

Abstract
Strong flavor baijiu (SFB), also called Luzhou-flavor liquor, is the most popular Chinese baijiu. It is manufactured via solid fermentation, with daqu as the starter. Microbial diversity of the SFB ecosystem and the synergistic effects of the enzymes and compounds produced by them are responsible for the special flavor and mouthfeel of SFB. The present review covers research studies focused on microbial community analysis of the SFB ecosystem, including the culturable microorganisms, their metabolic functions, microbial community diversity and their interactions. The review specifically emphasizes on the most recently conducted culture-independent analysis of SFB microbial community diversity. Furthermore, the possible application of systems biology approaches for elucidating the molecular mechanisms of SFB production were also reviewed and prospected.
Keywords: strong flavor baijiu, microbial community, pit mud, ethyl hexanoate, systems biology
Introduction
Strong-flavor baijiu (SFB), also called Luzhou-flavor liquor, is the most popular Chinese baijiu that is known to exist since the past many centuries (Zheng and Han, 2016; Jin et al., 2017; Xu et al., 2017). Latest data reveal that the total yield of SFB has reached 9.1 million tons per annum (Wang, 2016). SFB is usually produced with the help of a typical method of natural solid fermentation that uses daqu as the main saccharification agent (Figure 1). The materials of SFB are cereals, mostly sorghum or a mixture of corn, rice, millet, sticky rice, and wheat. The fermentation process is anaerobic, carried out in a mud pit (normally with a volume of 6–8 m3) and lasts for 60–90 days (Zheng and Han, 2016). SFB is known to contain over 1300 different kinds of flavoring compounds (Yao et al., 2015). It has a characteristic fragrant flavor, soft mouthfeel, and long-lasting aftertaste (Zheng and Han, 2016). Like beer and wine, the composition of the flavoring compounds in SFB is determined by its microbial diversity (Bokulich et al., 2012). However, the open fermentation environment and complex microbial composition of the procedure makes it difficult to elucidate the exact specifications of SFB production.
FIGURE 1.
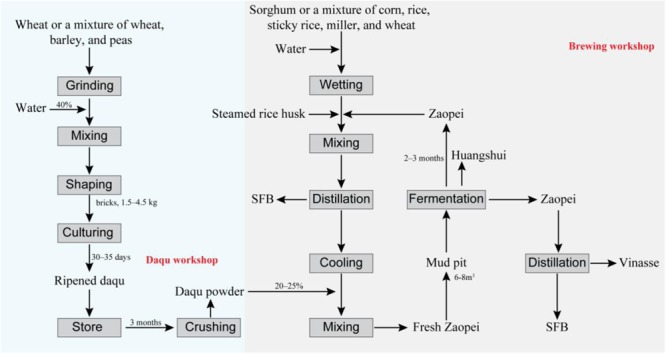
Workflow of the strong flavor baijiu (SFB) production. The technology used in SFB production is called “Back-slopping technique” (Zheng and Han, 2016). The raw material for SFB production are cereals, mostly sorghum or a mixture of corn, rice, millet, sticky rice, and wheat (Zheng and Han, 2016). Daqu is the saccharifying and fermentative agent used in the SFB production (Zheng et al., 2011). The production of daqu is a solid fermentation process with a natural inoculation of microorganisms originated from production environment and cereals materials, mainly involving three stages: material shaping, incubating, and drying (Zheng et al., 2011; Wu et al., 2014). The zaopei (fermented cereals) is mixed with sorghum, rice or other cereals and steamed rice husk and distilled for the SFB, then after cooling, the distilled solid residue was mixed with daqu powder, and fresh zaopei was obtained. The fresh zaopei was put into a mud pit (volume of 6–8 m3) and the top is covered with yellow mud so as to achieve and maintain an anaerobic environment (Jin et al., 2017). In mud pit, the saccharification and fermentation process occur simultaneously and last for 2–3 months (Xu et al., 2010; Zheng and Han, 2016). Then, the fermented zaopei is took out. The zaopei in the upper layer (also name mianzao) is distilled to obtain SFB. The distilled solid residue is vinasse. The other zaopei (also name niangzao) is mixed with cereals and enters into another cycle for SFB production. Huangshui (HS) is the brown viscous liquid which is formed by the liquid that permeates to the bottom of mud pit during the fermentation process (Feng et al., 2017).
At present, the SFB industry faces many constraints, such as long term fermentation time, unstable quality, lack of knowledge of the exact mechanism of formation of the flavoring compounds, low mechanization, and labor-intensive procedure (Jin et al., 2017). The key factors that can help in solving these problems can only be determined by procuring comprehensive understanding of the microbial community composition of the SFB ecosystem. Furthermore, it is crucial to understand the functional dynamics of the dominant microbial strains present in SFB ecosystem. In an effort to achieve the same many microbes have already been isolated and cultured and their physiological and metabolic functions studied. The information thus obtained provided some crucial insights into the chemical nature and mechanism of formation of SFB flavoring compounds. Recent introduction of application of culture-independent methods, such as polymerase chain reaction denaturing gradient gel electrophoresis (PCR-DGGE) and Illumina sequencing have definitely enhanced our knowledge regarding the diversity and structure of the microbial community of SFB ecosystem (Ding et al., 2014b; Sun et al., 2016; Huang et al., 2017a). In this review paper, recent findings pertaining to the diversity and function of microbial community of SFB ecosystem are discussed. The article most specifically emphasizes on the results discovered from culture-independent method based research studies.
Microbial Diversity of Cultured Microorganism
Isolation and culture of microorganisms from the SFB ecosystem was initially started in the 1960s (Wu et al., 1991). Since then, many microbial strains have been screened and identified (Table 1). As far as bacterial diversity is concerned, two elaborate systematic studies encompassing the isolation and culture of bacteria from the SFB ecosystem (including, workshop environment, daqu, pit mud, and zaopei) were performed (Zhou et al., 2010; Wang T. et al., 2011). A total of 34 genera of bacteria were identified, of which Bacillus, Streptomyces, Lysinibacillus, Staphylococcus, Rummeliibacillus, Brevibacillus, and Brachybacterium were the most dominant (Zhou et al., 2010; Wang T. et al., 2011). Other dominant genera included Weissella, Pediococcus (Yang J.-G. et al., 2017), Lactobacillus, Acetobacter (Ming et al., 2013) from daqu; and Sporolactobacillus, Clostridium, Mycobacterium, and Flavobacterium from pit mud (Yue et al., 2007). In addition, six new species of bacteria [Paenibacillus vini (Chen et al., 2015), Bacillus vini (Ma et al., 2016), Lysobacter zhanggongensis (Zhang et al., 2017), Clostridium swellfunianum (Liu et al., 2014), C. luticellarii (Wang et al., 2015), and C. liquoris (Yin et al., 2016)] were detected and identified from the SFB ecosystem.
Table 1.
Microorganisms isolated and identified from strong flavor baijiu ecosystem through culture-dependent methods.
| Samples | Places | Isolated species/genus | Reference |
|---|---|---|---|
| Mature daqu | Gansu | Bacillus licheniformis, Bacillus cereus, Bacillus subtilis, Bacillus sonorensis, Brevibacillus sp., Bacillus amyloliquefaciens, Bacillus atrophaeus | Lin et al., 2012 |
| Mature daqu | Luzhou, Sichuan | Aspergillus, Monascus, Rhizomucor, Lichtheimia, Penicillium, Paecilomyces, Saccharomyces cerevisiae, Debaryomyces, Wickerhamomyces anomala, Sporidiobolus pararoseus, Merimbla ingelheimensis, Talaromyces, Cercospora, Cladosporium, Acremonium impicatum, Neurospora | Luo et al., 2013 |
| Mature daqu | Luzhou, Sichuan | Dominant bacteria: Lactobacillus, Acetobacter, Bacillus, Brettanomyces. Dominant fungi: Candida, Dekkera, Mucor, Aspergillus, Rizopus | Ming et al., 2013 |
| Mature daqu | Hunan | Bacillus licheniformis, Bacillus subtilis Bacillus amyloliquefaciens, and Bacillus cereus were dominant bacteria those possessed high activities of a-amylase and glucoamylase | Li et al., 2014 |
| Mature daqu | Luzhou, Sichuan | Ester-producing yeasts: Candida sp., Hansenula sp., Brettanomyces sp., Dekkera sp. | Xu, 2016 |
| Daqu fermented for 5, 7, 10, 25, and 90 days | Luzhou, Sichuan | The dominant bacteria, yeast, and mold strains in mature daqu were Bacillus subtilis subsp. inaquosorum, Saccharomycopsis fibuligera, and Lichtheimia ramosa. Mucor circinelloides f. circinelloides was strongly correlated with protease, and saccharifying enzyme activity was mainly correlated with Rhizopus oryzae | Yang J.-G. et al., 2017 |
| Pit mud | Luzhou, Sichuan | Facultative anaerobes: Bacillus, Sporolactobacillus, Pseudomonas, Clostridium, Mycobacterium, Pseudomonas, Microbacterium, Corynebacterium, Flavobacterium | Yue et al., 2007 |
| Pit mud | He Bei, Bei Jing, Si Chuan | Pediococcus pentosaceus, P. acidilactici, P. stilesii, P. dextrinicus, P. parvulus, P. inopinatus, P. ethanolidurans, P. damnosus, P. cellicola | Liu et al., 2007 |
| Pit mud | –# | Schizosaccharomyces pombe, Kluyveromyces thermotolerans, Hansenula polymorpha, Saccharomyces cerevisiae, Zygosaccharomyces rouxil | Yang et al., 2011 |
| Pit mud | – | Caproic acid producing strains: B. megaterium, B. fusiformis, B. licheniformis | Zhao H. et al., 2012 |
| Pit mud from 1 and 10 years old pits | Sichuan | Bacillus, Rummeliibacillus, Clostridium, Paenibacillus | Wang C.-D. et al., 2014 |
| Pit mud | Luzhou, Sichuan, Suqian, Jiangsu, Yibin, Sichuan | C. celerecrescens, C. indolis, C. tyrobutyricum, C. amylolyticum, C. butyricum, C. sartagoforme, and C. kluyveri | Hu et al., 2015 |
| Pit mud from 45 and 65 years old pits | Sangqiu, Henan | C. celerecrescens, C. cochlearium, C. carboxidivorans, C. sporogenes, C. sartagoforme, C. thermopalmarium, C. aurantibutyricum, C. butyricum | He et al., 2017 |
| Pit mud from 100 years old pit | Luzhou, Sichuan | Lysinibacillus sphaerieus, Brevibacillus brevis, Paenibacillus larvae subsup. pulvifacies | Liu Y. et al., 2017 |
| Zaopei fermented for 1, 4, 7, 14, 24, 34, 44 days | – | Corynebacterium xerosis, Staphylococcus auricularis, Bacillus subtilis, Bacillus megaterium, Bacillus cereus group, and Paenibacillus macerans produced lactic acid | Yao et al., 2010 |
| Zaopei | Suqian, Jiangsu | Lactococcus garvieae, Bacillus amyloliquefaciens, Pediococcus acidilactici, Staphylococcus pasteuri produced citrulline from arginine in high efficacy | Qiu et al., 2016 |
| Zaopei fermented for 0, 4, 8, 12, 20, 30, 44 days | Luzhou, Sichuan | Acetobacter malorum, Acetobacter cerevisiae, Bacillus methylotrophicus, Acetobacter aceti, Acetobacter estunensis, Acetobacter pasteurianus subsp. pasteurianus, Bacillus vanillea were the main species in initial fermentation stage, Bacillus amyloliquefaciens subsp. plantarum, Bacillus methylotrophicus, Bacillus atrophaeus, Bacillus subtilis subsp. inaquosorum, Lactobacillus buchneri, Lactobacillus paracasei subsp. tolerans, Bacillus vanilla were the dominant species in medium fermentation stage. Bacillus amyloliquefaciens subsp. plantarum, Bacillus subtilis subsp. inaquosorum, Bacillus sonorensis, Bacillus methylotrophicus, Bacillus vanillea, Bacillus atrophaeus, Gluconobacter cerinus were dominant in late fermentation stage | Dou et al., 2017 |
| Zaopei fermented for 0, 4, 8, 12, 20, 30, 44 days | Luzhou, Sichuan | Candida rugopelliculosa, Pichia fermentans, Naumovozyma castellii, Torulaspora delbrueckii, Saccharomyces cerevisiae, Pichia membranifaciens were dominant in 0, 4 days; Saccharomyces cerevisiae, Candida humilis, and Kazachstania exigua were dominant in 8 days; Naumovozyma castellii and Saccharomyces cerevisiae were dominant in 12 days; Saccharomyces cerevisiae were dominant in 20, 30 days; Candida ethanolica was found in 44 days | Yang J. et al., 2017 |
| SFB ecosystem∗ | Yibin, Sichuan | Bacillus, Lysinibacillus, Staphylococcus, Rummeliibacillus, Brevibacillus, Brachybacterium | Zhou et al., 2010 |
| SFB ecosystem | Yibin Sichuan | Streptomyces, Massilia, Nocardiopsis | Zhang et al., 2011 |
| SFB ecosystem | Yibin, Sichuan | Dominate genera: Bacillus, Streptomyces, Lysinibacillus, Staphylococcus | Wang T. et al., 2011 |
| SFB ecosystem | Yibin, Sichuan | Streptomyces utabilis, S. vinaceusdrappus, S. coelicoflavus, S. violascens produced ethyl lactate, caproic acid as the main volatile products | You and Wang, 2012 |
| SFB ecosystem | Yibin, Sichuan | Bacillus methylotrophicus, B. cereus, B. megaterium, B. aryabhattai, B. thuringiensis, B. tequilensis, Rhodococcus ruber, Brevudimonas naejangsanensis, Pseudomonas koreensis, Lysinibacillus sphaericus produced soy-like aroma | You et al., 2014 |
#, not available. ∗SFB ecosystem includes Daqu and fermentation workshop air, daqu, pit mud, zaopei.
