

Abstract
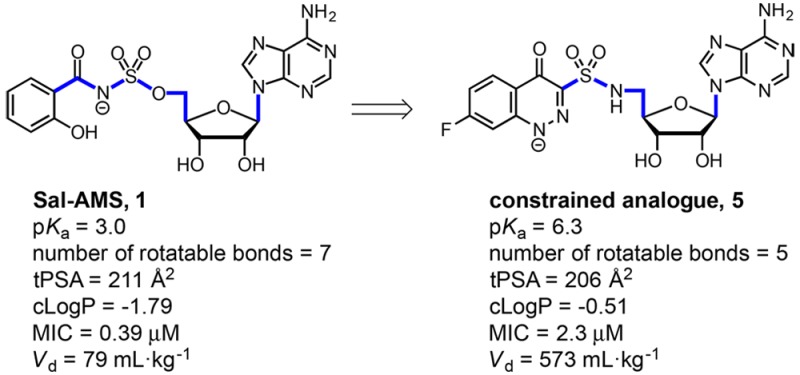
5′-O-[N-(Salicyl)sulfamoyl]adenosine (Sal-AMS, 1) is a nucleoside antibiotic that inhibits incorporation of salicylate into siderophores required for bacterial iron acquisition and has potent activity against Mycobacterium tuberculosis (Mtb). Cinnolone analogues exemplified by 5 were designed to replace the acidic acyl-sulfamate functional group of 1 (pKa = 3) by a more stable sulfonamide linkage (pKa = 6.0) in an attempt to address potential metabolic liabilities and improve membrane permeability. We showed 5 potently inhibited the mycobacterial salicylate ligase MbtA (apparent Ki = 12 nM), blocked production of the salicylate-capped siderophores in whole-cell Mtb, and exhibited excellent antimycobacterial activity under iron-deficient conditions (minimum inhibitor concentration, MIC = 2.3 μM). To provide additional confirmation of the mechanism of action, we demonstrated the whole-cell activity of 5 could be fully antagonized by the addition of exogenous salicylate to the growth medium. Although the total polar surface area (tPSA) of 5 still exceeds the nominal threshold value (140 Å) typically required for oral bioavailability, we were pleasantly surprised to observe introduction of the less acidic and conformationally constrained cinnolone moiety conferred improved drug disposition properties as evidenced by the 7-fold increase in volume of distribution in Sprague–Dawley rats.
Keywords: Tuberculosis, siderophores, mycobactin, conformationally constrained analogues, pharmacokinetics
Tuberculosis (TB) superseded HIV as the leading cause of infectious disease mortality worldwide in 2015, and TB shows no indication of relinquishing this notorious distinction.1 The obligate pathogen Mycobacterium tuberculosis (Mtb) has evolved over millennia to evade and co-opt host immune responses to establish a persistent infection. Consequently, antimicrobial therapy for even the simplest drug-susceptible TB involves prolonged treatment for six to nine months employing a combination regimen of four drugs: isoniazid, rifampicin, ethambutol, and pyrazinamide. Drug-resistant TB (DR-TB) or comorbidity with the chronic diseases diabetes and HIV further complicates the management of TB.2 Motivated by the need to combat DR-TB, shorten the treatment duration, and improve adherence, researchers have sought to identify vulnerable metabolic pathways for drug development.
M. tuberculosis, like almost all forms of life, has an absolute requirement for iron, an essential metal cofactor in many biochemical processes including respiration, central metabolism, and nucleic acid biosynthesis.3,4 Due to very low aqueous solubility of ferric salts as well as to prevent bacterial colonization and growth, the free Fe3+ concentrations in human serum and body fluids is maintained at an exceptionally low level (∼10–24 M).5M. tuberculosis overcomes host iron restriction through the synthesis, export, and reuptake of siderophores known as the mycobactins.6,7 Isogenic Mtb mutants deficient in production or transport of these high-affinity iron chelators have severe growth defects in vitro under iron limiting conditions and are rapidly eliminated in vivo.7−10 Based on this genetic validation, the bisubstrate inhibitor 5′-O-[N-(salicyl)sufamoyl]adenosine (Sal-AMS 1, Figure 1A) was designed to block mycobactin biosynthesis in Mtb at the first committed step catalyzed by the adenylating enzyme MbtA, which ligates salicylic acid onto the nonribosomal peptide synthetase MbtB at the expense of ATP.11−13 The nucleoside 1 possesses potent enzyme inhibition of MbtA (apparent Ki = 6.6 nM), selective iron-dependent antitubercular activity (minimum inhibitory concentration (MIC) = 0.39 μM), and in vivo efficacy in an acute murine TB infection model when dosed intraperitoneally (reducing the lung colony forming units (CFU) by 1.1 log10).13,14 Structure–activity relationship (SAR) studies of 1 indicate only conservative modifications are tolerated in the salicyl15 and acyl-sulfamate13,16 moieties, whereas the nucleoside exhibits substantial flexibility.17−21 Despite its promising activity, 1 suffers from nonoptimal drug disposition and pharmacokinetic (PK) properties, in part caused by the ionized acyl-sulfamate. Moreover, hydrolysis across the acyl-sulfamate linkage of 1 would liberate cytotoxic 5′-O-(sufamoyl)adenosine.22 Although hydrolysis has not been observed in cell culture and in vitro metabolic stability studies, removal of this potential liability is desirable.20
Figure 1.

