

Abstract
The anticancer effect of manumycin A (Man A) has been attributed to the inhibition of farnesyl transferase (FTase), an enzyme that is responsible for post-translational modification of Ras proteins. However, we have discovered that Man A inhibits mammalian cytosolic thioredoxin reductase 1 (TrxR-1) in a time-dependent manner, with an IC50 of 272 nM with preincubation and 1586 nM without preincubation. The inhibition of TrxR-1 by Man A is irreversible and is the result of a covalent interaction between Man A and TrxR-1. Evidence presented herein demonstrates that Man A forms a Michael adduct with the selenocysteine residue, which is located in the C-terminal redox center of TrxR-1. Inhibitors of TrxR-1, which act through this mechanism, convert TrxR-1 into a SecTRAP, which utilizes NADPH to reduce oxygen to superoxide radical anion (O2–•).
Keywords: Thioredoxin reductase, reactive oxygen, oxidative stress, SecTRAP
Man A (1) is a
bacterial secondary metabolite that was first isolated from Streptomyces parvulus as a result of a random screening
program for farnesyl transferase (FTase) inhibitors.1 FTase catalyzes the post-translational farnesylation of
proteins including the Ras family of proteins. Ras proteins regulate numerous functions, which are related
to cell growth, proliferation, and cell signaling. They function by
binding to and activating several effector proteins, which regulate
critical cellular processes including transcription, translation,
cell-cycle progression, and calcium signaling.2 Upon farnesylation, Ras associates with intracellular
membranes where it becomes active. FTase inhibitors block farnesylation
such that Ras remains in the cytosol and does not
stimulate its downstream targets. Ras is the most
frequently mutated oncogene in human cancers with as many as 25% of
known human tumors having mutated Ras.3 FTase inhibitors have been discovered and developed
for the treatment of cancers4,5 as well as progeria6 and parasitic infections.7−10 Man A inhibits rat brain FTase
with a Ki of 1.2 μM1 and has shown antitumor activity in a variety of cancer
cell types11,12 and tumor models.13,14
The tumoricidal activity of manumycin was initially attributed to the inhibition of FTase preventing activation of Ras. However, it later became apparent that Man A can stimulate tumor and cell death by pathways that are independent of FTase.15 Numerous studies report the induction of reactive oxygen species (ROS) or, more specifically, superoxide radical anion (O2–•) in Man A treated cells and tumors.15−24 However, the mechanism of O2–• induction has remained undetermined. Herein, we report that manumycin is a potent inhibitor of mammalian thioredoxin reductase-1 (TrxR-1) and inducer of NADPH oxidase activity.
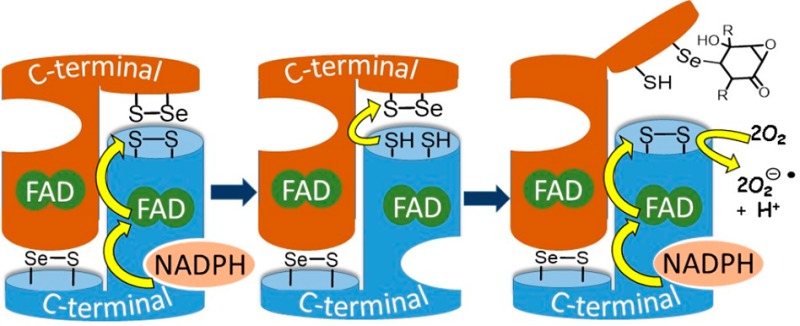
The thioredoxin system is a major regulatory system for the maintenance of redox homeostasis of the cell.25 It consists of thioredoxin (Trx) and thioredoxin reductase (TrxR). Trx reduces disulfide bridges in protein targets by thiol disulfide exchange. TrxR reduces the oxidized Trx back to its active disulfide form, with the reducing equivalents provided by NADPH. Mammalian TrxR-1 is a homodimer with the monomers aligned antiparallel to each other. NADPH initiates a cascade of redox reactions across three redox active sites: FAD is first reduced by NADPH, next FADH2 reduces an N-terminal disulfide (Cys59-Val-Asn-Val-Gly-Cys64 in human TrxR-1), which undergoes thiol disulfide exchange with a C-terminal seleno-sulfide (Cys497-Sec498-Gly in human TrxR-1) on the opposite chain of the dimer. The C-terminal redox center of TrxR-1 undergoes thiol–disulfide exchange with oxidized Trx (Figure 1).