On the other hand, Wickerhamomyces, Saccharomycopsis, Meyerozyma, Candida, Pichia, Cryptococcus, Brettanomyces, Dekkera, Issatchenkia, Debaryomyces, Saccharomyces, Rhodotorula, Schizosaccharomyces, Kluyveromyces, Hansenula, and Zygosaccharomyces were the yeast genera that were isolated and identified from the SFB ecosystem (Wu et al., 2006; Ming et al., 2013; Zhang X. et al., 2015; Yang J.-G. et al., 2017), among which Saccharomycopsis was the most dominant in daqu (Yang J.-G. et al., 2017); and Issatchenkia, Pichia, and Candida in zaopei (Wu et al., 2006; Zhang X. et al., 2015). Lichtheimia, Aspergillus, Penicillium, Rhizomucor, Mucor, Rhizopus, Monascus, Emericella, Cladosporium, and Gibberella were the mold genera isolated and identified from the ecosystem (Pu et al., 2012; Wang et al., 2012; Ming et al., 2013; Zhang X. et al., 2015; Yang J.-G. et al., 2017). It was also observed that among these mold genera, Penicillium and Aspergillus were the most dominant in the brewing workshop (Wang et al., 2012), while Lichtheimia was dominant in daqu (Yang J.-G. et al., 2017). Similarly, Aspergillus, Monascus, and Rhizopus were found to be most dominant in zaopei (Zhang X. et al., 2015). In addition, three archaea were also isolated. Two of them were identified as Methanobacterium bryantii (Wu et al., 1990) and Methanobacterium formicicum (Wang et al., 2010).
Physiological Traits of Cultured Microorganisms
The production of SFB involves three basic functional processes: saccharification, alcohol fermentation, and flavor compound formation. Saccharification is achieved through extracellular enzymes (amylase, glucoamylase, protease, cellulose, and lipase) that are secreted by the microorganisms present in the SFB ecosystem, mostly in daqu. For amylase, Bacillus and Staphylococcus are the main bacterial producers, and for glucoamylase, Bacillus, Enterobacter, and Weissella are the main bacterial producers (Lin et al., 2012; Li et al., 2014; Yang J.-G. et al., 2017). In addition, Rhizopus, and Lichtheimia were found to produce amylase; and Aspergillus, Mucor, Penicillium, Rhizopus, Gibberella, and Cladosporium, produced glucoamylase (Pu et al., 2012; Li et al., 2014; Yang J.-G. et al., 2017). Some of the chief microbial cellulase producers included, Bacillus, Penicillium, Aspergillus, and Alternaria (Zeng et al., 2016). In addition, Staphylococcus gallinarum, Mucor circinelloides f. circinelloides were found to secrete protease (Yang J.-G. et al., 2017); and Rhizopus chinensis lipase (Wang D. et al., 2013).
Yeasts are the main ethanol producers in SFB ecosystem. Saccharomyces cerevisiae was the main ethanol producer (Hu et al., 2017) and was dominant in the zaopei fermentation process (Yang J. et al., 2017). Ethyl hexanoate was identified as the most typical flavoring compound of SFB (Zheng and Han, 2016). In an effort to elucidate the process of its formation many hexanoate producing microorganisms have been identified and studied. Generally, hexanoate is produced by the members of the genus Clostridium by using glucose, lactate, ethanol and acetic acid as substrates (De Araújo Cavalcante et al., 2017). Some of the most common hexanoate producing Clostridium species include, C. kluyveri (Hu et al., 2015), C. lushun (Wu and Yi, 1986), Clostridium Sp. W1 (Xue et al., 1988), and C. celerecrescens (Zhao H. et al., 2012; Xue and Xue, 2016). In addition, Bacillus megaterium, Bacillus fusiformis, and Bacillus licheniformis are also known to produce hexanoate (Zhao H. et al., 2012). Ethyl lactate, ethyl acetate, and ethyl butanoate are another representative compounds in SFB. Lactic acid bacteria (LAB) were found as the dominant microorganisms in mature daqu (Ming et al., 2013) and zaopei (Dou et al., 2017), contributing to the lactic acid production. Other lactic acid producers in SFB ecosystem included: Corynebacterium xerosis, Staphylococcus auricularis, Bacillus subtilis, Bacillus megaterium, Bacillus cereus group, and Paenibacillus macerans (Yao et al., 2010). Acetic acid was produced via genus Acetobacter, which was found as one of the main genera in the initial stage of zaopei fermentation (Dou et al., 2017). Furthermore, specific members of the genus Clostridium were also identified to biosynthesize acetate, butanoate, and lactate from various substrates, such as sugar, starch, and cellulose (Dürre, 2016). In addition, Bacillus, Lysinibacillus, Sporosarcina, Staphylococcus isolated from SFB ecosystem were found to produce different types of organic acids (Tang et al., 2013). These organic acids, together with ethanol get esterified into corresponding ethyl esters via a reaction catalyzed by esterases. Further investigations revealed that species of Penicillium, Aspergillus, Emericella, Rhizopus, Cladosporium, Mucor, Hansenula, Candida, Hansenula, Brettanomyces, and Dekkera isolated from SFB ecosystem are capable of secreting esterases (Wang et al., 2012; Xu, 2016). Other flavoring compounds of SFB, such as ketones, pyrazine, and phenylethanol were also found to be produced by Bacillus, Wickerhamomyces anomalus (Ming et al., 2015; Zhou et al., 2016).
In addition to the beneficial microorganisms that were responsible for the characteristic taste and flavor of SFB production, certain other unpleasant flavoring compound producing strains were also detected. p-Cresol was identified as the major off-odor and toxic component present in SFB. It was later found to be produced by C. butyricum, C. tyrobutyricum, C. aminovalericum, C. ultunense, and C. purinilyticum (Du et al., 2017; Liu B. et al., 2017). Ethyl carbamate is another potential carcinogenic compound that was found to be present in SFB. Citrulline, one of the precursors of ethyl carbamate (Wang H. et al., 2014) that was found to be aacumulated (by using argine) by Lactococcus garvieae, Bacillus amyloliquefaciens, Pediococcus acidilactici, and Staphylococcus pasteuri (Qiu et al., 2016). On the other hand, geosmin that has an earthy off-flavor was found to be produced by Streptomyces isolated from SFB ecosystem (Wang T. et al., 2011; Du and Xu, 2012).
Microbial Diversity Revealed by Culture-Independent Methods
Although the research studies based on traditional microbial culture methods helped in gaining preliminary insights into the microbial diversity of SFB ecosystem, it was realized that these procedures were unable to study a large number of microbes whose isolation and culturing is difficult to achieve with the help of adept microbiological methods (Kaeberlein et al., 2002). This led to the implementation of various culture-independent methods that were directed toward attaining comprehensive understanding of microbial diversity of the SFB ecosystem (Table 2). PCR-DGGE and sequencing technology were mostly used to investigate the microbial diversity of the SFB ecosystem via the culture-independent approach.
Table 2.
Studies on microbial diversity of strong flavor baijiu ecosystem with culture-independent methods.
