

Abstract
Key points
The current theory behind matching blood flow to metabolic demand of skeletal muscle suggests redundant interactions between metabolic vasodilators.
Capillaries play an important role in blood flow control given their ability to respond to muscle contraction by causing conducted vasodilatation in upstream arterioles that control their perfusion.
We sought to determine whether redundancies occur between vasodilators at the level of the capillary by stimulating the capillaries with muscle contraction and vasodilators relevant to muscle contraction.
We identified redundancies between potassium and both adenosine and nitric oxide, between nitric oxide and potassium, and between adenosine and both potassium and nitric oxide. During muscle contraction, we demonstrate redundancies between potassium and nitric oxide as well as between potassium and adenosine.
Our data show that redundancy is physiologically relevant and involved in the coordination of the vasodilator response during muscle contraction at the level of the capillaries.
Abstract
We sought to determine if redundancy between vasodilators is physiologically relevant during active hyperaemia. As inhibitory interactions between vasodilators are indicative of redundancy, we tested whether vasodilators implicated in mediating active hyperaemia (potassium (K+), adenosine (ADO) and nitric oxide (NO)) inhibit one another's vasodilatory effects through direct application of pharmacological agents and during muscle contraction. Using the hamster cremaster muscle and intravital microscopy, we locally stimulated capillaries with one vasodilator in the absence and the presence of a second vasodilator (10−7 m S‐nitroso‐N‐acetylpenicillamine (SNAP), 10−7 m ADO, 10 mm KCl) applied sequentially and simultaneously, and observed the response in the associated upstream 4A arteriole controlling the perfusion of the stimulated capillary. We found that KCl significantly attenuated SNAP‐ and ADO‐induced vasodilatations by ∼49.7% and ∼128.0% respectively and ADO significantly attenuated KCl‐ and SNAP‐induced vasodilatations by ∼94.7% and ∼59.6%, respectively. NO significantly attenuated KCl vasodilatation by 93.8%. Further, during muscle contraction we found that inhibition of NO production using l‐N G‐nitroarginine methyl ester and inhibition of ADO receptors using xanthine amine congener was effective at inhibiting contraction‐induced vasodilatation but only in the presence of K+ release channel inhibition. Thus, only when the inhibiting vasodilator K+ was blocked was the second vasodilator, NO or ADO, able to produce effective vasodilatation. Therefore, we show that there are inhibitory interactions between specific vasodilators at the level of the capillary. Further, these inhibitions can be observed during muscle contraction indicating that redundancies between vasodilators are physiologically relevant and influence vasodilatation during active hyperaemia.
Keywords: redundancy, vasodilatation, skeletal muscle contraction, capillary
Key points
The current theory behind matching blood flow to metabolic demand of skeletal muscle suggests redundant interactions between metabolic vasodilators.
Capillaries play an important role in blood flow control given their ability to respond to muscle contraction by causing conducted vasodilatation in upstream arterioles that control their perfusion.
We sought to determine whether redundancies occur between vasodilators at the level of the capillary by stimulating the capillaries with muscle contraction and vasodilators relevant to muscle contraction.
We identified redundancies between potassium and both adenosine and nitric oxide, between nitric oxide and potassium, and between adenosine and both potassium and nitric oxide. During muscle contraction, we demonstrate redundancies between potassium and nitric oxide as well as between potassium and adenosine.
Our data show that redundancy is physiologically relevant and involved in the coordination of the vasodilator response during muscle contraction at the level of the capillaries.
Introduction
Skeletal muscle active hyperaemia describes the phenomenon whereby an increase in blood flow is recruited to the tissue during periods of heightened metabolism ensuring tissue oxygen demand and oxygen delivery are adequately matched. The locus of blood flow control during muscle contraction has been placed locally within the muscle tissue itself. Active skeletal muscle fibres signal to their supplying microvessels through production of vasodilators, which act directly on the local microvasculature to increase blood flow. This metabolic hypothesis has been extensively studied and although many important communicating molecules have been identified in mediating the active hyperaemic response, none have been found to be obligatory for facilitation of the process. Current thinking has shifted to a more integrated control paradigm in which groups of communicating molecules interact and influence one another (Boushel et al. 2002; Schrage et al. 2004; Mortensen et al. 2007; Markwald et al. 2011; Casey et al. 2013), leading to the hypothesis that these interactions create redundancies between vasodilators (Chilian & Koshida, 2001; Rowell, 2004; Joyner & Wilkins, 2007; Hellsten et al. 2012; Lamb & Murrant, 2015).
Redundancy, in the context of active hyperaemia, refers to one vasodilator being able to compensate for the loss of another. Recently, we have provided a proof‐in‐principle clearly demonstrating that vasodilators relevant to muscle contraction (adenosine (ADO), nitric oxide (NO) and potassium (K+)) interact at the arteriolar level, whereby K+ was shown to inhibit NO‐ and ADO‐induced vasodilatations and ADO inhibited NO‐induced vasodilatations (Lamb & Murrant, 2015). Inhibition between vasodilators provides a mechanism for redundancies between vasodilators whereby one vasodilator can take the place of another. In order to further understand and contextualize the physiological relevance of redundancy, we aimed to determine whether inhibition between vasodilators occur during skeletal muscle contraction. Given the potential importance of capillaries in the distribution of blood flow during active hyperaemia (Murrant et al. 2016), we would expect redundancy between vasodilators to occur at the level of the capillary if this signalling paradigm is, in fact, physiologically relevant; however, whether redundancy occurs between vasodilators at the level of the capillary is not known.
Therefore, we sought to establish whether inhibitory interactions between vasodilators relevant to muscle contraction occurs at the level of the capillary. Secondly, we sought to determine the physiological relevance of redundancy by establishing if redundancy between vasodilators occurred during muscle contraction. We have previously shown that NO was critical at producing arteriolar vasodilatation during muscle contraction at low contraction frequencies (6 contractions per minute (cpm)), but at high contraction frequencies (60 cpm) NO was not detectable as important (Dua et al. 2009). Further, during the increase from 6 to 60 cpm, the vasodilatory role of K+ changed from being insignificant at low contraction frequencies to significant at high contraction frequencies. Given this relationship between NO and K+, we hypothesized that the K+ release at high contraction frequencies may be exerting an inhibitory influence on the ability of NO to elicit vasodilatation. Further, we have previously demonstrated that ADO was a critical vasodilator of arterioles at low stimulus frequencies (4 Hz), but at high stimulus frequencies (40 Hz) ADO was not detectable as important (Dua et al. 2009). We hypothesized that K+ release at high stimulus frequencies was inhibiting the vasodilatory actions of ADO.
Methods
All experiments were approved by the Institutional Animal Care Committee Review Board at the University of Guelph and were conducted in accordance with the guidelines of the Canadian Council on Animal Care. All animals had continuous access to food and water. Following all experimental protocols animals were euthanized with an overdose of sodium pentobarbital (0.26 mg ml−1 i.v. to effect).
General protocol: preparing the cremaster muscle for experimentation
Adult male Golden Syrian hamsters (Charles River Laboratories, Canada) (100–140 g; n = 105) were anaesthetized with sodium pentobarbital (70 mg kg−1, i.p.), tracheotomized and catheterized using polyethylene catheters (outer tip diameter approx. 0.5 mm) placed in the left femoral artery to monitor mean arterial pressure and the left femoral vein for supplemental sodium pentobarbital infusion (10 mg ml−1 saline, 0.56 ml h−1) throughout the experimental protocol. To ensure that the hamsters were in the surgical plane of anaesthesia, the animals’ rate and depth of breathing and the lack of withdrawal from a toe pinch were monitored throughout the experimentation. Oesophageal temperature was maintained at 37°C via convective heat from a coiled water‐filled glass tube (42°C) secured under the hamster. The right cremaster was prepared for in situ microscopy as described originally (Baez, 1973) and modified (Murrant, 2005). Briefly, the cremaster muscle was surgically exposed and spread over a semicircular Lucite plate and the edges of the cremaster were secured using insect pins in order to maintain muscle tension. Throughout the cremaster isolation surgery and all experimental protocols, the cremaster muscle was constantly superfused with a physiological salt solution (PSS) containing (in mm): 131.9 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4, 30 NaHCO3 and 0.3 mg L−1 tubocurarine hydrochloride pentahydrate (Sigma‐Aldrich, St Louis, MO, USA). A physiological pH (7.35–7.45) of PSS was maintained by aeration with 5% CO2 and 95% N2 gas. Cremaster muscle temperature was maintained by heating the superfusion solution to 42°C and adjusting the drip rate to achieve 34 ± 0.5°C. Following surgery, the hamster was transferred onto the microscope stage and allowed to equilibrate for 45–60 min prior to data collection.