(A) Chemical structure of 1 and its biological profile. (B) Quinolone 2 and benzoxazinone 3. (C) Structure of cinnolinones 4–7 described in this study.
Design and Synthesis of Constrained Analogues
To simultaneously address these concerns we previously synthesized benzoxazinone 2 and quinolone 3 (Figure 1B) that mimic the MbtA-bound conformation of 1.23 Notably, both heterocyclic analogues remove the acidic acyl-sulfamate linker moiety of 1 and replace it with a more stable sulfonamide linkage incapable of releasing 5′-O-(sufamoyl)adenosine through hydrolysis or metabolism. Unfortunately, neither 2 nor 3 were active toward Mtb (Table 1). Biochemical evaluation against MbtA revealed quinolone 3 retained some enzyme inhibition (appKi = 120 nM) owing to its ionizable proton at N-1 (pKa ≈ 7.8) while benzoxazinone 2 lacking an acidic proton in the heterocycle was devoid of activity (appKi > 50 μM). These results suggested the formal negative charge of the acyl-sulfamate linker in 1 is critical for binding to MbtA, a finding supported by computational and crystallographic analyses. However, a formal negative charge is deleterious to passive diffusion of 1 across mammalian membranes. Thus, a delicate balance of the linker pKa is required to maintain potent antitubercular activity and acceptable membrane permeability. In the present work, we report on the design of new conformationally constrained analogues 4–7 (Figure 1C) containing a cinnolone as a closer isosteric mimic of 1 that possess a range of pKas at N-1 through strategic incorporation of fluorines at C-6 and C-7.
Table 1. Physicochemical Properties, Enzyme Inhibition, and Antimycobacterial Activity of Analogues 4–7 Compared to 1, 2, and 3.
| compd | pKaa | cLogPb | appKi (nM)c | MIC (μM)d | CC50 (μM)e |
|---|---|---|---|---|---|
| 1f | 2.8 | –1.79 | 6.6 ± 1.5 | 0.39 | >200 |
| 2g | –1.26 | >50,000 | >50 | >200 | |
| 3g | 7.8 | –0.57 | 120 ± 20 | >50 | >200 |
| 4 | 7.0 | –0.65 | 14.4 ± 1.8 | 4.7 | >200 |
| 5 | 6.0 | –0.51 | 11.6 ± 2.4 | 2.3 | >200 |
| 6 | 6.1 | –0.51 | 20.9 ± 4.5 | 4.7 | >200 |
| 7 | 5.7 | –0.44 | 14.0 ± 3.4 | 3.1 | >200 |
Calculated using pKa module of Jaguar.
Calculated using ChemBioDraw Ultra 14.0.
Assay performed with 7 nM MbtA, 10 mM ATP, 250 μM salicylic acid, 1 mM PPi.
Grown in glycerol-alanine salts (GAS) medium without ferric ammonium citrate at pH 6.6.
Cell cytotoxicity evaluated against Vero and HepG2 cell lines.
Ref (13).
Ref (23).
We devised a four-step synthesis of the cinnolones 4–7 as shown in Scheme 1. Claisen-like condensation of anthranilic acid methyl esters 8a–d and the dianion of N-Boc methansulfonamide 9 afforded the β-ketosulfonamides 10a–d. Utilization of unprotected anilines was preferred; although four equivalents of LDA were required to obtain complete conversion because the starting aniline and β-ketosulfonamide product each consumed one equivalent of LDA. Diazotization of the anilines 10a–d in a mixed AcOH–H2O–THF solvent system provided the cinnolin-4-one-3-sulfonamide derivatives 11a–d in yields ranging from 73% to 87%.24 The reaction presumably proceeds through intramolecular attack of the enol tautomer of the β-ketosulfonamide onto a diazonium intermediate to directly afford a cinnolin-4(3H)-one, which tautomerizes to the observed cinnolin-4(1H)-one. If the aniline is absent or protected, competitive nitrosation occurs at the active methylene furnishing oxime byproducts.23 Overall, this two-step route to cinnolones is exceptionally efficient compared to the reported five to seven step synthesis of the analogous cinnolin-4-one-3-carboxylates.24 Installation of the nucleoside was accomplished by regioselective Mitsunobu coupling of the acylsulfonamide NH over the N-1 cinnolone nitrogen atom in 11a–d with bis-Boc adenosine 12(25) to provide 13a–d.23 Global deprotection of 13a–d by 80% aqueous TFA afforded 4–7. The crude products were purified by reverse-phase preparative HPLC using aqueous triethylammonium bicarbonate (pH 7.5)/acetonitrile and isolated as the triethylammonium salts.
