4 signalling as evidenced by the phosphorylation of the NF‐kB subunit p65 was verified (Fig. 7 A). LPS induced ERK phosphorylation, DLL4/ FOXC2 RNA, and protein expression in mouse lung EC at 24 h (Fig. 7 B–D). Pre‐treatment with ERK‐I strongly suppressed LPS‐induced ERK phosphorylation, DLL4/FOXC2 RNA, and protein expression (Fig. 7 B–D). We also performed immunofluorescence studies in lung sections to determine the proportion of lung ECs showing DLL4 induction after i.p. LPS. The proportion of PECAM positive cells that were DLL4 positive increased >2.4‐fold with LPS in wild‐type mouse pups, but not in mice pre‐treated with ERK‐I (Fig. 7 E and F). These data demonstrate that activation of TLR4 signalling in lung ECs induces ERK‐dependent DLL4 and FOXC2 expression, known regulators of tip cell EC specification during angiogenesis.
LPS‐induced deviant angiogenesis and DLL4 expression in the retinal vasculature is attenuated with ERK inhibition (Fig. 8)
Figure 8. LPS‐induced stimulation of DLL4 expression during retinal angiogenesis.
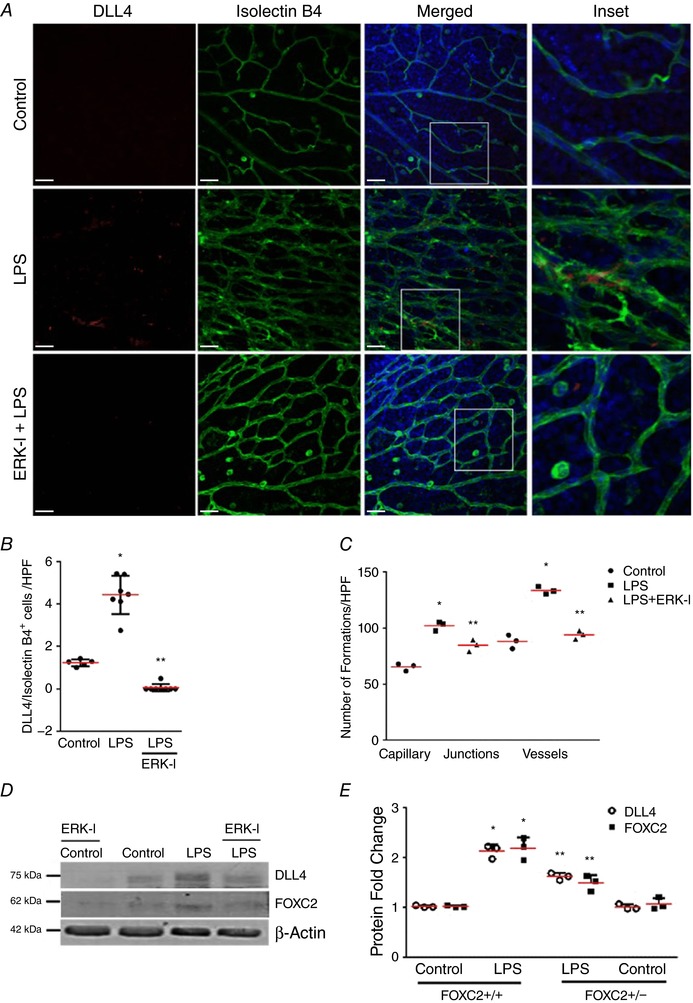
A–C, retinas were extracted from 4‐day‐old mouse pups 24 h following i.p. LPS treatment with or without ERK‐I. A, confocal microscope images of mouse retinas depicting immunofluorescent staining of Isolectin B4 and DLL4 antibody. Isolectin B4 is shown in green, DLL4 is shown in red, and Hoescht (nucleus) is blue. Images were captured at 25×, with the inset being at 63× magnification. B, graph summarizing percentage of DLL4+/IsolectinB4+ cells per high power field (HPF), * P < 0.01 (Control vs. LPS) and ** P < 0.05 (LPS vs. ERK‐I + LPS). C, graph summarizing the number of capillary junctions and vessel count per high power field (HPF), * P < 0.01 (Control vs. LPS) and ** P < 0.05 (LPS vs. ERK‐I + LPS), n ≥ 3 mice in each group. D and E, DLL4 and FOXC2 expression were examined in monkey retinal endothelial cells by immunoblotting lysates 24 h after LPS treatment with or without ERK‐I, with densitometry shown graphically (E), P < 0.01 (*Control vs. LPS; **LPS vs. LPS and ERK‐I), n = 3. Scale bar represents 20 μm.