The cremaster muscle microvasculature was visualized with an Olympus BX51WI microscope (Olympus Canada Inc., Richmond Hill, ON, Canada) with either a ×40 long working distance water immersion objective (numerical aperture 0.80) or dry objective (numerical aperture 0.95) depending on the experimental protocol. Microvasculature images were displayed on a video monitor using a video camera (DC220, Dage‐MTI, Michigan City, MI, USA) and digitized using EZ Grabber video compression software (Geniatech, Indianapolis, IN, USA). Final magnification of the site was approx. ×2000. Diameter measurements were reproducible to within ±0.3 μm (n = 10).
For all protocols capillaries were stimulated with either micropipette application of pharmacological agents directly over a capillary or via contraction of skeletal muscle fibres underlying a capillary module (Fig. 1). The capillary stimulation site was chosen as one that could be visually tracked back to the originating 4A arteriole. The originating 4A arteriole upstream of the stimulated capillary was used as the observation site (Fig. 1).
Figure 1. Schematic representation of the experimental site.
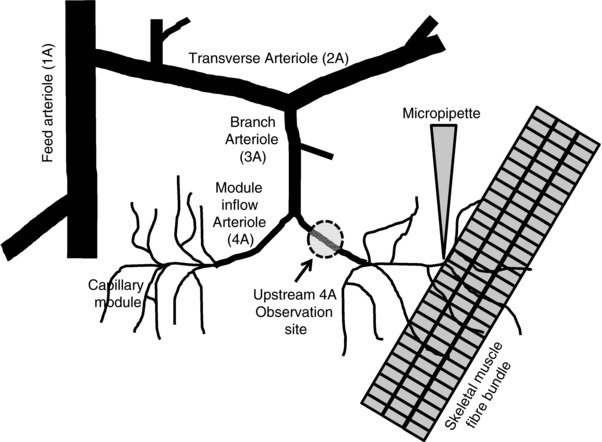
Schematic representation of part of the arteriolar microvascular network (feed arteriole, 1A; transverse arteriole, 2A; branch arteriole, 3A; module inflow arteriole, 4A) and associated capillaries. The diagram indicates the set‐up of different protocols that used capillary stimulation by either micropipette drug application or microelectrode stimulation and the modular inflow (4A) arteriolar observation site. Diagram not drawn to scale.
Micropipette drug application
Using a micromanipulator (Narishige, East Meadow, NY, USA), the tip of glass, drug‐filled micropipettes (tip diameter approx. 10 μm) were placed as close to the endothelial wall of a capillary as possible without touching the vessel or overlying tissue and positioned approximately 1000 μm from the upstream 4A arteriolar observation site. Pharmacological agents were ejected from the micropipette via pressure ejection through a water manometer (30 cmH2O) and applied to a small region of the capillary (approx. 200 μm) as previously described (Frame & Sarelius, 1995; Murrant & Sarelius, 2002). 100 μM fluorescein isothiocyanate‐dextran (FITC; 4 kDa; Sigma‐Aldrich) was added to the micropipette solution so brief epifluorescence could be used to verify that the pipette contents flowed away from the upstream 4A arteriolar observation site to avoid direct exposure. Control experiments demonstrated no significant changes in vessel diameter during a 2 min application of FITC alone (data not shown).
Skeletal muscle contraction
Muscle fibre bundles (3–5 fibres) were stimulated directly using a silver wire microelectrode (tip diameter approx. 100 μm) placed in close proximity to, but not touching, muscle fibres running underneath the capillary module (Fig. 1). The microelectrode was positioned approximately 1000 μm away from the stimulated capillaries and at least 1000 μm away from the 4A arteriole observation site. Tubocarine chloride was added to the superfusate to block nicotinic, cholinergic membrane receptors to ensure muscle fibres were stimulated to contract directly and not through the motor nerve. The ground electrode was placed in the superfusate around the outer rim of the tissue support pedestal. Each stimulus was a square wave pulse of 0.4 ms duration and 8–15 V (Grass S48 stimulator, Quincy, MA, USA). The voltage was adjusted to maximally stimulate four to five muscle fibres and then kept constant throughout the duration of the experiment. Muscle fibres were contracted tetanically and the stimulus frequency within a train and number of trains or contractions per minute (cpm) varied depending on the experimental protocol (see below).
Capillary stimulation control protocol
Since skeletal muscle fibres are long enough to be associated with many capillary units along their length, contracting skeletal muscle fibres will stimulate remote parts of the vasculature, which may also cause vasodilatation at the 4A arteriolar observation site. In order to confirm that the 4A arteriolar vasodilatation was originating solely from stimulation of the capillaries of interest, we used a light‐dye ablation methodology (Segal & Jacobs, 2001) to disrupt transmission of the conducted response pathway between the capillary stimulation site and the upstream 4A arteriolar observation site. Initially we stimulated the capillaries for 2 min with micropipette application of 10−5 m ACh and separately, 2 min of muscle contraction (20 Hz, 60 cpm, 250 ms train duration) and observed the vasodilatation at the upstream 4A arteriole (Fig. 1). We then ablated the cellular function of the vessel wall between the stimulation and observation site by infusing 0.3 ml FITC–saline solution (10% wt/v) via the femoral vein catheter and allowed the dye to circulate for 10 min. We chose a section of 4A arteriole approximately half way between the capillary stimulation site and the upstream 4A arteriolar observation site and exposed approx. 80 μm of vessel to excitation light of 480 nm (X‐CITE 120, Excelitas, Mississauga, ON, Canada) using a ×40 water immersion objective. The vessel was exposed for 6 min followed by a 5 min washout period, and this interval was repeated 3–4 times, until the site of exposure showed constriction and no longer vasodilated to ACh. These characteristics indicated that both endothelial cells and smooth muscle cells were damaged and no longer responsive. The light‐dye injury was limited to the 80 μm segment of arteriole exposed to light while the upstream 4A arteriolar observation site remained responsive to ACh applied directly. After light‐dye treatment, the ACh application and contraction protocols were repeated while observing the 4A arteriolar response.
Protocol 1. Sequential addition of vasodilators
To test whether NO can alter the vasodilator ability of K+, we micropipette‐applied 10 mm KCl (equimolar NaCl removed to preserve the osmolarity of the superfusate) to the capillaries for 10 min. Following a 20 min washout with PSS alone, 10−6 m S‐nitroso‐N‐acetylpenicillamine (SNAP; an NO donor; Sigma‐Aldrich) from a second micropipette was applied onto the KCl stimulation site for 5 min prior to and then during the re‐application of KCl. KCl was re‐applied at the same capillary site for 10 min during the continuous application of SNAP. To test for interactions between vasodilators, the above protocol was repeated with (i) SNAP in the absence and presence of KCl, (ii) 10−6 m ADO in the absence and presence of KCl, (iii) KCl in the absence and presence of ADO, (iv) SNAP in the absence and presence of ADO and (v) ADO in the absence and presence of SNAP.
Protocol 2. Simultaneous addition of vasodilators
Using the same drug concentrations as in protocol 1, we tested for simultaneous interactions between NO and K+, by first micropipette‐applying SNAP alone over the capillaries for 10 min. The effect of SNAP was then washed out with 20 min superfusion of PSS. Following washout, SNAP and KCl were micropipette‐applied together (from the same micropipette) for 10 min onto the same capillary site. To test for simultaneous interactions between other vasodilators, the above protocol was repeated (i) with an initial 10 min exposure of ADO alone and then in combination with KCl and (ii) with an initial exposure of ADO or SNAP for 10 min (chosen randomly) followed by capillary stimulation with ADO and SNAP applied together.
Protocol 3. Redundancy between NO and K+ during muscle contraction
Muscle fibres were contracted under capillaries at contraction frequencies of 6 and 60 cpm while stimulus frequency within a train (20 Hz) and train duration (250 ms) were held constant. Muscle fibres were contracted for 2 min in the absence of any blockers added to the superfusate and then the same muscle fibres were contracted again after a 30 min incubation with NO synthase inhibitor l‐N G‐nitroarginine methyl ester (l‐NAME; 10−6 m; Sigma‐Aldrich) with the addition of 10−7 m SNAP, to restore the tonic NO influence in the presence of NO synthase blockade. Separately, muscle fibres were contracted in the presence of the voltage‐gated K+ channel inhibitor 3,4‐diaminopyridine (DAP; 3 × 10−4 m; Sigma‐Aldrich) to inhibit K+ release from contracting muscle. Finally, muscle fibres were contracted in the presence of l‐NAME+SNAP and DAP to determine if inhibiting K+ (the presumed inhibitor of NO) would enable the detection of NO as an effective vasodilator at contraction frequencies where its role was previously undetectable. We have previously shown that neither 10−6 m l‐NAME + 10−7 m SNAP nor 3 × 10−4 m DAP affected the force of contraction of cremaster muscle (Dua et al. 2009).