Figure 1.
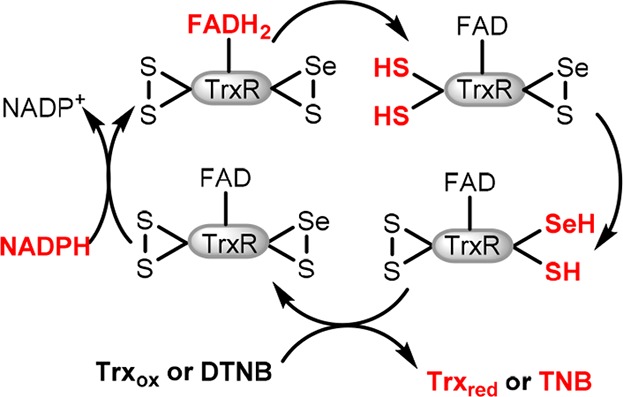
Electron flow in TrxR. Reduced sites shown in red.
We recently demonstrated that the algal toxin, brevetoxin, is a unique and potent inhibitor of TrxR-1.26 After making this discovery, we reasoned that other molecules, which are similar in size and functionality, might behave in a similar manner. We examined a series of compounds for their effect on TrxR-1, and while the effect of Man A is not identical to that of brevetoxin, we did discover that it is indeed a potent inhibitor of TrxR-1. Furthermore, through a series of enzyme assays and the use of a selective probe, we have established the mechanism of inhibition of TrxR-1 by Man A. These results should stimulate a reassessment of the tumoricidal activity of Man A in the context of our recent finding.
The effect of Man A on the thioredoxin system was examined using an assay that is based on the reduction of eosin-modified insulin by Trx.27 In this two-enzyme assay, oxidized Trx is continuously reduced by TrxR-1 with reducing equivalents ultimately provided by NADPH. Man A was incubated (30 min) at concentrations ranging from 0 to 1000 nM with a mixture of prereduced TrxR-1. In this assay, Man A exhibited a dose-dependent inhibition of TrxR-1/Trx activity with an IC50 of 572 (±88) nM (Figure 2A).
Figure 2.
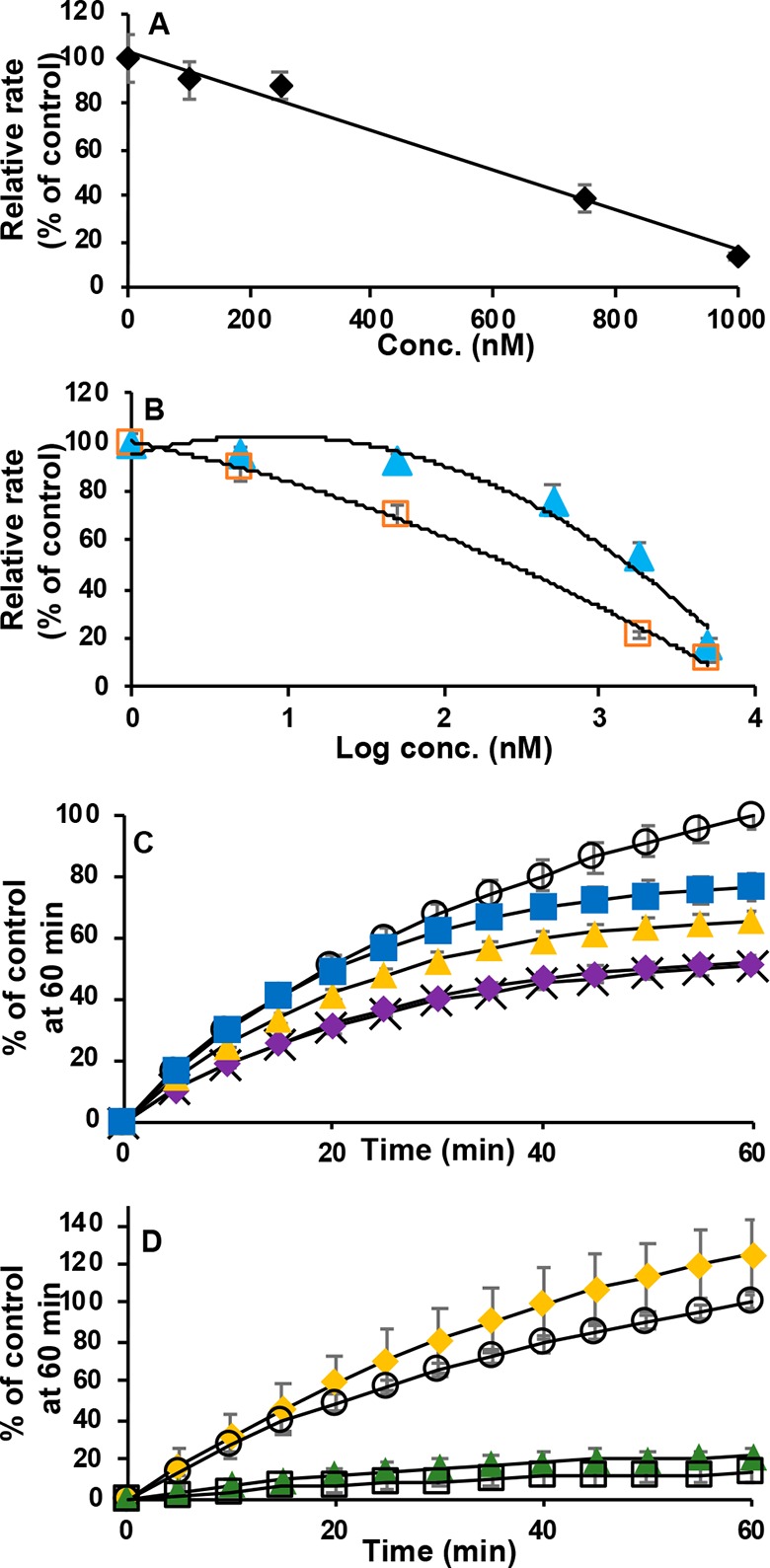
(A) TrxR-1/Trx insulin reduction assay. Percent inhibition by Man A vs concentration. Results are expressed as rate as % of control. (B) Inhibition of DTNB (2 mM) reduction by TrxR-1 (5.6 nM) in the presence of Man A (0–5000 nM). Sixty minute preincubation with Man A (orange □). No preincubation (light