To examine the effect of endothelial TLR4 signalling on in vivo angiogenesis, we interrogated the developing retinal vasculature, as this accessible vascular bed readily reflects acute angiogenic responses (Lobov et al. 2007; Rabinowitz et al. 2012). Systemic LPS induced a robust angiogenic response in retinal vessels evident as an increase in vessel branching and new vessel formation in the background of vessel leakiness at 24 h (Fig. 8 A–C). We also noted increased retinal DLL4 expression with i.p. LPS. LPS‐induced aberrant retinal angiogenic responses and DLL4 expression was attenuated with ERK inhibition (Fig. 8 A–C). To confirm the key signalling events underlying LPS‐induced angiogenic signalling in retinal endothelial cells, we performed experiments in monkey retinal endothelial cells (MREC, RF/6A). LPS‐induced DLL4 and FOXC2 expression in MREC was suppressed with pre‐treatment by ERK‐I (50 μm) at 24 h (Fig. 8 D and E). These data suggest that the TLR4 agonist LPS induces ERK‐dependent DLL4 expression and neo‐angiogenesis in vivo and in vitro in retinal endothelial cells.
FOXC2 regulates LPS‐induced DLL4 expression and in vitro angiogenesis (Fig. 9)
Figure 9. FOXC2 regulates LPS‐induced DLL4 expression in vivo and in vitro angiogenesis.
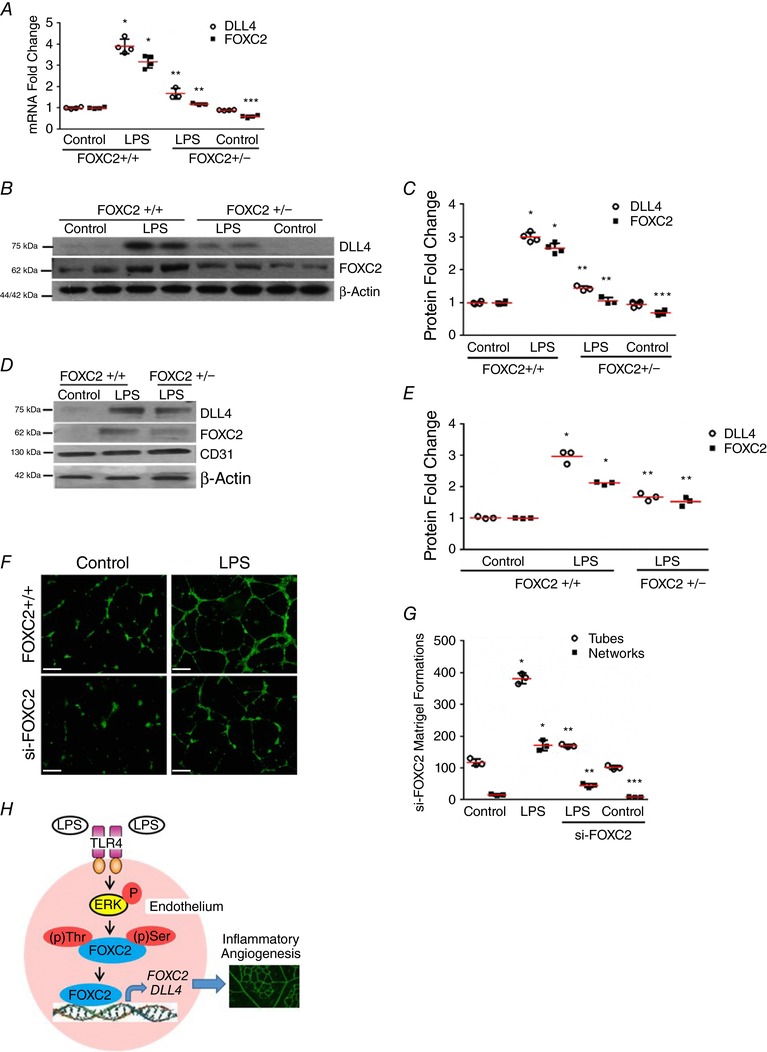
A–C, lung homogenates obtained from 7‐day mice 24 h after i.p. LPS were used. A, lung RNA was used to quantify DLL4 and FOXC2 expression by qRT‐PCR, P < 0.01 (*FOXC2+/+ vs. FOXC2+/− LPS; **FOXC2+/+ LPS vs. FOXC2+/− LPS; ***FOXC2+/+ Control vs. FOXC2+/− Control), n = 4. B and C, DLL4 and FOXC2 protein were quantified 24 h after LPS treatment, with densitometric quantification shown (C), P < 0.05 (*FOXC2+/+ vs. FOXC2+/+‐ LPS; **FOXC2+/+ LPS vs. FOXC2+/− LPS; ***FOXC2+/+ Control vs. FOXC2+/− Control), n = 4 mice/group. D and E, lung endothelial cells isolated from 7‐day mice 24 h after i.p. LPS were used. D, DLL4 and FOXC2 protein were assessed by immunoblotting, with densitometry quantification shown (E), P < 0.05 (*FOXC2+/+ vs. FOXC2+/+‐ LPS; **FOXC2+/+ LPS vs. FOXC2+/− LPS), n = 3 mice/group. F and G, in vitro angiogenesis was assessed 12 h after LPS in HPMECs treated with si‐FOXC2 or scrambled siRNA. F, fluorescent microscope images depicting angiogenic tube and network formation in Matrigel (4× magnification). G, graphical summary of data for tube and network formations, P < 0.01 (*Control vs. LPS; **LPS vs. siFOXC2 + LPS; ***Control vs. siFOXC2 Control), n = 3. H, cartoon depicting the proposed mechanism underlying LPS‐mediated angiogenesis through endothelial TLR4‐ERK‐FOXC2‐DLL4 signalling in lung EC. Scale bar represents 500 μm.