Protocol 4. Redundancy between ADO and K+ during muscle contraction
Muscle fibres were contracted under the capillaries at stimulus frequencies of 4 and 40 Hz while contraction frequency (15 cpm) and train duration (250 ms) were held constant. Muscle fibres were stimulated for 2 min in the absence of blockers added to the superfusate and then the same muscle fibres were contracted again after a 30 min incubation with xanthine amine congener (XAC; 10−6 m; a non‐specific ADO receptor antagonist; Tocris, Bristol, UK), to determine under which conditions there was a detectable contribution of ADO. Muscle fibres were also contracted in the presence of 3 × 10−4 m DAP to inhibit K+ release from contracting muscle. Finally, muscle fibres were contracted in the presence of both XAC and DAP to determine if inhibiting K+ (presumably the inhibitor of ADO) would enable ADO to be an effective vasodilator at stimulus frequencies where its role was previously undetectable. We have previously shown that neither 10−6 m XAC nor 3 × 10−4 m DAP affected the force of contraction of cremaster muscle (Dua et al. 2009).
Following each experiment within a protocol, maximal arteriolar diameters were recorded after 2 min superfusion with 10−2 m sodium nitroprusside (NO donor; Sigma‐Aldrich), considered to produce maximal vasodilatation (Murrant et al. 2014).
Data analysis and statistics
4A arteriolar diameter was continuously recorded for 1 min prior to drug application or muscle contraction, during stimulation and for 2 min following cessation of stimulation. Control baseline diameter was defined as the diameter of the arteriole immediately prior to the application of the first stimulus. Experimental baseline diameter was defined as arteriolar diameter in the presence of a blocker prior to the application of the stimulus. For protocols 3 and 4 a second experimental baseline was defined as the arteriolar diameter in the presence of a combination of blockers prior to muscle contraction.
In most preparations only one arteriole was observed per cremaster, but on a few occasions two arterioles were observed per preparation; n indicates the number of arterioles observed. All experiments were analysed offline. For each protocol, arteriolar diameter was measured every 10 s during the recording period. Still images from digitized recording were captured every 10 ± 1 s using FrameShot software (EoF Productions, Oakland, CA, USA) and arteriolar diameters were measured via calibrated ImageJ software (NIH, http://rsbweb.nih.gov/ij/).
Data are reported as the mean ± standard error. Where only two group means were compared (Figs 2 and 3), Student's t test was used. When more than two means were compared per data set (Figs 4, 5, 6), means were compared with a repeated‐measures ANOVA. When the ANOVA identified significant differences, a protected least square difference test was used post hoc to determine significant differences. Differences were considered significant when P < 0.05.
Figure 2. Ablation of cells between the capillary stimulation site and the upstream 4A arteriolar observation site eliminated upstream 4A arteriolar vasodilatation.
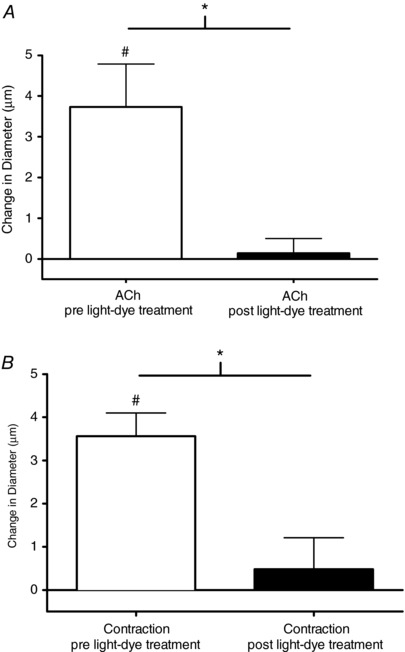
The change in diameter in response to capillary stimulation using micropipette application of ACh (A) or microelectrode stimulation of a bundle of skeletal muscle fibres (B) under control conditions (white bars) and after ablation of cells between the stimulation site and the 4A arteriolar observation site (black bars). #Vasodilatation significantly different from 0. *Significant differences between conditions.
Figure 3. When added sequentially, KCl attenuated the magnitude of vasodilatation produced by SNAP and ADO, and ADO attenuated the vasodilatation induced by SNAP and KCl.
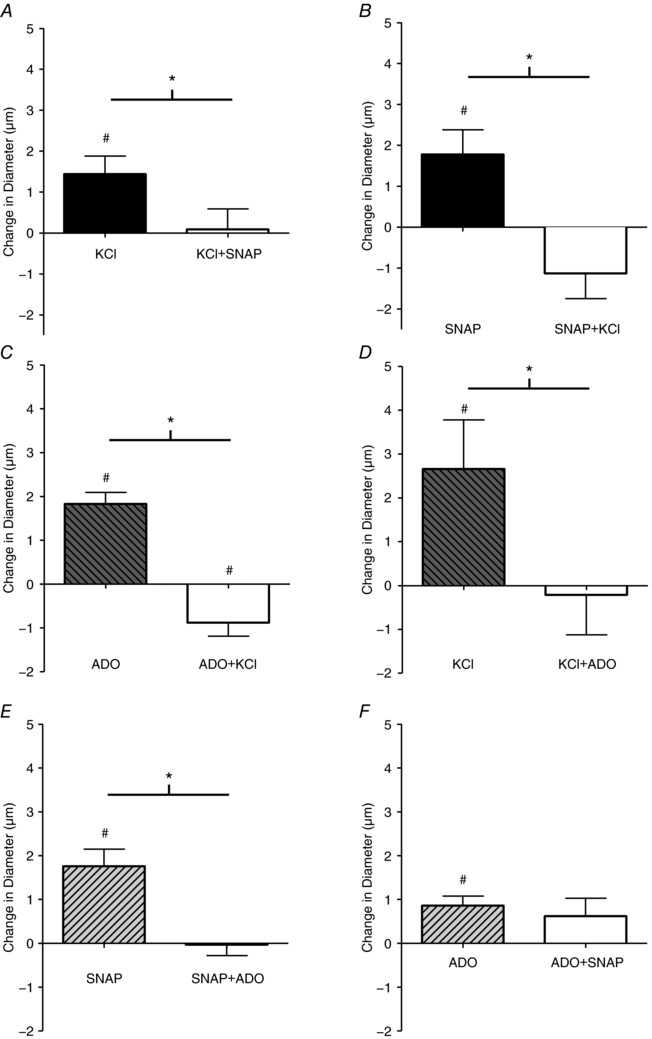
The change in diameter in of the 4A arteriole in the last minute of drug application in response to SNAP in the absence (■) and presence (□) of 10 mm KCl (A); KCl in the absence (■) and presence (□) of SNAP (B); ADO in the absence (hatched bar) and presence (□) of KCl (C) KCl in the absence (hatched bar) and presence (□) of ADO (D); SNAP in the absence (hatched bar) and presence (□) of ADO (E); and ADO in the absence (hatched bar) and presence (□) of SNAP (F). #Vasodilatation is significantly different from 0. *Significant differences between conditions. Protocol details can be found under Protocol 1 and raw baseline and maximal diameters can be found in Table 1.
Figure 4. Simultaneous addition of vasodilators demonstrates inhibition between vasodilators.
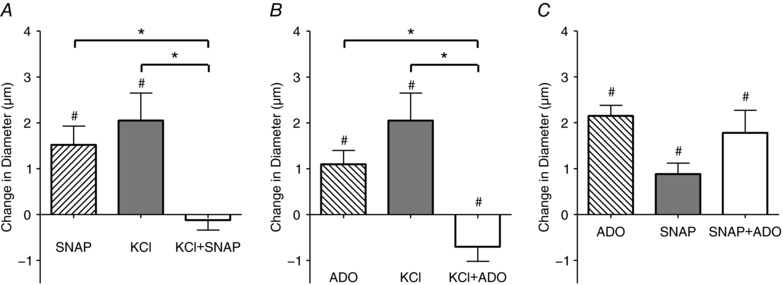
The change in diameter in of the 4A arteriole in the last minute of drug application in response to 10−7 m SNAP alone (□), 10 mm KCl alone (△) and 10−6 m SNAP+10 mm KCl added together simultaneously (■) (A); 10−6 m ADO alone (□), 10 mm KCl alone (△) and 10−6 m ADO+10 mm KCl added together simultaneously (■) (B); 10−6 m ADO alone (□),10−6 m SNAP alone (△) and 10−6 m ADO+10−6 m SNAP added together simultaneously (■) (C). #Vasodilatation is significantly different from 0. *Significant differences between conditions. Protocol details can be found under Protocol 2 and raw baseline and maximal diameters can be found in Table 2.