blue ▲). Results are expressed as initial rates relative to control (no Man A). (C) Time course inhibition of DTNB reduction by TrxR-1 (5.6 nM) in the presence of Man A (1 μM); control (DMSO only, ○). Incubation times 15 min (X); 10 min (purple ◆); 5 min (yellow ▲); 2 min (blue ■) after Man A addition. (D) Test of irreversibility of inhibition of DTNB reduction by TrxR-1 (5.2 nM) in the presence of Man A (2 μM). TrxR-1 reduced only after removal of Man A by gel filtration (yellow ◆); control (no Man A, ○); Man A not subjected to gel filtration (gray ▲); Man A removed by gel filtration after incubation with reduced TrxR-1 (□).
Mammalian TrxR-1 has a broad substrate specificity and reduces low molecular weight disulfides in addition to Trx. The effect of Man A on the reduction of the disulfide 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB or Ellman’s reagent) by rat recombinant TrxR-1 was examined at concentrations ranging from 0 to 5000 nM. When prereduced TrxR-1 was incubated with Man A for 1 h prior to the addition of substrate, the reduction of DTNB by TrxR-1 was inhibited with an IC50 of 272 (±29) nM. However, when Man A was added simultaneously with DTNB (in other words, without preincubation of TrxR-1 with Man A) TrxR-1 was inhibited with IC50 of 1586 (±128) nM (Figure 2B). At a concentration of 5 μM, the inhibition is similar with or without preincubation.
The differences in IC50 with and without preincubation suggest that the inhibition of DTNB reduction is time dependent. Figure 2C shows the time course of inhibition of TrxR-1 with 1 μM Man A. Inhibition is observed as early as 2 min of incubation and continues to increase over time.