| Samples | Locations | Methods | Main species or results | Reference |
|---|---|---|---|---|
| Mature daqu | Haozhou, Anhui | PCR-cloning | Lactobacillus, Pantoea, Enterobacter, Klebsiella, Leuconostoc, Erwinias, Pseudomonas, Bacillus licheniformis | Bin et al., 2011 |
| Mature daqu | Huaian, Jiangsu; Mianzhu, Sichuan | PCR-DGGE | Dominant bacteria: lactic acid bacteria and Staphylococcus xylosus. Dominant yeasts Saccharomycopsis fibuligera and Pichia anomala. Dominant molds: Rhizomucor miehei, Absidia blakesleeana and Aspergillus terreus | Wang H.Y. et al., 2011 |
| Mature daqu | Luzhou, Sichuan | Nested PCR-DGGE | Dominant bacteria: Lactic acid bacteria and Bacillus were. Dominant yeasts: Saccharomycopsis fibuligera, Wallemia sebi, Wallemia muriae, and Pichia subpelliculosa. Dominant molds: Aspergillus | Zhang et al., 2014 |
| Mature daqu | Sichuan | Cloning | Main bacteria: Thermoactinomyces sanguinis, Enterobacter cloacae, Pantoea agglomerans, and uncultured bacteria. Main molds: Aspergillus glaucus, Thermomyces lanuginosus, and Thermoascus crustaceus | Gou et al., 2015 |
| Mature daqu | 40.02°N; 28.88° N; 28.55°N | Gene clone libraries | Staphylococcus gallinarum, Staphylococcus saprophyticus were only found in southern daqu. Saccharomycopsis fibuligera and Lichtheimia ramosa were dominated fungi; Bacillus licheniformis, S. fibuligera and one uncultured bacterium were detected in all samples | Zheng X.W. et al., 2015 |
| Daqu fermented 0, 2, 4, 6, 8, 10, 12, 17, 27, and 32 days | Luzhou, Sichuan | 454 pyrosequencing and Illumina MiSeq sequencing | In the first 4 days of fermentation, most bacterial taxa, and several fungal taxa containing Candida, Wickerhamomyces, and unclassified Dipodascaceae, and Saccharomycetales, grow well. From day 4 to day 12, thermotolerant taxa including Bacillus, unclassified Streptophyta, Weissella, Thermoactinomyces, Thermoascus, and Thermomyces survived or kept on growing. Lactic acid bacteria related to Weissella, Leuconostoc, and Lactobacillus were dominant bacteria through fermentation, while Bacillus became a dominant genus after 10 days of fermentation. Thermoascus, Candida, Wickerhamomyces, and Thermomyces were dominant fungal genera through fermentation | Xiao et al., 2017 |
| Daqu fermented 0, 3, and 9 days, and mature daqu | Yibin, Sichuan | 454 pyrosequencing | Lactobacillales became dominant during the first 3 days and then decreased markedly. Bacillales became dominant in 9 days and mature daqu. Staphylococcus spp. and Chryseobacterium spp. were the most abundant genera in common across the 4 samples. Acetobacter and Lactobacillus increased quickly from 0 day to 3 days and decreased later. Saccharomycetales were predominant fungi after 3 days of incubation. Saccharomycetales and no-rank Eukaryota were dominant in 9 days. Eurotiales became the dominant in mature daqu. Pichia was the dominant genus. | Huang et al., 2017b |
| Pit mud from 20, 100 and over 300 years old pits | Luzhou, Sichuan | PLFA | The microbial community of pit mud was composed of bacteria, actinomycetes and fungi, with Gram-positive bacteria and anaerobic bacteria being dominant. As the pit age increased, pit mud biomass increased and the microbial community shifted to Gram positive bacteria | Zhao et al., 2012 |
| Pit mud from 20, 50, 100, 200, and 300 years old pits | Luzhou, Sichuan | PCR-DGGE and PLFA | Dominant bacteria: Clostridiales, Lactobacillales, and Bacillales; Dominant yeasts: Wickerhamomyces, Kluyveromyces, Pichia, and Pichia anomala | Zheng et al., 2013 |
| Pit mud from 1, 2, 3, and 4 years old pits | Luzhou, Sichuan | PCR-DGGE and FISH | Dominant bacteria: Leuconostocaceae, Clostridiaceae, Lactobacillaceae, Moraxellaceae, Enterococcaceae, Lachnospiraceae, Comamonadaceae, Sphingomonadaceae, and Ruminococcaceae; Dominant archaea: Methanobrevibacter, Methanobacterium, and Methanoculleus | Ding et al., 2014a |
| Pit mud from the wall and bottom of 200 years old pits | Luzhou, Sichuan | PCR-DGGE | Clostridium was the dominant eubacteria; Methanoculleus, and Methanosaeta were the main archaea; eubacteria and archaea community diversities in samples from the bottom were almost higher than that from the wall; Acinetobacter was found in all samples from the wall, but not the bottom | Ding et al., 2014b |
| Pit mud from 50, 140, 220, 440 years old pits | Luzhou, Sichuan | Metagenomics sequencing | The microbial communities in all the pits were dominated by Firmicutes. The youngest pit had the highest proportions of Gammaproteobacteria and opisthokonts. The abundances of Euryarchaeota and Bacteroidetes increased as the age of pit mud increasing | Guo et al., 2014 |
| Pit mud from the bottom of aged and aging pits | Anhui | 16S rRNA gene clone libraries and quantitative real time PCR | Firmicutes and Chloroflexi predominated in the aged pit mud while Firmicutes and Bacteroidetes predominated in the aging pit mud. Chloroflexi and Actinobacteria were only detected in the aged pit mud. The quantity of Actinobacteria in the aged pit mud was 29 times as much as in the aging pit mud | Luo et al., 2014a |
| Pit mud from aged and aging pits | Anhui | Gene clone libraries and amplified ribosomal DNA restriction analysis | Bacteroidetes and Firmicutes predominated in both the aged and aging pit mud, but Synergistetes and Actinobacteria were only detected in the aged pit mud. The Methanosaeta dominated in the aged pit mud, while the Methanosarcina predominated in the aging pit mud | Luo et al., 2014b |
| Pit mud from 1, 10, 25, and 50 years old pits | Mianzhu, Sichuan | Pyrosequencing | Dominant genera include Petrimonas, unclassified Clostridiaceae, Methanoculleus, Methanosarcina, Methanobacterium, Methanobrevibacter, Lactobacillus, Clostridium IV, Sedimentibacter, Syntrophomonas, Spirochaetes SHA-4, Methanobrevibacter, and unclassified Porphyromonadaceae, Anaerobrancaceae, and Ruminococcaceae | Tao et al., 2014 |
| Zaopei, pit mud, and huangshui from 2, 10, and 30 years old pits | Yibin, Sichuan | PCR-DGGE and PLFA | All the eubacteria belonged to Lactobacillaceae, Clostridiaceae, Porphyromonadaceae, Synergistaceae, and Acetobacteraceae. Lactobacillaceae was dominant eubacteria in the ZP, while Clostridiaceae was dominant eubacteria in the PM and HS, respectively. Methanosaeta, Methanocorpusculum, Methanobrevibacter, Methanobacterium, and Methanoculleus were the majority of archaea. Methanosaeta, increased gradually in the PM and HS with pit age, and decreased in the ZP. Pichia was dominated in fungal community | Ding et al., 2015 |
| Pit mud from aged and aging pits | Sichuan | PCR-DGGE and quantitative PCR | Clostridiales was dominant in aged pit mud while Bacillales and Lactobacillales were dominant in aging pit mud | Liang et al., 2015 |
| Pit mud from 1, 50, 100, and 300 years old pits | Luzhou, Sichuan | PCR-DGGE and FISH | Methanobacteriales dominated in low-age (1 and 50 years) pit mud. Methanomicrobiales dominated in old age (100 and 300 years) pit mud | Wu et al., 2015 |
| Pit mud from 1 and 2 years old pits | Yibin, Sichuan | nested PCR-DGGE, PLFA, PLEL, FISH | Dominated bacteria: Clostridiales, Lactobacillales, Bacteroidales, and Rhizobiales. Dominated archaea: Methanomicrobiales and Methanosarcinales. Dominated fungi: Saccharomycetales and Eurotiales | Zhang L. et al., 2015 |
| Pit mud from 30 and 300 years old pits | Luzhou, Sichuan | iTRAQ-based proteomic approach and high-throughput sequencing | The aroma-forming functional proteins in 300-year pit mud were highly expressed with much higher content than that of 30-year pit mud, Firmicutes and Methanobacterium, were important components of aroma-forming functional colonies in the pit muds | Zheng et al., 2015 |
| Degraded, normal, and high quality pit mud | Jiangsu | Illumina MiSeq sequencing | Core genera in all samples included: Lactobacillus, Ruminococcus, Caloramator, Clostridium, Sedimentibacter, Syntrophomonas, Sporanaerobacter, Pelotomaculum, T78, Prevotella, Blvii28 group, Methanobacterium, Methanobrevibacter, Methanosaeta, Methanoculleus, Methanosarcina, and Nitrososphaera. Clostridia, Bacteroidia, Methanobacteria, and Methanomicrobia, may play important roles in pit mud ecosystem stability, which may be destroyed with rapidly increased levels of lactic acid bacteria (Lactobacillus, Pediococcus, and Streptococcus) | Hu et al., 2016 |
| Matured and degenerated pit mud | Sichuan, Anhui | PCR-DGGE and qPCR | Bacterial community in the degenerated pit mud did not change with different regions. Bacterial community in the matured pit mud from different regions could be different. Actinobacteria could serve as an indicator to distinguish pit muds | Liang et al., 2016 |
| Pit mud from 5 and 100 years old pits | Luzhou, Sichuan | PCR-DGGE, illumina MiSeq sequencing | Rhizopus, Aspergillus, Phoma, Trichosporon, Candida, Thermoascus, Wickerhamomyces, Penicillium, Thermomyces, Debaryomyces, Saccharomyces, Malassezia, Mucor, Davidiella, Wallemia, Toxicocladosporium, Fusarium, Pichia, and Cladosporium were identified as core genera. Rhizopus, Phoma, and Trichosporon were relatively richer in the 5-year PM samples, and Aspergillus and Candida were rich in the 100-year PM samples | Liu M. et al., 2017 |
| Pit mud from 30 years old pits | Mianzhu, Sichuan | Illumina sequencing | The dominant prokaryotic phyla were Firmicutes, Euryarchaeota, Bacteroidetes, Actinobacteria, and Proteobacteria. Clostridial cluster IV, Lactobacillus, Caloramator, Clostridium, Sedimentibacter, Bacteroides and Porphyromonas were active populations in situ, in which Clostridial cluster IV and Clostridium were likely involved in the hexanoate production. | Tao et al., 2017 |
| Pit mud from 40 and 400 years old pits | Luzhou, Sichuan | Illumina MiSeq sequencing | Methanobrevibacter, Caproiciproducens, Petrimonas, Lactobacillus, Sedimentibacter, Proteiniphilum, Syntrophomonas, Aminobacterium, Christensenellaceae R-7, Caldicoprobacter, and Olsenella were the dominate genera. PM hosts a large number of novel taxa. The class Clostridia presented the highest proportion of novel OTUs. | Liu M.-K. et al., 2017 |
| Zaopei fermented 0, 1, 4, 7, and 10 weeks | Sichuan | DGGE and gene clone | Diversity of bacteria in Zaopei decreased and after 1 week, only one bacterium phenotype was dominant. Lactobacillus acetotolerans appeared to play a key role during Chinese liquor fermentation. | Zhang et al., 2005 |
| Zaopei fermented 0, 1, 4, 7 and 10 weeks, from the center and edge of the middle layer of the pit | Sichuan | DGGE and gene clone | Issatchenkia, Talaromyces, Aspergillus and Eurotium were the main dominant during the fermentation process. Talaromyces, and Issatchenkia were dominant fungal communities during the early stage of fermentation. After 4 weeks of fermentation, Talaromyces, Eurotium, and Aspergillus, became dominant. | Zhang et al., 2007 |
| Multiple grains or single grains zaopei collected from the top layer and bottom layer of pits | – | DGGE and culture method | Debaryomyces, Pichia and Candida were dominant in multiple-grains zaopei. Candida was dominant in single-grains zaopei. Thermophilic fungi (Thermomyces lanuginosus and Thermoascus aurantiacus) were detected. Fungi communities in the top layer were richer than those in the bottom | Shi et al., 2011 |
| Zaopei fermented 5, 20, and 40 days in summer or winter | Sichuan Province | Illumina Miseq sequencing | Bacterial population was mainly represented by Acetobacter and Lactobacillus both in winter and summer zaopei. The summer zaopei contained significantly higher proportions of LAB and lower proportions of Acetobacter than winter zaopei. Thermoactinomycetaceae, Prevotella, Alcaligenes, and Gluconacetobacter were identified | Sun et al., 2016 |
| Pit mud, zaopei, and huangshui from new, 5-year, and 20-year pits | Yibin, Sichuan | FISH, PLFA, PCR-DGGE | Lactobacillus, Clostridium, Sedimentibacter, Eubacterium, uncultured bacterium were dominated in pit mud. Lactobacillus, Clostridium, Sedimentibacter, and uncultured bacterium were dominated in zaopei. Methanobrevibacter, Methanocorpusculum, Methanoculleus, Saccharomycopsis, and Galactomyces were detected in all samples. | Li et al., 2017 |
| Mature daqu, pit mud, and zaopei fermented for 3, 15, and 45 days | Hunan | Illumina sequencing | Lactobacillus, Leuconostoc, Staphylococcus, Gluconobacter, Acetobacter, Petrimonas, Clostridium, Ruminococcus, Methanobacterium and Methanobrevibacter were dominant in 3 days’ zaopei. Lactobacillus was the predominant genus in 15 and 45 days’ zaopei. Methanobacterium, Methanobrevibacter, Methanoculleus, Methanosarcina, Petrimonas, Lactobacillus, Sedimentibacter, Clostridium, Ruminococcus, Syntrophomonas, and Symbiobacterium were dominant in pit mud. Micromonospora, Petrimonas, Staphylococcus, Thermoactinomyces, Pediococcus, Lactobacillus, Leuconostoc, Weissella, Lactococcus, Sedimentibacter, Clostridium, Ruminococcus, Pantoea, and Pseudomonas were dominant in daqu. | Wang X. et al., 2017 |
| Huangshui from 20-year old pit | Sichuan | SSU rRNA library | Proteobacteria, Firmicutes, Bacteroidetes, Lentisphaerae, Actinobacteria, Tenericutes, and an unclassfied domain, respectively. The Firmicutes and Proteobacteria were the dominant in yellow water. The Clostridium, Lactobacillus, and Serratia were the dominant genus. Archea community in yellow water mostly consisted of genera Methanosarcina and Methanoculleus. | Li K. et al., 2015 |
#, not available.
Microbial Diversity of Daqu
Daqu is the saccharification and fermentation agent used in the process of SFB production (Zheng et al., 2011). Daqu provides: microbial strains responsible for carrying out SFB fermentation. It also provides hydrolytic enzymes that can hydrolyze the macromolecules present in the fermented cereals; and flavoring compounds that act as precursors of SFB; as well as part of the fermentation material (Hu et al., 2004).
As far as bacterial diversity of daqu is concerned, Bacillus species have been detected in various SFB distilleries, which were analyzed with both culture-dependent as well as culture-independent methods. Among the various species identified, B. licheniformis was found to be the most common (Bin et al., 2011; Lin et al., 2012; Li et al., 2014; Zhang et al., 2014; Zheng X.W. et al., 2015). B. licheniformis present in daqu were found to secrete amylase (Li et al., 2014), protease, and some flavoring compound precursors (Yan et al., 2015). A recent study showed that inoculation of B. licheniformis in daqu fermentation changed the entire microbial community structure and metabolic profile of daqu (Wang P. et al., 2017).