Scheme 1. Synthesis of 5–8.

Enzyme Inhibition and Antitubercular Activity
Compounds 4–7 were evaluated for enzyme inhibition against recombinant MbtA using a [32P]PPi-ATP exchange assay under initial velocity condition employing physiologically relevant supersaturating substrate concentrations of salicylic acid and ATP.13 The concentration–response plots were fitted to the Morrison equation for tight binding inhibitors to determine the apparent inhibition constants (appKi) (Table 1). The cinnolones 4–7 maintained potent low nanomolar appKi ranging from 14–21 nM and were 2000-fold more potent than the isoelectronic benzoxazinone 2 highlighting the tremendous impact of the negative charge for MbtA binding. As expected from our previous SAR studies, introduction of a fluorine at either the C-6 or C-7 position of the cinnolone was well tolerated and had minimal effect on activity.15 Analogues 4–7 were approximately 1 order of magnitude more potent than quinolone 3, illustrating the better isosteric design of the cinnolone that contains a smaller nitrogen atom at the 2-position compared to the CH found in the quinolone. The pKa of the heterocycle is less important to discriminate activity in the in vitro biochemical assay since it was conducted at pH 8.0; however, the pH within the phagosomal compartment of activated macrophages, where Mtb resides, is around 4.5 while the intracellular pH of mycobacteria is near 7.26,27 Thus, we anticipated the lower cinnolone pKa would be manifested in improved antimycobacterial activity relative to quinolone 3.
The whole-cell activity of 4–7 was assessed with M. tuberculosis H37Rv in glycerol-alanine-salts (GAS) medium lacking supplemented iron at pH 6.6.13 The minimum inhibitory concentrations (MICs) required to inhibit 99% of bacterial growth of 4–7 were nearly uniform ranging from 2.3–4.7 μM, consistent with the biochemical data and validating our design strategy. Under these conditions quinolone 3 was completely inactive (MIC > 50 μM), demonstrating the critical impact of the inhibitor pKa for mycobacterial activity. Finally, to confirm selectivity, we evaluated 4–7 against two mammalian cell lines, but did not observe reduced cell viability at the highest concentration (200 μM).
Next, to provide support for the mechanism of action, we measured the effect of salicylate on Mtb susceptibility to 4–7. We predicted exogenous salicylate would antagonize 4–7 through direct competition with MbtA. As shown in Figure 2, supplemental salicylate relieved inhibition of 4–7 in a dose-dependent manner. Salicylate concentrations of 12.5 μM conferred high-level resistance to 4–7 as well as 1 (not shown), raising the MIC to greater than or equal to 50 μM. While the activity of competitive inhibitors can be overcome by increasing substrate concentration, we did not expect salicylate to cause such a dramatic effect and hypothesize other factors may be responsible for the observed strong antagonism.
Figure 2.
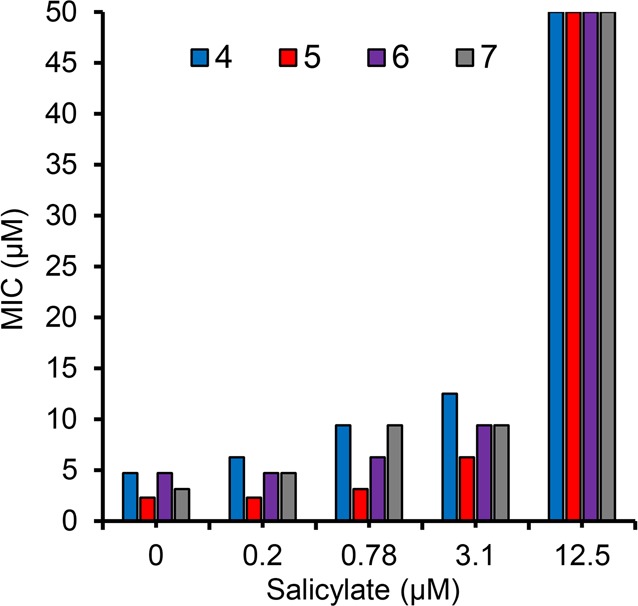
Effect of salicylate concentration on the sensitivity of Mtb to 4–7 that was grown in GAST medium supplemented with salicylate at 0, 0.2, 0.78, 3.1, and 12.5 μM.