In an effort to determine if the inhibition of TrxR-1 by Man A is reversible, the DTNB reduction assay was performed as before; however, after 1 h of incubation of prereduced TrxR-1 with 2.45 μM Man A, the mixture was passed through a gel filtration column (MW cutoff of 6000 amu) followed by a 30 min incubation with additional NADPH (Figure 2D). Gel filtration should remove all unbound Man A from the solution, and the subsequent 30 min incubation should allow for reestablishing equilibrium. As shown in Figure 2D, this treatment still resulted in complete inhibition of TrxR-1 demonstrating that the inhibition of TrxR-1 by Man A is irreversible. It is particularly important to note that Man A has no effect on the rate of DTNB reduction when incubated with oxidized TrxR-1. When oxidized TrxR-1 is incubated with Man A for 30 min, followed by gel filtration to remove Man A, TrxR-1 activity is restored completely upon reduction with NADPH. This observation demonstrates that either the N-terminal or C-terminal redox centers, or both, must be reduced for Man A to have an effect on TrxR-1. Furthermore, it suggests that one or both are sites of reactivity with Man A.
Numerous electrophiles inhibit mammalian TrxR-1 by alkylating the
C-terminal selenocysteine. Many of these electrophiles are α,β-unsaturated
carbonyl compounds that irreversibly react with TrxR-1.25,28−36 Any of the four (mono, di, and tri) unsaturated carbonyl groups
of Man A could serve as Michael acceptors and alkylate TrxR-1 at the
C-terminal selenocysteine to inhibit the enzyme. Additionally, the
epoxide may also act as an electrophile. Two Man A derivatives were
examined for their ability to inhibit TrxR-1/Trx toward insulin reduction:
deoxy-Man A (2) and dihydro-Man A (3). These
results are shown in Figure 3. Neither deoxy-Man A (2) nor dihydro-Man A (3) inhibits insulin reduction by the TrxR-1/Trx system at
concentrations of 20 μM. This demonstrates that while Man A
has several electrophilic sites, the site of reactivity with TrxR-1
must be the α,β-unsaturated cyclohexenone.
Figure 3.
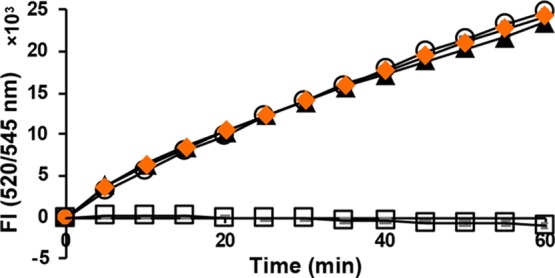
Insulin reduction by TrxR-1/Trx in the presence of 20 μM each of deoxy-Man A (▲); dihydro-Man A (orange ◆); Man A (□); and control (DMSO only, ○).
The selenol selective probe Sel-green37 was used to evaluate the reactivity of Man A and its derivatives with selenocysteine. The strongly nucleophilic, reduced selenol undergoes nucleophilic aromatic substitution with the probe, which then releases the fluorophore as shown in Scheme 1. When selenocysteine is alkylated, it will not react with the probe, and fluorescence will be suppressed. Figure 4 demonstrates that the release of fluorescent reporter by selenocysteine is completely inhibited in the presence of Man A. Under the same conditions, deoxy-Man A shows only slight inhibition, indicating that Man A reacts readily with selenocysteine, while its derivatives do not.
Scheme 1. Nucleophilic Substitution Reaction of Sel-green Probe by Reduced Selenocysteine.

Figure 4.
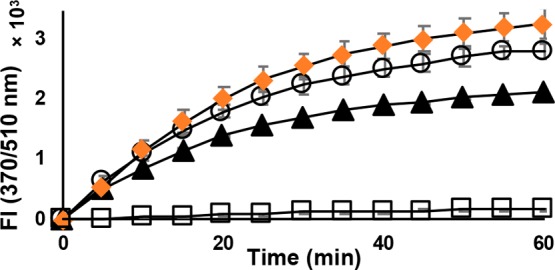
Reaction of reduced l-selenocysteine (20 μM) with Sel-green probe (20 μM) in the presence of Man A and derivatives (20 μM): Control (DMSO only, ○); deoxy-Man A (▲); dihydro-Man A (orange ◆); Man A (□).