To determine whether LPS‐induced DLL4 expression is FOXC2 dependent in vivo, we performed experiments in FOXC2+/− mice, as FOXC2−/− mice exhibit embryonic lethality (Iida et al. 1997; Kume, 2009). LPS‐induced lung DLL4 RNA and protein expression was attenuated in FOXC2+/− mice compared to litter‐mate FOXC2+/+ mouse pups (Fig. 9 A–C). We also observed a muted increase in FOXC2 expression after LPS treatment in FOXC2+/− mice. To confirm the role of endothelial FOXC2 in regulating LPS‐induced angiogenic signalling in vivo, we performed studies in lung ECs isolated from FOXC2+/− mice. LPS‐induced FOXC2 and DLL4 expression in lung ECs was attenuated in FOXC2+/− mice (Fig. 9 D and E). To evaluate the role of FOXC2 in regulating LPS‐induced angiogenesis we performed an in vitro angiogenesis assay (Akhtar et al. 2002) in fetal HPMECs. LPS‐induced tube and network formation was abrogated in HPMECs treated with siFOXC2 but not control siRNA (Fig. 9 F and G). These data demonstrate that endothelial FOXC2 regulates LPS‐mediated DLL4 expression in the developing mouse lung, and is required for in vitro angiogenic responses to LPS.
Discussion
Activation of immune signalling in EC is associated with deviant angiogenesis complicating various diseases, and yet the mechanisms remain unclear (Thebaud & Abman, 2007; Usman et al. 2015). In this study, we reveal the mechanisms by which bacteria can directly programme EC phenotypic changes that contribute to sepsis‐induced inflammatory angiogenesis. The major finding of this study is that endothelial TLR4 signalling induces expression and transcriptional activation of FOXC2, in an ERK‐dependent manner. EC TLR4‐ERK‐FOXC2 activation signals expression of the tip cell EC specification marker, DLL4, and inflammatory angiogenesis (Fig. 9 H). Interestingly, we noted that FOXC2 self‐regulates its expression in human lung endothelial cells in vitro during EC immune activation. Our studies suggest that TLR4‐dependent, ERK‐mediated activation of FOXC2 and DLL4 is conserved in human and mouse lung ECs, as well as monkey retinal ECs, and identify ERK2 as the isoform that regulates this conserved pathway in human lung ECs. These data uncover a novel signalling mechanism by which EC TLR signalling regulates cell‐fate decisions crucial for sprouting angiogenesis.