In addition to B. licheniformis, daqu was also found to contain LAB (predominantly Lactobacillus) (Bin et al., 2011; Wang H.Y. et al., 2011). Members of LAB are known to be the main producers of lactic acid, which subsequently helps in the synthesis of ethyl lactate (via esterification). Apart from these, other genera detected in daqu included Pseudomonas, Pantoea, Enterobacter, Klebsiella, Leuconostoc, Erwinia, Geobacillus, Weissella, and Staphylococcus (Wang H.Y. et al., 2011; Gou et al., 2015; Zheng X.W. et al., 2015). Furthermore, actinomycetes, mostly Thermoactinomyces were detected as the dominant bacteria in daqu (Gou et al., 2015; Huang et al., 2016).
Understandably, yeasts are necessary for the production of alcohol. The yeast species present in SFB daqu can be divided into two groups: (i) those responsible for the production of ethanol (Saccharomyces that convert glucose into ethanol); and (ii) those responsible for the production of different kinds of esters (flavoring compounds of SFB), e.g., Pichia. The most dominant yeast varieties identified from SFB daqu included Saccharomycopsis (Wang H.Y. et al., 2011; Zhang et al., 2014; Huang et al., 2016), Pichia (Wang H.Y. et al., 2011; Zhang et al., 2014), and Wickerhamomyces (Zhang et al., 2014). Saccharomycopsis fibuligera was the most dominant yeast in SFB daqu (Wang H.Y. et al., 2011; Zhang et al., 2014; Huang et al., 2016) and was found to secrete amylases, acid proteases, and β-glucosidases, which in turn helped in starch degradation and their subsequent alcoholic fermentation (Chi et al., 2009). Wickerhamomyces anomalus was found to produce intra- and extracellular glucoside hydrolases, arabinosidase, and xylosidase (Sabel et al., 2014). These enzymes are highly important for the wine aroma of SFB. Other non-Saccharomyces yeast species found in SFB daqu included Hanseniaspora, Issatchenkia, Trichosporon, Debaryomyces, and Sporidiobolus (Wang H.Y. et al., 2011; Luo et al., 2013). In addition to yeast, molds were also found to dominate SFB daqu. Aspergillus [from the surface of wheat (Xu et al., 2004) or brewing workshop (Wang et al., 2012)] and Lichtheimia [isolated via culture-based method (Yang J.-G. et al., 2017)], were detected to be the most commonly found species in different SFB daqu samples (Wang H.Y. et al., 2011; Luo et al., 2013; Gou et al., 2015; Zhang X. et al., 2015). Furthermore, Thermomyces, Thermoascus, Absidia, and Geotrichum were identified as the most commonly occurring mold species that could not be isolated via the culture-based methods.
Microbial Diversity of Pit Mud
SFB is usually produced by fermentation of cereals in an underground mud pit. The inside of these pit walls are covered with pit mud (PM), which significantly contributes toward maintaining the microbial diversity necessary for SFB fermentation. The microbial community of PM is most dominantly composed of eubacteria (especially, gram-positive and anaerobic bacteria; Zhao et al., 2012), archaea and fungi. The results obtained from these culture-independent methods indicated the dominant presence of Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria, and Synergistetes from the eubacteria domain and some Unclassified Bacteria (Ding et al., 2014b; Luo et al., 2014b; Liang et al., 2015). It is noteworthy that Firmicutes was found to predominate in many PM microbial communities (Guo et al., 2014; Luo et al., 2014a,b; Liang et al., 2015, 2016). Among Firmicutes, Clostridiales, Lactobacillales, and Bacillales were the main bacteria found in different SFB PMs (Zheng et al., 2013; Liang et al., 2015; Zhang L. et al., 2015). Furthermore, Clostridium (order Clostridiales) was detected as one of the most predominant bacteria in the PM microbial community and many specifies belonging to the genus Clostridium have already been isolated and identified from PM (Ding et al., 2014a; Tao et al., 2014; Zheng et al., 2015; Hu et al., 2016; Liang et al., 2016; Li et al., 2017). Some of the most common examples of such species include Clostridium kluyveri (Hu et al., 2015), C. swellfunianum (Liu et al., 2014), C. butyricum (Li et al., 2016), and C. liquoris (Yin et al., 2016). It was also proposed that these members contribute in the: (1) production of organic acids (acetic, butyric, and hexanoic (caproic) acid), which then gets esterified with ethyl alcohol via enzymatic and non-enzymatic catalysis to form ethyl butyrate and caproate; (2) production of H2 for the synergistic metabolism of methanogens. Meanwhile, Ruminococcus, Syntrophomonas, Desulfotomaculum, Anaerobrancaceae, Pelotomaculum, Eubacterium, and Butyrivibrio were identified as the most dominant members of Clostridiales (Ding et al., 2014a; Hu et al., 2014, 2016; Tao et al., 2014; Zheng et al., 2015; Liang et al., 2016; Li et al., 2017). On the other hand, Lactobacillus, especially Lactobacillus acetolerans, Lb. alimentarius and Lb. acetolerans (Zheng et al., 2013), followed by Lactococcus (Liang et al., 2016), were the most dominant of all Lactobacillales found in PMs (Ding et al., 2014a; Tao et al., 2014; Zheng et al., 2015; Hu et al., 2016; Liang et al., 2016; Li et al., 2017). Interestingly, Bacillales, especially those belonging to the genus Bacillus, were mostly detected in young PMs (Ding et al., 2014b; Liang et al., 2016). Virgibacillus is also detected (Ding et al., 2014b). Other commonly detected bacterial strains belonged to the phylum Firmicutes, and included members of Sedimentibacter (Ding et al., 2014b; Tao et al., 2014; Zheng et al., 2015; Hu et al., 2016; Li et al., 2017), Sporanaerobacter (Hu et al., 2016), and Tissierella (Liang et al., 2016).
Apart from Firmicutes, other commonly observed bacterial species found in PM included: Proteobacteria, Pseudomonas (Liang et al., 2016), Bacteroidetes, Petrimonas, Prevotella (Tao et al., 2014; Hu et al., 2016), Chloroflexi (Luo et al., 2014a), Actinobacteria, Rhodococcus, Microbacterium, Acinetobacter (Ding et al., 2014b; Liang et al., 2016), Synergistetes, Altererythrobacter (Liang et al., 2016), and Aminobacterium (Zheng et al., 2015; Liang et al., 2016). It is noteworthy that Chloroflexi, Synergistetes, and Actinobacteria were only detected in aged PMs (Luo et al., 2014a,b).
Among the various categories of fungal strains, those belonging to the order Saccharomycetales were found to be the most dominant (Zhang L. et al., 2015). Pichia, Wickerhamomyces, Saccharomyces, and Galactomyces were found to constitute a major portion of the core fungal strains isolated from different PM samples (Zheng et al., 2013, 2015; Ding et al., 2015; Li et al., 2017; Liu M. et al., 2017). In addition, Kluyveromyces (Zheng et al., 2013), Zygosaccharomyces, Geotrichum (Ding et al., 2015), Saccharomycopsis, Issatchenkia (Li et al., 2017), and Debaryomyces (Liu M. et al., 2017) were also detected. Aspergillus is another fungal genus that was categorized as the chief constituent of the fungal diversity of PMs (Ding et al., 2015; Zheng et al., 2015; Liu M. et al., 2017). Other genera that formed an integral part of core fungal diversity included Rhizopus, Phoma, Trichosporon, Thermoascus, Penicillium, Thermomyces, Malassezia, Mucor, Davidiella, Wallemia, Toxicocladosporium, Fusarium, and Cladosporium (Liu M. et al., 2017). Among them, Rhizopus, Phoma, and Trichosporon were found to be relatively richer in young PM samples, while Aspergillus and Candida were in older PM samples (Liu M. et al., 2017).
Methanobrevibacter, Methanobacterium, and Methanoculleus were found to be the most predominant genera of the domain archaea that were found in different PMs (Ding et al., 2014a,b, 2015; Luo et al., 2014b; Tao et al., 2014; Wu et al., 2015; Zheng et al., 2015; Hu et al., 2016; Li et al., 2017). Few other studies indicated the presence of members of Methanosaeta, Methanosarcina (Ding et al., 2014b, 2015; Luo et al., 2014b; Tao et al., 2014; Wu et al., 2015; Zheng et al., 2015; Hu et al., 2016)., Nitrososphaera (Hu et al., 2016), Methanocorpusculum (Ding et al., 2015; Li et al., 2017), Methanocorpusculum (Wu et al., 2015), Methanoplanus, Methanotorris, Methanolobus, Methanothermobacter, and Methanomethylovorans (Zheng et al., 2015).
Microbial Diversity of Zaopei
Zaopei represents the fermented cereals that are placed inside the pit cellar for alcohol fermentation and formation of flavoring compounds. Typically, fresh zaopei is essentially a mixture of steamed cereals, steamed rice husks, and daqu powder (Figure 1). Microorganisms enter into the zaopei via daqu powder, PM and the ambient brewing workshop environment. The primary role of the bacteria existing in zaopei is to produce varieties of flavoring compounds or precursor of those compounds, such as caproic acid, lactic acid, and butyric acid. It was found that these bacteria mostly belonged to the Lactobacillaceae and Acetobacteraceae families (Zhang et al., 2005; Sun et al., 2016; Li et al., 2017). Subsequent studies indicated that Lactobacillus acetotolerans was the most dominant strain present in zaopei (Zhang et al., 2005). Other members of LAB that were detected were Streptococcus, Lactococcus, Leuconostoc, and Weissella (Zhang et al., 2005; Sun et al., 2016). Apart from Lactobacillus, Acetobacter was found to be another chief microbial constituent of zaopei (Sun et al., 2016). In addition, members of Bacillus were found to be the main bacterial strains that were isolated and cultured from zaopei (Wang T. et al., 2011). Clostridium, which is a dominant bacterium in PM, was also identified in zaopei (Zhang et al., 2005; Sun et al., 2016; Li et al., 2017). Other not so dominant genera identified in zaopei were Erwinia, Kozakia, Staphylococcus, Granulicatella, Arthrobacter, Microbacterium, Shewanella, Sporolactobacillus, Thermoactinomyces, Desmospora, Alcaligenes, Gluconacetobacter, Prevotella, and Sedimentibacter (Zhang et al., 2005; Sun et al., 2016; Li et al., 2017).
As far as fungal diversity is concerned, members of the order Saccharomycetales were identified as the main fungal strains present in different zaopei samples (Shi et al., 2011; Li et al., 2017). Candida, Issatchenkia, Debaryomyces, and Pichia were reported as the main fungal genera identified in it. Other Saccharomycetales found in the zaopei included: Torulaspora, Zygosaccharomyces, Saccharomycopsis, Citeromyces, Galactomyces, Hyphopichia, Cyberlindnera, Geotrichum, Magnusiomyces, and Kluyveromyces (Zhang et al., 2007; Shi et al., 2011; Li et al., 2017). Some other dominantly present fungal species reported are: Talaromyces, Aspergillus, Eurotium (Zhang et al., 2007), Fomitopsis, Trichosporon, Thermomyces, and Thermoascus (Zhang et al., 2007; Shi et al., 2011).
In addition, five genera from archaea, namely Methanocorpusculum, Methanobrevibacter, Methanobacterium, Methanoculleus (most dominant; Ding et al., 2015), and Methanosaeta were also detected in zaopei (Sun et al., 2016; Li et al., 2017).
Microbial Diversity of Huangshui
Huangshui (HS) is the brown viscous liquid which is formed by the liquid that permeates to the bottom of pit during the fermentation process. It was found to be full of microbial strains that have evolved through long term domestication. Lactobacillus and Clostridium were identified as the dominant bacterial genera present in HS (Ding et al., 2015; Li K. et al., 2015; Li et al., 2017). Other commonly occurring genera detected in HS included Acetobacter, Proteiniphilum, and Caloramator (Ding et al., 2015; Li K. et al., 2015; Li et al., 2017). Methanocorpusculum, Methanoculleus, Methanosarcina, Methanobrevibacter, Methanobacterium, and Methanosaeta were the prevalently detected genera belonging to archaea in HS, out of which the former two genera were the most dominant (Ding et al., 2015; Li K. et al., 2015; Li et al., 2017). In addition, fungal genera viz. Aspergillus, Geotrichum, Galactomyces, Pichia, Zygosaccharomyces, and Candida were also detected in HS (Ding et al., 2015; Li et al., 2017), among which Pichia was found to be the most dominant (Ding et al., 2015).