The chemical modification of the C-terminal selenocysteine of TrxR-1 yields a SecTRAP (selenium compromised thioredoxin reductase-derived apoptotic proteins), which promotes both apoptosis and necrosis via oxidative stress and increased intracellular reactive oxygen species (ROS) production.38 Both curcumin and juglone modified TrxR-1 have demonstrated strongly induced NADPH oxidase activity producing O2–• in the presence of oxygen via the N-terminal (Cys59/Cys64) redox center.28,39,40 In addition to increased ROS, Man A treated cells have shown NADPH oxidase activity,21 decreased TrxR-1 activity,16 and decreased Trx expression.20 One reason for activation of NADPH oxidase activity may be due to the alkylation of TrxR-1, producing a SecTRAP. Transfection of cells with SOD and TrxR cDNA or pretreatment with ROS scavengers has been shown to block the adverse effects of Man A.15,41 The NADPH oxidase activity of TrxR-1 in the absence of a disulfide substrate was examined (Figure 5A). When preincubated with 62.5 μM Man A, the consumption of NADPH by TrxR-1 is increased 10-fold relative to the control.
Figure 5.
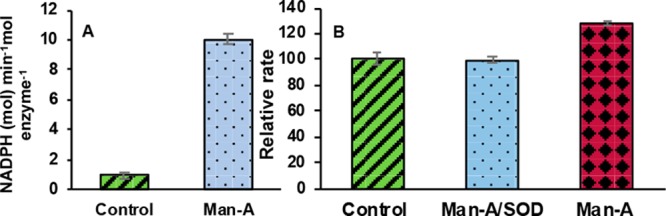
(A) NADPH oxidase activity of TrxR-1 (0.2 μM) induced by Man A (62.5 μM). (B) Reduction of cytochrome C (100 μM) by TrxR-1 (100 nM) in the presence of Man A (62.5 μM); Man A and SOD (62.5 μM and 6 units/well); control (DMSO only).
The reduction of cytochrome C in the presence of Man A treated TrxR-1 was monitored over time with and without the addition of SOD (Figure 5B). Superoxide can reduce cytochrome C. If superoxide is produced by Man A treated TrxR-1, then the addition of SOD should inhibit cytochrome C reduction. This was indeed the case. The rate of cytochrome C reduction was decreased by 28% in the presence of SOD when compared to the Man A treated sample alone.
Finally, Man A has numerous electrophilic sites that may serve as sites of reactivity. Given that living cells contain numerous endogenous nucleophiles, it may be difficult for Man A to reach its target TrxR-1 in living cells. The TrxR-1 activity of human lymphoblast (GM02152) cell lysate in the presence of Man A (1 μM) was analyzed using the insulin reduction assay (Figure 6). The ratio of initial rates for control/Man A treated sample is 2.3:1. There appears to be a longer induction time in the Man A treated sample. However, the manufacturer of the assay kit recommends comparing rates between 15 and 45 min. Here, the relative rate for the control/Man A treated sample is 1.2:1.
Figure 6.

TrxR-1/Trx insulin reduction assay with cell homogenate (GM02152, 15.6 μg) in the presence of Man A (1 μM): control (DMSO only, ○); Man A (□).
In conclusion, we have demonstrated that Man A is an irreversible inhibitor of human TrxR-1. The mechanism of inhibition of TrxR-1 by Man A is very likely by acting as a Michael acceptor to the nucleophilic Sec residue in the C-terminal redox center of the enzyme. These findings should stimulate a reconsideration of the mechanism for the antitumor activity of Man A.
Acknowledgments
The authors are grateful to Dr. Yuan Liu and Shantelle Rolle for providing untreated human lymphoblast cells.
Glossary
ABBREVIATIONS
- Man A
manumycin A
- Deoxy-Man A
deoxymanumycin A
- Dihyro-Man A
dihydromanumycin A
- FTase
farnesyl transferase
- TrxR1
mammalian cytosolic thioredoxin reductase 1
- NADPH
nicotinamide adenine dinucleotide phosphate
- SecTRAP
selenium compromised thioredoxin reductase apoptotic proteins
- Trx
thioredoxin
- FAD
flavin adenine dinucleotide
- ROS
reactive oxygen species
- DTNB
5,5′-dithio-bis(2-nitrobenzoic acid)
- amu
atomic mass unit
- SOD
superoxide dismutase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00489.
Experimental procedures for all enzyme and biochemical assays, cell culture, and harvesting and lysis conditions (PDF)
Author Contributions
The manuscript was written through contributions of all authors who have given approval to the final version. K.R. designed the research project. A.T. conducted all experiments. K.R. wrote the manuscript. K.R. and A.T. revised the manuscript. A.T. wrote and K.R. revised the Supporting Information.