We targeted ERK as it is known to regulate vascular development and is activated by TLR signalling (Takahashi et al. 2001; Akira, 2006; Fatima et al. 2016). Using in vitro and in vivo studies we show that ERK inhibition suppresses TLR4‐mediated ERK‐FOXC2 binding, FOXC2 phosphorylation, FOXC2 binding to the DLL4 promoter, and DLL4 expression in mouse and human lung ECs. We identified ERK2 as the isoform that mediates TLR4‐dependent FOXC2 and DLL4 expression in human lung ECs. While we reveal cross‐talk between ERK and FOXC2 in the context of EC TLR4 signalling, other investigators have reported interactions between ERK and FOXC2 in the context of developmental signalling (Hayashi & Kume, 2008; Kume, 2009; Fatima et al. 2016). Hayashi & Kume (2008) showed that chemical inhibition of ERK promoted VEGF‐A‐dependent FOXC2 transcriptional activity in mouse lung ECs. In contrast, our data suggest that ERK positively regulates FOXC2‐transcriptional activity. Studies with the ERK wild‐type and dominant negative plasmids confirmed our ERK inhibitor studies demonstrating that ERK2 mediates FOXC2 activation in HPMECs. Differences in signalling cascades activated by VEGF‐A and TLR4 may have contributed to discordant results seen with ERK inhibition on FOXC2 activation. Our results are, however, consistent with a study in zebrafish, which showed that ERK activation in dorsal angioblasts causes Notch activation via the transcriptional factors FOXC1/2 (Lawson et al. 2002). Interestingly, LPS‐mediated, ERK‐dependent FOXC2 and DLL4 expression was apparent in human fetal lung ECs, mouse lung and retinal ECs, and monkey retinal ECs, suggesting conservation of TLR4‐ERK‐FOXC2‐DLL4 signalling. Importantly, ERK‐I did not induce HPMEC apoptosis, suggesting its effects on suppressing DLL4 expression and in vitro angiogenesis were not associated with increased cell death. Another facet of ERK‐FOXC2 interactions was reported by Fatima et al. (2016), who noted hyper‐activation of ERK in lymphatic endothelial cells in mice with a combined deletion of FOXC1 and FOXC2. Whether ERK hyper‐activation was a compensatory response to inactivation of FOXC transcription factors or whether FOXC2 suppresses ERK activation was not delineated in this study. While previous work has shown that ERK‐FOXC2 signalling regulates developmental processes across species, our data support a non‐developmental role for this pathway during TLR4‐mediated lung EC immune activation.
FOXC2 plays important roles in lymphangiogenesis and vascular development (Hayashi & Kume, 2008; Kume, 2009; Fatima et al. 2016), but its role in endothelial innate immune signalling is unknown. Our data reveal a novel link between endothelial TLR4 signalling and angiogenic signalling through induction of FOXC2 transcriptional activation and FOXC2 expression. Studies in HPMECs and in the mouse lung ECs demonstrate that ERK phosphorylates FOXC2, resulting in transcriptional activation of FOXC2 and expression of its downstream target, DLL4. Our results are consistent with those of Ivanov et al. (2013), who showed that FOXC2 phosphorylation‐deficient mutants lose their ability to induce vascular remodelling. FOXC2 regulation of DLL4 was initially described by Hayashi & Kume (2008), who showed that VEGF‐dependent DLL4 expression was augmented by over‐expression of FOXC1 or FOXC2, and FOXC1 and FOXC2 bound to the DLL4 promoter. ChIP analysis revealing that FOXC2 constitutively binds to the DLL4 promoter at baseline, and increases after LPS in HPMECs and in the lung, is consistent with the above study. In contrast with their study, our data indicate a non‐developmental role for FOXC2 in regulating DLL4 expression during EC TLR4 signalling. Interestingly, we also noted that FOXC2 expression increased in association with FOXC2 phosphorylation after LPS in an ERK‐dependent manner. Studies with FOXC2‐CA, consisting of VP16 fused to the nuclear binding domain of FOXC2, revealed that FOXC2 regulates its own expression and further confirmed FOXC2 regulation of DLL4 (Gerin et al. 2009). Previously, Notch1 and MiR‐520h have been shown to induce FOXC2 expression in the context of haematopoiesis and lung cancer suppression, but FOXC2 auto‐regulation has not been shown before (Yu et al. 2013; Jang et al. 2015). While the mechanisms and ramifications of FOXC2 auto‐regulation during EC immune signalling need to be determined, our data reveal that EC TLR signalling activates a key transcription factor regulating embryonic vascular development and EC specification.