Microbial Community Dynamics in SFB Ecosystem
For the microbial community dynamics of daqu, the rapid propagation of most bacterial taxa (especially Lactobacillales), and several fungal taxa containing Candida, Wickerhamomyces, unclassified Dipodascaceae, and Saccharomycetales significantly enhanced the temperature at the initial fermentation stage (Xiao et al., 2017). Wickerhamomyces anomalus, Candida metapsilosis were isolated and identified as the main yeasts, and Rhizopus oryzae as the main molds in this stage (Yang J.-G. et al., 2017). When the fermented temperature increased to highest values (about 55°C), thermotolerant taxa including Bacillus, unclassified Streptophyta, Weissella, Thermoactinomyces, Thermoascus, and Thermomyces, Saccharomycetales, and no-rank Eukaryota were dominant (Huang et al., 2017b; Xiao et al., 2017). For mature daqu, Bacillales and Eurotiales became the dominant bacterial and fungal taxon, respectively (Huang et al., 2017b). After 10 days fermentation, Saccharomycopsi fibuligera, Bacillus subtilis subsp. inaquosorum, Lichtheimia ramose were isolated and identified as the dominant yeast, bacterium, and mold, respectively (Yang J.-G. et al. 2017). In addition, Lactic acid bacteria related to Weissella, Leuconostoc, and Lactobacillus were dominant bacteria through fermentation (Xiao et al., 2017). Thermoascus, Candida, Wickerhamomyces, and Thermomyces were dominant fungal genera through fermentation (Xiao et al., 2017).
For the microbial community dynamics in zaopei, Lactobacillus, Leuconostoc, Methanobacterium, Clostridium, Acetobacter, Gluconobacter, Staphylococcus, Petrimonas, Methanobrevibacter, and Ruminococcus were the main prokaryotic genera at the beginning of fermentation (Wang X. et al., 2017). After 2 weeks fermentation, Lactobacillus became dominant, and other bacterial genera decreased (Wang X. et al., 2017). At the end of zaopei fermentation, only Lactobacillus was absolutely dominant (Zhang et al., 2005; Wang X. et al., 2017). However, culture method identified that Bacillus and Lactobacillus were both the dominant bacterial genera during the zaopei fermentation (Dou et al., 2017). Talaromyces and Issatchenkia are the dominant fungus at the beginning stage, Eurotium and Aspergillus became the dominant genera after 4 weeks fermentation (Zhang et al., 2007). However, Saccharomyces cerevisiae, the main ethanol producer, was not identified as the dominant species during the fermentation by the sequencing method, which was not in accordance with the result obtained by traditional culture methods (Yang J. et al., 2017).
Systems Biology Based Analysis of SFB Microbial Community
The diversity of microbial community of the SFB ecosystem has been widely studied in the past few years (Tables 1, 2). However, a comprehensive understanding of such complex microbial communities necessitates establishing links between the active microbial diversity and their functional aspects (Maukonen and Saarela, 2009). Until now, the metabolic function of microbial community of SFB ecosystem was mainly studied by analyzing the physiological and biochemical features of the isolated microorganisms. This method is not only labor intensive and time consuming, the results obtained are also limited to the number of culturable microbes. Fortunately, the advent of highly efficient contemporary sequencing technology, omics based technologies as well as corresponding bioinformatics software and database have greatly promoted the study on the functions of even the uncultured microbial communities present in traditional food ecosystems, such as SFB ecosystem (Franzosa et al., 2015; Chen et al., 2017).
Several omics based research studies were carried out to elucidate the microbial community in SFB ecosystem. On the species level, genomes of Rhizopus chinensis from daqu, C. kluyveri and C. butyricum from PM have already been sequenced (Wang D. et al., 2013; Li et al., 2016). It is proposed that the genomic sequence analysis of these three microorganisms can help in achieving better understanding of their genetic background and potential functions. On the microbial community level, Tao et al. (2017) applied the metagenomic approach and MiSeq-sequencing analyses of 16S rDNA and 16S rRNA genes to identify the hexanoate producing microorganisms (Clostridial cluster IV and Clostridium) in the SFB ecosystem. They also achieved the detailed elucidation of the interspecies hydrogen transfer mechanisms between hexanoate-producing bacteria and methanogens in PM microbiome. Zheng et al. (2015) investigated the aroma-forming functional proteins in PM samples by using the iTRAQ-based proteomic technology. The results thus obtained indicated that most proteins were involved in the process of methanogenesis, and caproic and butyric acid formation. Furthermore, it was found that these results were in accordance with the data obtained from metagenomic analysis (Tao et al., 2017).
Huang et al. (2017a) studied the metabolism and functional enzymes of the active microbial communities in SFB daqu via the metatranscriptomics approach. It was found that key enzymes involved in glycolysis and starch as well as pyruvate and ethanol metabolism were over-expressed at 50 and 62°C. Furthermore, the citrate cycle was up-regulated at 62°C and all the up-regulated genes in the glycolysis pathways mainly belonged to Saccharomycetales and Mucorales (Huang et al., 2017a). Liu J. et al. (2017) implemented the metatranscriptomic analysis approach to identify that Saccharomyces and Lactobacillus were the core microbiota responsible in the sulfur compound production mechanism in zaopei. These studies helped in providing a detailed description of the biological components of an active SFB ecosystem. It was then proposed that correlating these components with the functional aspects of the microbial community needs to be accomplished, which can be achieved by integrating these multi-omics data with systems biology approaches (Franzosa et al., 2015). Data so obtained (on various functional levels) can be implemented for the construction and analysis of a community-level metabolic model with predictable capability (Biggs et al., 2015; Cardona et al., 2016).
Interactions in Microbial Community of SFB Ecosystem
It was found that there existed extensive interspecies interactions among microbial communities of SFB ecosystem (Li et al., 2017). Cooccurrence pattern analysis suggested the presence of potential synergetic relationships between members belonging to the genera: Clostridia, Bacteroidia, Methanobacteria, and Methanomicrobia (Hu et al., 2016). It was also suggested that these relationships may be beneficial for the stability of the PM ecosystem. However, LAB (Lactobacillus, Pediococcus, and Streptococcus) may destroy this stability by producing lactate or various bacteriocins (Hu et al., 2016). Moreover, in zaopei ecosystem, Lactobacillus was found to be negatively related with the occurrence of Clostridium, Ruminococcus, Sedimentibacter, Syntrophomonas, Thermoactinomyces, Leuconostoc, Pediococcus, Staphylococcus, Bacillus, and Lactococcus (Wang X. et al., 2017). Synergistic interactions between the hexanoate producing strains (mostly, genus Clostridium) and methanogenic archaea (Methanobacterium) were also detected (Barker and Taha, 1942; Wu et al., 1990; Tao et al., 2017) and the main mechanism was the interspecies hydrogen transfer (Thauer et al., 2008). The caproic acid biosynthesis and hydrogenotrophic and acetoclastic methanogenesis pathways were also detected in a recent metagenomics analysis of PM microbial community (Tao et al., 2017). In addition, co-culture of Clostridium sp. W1 and Methanobacterium bryantii had been used for the cultivation of PM in new pit cellar, which resulted in a higher caproate and ethylcaproate production capacity (Wu et al., 1990). Another example is that the production of caproic acid in Clostridium was enhanced by the melanin secreted by Streptomyces avicenniae GW01 (Guo et al., 2016).
Recently, a novel synergistic effect between Saccharomyces and Lactobacillus in the production of sulfur compounds via methionine recycling was identified (Liu J. et al., 2017). It was found that the presence of L. buchneri up-regulated the expression of genes responsible for the generation of 3-(Methylthio)-1-propanol and dimethyl disulfide in S. cerevisiae, which further regenerated the precursor of methionine catabolism (Liu J. et al., 2017). Though, several examples of interspecies interactions have been identified, but the detailed mechanisms of the same are still unclear.
It has also been observed that some Aminobacterium strains and Methanosarcina barkeri can enhance the generation of VFAs, which further contributes to the aroma composition of Chinese liquor (Nadell et al., 2016). Furthermore, Bacillus species were found to inhibit the growth of Streptomyces sampsonii, which is one of the most dominant geosmin producers (Zhi et al., 2016).
Flavor Contributions of Microbial Community in SFB Ecosystem
To full exploration of SFB ecosystem, one key is to understand function of microbial community in SFB ecosystem attributing to flavor compounds formation. Because flavor compounds of SFB are complex, the first thing is to identify the characteristic flavor compounds of SFB. Ethyl esters (ethyl hexanoate, ethyl lactate, ethyl acetate, and ethyl butanoate), acids (lactic acid, acetic acid), alcohols (n-propanol, iso-butanol), aldehyde had been reported as the main flavor compounds in SFB (Zhou et al., 2012; Li J. et al., 2015; Yao et al., 2015). At present, studies on the flavor contributions of microbial community in SFB ecosystem can be divided into three groups: (i) isolation and identification of microorganism with high flavor compound producing capacity under pure liquid culture, (ii) regulation of SFB ecosystem with addition of microorganisms during fermentation, and (iii) detection and analysis of biological components of SFB ecosystem on a certain level with the systems biology approaches. The first and third groups have been discussed in the above sections: “Physiological traits of cultured microorganisms” and “Systems biology based analysis of SFB microbial community.” For the second group, addition of yeast strains (Debaryomyces hansenii, Issatchenkia orientalis, Zygosaccharomyces bailii, Trichosporon coremiiforme) to zaopei showed an increase of ethyl hexanoate (Wang T. et al., 2013). Inoculation of Wickerhamomyces anomalus showed an increase of amino nitrogen, n-butyl alcohol, and sec-butyl alcohol in zaopei (Jian et al., 2017). However, inoculation of Saccharomyces cerevisiae showed a decrease of esters compounds and bacterial and fungal diversity in zaopei (Wang S. et al., 2017). For further study, flavor–oriented technology, which integrates the identification of flavor compounds profile and characteristic flavor compounds, critical microbes and their metabolic features, regulation of flavor production of ecosystem by the critical microbes, has been carried out and showed great potential (Xu, 2015).
Conclusion and Perspectives
SFB are produced by the synergistic effect of various microorganisms present in the SFB ecosystem. Due to the rich microbial diversity of the SFB ecosystem, it can be considered as a potential resource for the isolation of many potential industrial microorganisms that are capable of producing organic acids, novel enzymes and other high-value products. In addition, the SFB ecosystem could be a good example for the study of microbial community formation (Wolfe and Dutton, 2015) and microbial adapt evolution. Up until now, most of the microbial diversity studies on the SFB ecosystem were focused on the isolation and culture of the functional microbes and identification of microbial diversity of SFB ecosystem. However, the results obtained from such studies fail to provide comprehensive overview of the function and interspecies interactions of the microbial community of the SFB ecosystem. It was then observed that such culture based methods fail to study many un-cultured microorganisms, due to which their potential roles remain ambiguous (Liu M.-K. et al., 2017). Furthermore, the isolated microorganisms may behave differently under axenic liquid and solid culture conditions and fail to provide crucial information on the interspecies interactions associated with them (Zhou et al., 2016). New cultivation methods should be developed to identify phenotypes of specific species and elucidate physiological interactions of these microbes and their functions in SFB ecosystem (Sommer, 2015; Liu J. et al., 2017).
The contemporary application of systems biology approaches has boosted our understanding of SFB ecosystem, not only the microbial diversity, but also the potential metabolic functions (Huang et al., 2017a; Tao et al., 2017). It is therefore proposed that a detailed explanation of microbial community of SFB ecosystem calls for a combination of prevalent omics data, results obtained from physiological experiments, application of molecular analysis methods, systems biology approaches as well as bioinformatics tools. In the near future, we believe that the persistent investigation on the molecular mechanisms of microbial constituents of the SFB ecosystem will help in accelerating the improvement of SFB quality and stability.