Figure 5. A role for NO in vasodilatation induced by muscle contraction at 60 cpm can be observed only after K+ release channels are inhibited.
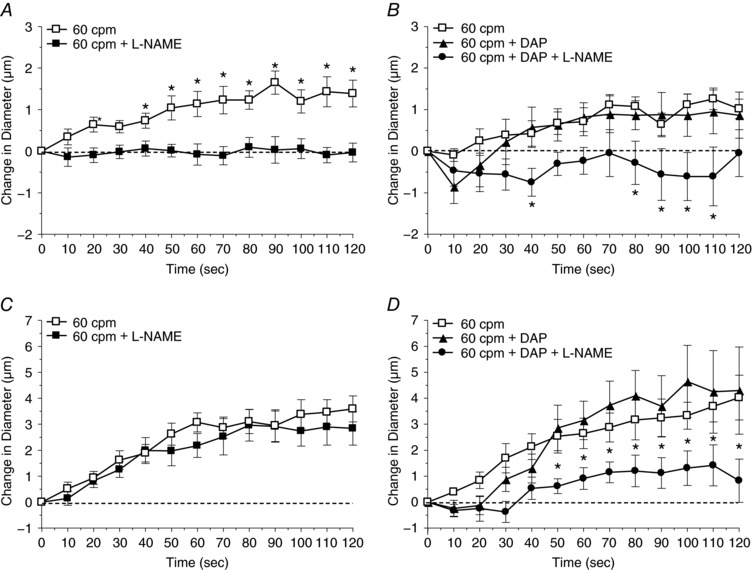
A, the change in diameter in response to muscle contraction under the capillaries at 6 cpm in the absence (□) and presence (■) of l‐NAME (nitric oxide synthase activity inhibitor). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of l‐NAME. B, the change in diameter in response to muscle contraction under the capillaries at 6 cpm in the absence (□) and presence of DAP (voltage dependent K+ channel inhibitor) (▲) and DAP+l‐NAME (●). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of DAP+l‐NAME. C, the change in diameter in response to muscle contraction under the capillaries at 60 cpm in the absence (□) and presence (■) of l‐NAME. D, the change in diameter in response to muscle contraction under the capillaries at 60 cpm in the absence (□) and presence of DAP (▲) and DAP+l‐NAME (●). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of DAP+l‐NAME. Protocol details can be found under Protocol 3 and raw baseline and maximal diameters can be found in Table 3.
Figure 6. A role for ADO in vasodilatation induced by muscle contraction at 40 Hz can be observed only after K+ release channels are inhibited.
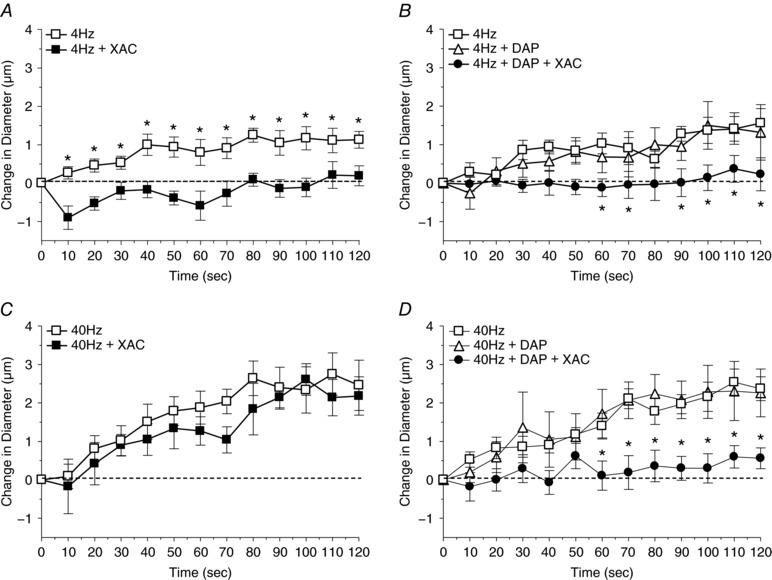
A, the change in diameter in response to muscle contraction under the capillaries at 4 Hz in the absence (□) and presence (■) of the adenosine receptor antagonist XAC (purinergic membrane receptor antagonist). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of XAC. B, the change in diameter in response to muscle contraction under the capillaries at 4 Hz in the absence (□) and presence of DAP (voltage‐dependent K+ channel inhibitor) (▲) and DAP+XAC (●). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of DAP+XAC. C, the change in diameter in response to muscle contraction under the capillaries at 40 Hz in the absence (□) and presence (■) of XAC. D, the change in diameter in response to muscle contraction under the capillaries at 40 Hz in the absence (□) and presence of DAP (▲) and DAP+XAC (●). *Vasodilatation induced by muscle contraction alone differed significantly from muscle contraction in the presence of DAP+XAC. Protocol details can be found under Protocol 4 and raw baseline and maximal diameters can be found in Table 3.
Results
During all protocols there were no significant differences between baseline diameters of control and experimental groups within each set of experiments (Tables 1, 2, 3).
Table 1.
The average baseline and maximum arteriolar diameter for experiments where vasodilators were added in sequence (protocol 1)
| Figure | Protocol | Baseline diameter (μm) | Maximum diameter (μm) | n |
|---|---|---|---|---|
| 3A | SNAP | 6.0 ± 0.7 | 14.2 ± 1.0 | 6 |
| 3A | SNAP + KCl | 8.2 ± 1.0 | ||
| 3B | KCl | 6.8 ± 1.0 | 16.7 ± 1.4 | 6 |
| 3B | KCl + SNAP | 8.5 ± 1.5 | ||
| 3C | ADO | 8.6 ± 0.7 | 15.9 ± 1.3 | 7 |
| 3C | ADO + KCl | 10 ± 0.8 | ||
| 3D | KCl | 7.0 ± 0.7 | 21.3 ± 2.3 | 6 |
| 3D | KCl + ADO | 11.4 ± 1.0 | ||
| 3E | ADO | 6.0 ± 0.4 | 14.9 ± 1.3 | 6 |
| 3E | ADO + SNAP | 5.7 ± 0.8 | ||
| 3F | SNAP | 6.9 ± 1.0 | 14.2 ± 1.2 | 6 |
| 3F | SNAP + ADO | 8.7 ± 1.6 |
Values are means ± SE.
Table 2.
The average baseline and maximum arteriolar diameter for experiments where vasodilators were added simultaneously (protocol 2)
| Figure | Protocol | Baseline diameter (μm) | Maximum diameter (μm) | n |
|---|---|---|---|---|
| 4A | SNAP | 5.1 ± 0.2 | 13.6 ± 1.0 | 10 |
| 4A | SNAP + KCl | 6.0 ± 0.9 | ||
| 4B | ADO | 6.4 ± 0.6 | 15.1 ± 1.3 | 9 |
| 4B | ADO + KCl | 6.1 ± 0.8 | ||
| 4C | ADO or SNAP | 6.7 ± 1.0 | 14.3 ± 1.0 | 9 |
| 4C | ADO + SNAP | 7.5 ± 0.8 |
Values are means ± SE.
Table 3.
The average baseline and maximum arteriolar diameter for experiments involving muscle contraction (protocol 3)
| Figure | Protocol | Baseline diameter (μm) | Maximum diameter (μm) | n |
|---|---|---|---|---|
| 5A | 6 cpm | 6.8 ± 0.5 | 14.2 ± 0.7 | 12 |
| 5A | 6 cpm + l‐NAME | 7.9 ± 0.6 | ||
| 5B | 6 cpm | 8.2 ± 0.6 | 15.5 ± 0.7 | 8 |
| 5B | 6 cpm + DAP | 7.2 ± 0.8 | ||
| 5B | 6 cpm + DAP + l‐NAME | 6.8 ± 1.0 | ||
| 5C | 60 cpm | 7.6 ± 0.6 | 14.1 ± 0.7 | 12 |
| 5C | 60 cpm + l‐NAME | 6.8 ± 0.6 | ||
| 5D | 60 cpm | 7.8 ± 0.4 | 14.8 ± 1.0 | 8 |
| 5D | 60 cpm + DAP | 6.5 ± 0.8 | ||
| 5D | 60 cpm + DAP + l‐NAME | 7.4 ± 0.9 | ||
| 6A | 4 Hz | 8.6 ± 1.2 | 17.4 ± 1.5 | 9 |
| 6A | 4 Hz + XAC | 9.9 ± 1.4 | ||
| 6B | 4 Hz | 7.5 ± 0.6 | 15.4 ± 1.4 | 8 |
| 6B | 4 Hz + DAP | 8.4 ± 1.3 | ||
| 6B | 4 Hz + DAP + XAC | 5.6 ± 0.5 | ||
| 6C | 40 Hz | 10.3 ± 1.5 | 18.8 ± 1.5 | 7 |
| 6C | 40 Hz + XAC | 10.6 ± 1.7 | ||
| 6D | 40 Hz | 7.9 ± 1.0 | 15.4 ± 1.5 | 8 |
| 6D | 40 Hz + DAP | 7.8 ± 1.3 | ||
| 6D | 40 Hz + DAP + XAC | 6.9 ± 1.0 |
Values are means ± SE.