DLL4, an EC Notch ligand, regulates tip versus stalk cell endothelial specification during angiogenesis (Eilken & Adams, 2010). We examined DLL4 in EC TLR4 signalling as a FOXC2‐regulated target gene that directs sprouting angiogenesis. Our data show that DLL4 RNA expression was induced early after LPS in vitro and in vivo (3 and 6 h), and LPS‐induced DLL4 protein was suppressed in the TLR4−/− and FOXC2+/− mouse lung. Further, treatment with ERK2‐DN or ERK‐I suppressed LPS‐induced DLL4 expression in conjunction with decreased binding of FOXC2 to DLL4 promoter, demonstrating that the TLR4‐ERK‐FOXC2 axis regulates inflammatory DLL4 expression. Fung et al. (2007) showed that LPS‐induced DLL4 expression in macrophages resulted in activation of Notch signalling but the underlying mechanisms were not elucidated. Recent work from our group has shown that systemic LPS induces lung DLL4 expression but neither the mechanisms nor the endothelial specificity of this response was shown (Li et al. 2015). Studies to evaluate the role of FOXC2 in TLR4‐mediated angiogenesis in vitro showed abrogation of LPS‐mediated angiogenic sprouting in cells treated with FOXC2‐siRNA (Akhtar et al. 2002; Menden et al. 2015). While these data are consistent with Pollet et al. (2003), who showed that LPS induces angiogenesis in dermal ECs, we identify FOXC2 as the mediator of TLR4‐dependent DLL4 expression and angiogenic responses. Further, unlike Sun et al. (2016), who suggested that LPS indirectly induces angiogenesis in pancreatic carcinoma through macrophage‐dependent VEGF signalling, our results reveal an intrinsic TLR4‐ERK‐FOXC2 axis in ECs that regulates angiogenesis. Although the focus of our study was lung ECs, we also performed experiments in the retina, as postnatal retinal microvascular arborization serves as a sensitive window to examine acute angiogenic changes (Lobov et al. 2007; Rabinowitz et al. 2012). These studies suggest that lung and retinal ECs are phenotypically similar with respect to LPS‐ERK‐DLL4 signalling. Interestingly, noxious insults like hyperoxia or systemic sepsis in premature babies injure the developing retinal and lung ECs, disrupting vascular development, suggesting phenotypic congruence in inflammatory angiogenic signalling (Coalson, 2006; Thomas et al. 2015; Choi et al. 2017). We discovered that systemic LPS mediated ERK‐dependent increase in angiogenic sprouting and new vessel formation in association with increased DLL4. In conjunction with our data showing ERK‐dependent, LPS‐mediated increases in DLL4+/PECAM+ cells in the developing lung, these results suggest that TLR4‐ERK‐FOXC2 signalling is a conserved, tightly regulated pathway that mediates TLR4‐induced angiogenesis and DLL4 expression in lung ECs.
In summary, we reveal that lung EC TLR4 signalling activates ERK‐FOXC2‐DLL4 signalling in human lung ECs in vitro and mouse lung ECs in a neonatal model of systemic sepsis. To the best of our knowledge, this is the first study to demonstrate that EC TLR4 signalling regulates FOXC2 activation, resulting in increased expression of FOXC2 and its downstream target, DLL4. Our data suggest an unrecognized role of EC immune signalling in activating conserved pathways known to regulate EC specification, angiogenesis and embryonic vascular development (Kume, 2009; Eilken & Adams, 2010; Fatima et al. 2016). Studies from various investigators have suggested that endothelial injury in the developing lung contributes to abnormal vascular development complicating chronic lung disease in premature infants (Abman, 2001; De Paepe et al. 2006). In the context of these studies, our data suggest that sepsis‐induced FOXC2 and DLL4 activation in lung ECs might contribute to deviant angiogenesis and dysmorphic microvascular development in bronchopulmonary dysplasia. The ramifications of EC‐centric TLR‐ERK‐FOXC2‐DLL4‐activation for human diseases such as rheumatoid arthritis and atherosclerosis characterized by abnormal inflammatory angiogenesis are points of broader interest to the research community (Usman et al. 2015; Tas et al. 2016).