A.T. is grateful for a Graduate Assistantship from the FIU College of Arts Sciences and Education.
The authors declare no competing financial interest.
Supplementary Material
References
- Hara M.; Akasaka K.; Akinaga S.; Okabe M.; Nakano H.; Gomez R.; Wood D.; Uh M.; Tamanoi F. Identification of Ras farnesyltransferase inhibitors by microbial screening. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2281–2285. 10.1073/pnas.90.6.2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Papke B.; Der C. J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- Fletcher S.; Keaney E. P.; Cummings C. G.; Blaskovich M. A.; Hast M. A.; Glenn M. P.; Chang S. Y.; Bucher C. J.; Floyd R. J.; Katt W. P. Structure-based design and synthesis of potent, ethylenediamine-based, mammalian farnesyltransferase inhibitors as anticancer agents. J. Med. Chem. 2010, 53, 6867–6888. 10.1021/jm1001748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa S. F.; Fernandes P. A.; Ramos M. J. Farnesyltransferase inhibitors: a detailed chemical view on an elusive biological problem. Curr. Med. Chem. 2008, 15, 1478–1492. 10.2174/092986708784638825. [DOI] [PubMed] [Google Scholar]
- Capell B. C.; Erdos M. R.; Madigan J. P.; Fiordalisi J. J.; Varga R.; Conneely K. N.; Gordon L. B.; Der C. J.; Cox A. D.; Collins F. S. Inhibiting farnesylation of progerin prevents the characteristic nuclear blebbing of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 12879–12884. 10.1073/pnas.0506001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner F. S.; Eastman R. T.; Nepomuceno-Silva J. L.; Speelmon E. C.; Myler P. J.; Van Voorhis W. C.; Yokoyama K. Cloning, heterologous expression, and substrate specificities of protein farnesyltransferases from Trypanosoma cruzi and Leishmania major. Mol. Biochem. Parasitol. 2002, 122, 181–188. 10.1016/S0166-6851(02)00099-3. [DOI] [PubMed] [Google Scholar]
- Esteva M. I.; Kettler K.; Maidana C.; Fichera L.; Ruiz A. M.; Bontempi E. J.; Andersson B.; Dahse H.-M.; Haebel P.; Ortmann R. Benzophenone-based farnesyltransferase inhibitors with high activity against Trypanosoma cruzi. J. Med. Chem. 2005, 48, 7186–7191. 10.1021/jm050456x. [DOI] [PubMed] [Google Scholar]
- Glenn M. P.; Chang S. Y.; Hucke O.; Verlinde C. L.; Rivas K.; Hornéy C.; Yokoyama K.; Buckner F. S.; Pendyala P. R.; Chakrabarti D. Structurally simple farnesyltransferase inhibitors arrest the growth of malaria parasites. Angew. Chem., Int. Ed. 2005, 44, 4903–4906. 10.1002/anie.200500674. [DOI] [PubMed] [Google Scholar]
- Ibrahim M.; Azzouz N.; Gerold P.; Schwarz R. T. Identification and characterisation of Toxoplasma gondii protein farnesyltransferase. Int. J. Parasitol. 2001, 31, 1489–1497. 10.1016/S0020-7519(01)00268-5. [DOI] [PubMed] [Google Scholar]
- Kainuma O.; Asano T.; Hasegawa M.; Kenmochi T.; Nakagohri T.; Tokoro Y.; Isono K. Inhibition of growth and invasive activity of human pancreatic cancer cells by a farnesyltransferase inhibitor, manumycin. Pancreas 1997, 15, 379–383. 10.1097/00006676-199711000-00008. [DOI] [PubMed] [Google Scholar]
- Nagase T.; Kawata S.; Tamura S.; Matsuda Y.; Inui Y.; Yamasaki E.; Ishiguro H.; Ito T.; Miyagawa J.; Mitsui H. Manumycin and gliotoxin derivative KT7595 block Ras farnesylation and cell growth but do not disturb lamin farnesylation and localization in human tumour cells. Br. J. Cancer 1997, 76, 1001–1010. 10.1038/bjc.1997.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier M.; Kwon Y. K.; Pandey S. K.; Zhu T. N.; Zhao R. J.; Maciuk A.; He H. J.; DeCabo R.; Kole S. Binding of manumycin A inhibits IκB kinase β activity. J. Biol. Chem. 2006, 281, 2551–2561. 10.1074/jbc.M511878200. [DOI] [PubMed] [Google Scholar]