Capillary stimulation control protocol
Vasodilatation at the 4A arteriole was observed during capillary stimulation with 2 min application of ACh and 2 min bout of high intensity contraction (60 cpm) (Fig. 2). Following light‐dye ablation of the cells between the capillary stimulation site and 4A arteriolar observation site, the magnitude of vasodilatation elicited by ACh and contraction was not significantly different from 0 (Fig. 2). We confirmed that cremaster muscle contractility was not affected by FITC exposure in vitro (data not shown).
Control experiments: capillary responsiveness over time
We used multiple micropipette applications of the same vasodilator on the capillaries over time and found that the magnitude of 4A arteriole vasodilatation was similar with each application. The peak change in 4A arteriolar diameter with ADO application on the capillaries was 1.9 ± 0.1 μm initially and 1.7 ± 0.2 μm 1 h later (n = 6), with KCl application was 1.2 ± 0.1 μm initially and 1.6 ± 0.4 μm 1 h later, and with NO application was 1.4 ± 0.1 μm initially and 1.4 ± 0.4 μm 1 h later (n = 6).
Protocol 1: sequential addition of vasodilators
When KCl, SNAP or ADO were applied separately to capillaries, they each evoked dilatation of the 4A arteriole. However, when any vasodilator was applied in the presence of another vasodilator, no significant vasodilatation was detected, with the exception that NO did not affect ADO‐induced vasodilatations (Fig. 3).
Protocol 2: simultaneous addition of vasodilators
As in the sequential addition experiments, when each of SNAP, KCl and ADO were applied separately to the capillaries, each induced significant 4A arteriolar vasodilatation. When SNAP and KCl were applied together, and when ADO and KCl were applied simultaneously, the vasodilatation was abolished (Fig. 4). When ADO and SNAP were applied simultaneously the vasodilatation was not different from the response elicited by each vasodilator alone.
Protocol 3: redundancy between NO and K+ during muscle contraction
Muscle contraction at 6 cpm under capillaries evoked a significant upstream 4A vasodilatation that was inhibited in the presence of l‐NAME and not affected by the inhibition of K+ efflux by DAP. Muscle contraction at 60 cpm produced a vasodilatation that was not inhibited by either NOS inhibition by l‐NAME or K+ efflux inhibition by DAP, but when K+ efflux was inhibited, l‐NAME abolished the contraction‐induced vasodilatation (Fig. 5).
Protocol 4: redundancy between ADO and K+ during muscle contraction
Muscle contraction at 4 Hz under capillaries evoked a significant upstream 4A vasodilatation that was inhibited by ADO receptor inhibition using XAC but not affected by the inhibition of K+ efflux by DAP. Muscle contraction at 40 Hz produced a vasodilatation that was not inhibited by either XAC or DAP, but when K+ efflux was inhibited, XAC was able to significantly inhibit the contraction‐induced vasodilatation (Fig. 6).
Discussion
Our data demonstrate that vasodilators that stimulate capillaries interact in a complex and integrated manner whereby some vasodilators can inhibit the effectiveness of others. Further, we demonstrated this inhibition in the context of muscle contraction whereby adding an antagonist of an inhibitory vasodilator resulted in the increased effectiveness of a second vasodilator, presumably functioning to compensate for the loss of the inhibited vasodilator. The inhibitory interactions that exist between vasodilators provide strong support for the proposal that redundancy exists between vasodilators. Taken together, our data demonstrate that redundancy is a physiologically relevant phenomenon when stimulating the capillaries during contraction and implicate redundancy in the coordination of the distribution of blood flow during the active hyperaemic response.
Experimental considerations
We used muscle contraction as a stimulus for capillaries to initiate an upstream vasodilator response in the 4A arterioles, but given that the length of the contracting skeletal muscle fibres can far exceed the length of the capillary module being stimulated, contracting muscle fibres will stimulate other parts of the vascular network. If the contracting muscle fibres stimulate part of the microvascular network upstream of the 4A arteriole (i.e. the parent 2A arteriole) then a conducted response could be initiated that will spread downstream to the 4A arteriole resulting in 4A vasodilatation that may not be representative of capillary stimulation. We show that light/dye ablation of vascular cells between the capillary stimulation site and the upstream 4A arteriolar observation site abolished the vasodilatation resulting from muscle contraction and the micropipette application of ACh (Fig. 2), indicating that 4A arteriolar vasodilatation was the result of conducted responses coming upstream from the stimulated capillaries and not downstream from other vessels in the network. Therefore, 4A vasodilatation is a representative assay for capillary stimulation during muscle contraction and micropipette application protocols.
Protocol 3 was developed to test the hypothesis that the vasodilatory role of newly generated NO formed during muscle contraction was inhibited by the presence of K+. We used l‐NAME to inhibit NO formation during muscle contraction; however, addition of this inhibitor also inhibited the basal production of NO by resting skeletal muscle (Balon & Nadler, 1994; Kobzik et al. 1994; Reid et al. 1998; Tidball et al. 1998), and results in a decrease in arteriolar resting baseline diameter. In order to maintain basal levels of NO at rest while inhibiting newly formed NO generated by muscle contraction, we supplemented l‐NAME with an NO donor, SNAP, at levels that maintained resting baseline diameter (Table 3). This is a critical strength in our experimental design given that interactions between vasodilators may occur at the intracellular level between signalling pathways and consequently care must be taken not to disrupt these pathways at rest. Furthermore, this design allows us to conclude that newly formed, contraction‐dependent NO contributes to stimulation of the capillaries during muscle contraction.
Within each protocol, capillaries were stimulated multiple times (with either agonists/antagonists or skeletal muscle contraction). Our control experiments show that the magnitude of the vasodilatation induced by each agonist did not significantly differ over time or over repeated applications. Our data also show that stimulating capillaries with multiple bouts of contraction caused a similar vessel response with respect to the magnitude of vasodilatation produced (6 and 60 cpm (Fig. 5 B–D) and 4 and 40 Hz (Fig. 5 B–D)).
Vasodilator interactions at the capillary level
We have demonstrated inhibitory interactions between vasodilators at the capillary level that manifest in two patterns of vascular response as a consequence of whether the inhibition between two vasodilators was reciprocal or not. Reciprocal or mutual inhibition between vasodilators, as occurred between NO and K+, and ADO and K+, resulted in no net vasodilatation when both vasodilators were present. When the inhibition was not mutual – when ADO inhibited NO effects but NO did not inhibit ADO vasodilatation – the result was net vasodilatation such that the magnitude of the vasodilatation was not significantly different from the magnitude of each individual vasodilator. It is non‐reciprocal inhibition that more readily fits the proposed role of redundancy, when one vasodilator can compensate for the loss of another vasodilator. For example, given that ADO inhibits the effects of NO, during contraction, when both are present, there will be vasodilatation. If ADO were inhibited, the inhibition of ADO on NO would be removed and the effects of NO would take its place. Mutual inhibition, on the other hand, does not easily fit the definition of redundancy. If K+ and ADO were present during contraction there would be no net vasodilatation; if ADO were to be inhibited, the inhibition of ADO on K+ will be removed and result in a significant vasodilatation. Given that muscle contraction always produces vasodilatation, the presence of reciprocal inhibition would lead us to believe that there must be more vasodilators present, more than K+ and ADO, to produce vasodilatation. The consequences of reciprocal inhibition indicate that vasodilatation during muscle contraction involves a more complex network of vasodilators with a potentially complex network of redundant interactions.