Additional information
Competing interests
The authors have no competing interests to disclose.
Author contributions
Conception and design: V.S., S.X. and H.M. Data collection: H.M. and S.X. Analysis and interpretation: V.S., H.M., S.X., T.R.K. and T.K. Drafting and editing the manuscript: V.S., H.M., S.X., T.R.K. and T.K. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
H.M., S.X. and V.S. were supported by 1R01HL128374‐01 (V.S.).
Acknowledgements
The authors thank Ramani Ramchandran, PhD and Stephanie Cossette, PhD from Medical College of Wisconsin for their scientific discussion and insight on the delta like 4 staining protocol in the lung sections.
Biographies
Sheng Xia studied in his native China and then abroad for most of his training as a PhD scientist, before settling down in Kansas City, Missouri to pursue life as a research scientist. Throughout Sheng's career, he has focused on TLR signalling. A highlight of Sheng's career has been receiving fellowship awards while at the Karolinska Institute from EMBO and IBRO.

Heather L. Menden previously studied and researched in Wisconsin until relocating to Kansas City, Missouri to continue her work with TLR signalling and angiogenesis. A highlight of Heather's career has been giving a platform presentation at the Pediatric Academic Societies meeting in San Francisco.
S. Xia and H. L. Menden contributed equally to this work.
Edited by: Kim Barrett & Fernando Santana
References
- Abman SH (2001). Bronchopulmonary dysplasia: “a vascular hypothesis.” Am J Respir Crit Care Med 164, 1755–1756. [DOI] [PubMed] [Google Scholar]
- Akhtar N, Dickerson EB & Auerbach R (2002). The sponge/Matrigel angiogenesis assay. Angiogenesis 5, 75–80. [DOI] [PubMed] [Google Scholar]
- Akira S (2006). TLR signaling. Curr Top Microbiol Immunol 311, 1–16. [DOI] [PubMed] [Google Scholar]
- Andonegui G, Bonder CS, Green F, Mullaly SC, Zbytnuik L, Raharjo E & Kubes P (2003). Endothelium‐derived Toll‐like receptor‐4 is the key molecule in LPS‐induced neutrophil sequestration into lungs. J Clin Invest 111, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andonegui G, Zhou H, Bullard D, Kelly MM, Mullaly SC, McDonald B, Long EM, Robbins SM & Kubes P (2009). Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram‐negative bacterial infection. J Clin Invest 119, 1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M & Roux PP (2011). Activation and function of the MAPKs and their substrates, the MAPK‐activated protein kinases. Microbiol Mol Biol Rev 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chialda L, Zhang M, Brune K & Pahl A (2005). Inhibitors of mitogen‐activated protein kinases differentially regulate costimulated T cell cytokine production and mouse airway eosinophilia. Respir Res 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y‐B, Lee J, Park J & Jun YH (2017). Impact of prolonged mechanical ventilation in very low birth weight infants: results from a national cohort study. J Pediatr 194, 34–39.e3. [DOI] [PubMed] [Google Scholar]
- Clark JS, Faisal A, Baliga R, Nagamine Y & Arany I (2010). Cisplatin induces apoptosis through the ERK‐p66shc pathway in renal proximal tubule cells. Cancer Lett 297, 165–170. [DOI] [PubMed] [Google Scholar]
- Coalson JJ (2006). Pathology of bronchopulmonary dysplasia. Semin Perinatol 30, 179–184. [DOI] [PubMed] [Google Scholar]
- Dauphinee SM & Karsan A (2006). Lipopolysaccharide signaling in endothelial cells. Lab Investig J Tech Methods Pathol 86, 9–22. [DOI] [PubMed] [Google Scholar]
- De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, Dixon M & Gundogan F (2006). Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med 173, 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Chan JHP, Wong CH, Leung BP & Wong WSF (2004). Anti‐inflammatory effects of mitogen‐activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J Immunol Baltim Md 1950 172, 7053–7059. [DOI] [PubMed] [Google Scholar]
- Eilken HM & Adams RH (2010). Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol 22, 617–625. [DOI] [PubMed] [Google Scholar]