Author Contributions
WZ carried out the initial literature review and wrote the initial manuscript. HL provided expertise and insight relating to baijiu microbiology. WZ and CZ revised the text. All authors read and approved the final manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by grants from Research Center for International Transmission of Sichuan Liquor Culture (CJCB2016-04), Luzhou Laojiao Chinese National Engineering Research Center for Solid Fermentation (HX2015156), and Sichuan University of Science and Engineering (Item No. 2013RC12).
References
- Barker H. A., Taha S. M. (1942). Clostridium kluyveri, an organism concerned in the formation of caproic acid from ethyl alcohol. J. Bacteriol. 43 347–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs M. B., Medlock G. L., Kolling G. L., Papin J. A. (2015). Metabolic network modeling of microbial communities. Wiley Interdiscip. Rev. Syst. Bio. Med. 7 317–334. 10.1002/wsbm.1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bin T., Jin-ying L., Qing-wu Z., An-jun L., Chun-huan W., You-hong T. (2011). Phylogenetic diversity analyse of bacteria in Gujing-flavor liquor daqu using culture independent method. Food Ferment. Ind. 37 36–40. [Google Scholar]
- Bokulich N. A., Bamforth C. W., Mills D. A. (2012). A review of molecular methods for microbial community profiling of beer and wine. J. Am. Soc. Brew. Chem. 70 150–162. 10.1094/ASBCJ-2012-0709-01 [DOI] [Google Scholar]
- Cardona C., Weisenhorn P., Henry C., Gilbert J. A. (2016). Network-based metabolic analysis and microbial community modeling. Curr. Opin. Microbiol. 31 124–131. 10.1016/j.mib.2016.03.008 [DOI] [PubMed] [Google Scholar]
- Chen G., Chen C., Lei Z. (2017). Meta-omics insights in the microbial community profiling and functional characterization of fermented foods. Trends Food Sci. Technol. 65(Suppl. C) 23–31. 10.1016/j.tifs.2017.05.002 [DOI] [Google Scholar]
- Chen X. R., Shao C. B., Wang Y. W., He M. X., Ma K. D., Wang H. M., et al. (2015). Paenibacillus vini sp. nov., isolated from alcohol fermentation pit mud in Sichuan Province, China. Antonie Van Leeuwenhoek 107 1429–1436. 10.1007/s10482-015-0438-y [DOI] [PubMed] [Google Scholar]
- Chi Z., Liu G., Wang F., Ju L., Zhang T. (2009). Saccharomycopsis fibuligera and its applications in biotechnology. Biotechnol. Adv. 27 423–431. 10.1016/j.biotechadv.2009.03.003 [DOI] [PubMed] [Google Scholar]
- De Araújo Cavalcante W. A., Leitão R. C., Gehring T. A., Angenent L. T., Santaella S. T. (2017). Anaerobic fermentation for n-caproic acid production: a review. Process Biochem. 54 106–119. 10.1016/j.procbio.2016.12.024 [DOI] [Google Scholar]
- Ding X. F., Wu C., Huang J., Li H., Zhou R. (2014a). Eubacterial and archaeal community characteristics in the man-made pit mud revealed by combined PCR-DGGE and FISH analyses. Food Res. Int. 62 1047–1053. 10.1016/j.foodres.2014.05.045 [DOI] [Google Scholar]
- Ding X. F., Wu C. D., Zhang L. Q., Zheng J., Zhou R. Q. (2014b). Characterization of eubacterial and archaeal community diversity in the pit mud of Chinese Luzhou-flavor liquor by nested PCR-DGGE. World J. Microbiol. Biotechnol. 30 605–612. 10.1007/s11274-013-1472-4 [DOI] [PubMed] [Google Scholar]
- Ding X., Wu C., Huang J., Zhou R. (2015). Interphase microbial community characteristics in the fermentation cellar of Chinese Luzhou-flavor liquor determined by PLFA and DGGE profiles. Food Res. Int. 72 16–24. 10.1016/j.foodres.2015.03.018 [DOI] [Google Scholar]
- Dou X., Han P., Liu L., Zhang Y., He J., Zhuo X., et al. (2017). Study on isolation and identification and population succession law of bacterial in fermented grains during the brewing of Luzhou-flavor Liquor. Sci. Technol. Food Ind. 38 169–174. 10.13386/j.issn1002-0306.2017.01.025 [DOI] [Google Scholar]
- Du H., Liu B., Wang X., Xu Y. (2017). Exploring the microbial origins of p-cresol and its co-occurrence pattern in the Chinese liquor-making process. Int. J. Food Microbiol. 260 27–35. 10.1016/j.ijfoodmicro.2017.08.016 [DOI] [PubMed] [Google Scholar]
- Du H., Xu Y. (2012). Determination of the microbial origin of geosmin in Chinese liquor. J. Agric. Food Chem. 60 2288–2292. 10.1021/jf204648e [DOI] [PubMed] [Google Scholar]
- Dürre P. (2016). “Physiology and sporulation in Clostridium,” in The Bacterial Spore: from Molecules to Systems eds Driks A., Eichenberger P. (Washington, DC: ASM Press; ) 315–329. [Google Scholar]
- Feng X., Deng J., Xie J., Wei C., Luo H., Huang Z., et al. (2017). Brief analysis on current situation of comprehensive utilization of by-products yellow water from Baijiu-making. China Brew. 36 7–9. [Google Scholar]
- Franzosa E. A., Hsu T., Sirota-Madi A., Shafquat A., Abu-Ali G., Morgan X. C., et al. (2015). Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 13 360–372. 10.1038/nrmicro3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gou M., Wang H., Yuan H., Zhang W., Tang Y., Kida K. (2015). Characterization of the microbial community in three types of fermentation starters used for Chinese liquor production. J. Inst. Brew. 121 620–627. 10.1002/jib.272 [DOI] [Google Scholar]
- Guo M. Y., Huo D. Q., Ghai R., Rodriguez-Valera F., Shen C. H., Zhang N., et al. (2014). Metagenomics of ancient fermentation pits used for the production of Chinese strong-aroma liquor. Genome Announc. 2:e1045–14. 10.1128/genomeA.01045-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W., Guan J., Chen M., Xie Y., Zhang Y., Fang S. (2016). Mechanism of Actinomycetes promoting caproic acid bacteria to produce caproic acid. Liquor Mak. Sci. Technol. 10 48–52. 10.13746/j.njkj.2016187 [DOI] [Google Scholar]
- He P., Li F., Zheng Y., Zhang Y., Hu X., Sun X., et al. (2017). Isolation and volatile metabolites of Clostridium spp. in pit mud of strong-flavor Baijiu. China Brew. 36 45–49. [Google Scholar]
- Hu B., Liu G., Dong Q., Tang Y., Li A., Li X., et al. (2017). Research and application of functional yeast in Chinese liquor production. Liquor Mak. 44 13–18. [Google Scholar]
- Hu C., Wu J., Shen C., Wang Z., Hu Y. (2004). Research on Luzhou-flavor Daqu starter and its application. Liquor Mak. Sci. Technol. 1 33–36. [Google Scholar]
- Hu X., Du H., Ren C., Xu Y. (2016). Illuminating anaerobic microbial community and cooccurrence patterns across a quality gradient in Chinese Liquor fermentation pit muds. Appl. Environ. Microbiol. 82 2506–2515. 10.1128/AEM.03409-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X. L., Du H., Xu Y. (2015). Identification and quantification of the caproic acid-producing bacterium Clostridium kluyveri in the fermentation of pit mud used for Chinese strong-aroma type liquor production. Int. J. Food Microbiol. 214 116–122. 10.1016/j.ijfoodmicro.2015.07.032 [DOI] [PubMed] [Google Scholar]
- Hu X. L., Wang H. Y., Wu Q., Xu Y. (2014). Development, validation and application of specific primers for analyzing the clostridial diversity in dark fermentation pit mud by PCR-DGGE. Bioresour. Technol. 163 40–47. 10.1016/j.biortech.2014.04.008 [DOI] [PubMed] [Google Scholar]
- Huang X., Huang J., Li Z., Han B. (2016). Microbial diversity analysis in strong-flavor and sauce-flavor Daqu. China Brew. 35 33–37. 10.1021/jf902881p [DOI] [PubMed] [Google Scholar]
- Huang Y., Yi Z., Jin Y., Huang M., He K., Liu D., et al. (2017a). Metatranscriptomics reveals the functions and enzyme profiles of the microbial community in Chinese Nong-Flavor liquor starter. Front. Microbiol. 8:1747. 10.3389/fmicb.2017.01747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Yi Z., Jin Y., Zhao Y., He K., Liu D., et al. (2017b). New microbial resource: microbial diversity, function and dynamics in Chinese liquor starter. Sci. Rep. 7:14577. 10.1038/s41598-017-14968-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian X., Jian Z., Yu L., Deng W., You L. (2017). Effects of a Wickerhamomyces strain on the fermentation of Nongxiang Baijiu. Liquor Mak. Sci. Technol. 9 33–38. 10.13746/j.njkj.2017194 [DOI] [Google Scholar]
- Jin G., Zhu Y., Xu Y. (2017). Mystery behind Chinese liquor fermentation. Trends Food Sci. Technol. 63 18–28. 10.1016/j.tifs.2017.02.016 [DOI] [Google Scholar]
- Kaeberlein T., Lewis K., Epstein S. S. (2002). Isolating “uncultivable” microorganisms in pure culture in a simulated natural environment. Science 296 1127–1129. 10.1126/science.1070633 [DOI] [PubMed] [Google Scholar]
- Li C., Wang Y., Xie G., Peng B., Zhang B., Chen W., et al. (2016). Complete genome sequence of Clostridium butyricum JKY6D1 isolated from the pit mud of a Chinese flavor liquor-making factory. J. Biotechnol. 220 23–24. 10.1016/j.jbiotec.2016.01.003 [DOI] [PubMed] [Google Scholar]
- Li H., Huang J., Liu X., Zhou R., Ding X., Xiang Q., et al. (2017). Characterization of interphase microbial community in Luzhou-flavored liquor manufacturing pits of various ages by polyphasic detection methods. J. Microbiol. Biotechnol. 27 130–140. 10.4014/jmb.1605.05036 [DOI] [PubMed] [Google Scholar]
- Li J., Guo W., Luo Y., Wu J., Jiang W., Zhang M. (2015). Analysis of volatile compounds of Luzhou-flavor liquor from different pit ages by GC-MS. China Brew. 34 141–144. 10.11882/j.issn.0254-5071.2015.09.032 [DOI] [Google Scholar]
- Li K., Fan Z., Wang J., Lin K., Xiang W. (2015). Microbial diversity in fermented yellow water of traditional intense flavor liquor. J. Food Sci. Biotechnol. 34 1155–1161. 10.3969/j.issn.1673-1689.2015.11.006 [DOI] [Google Scholar]
- Li Z., Bai Z., Wang D., Zhang W., Zhang M., Lin F., et al. (2014). Cultivable bacterial diversity and amylase production in three typical Daqus of Chinese spirits. Int. J. Food Sci. Technol. 49 776–786. 10.1111/ijfs.12365 [DOI] [Google Scholar]
- Liang H., Li W., Luo Q., Liu C., Wu Z., Zhang W. (2015). Analysis of the bacterial community in aged and aging pit mud of Chinese Luzhou-flavour liquor by combined PCR-DGGE and quantitative PCR assay. J. Sci. Food Agric. 95 2729–2735. 10.1002/jsfa.7013 [DOI] [PubMed] [Google Scholar]
- Liang H., Luo Q., Zhang A., Wu Z., Zhang W. (2016). Comparison of bacterial community in matured and degenerated pit mud from Chinese Luzhou-flavour liquor distillery in different regions. J. Inst. Brew. 122 48–54. 10.1002/jib.296 [DOI] [Google Scholar]