Inhibition between vasodilators occurred regardless of whether vasodilators were added sequentially or simultaneously implicating redundancy as important throughout an entire contraction bout. At the onset of muscle contraction, there are multiple vasodilators present simultaneously, and thus redundancy between vasodilators has the potential to occur at the onset of exercise. If muscle contraction persists, additional vasodilators may be released as metabolism changes over time. In these instances, new vasodilators will be introduced sequentially, in the presence of existing vasodilators. Our data show that inhibitory interactions can form under these sequential conditions as well. Therefore, redundancies between vasodilators can occur at all time points over the course of a contraction bout. The assumption of the above discussion is that skeletal muscle is the source of the vasodilatory products, but there are other potential sources of vasodilators, especially over time, that may contribute to the redundant interactions between the vasodilators. For example, an increase in blood flow increases wall shear stress, which stimulates endothelial cells’ production of NO at the arteriolar level (Ungvari et al. 2001; Watanabe et al. 2005) resulting in wall shear stress‐induced vasodilatation. Therefore, once blood flow has increased, there may be a contribution of endothelial cell‐derived NO sequentially added to the extracellular mileau of vasodilators, which may then contribute to redundant interactions between vasodilators. Currently it is not known whether capillary endothelial cells respond to wall shear stress in a similar manner, so whether this specific example is relevant to the capillaries is unknown. Regardless of the source of the vasodilators, the observation that redundant interactions can form during simultaneous and sequential addition of vasodilators indicates that redundancies between vasodilators could be relevant throughout the duration of the hyperaemic response.
Determining the physiological relevance of redundancy required a demonstration of inhibition between vasodilators during muscle contraction. However, muscle contraction‐induced vasodilatation is derived from a complexity of multiple vasodilators, each of which may have their own inherent redundant interactions. To demonstrate redundancy amid this complexity we relied heavily on our understanding of how vasodilators change in relation to muscle contraction parameters. Firstly, twitch contractions can stimulate capillaries (Berg et al. 1997; Cohen et al. 2000; Cohen & Sarelius, 2002), but the vasodilators resulting from twitch and tetanic contractions differ (Mihok & Murrant, 2004; Twynstra et al. 2012; Ross et al. 2013). Therefore, using tetanic contractions was critical to producing physiologically relevant vasodilators. Secondly, the stimulation parameters involved in generating a tetanic contraction (i.e. train duration, stimulus frequency within a train, contraction frequency and total contraction time) are important in determining the vasodilators produced (Mihok & Murrant, 2004; Dua et al. 2009; Ray & Marshall, 2009; Ross et al. 2013). This observation is supported by our current data showing that vasodilatations induced by contractions at low contraction frequencies were NO‐dependent but those evoked by high contraction frequencies were not (Fig. 5 A and C) and vasodilatations induced by low stimulus frequencies were ADO‐dependent, while those evoked by high stimulus frequencies were not (Fig. 6 A and C). We chose contraction parameters that allowed a given vasodilator to produce significant vasodilatation at low stimulus or contraction frequencies but whose vasodilator role became insignificant as frequency was increased; a vasodilator becoming ineffective provided an indication that it may be being inhibited by another vasodilator. We found two combinations that displayed these characteristics: contraction frequency and the role of NO and stimulus frequency and the role of ADO (Dua et al. 2009). Using these contractile conditions, we show that the vasodilatation developed at high contraction frequencies was not affected by NO synthase inhibition with l‐NAME or inhibition of K+ release with DAP, when applied independently (Fig. 5 D). But when K+ release was inhibited, l‐NAME inhibited vasodilatation, indicating that with K+ blocked NO becomes critical in mediating vasodilatation. Similar results were found using stimulus frequency and the role of ADO; only when K+ release was blocked could the vasodilatory effects of ADO be detected. Therefore, using specific contractile parameters we were able to demonstrate inhibition between vasodilators during muscle contraction and show the physiological relevance of redundancy.
The demonstration that redundancy between vasodilators occurs at the level of the capillary under physiological conditions further solidifies the importance of capillaries in the vascular response to muscle contraction. The close proximity of the capillaries to all muscle fibres (or example see Plyley & Groom, 1975; Poole et al. 1989; Hargreaves et al. 1990; Mathieu‐Costello et al. 2005; Zoladz et al. 2005) and the ability of capillaries to be stimulated by contraction, initiate a conducted upstream signal to vasodilate arterioles from which they arise (Berg et al. 1997; Cohen et al. 2000; Cohen & Sarelius, 2002), and increase their own perfusion implicates capillaries as a vital vascular component in the coordination of skeletal muscle blood flow during contraction. That vasodilators interact in the inhibitory manner suggestive of redundant interactions between vasodilators at the level of the capillary further solidifies the importance of capillaries in the active hyperaemic response.
Mechanisms of inhibitory interactions between vasodilators
Delineating the mechanisms which produce inhibition between vasodilators will be a critical next step in understanding how redundancy functions during active hyperaemia. There are many potential levels at which vasodilators can interact. They can interact biochemically in the extracellular space, at the cell surface (i.e. with one vasodilator interfering with another's ability to bind to membrane receptors), or their intracellular signalling cascades may interact. The latter can manifest itself in either the inhibition of the effectiveness of a specific vasodilator or the inhibition of the release of a vasodilator (i.e. K+ inhibition of NO release from capillary endothelial cells). Unfortunately, there is little information regarding cell signalling cascades during acute capillary endothelial cell stimulation in skeletal muscle. What we do know about endothelial cell responses stems from research at the arteriolar level, but care must be taken in extrapolating arteriolar endothelial cell responses to capillary endothelial cell responses as significant differences between these two levels of the vasculature exist. Most notably, NO and ADO induce conducted responses when applied to capillaries (Song & Tyml, 1993; Tyml et al. 1997; Cohen & Sarelius, 2002), but do not consistently cause conducted responses when applied to arterioles (ADO: Delashaw & Duling, 1991; Rivers & Frame, 1999; Frame, 2000; Wölfle et al. 2007; NO: Kurjiaka & Segal, 1995; Doyle & Duling, 1997; Rivers & Frame, 1999; Chen & Rivers, 2001; Hoepfl et al. 2002; Budel et al. 2003; Tallini et al. 2007). Arteriolar conducted responses are facilitated by gap‐junction connections between vascular smooth muscle cells and endothelial cells allowing electrical (change in membrane potential) and Ca2+‐based signals to be shared between cells (for review see Bagher & Segal, 2011). Capillaries share some of these signalling properties: conducted responses initiated at the capillaries can be blocked using gap junction uncouplers (Berg et al. 1997; Cohen et al. 2000; Yu et al. 2000) and capillaries are electrically connected to arterioles (Beach et al. 1998; McGahren et al. 1998). That NO and ADO can cause a conducted response when applied to a capillary but not when applied to an arteriole indicates that NO and ADO stimulate fundamentally different hyperpolarization‐dependent or Ca2+‐dependent intracellular signalling pathways in capillary endothelial cells that are not stimulated in arteriolar endothelial cells. There may be similar signalling pathways in arterioles and capillaries with regards to K+ stimulation as K+ can initiate a conducted response at both vascular levels (current data; Dora, 2017). On arterioles, the vasodilatory response to low concentrations of K+ (<20 mm) has been attributed to hyperpolarization secondary to stimulation of inward rectifying K+ (KIR) channels (Loeb et al. 2000; Burns et al. 2004; McSherry et al. 2006; Dora, 2017) and the sodium–potassium pump (Haddy, 1983; Burns et al. 2004; McSherry et al. 2006). Capillaries may share a similar K+ signalling pathways. In skeletal muscle, K+ stimulates capillary endothelial cells (Song & Tyml, 1993; Tyml et al. 1997; McGahren et al. 1998) and K+ channels sensitive to BaCl have been shown to reside in capillaries in frog skeletal muscle (Tyml et al. 1997) and porcine capillary endothelial cells (Hoyer et al. 1991) indicating the presence of KIR channels. Therefore, while the signalling mechanisms for K+ in skeletal muscle capillary endothelial cells have not been characterized, skeletal muscle capillaries may respond to K+ by stimulating KIR channels resulting in endothelial cell hyperpolarization. Although there may be similarities in signalling pathways between capillary and arteriolar endothelial cells, there are also stark differences. Thus, understanding mechanisms of vasodilator interaction at the level of the capillary will require that capillary endothelial cell‐specific studies be performed.
Implications of redundancy
Redundancy between vasodilators in active hyperaemia may explain why some vasodilators are contentious with respect to their role in active hyperaemia and additionally may have led to the premature classification of some vasodilators as insignificant to the active hyperaemic process. For example, NO is thought to contribute only modestly to active hyperaemia (for reviews see Radegran & Saltin, 1999; Tschakovsky & Joyner, 2008), a conclusion derived predominantly from experiments where blocking NO production had little effect on the blood flow response to muscle contraction. In light of our findings with regards to redundancy, there are two explanations for why blocking NO would have seemingly no effect on blood flow: (i) NO is the primary vasodilator and when its production is blocked other vasodilators compensate for its loss, or (ii) NO is present but its vasodilatory effects are being inhibited by another vasodilator but are available to cause vasodilatation should this inhibition be removed. In either case NO would actually be important to the hyperaemic processes. Thus, adopting redundancy between vasodilators as a mechanism to integrate vasodilatory signals during active hyperaemia will require a re‐examination of the literature and reconsideration of the vasodilators considered to be involved in the active hyperaemic process.