- Fang J, Dagenais SL, Erickson RP, Arlt MF, Glynn MW, Gorski JL, Seaver LH & Glover TW (2000). Mutations in FOXC2 (MFH‐1), a forkhead family transcription factor, are responsible for the hereditary lymphedema‐distichiasis syndrome. Am J Hum Genet 67, 1382–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatima A, Wang Y, Uchida Y, Norden P, Liu T, Culver A, Dietz WH, Culver F, Millay M, Mukouyama Y‐S & Kume T (2016). Foxc1 and Foxc2 deletion causes abnormal lymphangiogenesis and correlates with ERK hyperactivation. J Clin Invest 126, 2437–2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung E, Tang S‐MT, Canner JP, Morishige K, Arboleda‐Velasquez JF, Cardoso AA, Carlesso N, Aster JC & Aikawa M (2007). Delta‐like 4 induces Notch signaling in macrophages: implications for inflammation. Circulation 115, 2948–2956. [DOI] [PubMed] [Google Scholar]
- Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G & Yancopoulos GD (2004). Haploinsufficiency of delta‐like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA 101, 15949–15954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerin I, Bommer GT, Lidell ME, Cederberg A, Enerback S & Macdougald OA (2009). On the role of FOX transcription factors in adipocyte differentiation and insulin‐stimulated glucose uptake. J Biol Chem 284, 10755–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H & Kume T (2008). Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF‐Notch signaling pathways in endothelial cells. PloS One 3, e2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida K, Koseki H, Kakinuma H, Kato N, Mizutani‐Koseki Y, Ohuchi H, Yoshioka H, Noji S, Kawamura K, Kataoka Y, Ueno F, Taniguchi M, Yoshida N, Sugiyama T & Miura N (1997). Essential roles of the winged helix transcription factor MFH‐1 in aortic arch patterning and skeletogenesis. Development 124, 4627–4638. [DOI] [PubMed] [Google Scholar]
- Ivanov KI, Agalarov Y, Valmu L, Samuilova O, Liebl J, Houhou N, Maby‐El Hajjami H, Norrmén C, Jaquet M, Miura N, Zangger N, Ylä‐Herttuala S, Delorenzi M & Petrova TV (2013). Phosphorylation regulates FOXC2‐mediated transcription in lymphatic endothelial cells. Mol Cell Biol 33, 3749–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang IH, Lu Y‐F, Zhao L, Wenzel PL, Kume T, Datta SM, Arora N, Guiu J, Lagha M, Kim PG, Do EK, Kim JH, Schlaeger TM, Zon LI, Bigas A, Burns CE & Daley GQ (2015). Notch1 acts via Foxc2 to promote definitive hematopoiesis via effects on hemogenic endothelium. Blood 125, 1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T (2009). The cooperative roles of Foxc1 and Foxc2 in cardiovascular development. Adv Exp Med Biol 665, 63–77. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Vogel AM & Weinstein BM (2002). sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3, 127–136. [DOI] [PubMed] [Google Scholar]
- Li K, Chowdhury T, Vakeel P, Koceja C, Sampath V & Ramchandran R (2015). Delta‐like 4 mRNA is regulated by adjacent natural antisense transcripts. Vasc Cell 7, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD & Wiegand SJ (2007). Delta‐like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104, 3219–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Virtue A, Shen J, Wang H & Yang X‐F (2013). An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol OncolJ Hematol Oncol 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis NA & Orfanos SE (2008). The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care 14, 22–30. [DOI] [PubMed] [Google Scholar]
- Mattsby‐Baltzer I, Jakobsson A, Sorbo J & Norrby K (1994). Endotoxin is angiogenic. Int J Exp Pathol 75, 191–196. [PMC free article] [PubMed] [Google Scholar]
- Mavria G, Vercoulen Y, Yeo M, Paterson H, Karasarides M, Marais R, Bird D & Marshall CJ (2006). ERK‐MAPK signaling opposes Rho‐kinase to promote endothelial cell survival and sprouting during angiogenesis. Cancer Cell 9, 33–44. [DOI] [PubMed] [Google Scholar]