- Lin C., Di-Feng R., Zu-Ming L., Zhi-Rui Z., Zhi-Hui B. (2012). Study on the cultivable bacterial community structure and amylase in Huangtai Liquor Daqu. Sci. Technol. Food Ind. 33 232–235, 238 10.13386/j.issn1002-0306.2012.08.080 [DOI] [Google Scholar]
- Liu B., Du H., Wang X.-S., Xu Y. (2017). Detecting source of p-cresol in strong flavor Chinese liquor by high throughput sequencing. Microbiol. China 44 108–117. 10.1186/s40168-017-0316-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Wu Q., Wang P., Lin J., Huang L., Xu Y. (2017). Synergistic effect in core microbiota associated with sulfur metabolism in spontaneous Chinese liquor fermentation. Appl. Environ. Microbiol. 10.1128/AEM.01475-17 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.-K., Tang Y.-M., Guo X.-J., Zhao K., Tian X.-H., Liu Y., et al. (2017). Deep sequencing reveals high bacterial diversity and phylogenetic novelty in pit mud from Luzhou Laojiao cellars for Chinese strong-flavor Baijiu. Food Res. Int. 102(Suppl. C) 68–76. 10.1016/j.foodres.2017.09.075 [DOI] [PubMed] [Google Scholar]
- Liu M., Tang Y., Zhao K., Liu Y., Guo X., Ren D., et al. (2017). Determination of the fungal community of pit mud in fermentation cellars for Chinese strong-flavor liquor, using DGGE and Illumina MiSeq sequencing. Food Res. Int. 91 80–87. 10.1016/j.foodres.2016.11.037 [DOI] [PubMed] [Google Scholar]
- Liu Y., Wang Y., Wang W., Li Y., Wu S., Liu M., et al. (2017). Screening, identification and metabolites analyses of Bacillus in pit mud of Luzhou-flavor Baijiu. China Brew. 36 76–79. 10.11882/j.issn.0254-5071.2017.07.017 [DOI] [Google Scholar]
- Liu C., Huang D., Liu L., Zhang J., Deng Y., Chen L., et al. (2014). Clostridium swellfunianum sp. nov., a novel anaerobic bacterium isolated from the pit mud of Chinese Luzhou-flavor liquor production. Antonie Van Leeuwenhoek 106 817–825. 10.1007/s10482-014-0251-z [DOI] [PubMed] [Google Scholar]
- Liu L. M., Zhang B., Dong X.-Z., Zhang L.-p. (2007). Separation & identification of Pediococci in the fermentation pits for Luzhou-flavor liquor. Liquor Mak. Sci. Technol. 2 22–24. 10.3969/j.issn.1001-9286.2007.02.003 [DOI] [Google Scholar]
- Luo H.-b., Yang X.-d., Yang Y.-h., Ye G.-b., Li D.-y. (2013). Isolation, identification and phylogenetic analysis of culturable fungi in Luzhou-flavor daqu. Mod. Food Sci. Technol. 29 2047–2052. [Google Scholar]
- Luo Q., Liu C., Li W., Wu Z., Zhang W. (2014a). Comparison between bacterial diversity of aged and aging pit mud from Luzhou-flavor liquor distillery. Food Sci. Technol. Res. 20 867–873. 10.3136/fstr.20.867 [DOI] [Google Scholar]
- Luo Q., Liu C., Wu Z., Wang H., Li W., Zhang K., et al. (2014b). Monitoring of the prokaryotic diversity in pit mud from a Luzhou-flavour liquor distillery and evaluation of two predominant archaea using qPCR assays. J. Inst. Brew. 120 253–261. 10.1002/jib.132 [DOI] [Google Scholar]
- Ma K., Chen X., Guo X., Wang Y., Wang H., Zhou S., et al. (2016). Bacillus vini sp. nov. isolated from alcohol fermentation pit mud. Arch. Microbiol. 198 559–564. 10.1007/s00203-016-1218-4 [DOI] [PubMed] [Google Scholar]
- Maukonen J., Saarela M. (2009). Microbial communities in industrial environment. Curr. Opin. Microbiol. 12 238–243. 10.1016/j.mib.2009.04.002 [DOI] [PubMed] [Google Scholar]
- Ming H., Dong R., Xu D., Guo Z., Zeng H., Shen C., et al. (2013). Separation and preliminary identification of dominant microbes in nong-flavor daqu. Liquor Mak. Sci. Technol. 12 57–60. [Google Scholar]
- Ming H.-M., Guo Z., Zhou J., Chen M.-e., Xu D.-f., Yao X. (2015). Screening and identification of aroma-producing microorganisms in Luzhou-flavor daqu. Mod. Food Sci. Technol. 31 186–191. 10.13982/j.mfst.1673-9078.2015.4.030 [DOI] [Google Scholar]
- Nadell C. D., Drescher K., Foster K. R. (2016). Spatial structure, cooperation and competition in biofilms. Nat. Rev. Microbiol. 14 589–600. 10.1038/nrmicro.2016.84 [DOI] [PubMed] [Google Scholar]
- Pu L., Wang T., Zhou Y., Yao T., You L., Wang S., et al. (2012). Research on molds producing glucoamylase in the air of fermentation workshop of multi-grains Luzhou-flavor liquor in Yibin. Liquor Mak. Sci. Technol. 9 29–31. 10.13746/j.njkj.2012.09.015 [DOI] [Google Scholar]
- Qiu Y., Fang F., Zhou X., Chen X., Zhang L., Du G., et al. (2016). Characterization of arginine utilization strains from fermented grains and evaluation of their contribution to citrulline accumulation in Chinese Luzhou-flavor spirits. Acta Microbiol. Sin. 56 1638–1646. 10.13343/j.cnki.wsxb.20160007 [DOI] [PubMed] [Google Scholar]
- Sabel A., Martens S., Petri A., König H., Claus H. (2014). Wickerhamomyces anomalus AS1: a new strain with potential to improve wine aroma. Ann. Microbiol. 64 483–491. 10.1007/s13213-013-0678-x [DOI] [Google Scholar]
- Shi S., Zhang L., Wu Z.-y., Zhang W.-x., Deng Y., Zhong F.-d., et al. (2011). Analysis of the fungi community in multiple- and single-grains Zaopei from a Luzhou-flavor liquor distillery in western China. World J. Microbiol. Biotechnol. 27 1869–1874. 10.1007/s11274-010-0645-7 [DOI] [Google Scholar]
- Sommer M. O. (2015). Advancing gut microbiome research using cultivation. Curr. Opin. Microbiol. 27 127–132. 10.1016/j.mib.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Sun W., Xiao H., Peng Q., Zhang Q., Li X., Han Y. (2016). Analysis of bacterial diversity of Chinese Luzhou-flavor liquor brewed in different seasons by Illumina Miseq sequencing. Ann. Microbiol. 66 1293–1301. 10.1007/s13213-016-1223-5 [DOI] [Google Scholar]
- Tang B., Wang H.-T., Zhou Q.-W., Li A.-j., Wan C.-h., Tang Y.-h. (2013). Isolation and identification of cultivable bacteria from pit mud of Gujing-flavor liquor and their production of organic acids. Ind. Microbiol. 43 68–73. [Google Scholar]
- Tao Y., Li J., Rui J., Xu Z., Zhou Y., Hu X., et al. (2014). Prokaryotic communities in pit mud from different-aged cellars used for the production of Chinese strong-flavored liquor. Appl. Environ. Microbiol. 80 2254–2260. 10.1128/aem.04070-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y., Wang X., Li X., Wei N., Jin H., Xu Z., et al. (2017). The functional potential and active populations of the pit mud microbiome for the production of Chinese strong-flavour liquor. Microb. Biotechnol. 10 1603–1615. 10.1111/1751-7915.12729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauer R. K., Kaster A. K., Seedorf H., Buckel W., Hedderich R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6 579–591. 10.1038/nrmicro1931 [DOI] [PubMed] [Google Scholar]
- Wang C.-D., Chen Q., Wang Q., Li C.-h., Leng Y.-y., Li S.-g., et al. (2014). Long-term batch brewing accumulates adaptive microbes, which comprehensively produce more flavorful Chinese liquors. Food Res. Int. 62 894–901. 10.1016/j.foodres.2014.05.017 [DOI] [Google Scholar]
- Wang H., Hu F., Hu J., Peng X., Chen M., Liu M. (2014). Advancement of the research in ethyl carbamate in Baijiu(Liquor). Liquor Mak. Sci. Technol. 9 88–91. 10.13746/j.njkj.2014.0212 [DOI] [Google Scholar]
- Wang D., Wu R., Xu Y., Li M. (2013). Draft genome sequence of Rhizopus chinensis CCTCCM201021, used for brewing traditional Chinese alcoholic beverages. Genome Announc. 1:e0019512. 10.1128/genomeA.00195-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T., Yao T., Li T., You L., Zhou R.-p., Wang S., et al. (2013). The role of Luzhou-flavor liquor correlated yeasts in promoting caproic acid ethyl ester generating in fermentative grain. Food Ferment. Ind. 39 41–45. 10.13995/j.cnki.11-1802/ts.2013.01.038 [DOI] [Google Scholar]
- Wang H. Y., Gao Y. B., Fan Q. W., Xu Y. (2011). Characterization and comparison of microbial community of different typical Chinese liquor Daqus by PCR–DGGE. Lett. Appl. Microbiol. 53 134–140. 10.1111/j.1472-765X.2011.03076.x [DOI] [PubMed] [Google Scholar]
- Wang T., Zhao D., Tian S., You L., Wang S., Feng R., et al. (2011). Phylogenetic diversity of cultivable bacteria during the brewing process of the Luzhou-flavor liquor in Yibin, Sichuan province, China. Wei Sheng Wu Xue Bao 51 1351–1357. [PubMed] [Google Scholar]
- Wang L., Zhang L., Liu L., Zhang S., Xu D., Liu G. (2010). Comparative study of two Methanobacterium strains in the ancient fermentation pits of Luzhou. Chin. J. Appl. Environ. Biol. 16 840–844. 10.3724/SP.J.1145.2010.00840 [DOI] [Google Scholar]
- Wang P., Wu Q., Jiang X., Wang Z., Tang J., Xu Y. (2017). Bacillus licheniformis affects the microbial community and metabolic profile in the spontaneous fermentation of Daqu starter for Chinese liquor making. Int. J. Food Microbiol. 250 59–67. 10.1016/j.ijfoodmicro.2017.03.010 [DOI] [PubMed] [Google Scholar]
- Wang S., Tang H.-l., Wang Z.-y., Jiang Y.-t., Mao J.-j., You L., et al. (2017). Influences of Saccharomyces cerevisiae on the fermentation of strong-flavoured liquor. Sci. Technol. Food Ind. 21 142–146, 150. 10.13386/j.issn1002-0306.2017.21.029 [DOI] [Google Scholar]
- Wang X., Du H., Xu Y. (2017). Source tracking of prokaryotic communities in fermented grain of Chinese strong-flavor liquor. Int. J. Food Microbiol. 244 27–35. 10.1016/j.ijfoodmicro.2016.12.018 [DOI] [PubMed] [Google Scholar]
- Wang Q., Wang C.-d., Li C.-h., Li J.-g., Chen Q., Li Y.-z. (2015). Clostridium luticellarii sp. nov., isolated from a mud cellar used for producing strong aromatic liquors. Int. J. Syst. Evol. Microbiol. 65 4730–4733. 10.1099/ijsem.0.000641 [DOI] [PubMed] [Google Scholar]
- Wang T., Zhao D., You L., Zhou Y.-k., Wang S., Feng R.-z., et al. (2012). Diversity of molds producing esterifying enzyme during fermentation of strong-flavor liquor. Food Ferment. Ind. 38 37–41. [Google Scholar]
- Wang Y. (2016). 2015 annual report on alcoholic drinks industry at the 3rd expanding meeting of the 5th council of China Alcoholic Drinks Association. Liquor Mak. Sci. Technol. 4–15. [Google Scholar]
- Wolfe B. E., Dutton R. J. (2015). Fermented foods as experimentally tractable microbial ecosystems. Cell 161 49–55. 10.1016/j.cell.2015.02.034 [DOI] [PubMed] [Google Scholar]