- Ito T.; Kawata S.; Tamura S.; Igura T.; Nagase T.; Miyagawa J. I.; Yamazaki E.; Ishiguro H.; Matsuzawa Y. Suppression of human pancreatic cancer growth in BALB/c nude mice by manumycin, a farnesyl: protein transferase inhibitor. Jpn. J. Cancer Res. 1996, 87, 113–116. 10.1111/j.1349-7006.1996.tb03146.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears K. T.; Daino H.; Carey G. B. Reactive oxygen species-dependent destruction of MEK and Akt in Manumycin stimulated death of lymphoid tumor and myeloma cell lines. Int. J. Cancer 2008, 122, 1496–1505. 10.1002/ijc.23207. [DOI] [PubMed] [Google Scholar]
- Ahmad F.; Ghosh S.; Sinha S.; Joshi S. D.; Mehta V. S.; Sen E. TGF-β-induced hCG-β regulates redox homeostasis in glioma cells. Mol. Cell. Biochem. 2015, 399, 105–112. 10.1007/s11010-014-2237-6. [DOI] [PubMed] [Google Scholar]
- Ahmad R.; Sylvester J.; Ahmad M.; Zafarullah M. Involvement of H-Ras and reactive oxygen species in proinflammatory cytokine-induced matrix metalloproteinase-13 expression in human articular chondrocytes. Arch. Biochem. Biophys. 2011, 507, 350–355. 10.1016/j.abb.2010.12.032. [DOI] [PubMed] [Google Scholar]
- Carey G. B.; Roy S. K.; Daino H. The natural tumorcide Manumycin-A targets protein phosphatase 1α and reduces hydrogen peroxide to induce lymphoma apoptosis. Exp. Cell Res. 2015, 332, 136–145. 10.1016/j.yexcr.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M. C.; Chen Y. J.; Chang H. H.; Chan C. P.; Yeh C. Y.; Wang Y. L.; Cheng R. H.; Hahn L. J.; Jeng J. H. Areca nut components affect COX-2, cyclin B1/cdc25C and keratin expression, PGE2 production in keratinocyte is related to reactive oxygen species, CYP1A1, Src, EGFR and Ras signaling. PLoS One 2014, 9, e101959. 10.1371/journal.pone.0101959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit D.; Sharma V.; Ghosh S.; Koul N.; Mishra P. K.; Sen E. Manumycin inhibits STAT3, telomerase activity, and growth of glioma cells by elevating intracellular reactive oxygen species generation. Free Radical Biol. Med. 2009, 47, 364–374. 10.1016/j.freeradbiomed.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Pan J.; She M.; Xu Z. X.; Sun L.; Yeung S. C. J. Farnesyltransferase inhibitors induce DNA damage via reactive oxygen species in human cancer cells. Cancer Res. 2005, 65, 3671–3681. 10.1158/0008-5472.CAN-04-2744. [DOI] [PubMed] [Google Scholar]
- She M. R.; Yang H.; Sun L.; Yeung S. C. J. Redox control of manumycin A-induced apoptosis in anaplastic thyroid cancer cells: involvement of the xenobiotic apoptotic pathway. Cancer Biol. Ther. 2006, 5, 275–280. 10.4161/cbt.5.3.2383. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Jiang H.; Xie L.; Hu J.; Li L.; Yang M.; Cheng L.; Liu B.; Qian X. Antitumor effect of manumycin on colorectal cancer cells by increasing the reactive oxygen species production and blocking PI3K-AKT pathway. OncoTargets Ther. 2016, 9, 2885–2895. 10.2147/OTT.S102408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Fan S. M.; Ye Y. C.; Tashiro S. I.; Onodera S.; Ikejima T. The tyrphostin AG1478 augments oridonin-induced A431 cell apoptosis by blockage of JNK MAPK and enhancement of oxidative stress. Free Radical Res. 2012, 46, 1393–1405. 10.3109/10715762.2012.720017. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Li X.; Han X.; Liu R.; Fang J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. 10.1016/j.tips.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Chen W.; Tuladhar A.; Rolle S.; Lai Y.; Rodriguez del Rey F.; Zavala C. E.; Liu Y.; Rein K. S. Brevetoxin-2, is a unique inhibitor of the C-terminal redox center of mammalian thioredoxin reductase-1. Toxicol. Appl. Pharmacol. 2017, 329, 58–66. 10.1016/j.taap.2017.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano S. J.; Lu J.; Gustafsson T. N.; Holmgren A. Activity assays of mammalian thioredoxin and thioredoxin reductase: fluorescent disulfide substrates, mechanisms, and use with tissue samples. Anal. Biochem. 2014, 449, 139–146. 10.1016/j.ab.2013.12.025. [DOI] [PubMed] [Google Scholar]