Redundancy between vasodilators adds another variable in the growing complexity of the regulation of active hyperaemia. We have identified here and elsewhere (Dua et al. 2009) that contraction parameters are an important variable at dictating what vasodilators are present to promote vasodilatation. We also have shown that redundancy between vasodilators exists and may change temporally (Lamb & Murrant, 2015). Finally we know that capillaries and arterioles differ in their redundant relationships (current data compared to Lamb & Murrant, 2015). These variables must be considered when thinking about the role of a vasodilator in active hyperaemia. For example, Armstrong et al. (2007) showed that K+ was involved in mediating arteriolar vasodilatation over a range of stimulus frequencies within a train at the onset of contraction, but under similar contractile conditions over 2 min, a role for K+ could not be identified (Dua et al. 2009). Therefore, K+ is an important vasodilator at the onset of contraction but over time its effects may be minimized by an inhibitor. Further, Dua et al. (2009) showed a role for K+ in contraction‐induced arteriolar vasodilatation over 2 min at 60 cpm, whereas in the current data there was no detectable role for K+ during 60 cpm at the level of the capillary. These differences may be due to the vascular level being studied. Finally, Crecelius et al. (2014) implicated K+ as significant in mediating the increase in forearm blood flow at the onset of contraction and over a 5 min contraction bout (10% maximal voluntary contraction at 20 cpm). At first glance these results oppose the results observed here where no significant role for K+ was found, but this comparison is too simplistic given that contraction parameters differ between the experiments and the specific vascular level being measured differs. It may be too simplistic to ask if K+ is involved in active hyperaemia, given the complexity of the factors. These factors include consideration of the contraction parameters used, vascular levels involved, and potential temporal changes in vasodilator production. It may be more appropriate to ask under what conditions will K+ be important. In order to further our understanding of active hyperaemia we must start to consider the complexity of the variables that integrate to result in a coordinated vascular response. It will be a critical next step to formulate these complexities into a unified understanding of how blood flow is ultimately matched to metabolic demand.
We have implicated redundancy as important physiologically, but the principles of redundancy may also have potential clinical relevance. Redundancy involving intracellular signalling pathways indicates that there may be multiple intracellular pathways that can interact to produce the same vasodilatory outcome and that pathways to the same outcome can be altered by inhibiting one part of the signalling pathway, thereby allowing another to be effective. Potentially defective pathways may be circumvented by attenuating an inhibitory influence and allowing the expression of secondary cell signalling pathways that produce the same outcome. Understanding how redundancy between vasodilators occurs mechanistically may open up new avenues in how we research and potentially treat clinical manifestations of blood flow dysregulation.
Summary
We have demonstrated that vasodilators relevant to muscle contraction can interact with each other at the level of the capillary to inhibit their ability to evoke vasodilatation. These inhibitory interactions provide the mechanism necessary to produce redundancies between vasodilators. Further, we have advanced the idea of redundancy as an important physiological phenomenon by showing that, during skeletal muscle contraction, inhibition of one vasodilator can enhance the effectiveness of another. Redundancy between vasodilators introduces a new complexity in how blood flow is matched to metabolic demand during active hyperaemia. Understanding of the specific vasodilator environment produced by contracting skeletal muscle and how each vasodilator interacts with others will be necessary in order to understand how coordination of vasodilators in the intercellular space results in matching blood flow to metabolic demand.
Additional information
Competing interests
None.
Author contributions
I.L. was responsible for conception and design of the experiments, data collection, analysis and interpretation of the data and writing of the manuscript. N.N. was responsible for data collection, analysis and interpretation of the data and reviewing the manuscript. C.M. was responsible for conception and experimental design, data analysis, data interpretation and writing the manuscript. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by NSERC, Canada (RGPIN‐2014‐05184).
Acknowledgements
The authors would like to acknowledge the FITC in vitro cremaster contraction experiments completed by Nicole Fletcher that contributed to the results and interpretation of our data.
Biography
Iain R. Lamb is a senior PhD at the University of Guelph under the mentorship of Dr. Coral Murrant. His research focuses on redundancy in skeletal muscle blood flow regulation. Specifically, if antagonism of one vasodilator can be compensated for by the augmented effect of another, safeguarding the coupling between flow and muscle metabolism. Rapidly approaching the end of his degree, he has more questions regarding flow regulation than he has time left to answer, so he hopes to continue his academic journey by finding a post‐doctoral fellowship that will help him approach blood flow regulation from a new perspective.

Edited by: Laura Bennet & Ylva Hellsten
References
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez S ( 1973). An open cremaster muscle preparation for the study of blood vessels by in vivo microscopy. Microvasc Res 5, 384–394. [DOI] [PubMed] [Google Scholar]
- Bagher P & Segal SS (2011). Regulation of blood flow in the microcirculation: role of conducted vasodilation. Acta Physiol (Oxf) 202, 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balon TW & Nadler JL (1994). Nitric oxide release is present from incubated skeletal muscle preparations. J Appl Physiol (1985) 77, 2519–2521. [DOI] [PubMed] [Google Scholar]
- Beach JM, McGahren ED & Duling BR (1998). Capillaries and arterioles are electrically coupled in hamster cheek pouch. Am J Physiol 275, H1489–H1496. [DOI] [PubMed] [Google Scholar]
- Berg BR, Cohen KD & Sarelius IH (1997). Direct coupling between blood flow and metabolism at the capillary level in striated muscle. Am J Physiol 272, H2693–H2700. [DOI] [PubMed] [Google Scholar]
- Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M & Kjaer M (2002). Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543, 691–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budel S, Bartlett IS & Segal SS (2003). Homocellular conduction along endothelium and smooth muscle of arterioles in hamster cheek pouch: unmasking an NO wave. Circ Res 93, 61–68. [DOI] [PubMed] [Google Scholar]
- Burns WR, Cohen KD & Jackson WF (2004). K+‐induced dilation of hamster cremasteric arterioles involves both the Na+/K+‐ATPase and inward‐rectifier K+ channels. Microcirc 11, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP, Mohamed EA & Joyner MJ (2013). Role of nitric oxide and adenosine in the onset of vasodilation during dynamic forearm exercise. Eur J Appl Physiol 113, 295–303. [DOI] [PubMed] [Google Scholar]
- Chen Y & Rivers RJ (2001). Nonvasomotor influence of sodium nitroprusside on arteriolar remote response to methacholine. J Vasc Res 38, 219–227. [DOI] [PubMed] [Google Scholar]
- Chilian WM & Koshida R (2001). EDHF and NO: different pathways for production–similar actions. Circ Res 89, 648–649. [PubMed] [Google Scholar]
- Cohen KD, Berg BR & Sarelius IH (2000). Remote arteriolar dilations in response to muscle contraction under capillaries. Am J Physiol Heart Circ Physiol 278, H1916–H1923. [DOI] [PubMed] [Google Scholar]
- Cohen KD & Sarelius IH (2002). Muscle contraction under capillaries in hamster muscle induces arteriolar dilatation via K(ATP) channels and nitric oxide. J Physiol 539, 547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2014). KIR channel activation contributes to onset and steady‐state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol 307, H782–H791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delashaw JB & Duling BR (1991). Heterogeneity in conducted arteriolar vasomotor response is agonist dependent. Am J Physiol 260, H1276–H1282. [DOI] [PubMed] [Google Scholar]
- Dora KA (2017). Conducted dilatation to ATP and K+ in rat skeletal muscle arterioles. Acta Physiol (Oxf) 219, 202–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle MP & Duling BR (1997). Acetylcholine induces conducted vasodilation by nitric oxide‐dependent and ‐independent mechanisms. Am J Physiol 272, H1364–H1371. [DOI] [PubMed] [Google Scholar]