- Menden H, Tate E, Hogg N & Sampath V (2013). LPS mediated endothelial activation in pulmonary endothelial cells; role of Nox2‐dependent IKK‐β phosphorylation. Am J Physiol Lung Cell Mol Physiol 304, L445–L455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menden H, Welak S, Cossette S, Ramchandran R & Sampath V (2015). LPS‐mediated Angiopoietin‐2 dependent autocrine angiogenesis is regulated by Nox2 in human pulmonary microvascular endothelial cells. J Biol Chem 290, 5449–5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DA, Makonnen S, Lassoued W, Feldman MD, Carter C & Lee WMF (2006). Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43‐9006). Am J Pathol 169, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollet I, Opina CJ, Zimmerman C, Leong KG, Wong F & Karsan A (2003). Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6‐mediated activation of NF‐kappaB and c‐Jun N‐terminal kinase. Blood 102, 1740–1742. [DOI] [PubMed] [Google Scholar]
- Rabinowitz R, Priel A, Rosner M, Pri‐Chen S & Spierer A (2012). Avastin treatment reduces retinal neovascularization in a mouse model of retinopathy of prematurity. Curr Eye Res 37, 624–629. [DOI] [PubMed] [Google Scholar]
- Ridgway J, Zhang G, Wu Y, Stawicki S, Liang W‐C, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo J‐AS, de Sauvage F, Plowman G & Yan M (2006). Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444, 1083–1087. [DOI] [PubMed] [Google Scholar]
- Ryan US (1986). Pulmonary endothelium: a dynamic interface. Clin Investig Med Med Clin Exp 9, 124–132. [PubMed] [Google Scholar]
- Scholl FA, Dumesic PA, Barragan DI, Harada K, Bissonauth V, Charron J & Khavari PA (2007). Mek1/2 MAPK kinases are essential for Mammalian development, homeostasis, and Raf‐induced hyperplasia. Dev Cell 12, 615–629. [DOI] [PubMed] [Google Scholar]
- Seo S, Fujita H, Nakano A, Kang M, Duarte A & Kume T (2006). The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol 294, 458–470. [DOI] [PubMed] [Google Scholar]
- Shin M, Beane TJ, Quillien A, Male I, Zhu LJ & Lawson ND (2016). Vegfa signals through ERK to promote angiogenesis, but not artery differentiation. Development 143, 3796–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczak M, Dargatz J & Chrzanowska‐Wodnicka M (2010). Isolation and culture of pulmonary endothelial cells from neonatal mice. J Vis Exp 46, 2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wu C, Ma J, Yang Y, Man X, Wu H & Li S (2016). Toll‐like receptor 4 promotes angiogenesis in pancreatic cancer via PI3K/AKT signaling. Exp Cell Res 347, 274–282. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Yamaguchi S, Chida K & Shibuya M (2001). A single autophosphorylation site on KDR/Flk‐1 is essential for VEGF‐A‐dependent activation of PLC‐gamma and DNA synthesis in vascular endothelial cells. EMBO J 20, 2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tas SW, Maracle CX, Balogh E & Szekanecz Z (2016). Targeting of proangiogenic signalling pathways in chronic inflammation. Nat Rev Rheumatol 12, 111–122. [DOI] [PubMed] [Google Scholar]
- Thebaud B & Abman SH (2007). Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175, 978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas K, Shah PS, Canning R, Harrison A, Lee SK & Dow KE (2015). Retinopathy of prematurity: risk factors and variability in Canadian neonatal intensive care units. J Neonatal‐Perinat Med 8, 207–214. [DOI] [PubMed] [Google Scholar]
- Tual‐Chalot S, Allinson KR, Fruttiger M & Arthur HM (2013). Whole mount immunofluorescent staining of the neonatal mouse retina to investigate angiogenesis in vivo. J Vis Exp 77, 50546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usman A, Ribatti D, Sadat U & Gillard JH (2015). From lipid retention to immune‐mediate inflammation and associated angiogenesis in the pathogenesis of atherosclerosis. J Atheroscler Thromb 22, 739–749. [DOI] [PubMed] [Google Scholar]
- Yu P, Tung JK & Simons M (2014). Lymphatic fate specification: an ERK‐controlled transcriptional programme. Microvasc Res 96, 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y‐H, Chen HA, Chen PS, Cheng YJ, Hsu WH, Chang YW, Chen YH, Jan Y, Hsiao M, Chang TY, Liu YH, Jeng YM, Wu CH, Huang MT, Su YH, Hung MC, Chien MH, Chen CY, Kuo ML, Su JL (2013). MiR‐520h‐mediated FOXC2 regulation is critical for inhibition of lung cancer progression by resveratrol. Oncogene 32, 431–443. [DOI] [PubMed] [Google Scholar]