- Cenas N.; Nivinskas H.; Anusevicius Z.; Sarlauskas J.; Lederer F.; Arnér E. S. Interactions of quinones with thioredoxin reductase a challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 2004, 279, 2583–2592. 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- Chew E. H.; Lu J.; Bradshaw T. D.; Holmgren A. Thioredoxin reductase inhibition by antitumor quinols: a quinol pharmacophore effect correlating to antiproliferative activity. FASEB J. 2008, 22, 2072–2083. 10.1096/fj.07-101477. [DOI] [PubMed] [Google Scholar]
- Fang J.; Holmgren A. Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 2006, 128, 1879–1885. 10.1021/ja057358l. [DOI] [PubMed] [Google Scholar]
- Powis G.; Wipf P.; Lynch S. M.; Birmingham A.; Kirkpatrick D. L. Molecular pharmacology and antitumor activity of palmarumycin-based inhibitors of thioredoxin reductase. Mol. Cancer Ther. 2006, 5, 630–636. 10.1158/1535-7163.MCT-05-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yao J.; Peng S.; Li X.; Fang J. Securinine disturbs redox homeostasis and elicits oxidative stress-mediated apoptosis via targeting thioredoxin reductase. Biochim. Biophys. Acta, Mol. Basis Dis. 2017, 1863, 129–138. 10.1016/j.bbadis.2016.10.019. [DOI] [PubMed] [Google Scholar]
- Duan D.; Zhang J.; Yao J.; Liu Y.; Fang J. Targeting thioredoxin reductase by parthenolide contributes to inducing apoptosis of HeLa cells. J. Biol. Chem. 2016, 291, 10021–10031. 10.1074/jbc.M115.700591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Li Y.; Duan D.; Yao J.; Gao K.; Fang J. Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress-mediated apoptosis of HeLa cells. Biochem. Pharmacol. 2016, 102, 34–44. 10.1016/j.bcp.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Duan D.; Ge C.; Yao J.; Liu Y.; Li X.; Fang J. Synthesis of xanthohumol analogues and discovery of potent thioredoxin reductase inhibitor as potential anticancer agent. J. Med. Chem. 2015, 58, 1795–1805. 10.1021/jm5016507. [DOI] [PubMed] [Google Scholar]
- Seki H.; Xue S.; Pellett S.; Šilhár P.; Johnson E. A.; Janda K. D. Cellular protection of SNAP-25 against botulinum neurotoxin/A: Inhibition of thioredoxin reductase through a suicide substrate mechanism. J. Am. Chem. Soc. 2016, 138, 5568–5575. 10.1021/jacs.5b12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B.; Ge C.; Yao J.; Liu Y.; Xie H.; Fang J. Selective selenol fluorescent probes: design, synthesis, structural determinants, and biological applications. J. Am. Chem. Soc. 2015, 137, 757–769. 10.1021/ja5099676. [DOI] [PubMed] [Google Scholar]
- Anestål K.; Prast-Nielsen S.; Cenas N.; Arnér E. S. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS One 2008, 3, e1846. 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J.; Lu J.; Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin a novel molecular mechanism for its anticancer activity. J. Biol. Chem. 2005, 280, 25284–25290. 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- Cheng Q.; Antholine W. E.; Myers J. M.; Kalyanaraman B.; Arnér E. S.; Myers C. R. The selenium-independent inherent pro-oxidant NADPH oxidase activity of mammalian thioredoxin reductase and its selenium-dependent direct peroxidase activities. J. Biol. Chem. 2010, 285, 21708–21723. 10.1074/jbc.M110.117259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Tsukahara F.; Maru Y. N-acetyl-cysteine enhances growth in BCR-ABL-transformed cells. Cancer Sci. 2005, 96, 240–244. 10.1111/j.1349-7006.2005.00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.