- Dua AK, Dua N & Murrant CL (2009). Skeletal muscle contraction‐induced vasodilator complement production is dependent on stimulus and contraction frequency. Am J Physiol Heart Circ Physiol 297, H433–H442. [DOI] [PubMed] [Google Scholar]
- Frame MD ( 2000). Increased flow precedes remote arteriolar dilations for some microapplied agonists. Am J Physiol Heart Circ Physiol 278, H1186–H1195. [DOI] [PubMed] [Google Scholar]
- Frame MD & Sarelius IH (1995). L‐arginine‐induced conducted signals alter upstream arteriolar responsivity to L‐arginine. Circ Res 77, 695–701. [DOI] [PubMed] [Google Scholar]
- Haddy FJ ( 1983). Potassium effects on contraction in arterial smooth muscle mediated by Na+,K+‐ATPase. Fed Proc 42, 239–245. [PubMed] [Google Scholar]
- Hargreaves D, Egginton S & Hudlicka O (1990). Changes in capillary perfusion induced by different patterns of activity in rat skeletal muscle. Microvasc Res 40, 14–28. [DOI] [PubMed] [Google Scholar]
- Hellsten Y, Nyberg M, Jensen LG & Mortensen SP (2012). Vasodilator interactions in skeletal muscle blood flow regulation. J Physiol 590, 6297–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepfl B, Rodenwaldt B, Pohl U & De Wit C (2002). EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 283, H996–H1004. [DOI] [PubMed] [Google Scholar]
- Hoyer J, Popp R, Meyer J, Galla HJ & Gogelein H (1991). Angiotensin II, vasopressin and GTP[gamma‐S] inhibit inward‐rectifying K+ channels in porcine cerebral capillary endothelial cells. J Membr Biol 123, 55–62. [DOI] [PubMed] [Google Scholar]
- Joyner MJ & Wilkins BW (2007). Exercise hyperaemia: is anything obligatory but the hyperaemia? J Phsyiol 583, 855–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobzik L, Reid MB, Bredt DS & Stamler JS (1994). Nitric oxide in skeletal muscle. Nature 372, 546–548. [DOI] [PubMed] [Google Scholar]
- Kurjiaka DT & Segal SS (1995). Conducted vasodilation elevates flow in arteriole networks of hamster striated muscle. Am J Physiol 269, H1723–H1728. [DOI] [PubMed] [Google Scholar]
- Lamb IR & Murrant CL (2015). Potassium inhibits nitric oxide and adenosine arteriolar vasodilatation via KIR and Na+/K+ ATPase: implications for redundancy in active hyperaemia. J Physiol 593, 5111–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb AL, Godeny I & Longnecker DE (2000). Functional evidence for inward‐rectifier potassium channels in rat cremaster muscle arterioles. Microvasc Res 59, 1–6. [DOI] [PubMed] [Google Scholar]
- Markwald RR, Kirby BS, Crecelius AR, Carlson RE, Voyles WF & Dinenno FA (2011). Combined inhibition of nitric oxide and vasodilating prostaglandins abolishes forearm vasodilatation to systemic hypoxia in healthy humans. J Physiol 589, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu‐Costello O, Ju Y, Trejo‐Morales M & Cui L (2005). Greater capillary‐fiber interface per fiber mitochondrial volume in skeletal muscles of old rats. J Appl Physiol 99, 281–289. [DOI] [PubMed] [Google Scholar]
- McGahren ED, Beach JM & Duling BR (1998). Capillaries demonstrate changes in membrane potential in response to pharmacological stimuli. Am J Physiol 274, H60–H65. [DOI] [PubMed] [Google Scholar]
- McSherry IN, Sandow SL, Campbell WB, Falck JR, Hill MA & Dora KA (2006). A role for heterocellular coupling and EETs in dilation of rat cremaster arteries. Microcirc 13, 119–130. [DOI] [PubMed] [Google Scholar]
- Mihok ML & Murrant CL (2004). Rapid biphasic arteriolar dilations induced by skeletal muscle contraction are dependent on stimulation characteristics. Can J Physiol Pharmacol 82, 282–287. [DOI] [PubMed] [Google Scholar]
- Mortensen SP, Gonzalez‐Alonso J, Damsgaard R, Saltin B & Hellsten Y (2007). Inhibition of nitric oxide and prostaglandins, but not endothelial‐derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Phsyiol 581, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL (2005). Stimulation characteristics that determine arteriolar dilation in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 289, R505–R513. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Dodd JD, Foster AJ, Inch KA, Muckle FR, Ruiz DA, Simpson JA & Scholl, JH (2014). Prostaglandins induce vasodilatation of the microvasculature during muscle contraction and induce vasodilatation independent of adenosine. J Physiol 592, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL, Lamb IR & Novielli NM (2016). Capillary endothelial cells as coordinators of skeletal muscle blood flow during active hyperaemia. Microcirc 24, e12348. [DOI] [PubMed] [Google Scholar]
- Murrant CL & Sarelius IH (2002). Multiple dilator pathways in skeletal muscle contraction‐induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol 282, R969–R978. [DOI] [PubMed] [Google Scholar]
- Plyley MJ & Groom AC (1975). Geometrical distribution of capillaries in mammalian striated muscle. Am J Physiol 228, 1376–1383. [DOI] [PubMed] [Google Scholar]
- Poole DC, Mathieu‐Costello O & West JB (1989). Capillary tortuosity in rat soleus muscle is not affected by endurance training. Am J Physiol 256, H1110–H1116. [DOI] [PubMed] [Google Scholar]
- Radegran G & Saltin B (1999). Nitric oxide in the regulation of vasomotor tone in human skeletal muscle. Am J Physiol 276, H1951–H1960. [DOI] [PubMed] [Google Scholar]
- Ray CJ & Marshall JM (2009). Elucidation in the rat of the role of adenosine and A2A‐receptors in the hyperaemia of twitch and tetanic contractions. J Physiol 587, 1565–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MB, Kobzik L, Bredt DS & Stamler JS (1998). Nitric oxide modulates excitation‐contraction coupling in the diaphragm. Comp Biochem Physiol Mol Integr Physiol 119, 211–218. [DOI] [PubMed] [Google Scholar]
- Rivers RJ & Frame MD (1999). Network vascular communication initiated by increases in tissue adenosine. J Vasc Res 36, 193–200. [DOI] [PubMed] [Google Scholar]
- Ross GA, Mihok ML & Murrant CL (2013). Extracellular adenosine initiates rapid arteriolar vasodilation induced by a single skeletal muscle contraction in hamster cremaster muscle. Acta Physiol (Oxf) 208, 74–87. [DOI] [PubMed] [Google Scholar]
- Rowell LB ( 2004). Ideas about control of skeletal and cardiac muscle blood flow (1876‐2003): cycles of revision and new vision. J Appl Physiol (1985) 97, 384–392. [DOI] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS & Jacobs TL (2001). Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H & Tyml K (1993). Evidence for sensing and integration of biological signals by the capillary network. Am J Physiol 265, H1235–H1242. [DOI] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang SM, Nakai J, Salama G, Segal SS & Kotlikoff MI (2007). Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res 101, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Tidball JG, Lavergne E, Lau KS, Spencer MJ, Stull JT & Wehling M (1998). Mechanical loading regulates NOS expression and activity in developing and adult skeletal muscle. Am J Physiol 275, C260–C266. [DOI] [PubMed] [Google Scholar]
- Tschakovsky ME & Joyner MJ (2008). Nitric oxide and muscle blood flow in exercise. Appl Physiol Nutr Metab 33, 151–161. [DOI] [PubMed] [Google Scholar]
- Twynstra J, Ruiz DA & Murrant CL (2012). Functional coordination of the spread of vassdilations through skeletal muscle microvasculature: implications for blood flow control. Acta Physiol (Oxf) 206, 229–241. [DOI] [PubMed] [Google Scholar]
- Tyml K, Song H, Munoz P & Ouellette Y (1997). Evidence for K+ channels involvement in capillary sensing and for bidirectionality in capillary communication. Microvasc Res 53, 245–253. [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Sun D, Huang A, Kaley G & Koller A (2001). Role of endothelial [Ca2+]i in activation of eNOS in pressurized arterioles by agonists and wall shear stress. Am J Physiol Heart Circ Physiol 281, H606–H612. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Yashiro Y, Mizuno R & Ohhashi T (2005). Involvement of NO and EDHF in flow‐induced vasodilation in isolated hamster cremasteric arterioles. J Vasc Res 42, 137–147. [DOI] [PubMed] [Google Scholar]
- Wölfle SE, Schmidt VJ, Hoepfl B, Gebert A, Alcoléa S, Gros D & de Wit C (2007). Connexin45 cannot replace the function of connexin40 in conducting endothelium‐dependent dilations along arterioles. Circ Res 101, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Yu J, Bihari A, Lidington D & Tyml K (2000). Gap junction uncouplers attenuate arteriolar response to distal capillary stimuli. Microvasc Res 59, 162–168. [DOI] [PubMed] [Google Scholar]
- Zoladz JA, Semik D, Zawadowska B, Majerczak J, Karasinski J, Kolodziejski L, Duda K & Kilarski WM (2005). Capillary density and capillary‐to‐fibre ratio in vastus lateralis muscle of untrained and trained men. Folia Histochem Cytobiol 43, 11–17. [PubMed] [Google Scholar]