- Wu C., Ding X., Huang J., Zhou R. (2015). Characterization of archaeal community in Luzhou-flavour pit mud. J. Inst. Brew. 121 597–602. 10.1002/jib.255 [DOI] [Google Scholar]
- Wu C., Qin Z., Huang J., Zhou R. (2014). Characterization of microbial community in Daqu by PLFA method. Food Sci. Technol. Res. 20 147–154. 10.3136/fstr.20.147 27713211 [DOI] [Google Scholar]
- Wu F., Chen H., Hu C., Zhang W., Shen C., Xu Y., et al. (2006). Systematic analysis of 18S rDNA total sequence of cultivable yeast in Luzhou-flavor fermented grains. Liquor Mak. Sci. Technol. 4 23–25. 10.3969/j.issn.1001-9286.2006.04.003 [DOI] [Google Scholar]
- Wu Y., Lu S., Liu G., Xue T., Hou M., Jiang Y. (1990). A study of enhancing the quality of Luzhou type Qujiu by using simultaneous fermentation of caproic acid bacteria and methane bacteria. Food Ferment. Ind. 6 1–6 8. [Google Scholar]
- Wu Y., Xue T., Chen Z., Lu S., Liu A., Lin S. (1991). Study on distribution and action of anaerobic bacteria in Wuliangye old fermented-pits. Acta Microbiol. Sin. 31 299–307. [Google Scholar]
- Wu Y., Yi W. (1986). Isolation and identification of the carproic acid-producing bacterium and its carproic acid-producing conditions in Lozhou Qu-jiu old pit-slime. Food Ferment. Ind. 5 4–9. [Google Scholar]
- Xiao C., Lu Z.-M., Zhang X.-J., Wang S.-T., Ao L., Shen C.-H., et al. (2017). Bio-heat is a key environmental driver shaping the microbial community of medium-temperature daqu. Appl. Environ. Microbiol. 83:e1550-17. 10.1128/aem.01550-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L. (2016). Screening and identification of ester-producing yeast in Luzhou Daqu and optimization of ester production conditions. China Brew. 35 74–78. 10.11882/j.issn.0254-5071.2016.02.017 [DOI] [Google Scholar]
- Xu S., Jin S., Wu K., Jia X. (2004). Studies on diversities of the microorganisms on the surface of wheat. Grain Storage 32 41–43. [Google Scholar]
- Xu Y. (2015). Study on liquor-making microbes and the regulation & control of their metabolism based on flavor-oriented technology. Liquor Mak. Sci. Technol. 2 1–11, 16 10.13746/j.njkj.2014533 [DOI] [Google Scholar]
- Xu Y., Sun B., Fan G., Teng C., Xiong K., Zhu Y., et al. (2017). The brewing process and microbial diversity of strong flavour Chinese spirits: a review. J. Inst. Brew. 123 5–12. 10.1002/jib.404 [DOI] [Google Scholar]
- Xu Y., Wang D., Fan W. L., Mu X. Q., Chen J. (2010). “Traditional Chinese biotechnology,” in Biotechnology in China II: Chemicals, Energy and Environment eds Tsao T. G., Ouyang P., Chen J. (Berlin: Springer; ) 189–233. [Google Scholar]
- Xue T., Chen Z., Lu S., Liu A., Wu Y. (1988). Isolation characterization and the culture conditions of Clostridium sp. W1. Food Ferment. Ind. 4 4–9. [Google Scholar]
- Xue Z., Xue Y. (2016). Polyphasic identification of a hexanoate acid-producing bacteria strain from Luzhou-flavor Liquor pit mud. Liquor Mak. Sci. Technol. 6 65–71. [Google Scholar]
- Yan Z., Zheng X.-W., Han B.-Z., Yan Y.-Z., Zhang X., Chen J.-Y. (2015). 1H NMR-based metabolomics approach for understanding the fermentation behaviour of Bacillus licheniformis. J. Inst. Brew. 121 425–431. 10.1002/jib.238 [DOI] [Google Scholar]
- Yang J.-G., Dou X., Han P.-J., Bai F.-Y., Zhou J., Zhang S.-Y., et al. (2017). Microbial diversity in Daqu during production of Luzhou-flavored liquor. J. Am. Soc. Brew. Chem. 75 136–144. [Google Scholar]
- Yang J., Su C., Dou X., Guo J., Zhang Q., Zhang S., et al. (2017). Isolation, identification and succession of yeast from fermented grains of Luzhou-flavor liquor based on 26S rDNA D1/D2 sequence analysis. Food Sci. 1–12. Available at: http://kns.cnki.net/kcms/detail/11.2206.TS.20171212.1612.082.html [Google Scholar]
- Yang S.-w., Zhou Z.-j., Han Y., Gao X.-l. (2011). Study on varieties of yeast in cellared mud used for brewing white alcohol. Sci. Technol. Food Ind. 32 138–141. [Google Scholar]
- Yao F., Yi B., Shen C., Tao F., Liu Y., Lin Z., et al. (2015). Chemical analysis of the Chinese liquor Luzhoulaojiao by comprehensive two-dimensional gas chromatography/time-of-flight mass spectrometry. Sci. Rep. 5:9553. 10.1038/srep09553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao W., Chen M., Zhen D., Guo Y. (2010). Isolation of lactate-producing microbes from fermented grains of Luzhou-flavor liquor and their effect on simulative solid-state fermentation. Liquor Mak. 37 37–41. 10.3969/j.issn.1002-8110.2010.03.013 [DOI] [Google Scholar]
- Yin Q., Tao Y., Zhu X., Zhou Y., He X., Cheng L., et al. (2016). Clostridium liquoris sp. nov., isolated from a fermentation pit used for the production of Chinese strong-flavoured liquor. Int. J. Syst. Evol. Microbiol. 66 749–754. 10.1099/ijsem.0.000787 [DOI] [PubMed] [Google Scholar]
- You L., Wang S., Nie L., Zhang Y., Yuan H., Wang T. (2014). Screening of bacteria strains producing soy-like aroma and study on their growth characteristics. Liquor Mak. Sci. Technol. 11 23–27. 10.13746/j.njkj.2014.0220 [DOI] [Google Scholar]
- You L., Wang T. (2012). Volatile products of 4 Streptomyces strains isolated from strong-flavor liquor factories. Food Ind. 33 118–120. [Google Scholar]
- Yue Y.-Y., Zhang W.-X., Liu X., Hu C., Zhang S.-Y. (2007). Isolation and identification of facultative anaerobes in the pit mud of Chinese Luzhou-flavor liquor. Microbiology 34 251–255. 10.3969/j.issn.0253-2654.2007.02.013 [DOI] [Google Scholar]
- Zeng L., Tan X., Yuan C., Zhang Q., Yang Y., Zhao T., et al. (2016). Isolation and identification of cellulose-degrading strains from yellow water of Baijiu (Chinese liquor) and its cellulase activity. China Brew. 35 59–63. 10.11882/j.issn.0254-5071.2016.11.012 [DOI] [Google Scholar]
- Zhang C., Zhao D., Wang T., You L., Feng R.-z., Wang S., et al. (2011). Diversity of Actinomycetes in the brewing process of Luzhou-flavor multiple-grains liquor. Food Sci. 32 192–196. [Google Scholar]
- Zhang L., Wu C., Ding X., Zheng J., Zhou R. (2014). Characterisation of microbial communities in Chinese liquor fermentation starters Daqu using nested PCR-DGGE. World J. Microbiol. Biotechnol. 30 3055–3063. 10.1007/s11274-014-1732-y [DOI] [PubMed] [Google Scholar]
- Zhang L., Zhou R., Niu M., Zheng J., Wu C. (2015). Difference of microbial community stressed in artificial pit muds for Luzhou-flavour liquor brewing revealed by multiphase culture-independent technology. J. Appl. Microbiol. 119 1345–1356. 10.1111/jam.12943 [DOI] [PubMed] [Google Scholar]
- Zhang X., Xia S., Xu K., Feng H., Feng X., Cao Z. (2015). Isolation, identification and phylogenetic analysis of culturable fungi in fermented grains of Nongxiang Baijiu(Liquor). Liquor Mak. Sci. Technol. 7 28–33. 10.13746/j.njkj.2015158 [DOI] [Google Scholar]
- Zhang W. X., Qiao Z.-w., Shigematsu T., Tang Y.-q., Hu C., Morimura S., et al. (2005). Analysis of the bacterial community in Zaopei during production of Chinese Luzhou-flavor liquor. J. Inst. Brew. 111 215–222. 10.1002/j.2050-0416.2005.tb00669.x [DOI] [Google Scholar]
- Zhang W.-x., Qiao Z.-w., Tang Y.-q., Hu C., Sun Q., Morimura S., et al. (2007). Analysis of the fungal community in Zaopei during the production of Chinese Luzhou-flavour liquor. J. Inst. Brew. 113 21–27. 10.1002/j.2050-0416.2007.tb00251.x [DOI] [Google Scholar]
- Zhang X.-F., Wang H.-H., Sun X.-Y., Pan C.-M. (2017). Lysobacter zhanggongensis sp. nov. isolated from a pit mud. Curr. Microbiol. 74 1389–1393. 10.1007/s00284-017-1330-y [DOI] [PubMed] [Google Scholar]
- Zhao H., Chang Y., Wang W., Ling H.-z., Ping W.-x., Zhao Z.-c., et al. (2012). Isolation and identification of facultative anaerobic strains with high yield of hexanoic acid from Luzhou-flavor liquor pit mud. Food Sci. 33 177–182. [Google Scholar]
- Zhao J.-S., Zheng J., Zhou R.-Q., Shi B. (2012). Microbial community structure of pit mud in a Chinese strong aromatic liquor fermentation pit. J. Inst. Brew. 118 356–360. 10.1002/jib.52 [DOI] [Google Scholar]
- Zheng J., Liang R., Zhang L., Wu C., Zhou R., Liao X. (2013). Characterization of microbial communities in strong aromatic liquor fermentation pit muds of different ages assessed by combined DGGE and PLFA analyses. Food Res. Int. 54 660–666. 10.1016/j.foodres.2013.07.058 [DOI] [Google Scholar]
- Zheng Q., Lin B., Wang Y., Zhang Q., He X., Yang P., et al. (2015). Proteomic and high-throughput analysis of protein expression and microbial diversity of microbes from 30- and 300-year pit muds of Chinese Luzhou-flavor liquor. Food Res. Int. 75 305–314. 10.1016/j.foodres.2015.06.029 [DOI] [PubMed] [Google Scholar]
- Zheng X. W., Yan Z., Nout M. J., Boekhout T., Han B. Z., Zwietering M. H., et al. (2015). Characterization of the microbial community in different types of Daqu samples as revealed by 16S rRNA and 26S rRNA gene clone libraries. World J. Microbiol. Biotechnol. 31 199–208. 10.1007/s11274-014-1776-z [DOI] [PubMed] [Google Scholar]
- Zheng X.-W., Han B.-Z. (2016). Baijiu, Chinese liquor: history, classification and manufacture. J. Ethn. Foods 3 19–25. 10.1016/j.jef.2016.03.001 [DOI] [Google Scholar]
- Zheng X.-W., Tabrizi M. R., Nout M. J. R., Han B.-Z. (2011). Daqu-A traditional Chinese liquor fermentation starter. J. Inst. Brew. 117 82–90. 10.1002/j.2050-0416.2011.tb00447.x [DOI] [Google Scholar]
- Zhi Y., Wu Q., Du H., Xu Y. (2016). Biocontrol of geosmin-producing Streptomyces spp. by two Bacillus strains from Chinese liquor. Int. J. Food Microbiol. 231 1–9. 10.1016/j.ijfoodmicro.2016.04.021 [DOI] [PubMed] [Google Scholar]
- Zhou P., Luo H.-b., Huang D., Deng B., Wang Q., Huang Z.-g., et al. (2016). Separation and identification of thermoduric bacteria strains in medium/high temperature Daqu and the analysis of the flavor metabolites. Sci. Technol. Food Ind. 37 215–220. 10.13386/j.issn1002-0306.2016.24.033 [DOI] [Google Scholar]
- Zhou R.-P., Chen Y.-z., Tang D.-y. (2010). Diversity and distribution of bacteria in Luzhou-flavor wine factory. Food Sci. 31 209–213. [Google Scholar]
- Zhou X., Cheng X., Yang Y., Luo H., Huang Z. (2012). Study on the change rules of parameters of Luzhou-flavor liquor in pits and their correlations (III): flavoring compositions. Liquor Mak. Sci. Technol. 6 47–51. 10.13746/j.njkj.2012.06.032 [DOI] [Google Scholar]