Salicylate, typically at millimolar concentrations, induces a multiple antibiotic resistance (MAR) phenotype in E. coli by binding to a MarR transcriptional regulator.28,29 The MAR phenotype has also been observed in Mtb where 0.125–1 mM salicylate mildly reduced susceptibility of Mtb to first- and second-line TB drugs.30 Several members of the MarR family of transcriptional factors have now been identified in Mtb including Rv2887, which regulates expression of the methyltransferase Rv0560c in a salicylate-dependent manner.31−33 Overexpression of Rv0560c, caused by spontaneous mutation to its transcriptional repressor Rv2887, was recently shown to confer resistance to a pyrido-benzimidazole drug candidate.32 Based on these findings, we hypothesized salicylate-mediated induction of Rv0560c may similarly confer resistance to 1 and 4–7. The susceptibility of 1 was therefore evaluated against wild type Mtb and an Mtb Rv0560c-overexpression stain (Mtb Rv0560c-OE). Both strains exhibited identical susceptibility to 1 indicating Rv0560c is not responsible for the powerful salicylate-mediated antagonism of 1 (Supporting Information Table S1).
Quantitation of Mycobactin
To verify that cinnolones inhibit mycobactin production, we selected compound 5 for further whole-cell studies. Compound 1 was shown to inhibit mycobactin biosynthesis by a radiometric assay employing [7-14C]-salicylic acid that is incorporated into the mycobactins.11,13 This method enables mycobactin quantitation by autoradiographic-thin layer chromatography (radio-TLC), but suffers from the requirement to maintain dual biosafety level three (BSL-3) and 14C-radioisotope certification.11,13 We thus developed a complementary liquid chromatography tandem-mass spectrometry (LC–MS/MS) assay in multiple reaction monitoring (MRM) mode employing authentic synthetic standards of two of the most abundant mycobactins (a lipophilic mycobactin T and a water-soluble carboxymycobactin, see Supporting Information Figure S1). We quantified both cell-wall associated and secreted mycobactins in the samples obtained from Mtb cells grown in iron-deficient GAST medium either in the presence or absence of inhibitor 5.
The total concentrations of mycobactins determined at the indicated time after treatment with or without 5 are shown in Figure 3 normalized to the cell density. In iron-deficient GAST medium, robust production of the cell-wall associated lipophilic mycobactin-T was observed, whereas in the presence of 5 at 5 × MIC, mycobactin-T production was reduced more than 2-fold at 27 h and 5-fold at 70 h (Figure 3). Similarly, production of the water-soluble carboxymycobactins was attenuated by 1.5-fold. From this experiment, we conclude 5 is able to block mycobactin production providing further support for the designed mechanism action.
Figure 3.
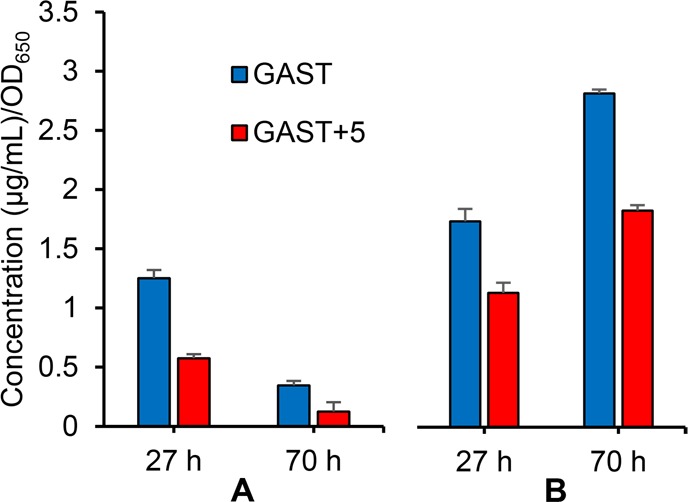
Normalized concentration of Mtb siderophores from cells (initial inoculum of OD650 = 0.2) grown in iron deficient (GAST) medium for 27 and 70 h with or without 5. (A) Concentration of cell-wall associated lipophilic mycobactin-T. (B) Concentration of water-soluble carboxymycobactin as a sum of secreted and cell-wall associated levels.
Pharmacokinetic Analysis of Analogue 5
To assess the impact of the cinnolone bioisostere on the pharmacokinetic (PK) properties, we selected compound 5 for single dose in vivo PK studies in female Sprague–Dawley rats (Table 2). The plasma concentrations of compound 5 at various time points after a single dose were determined by LC–MS/MS (Figure 4). Following intravenous (i.v.) administration, compound 5 (2.5 mg/kg) exhibited monoexponential elimination with an intrinsic elimination half-life of 40 min, which was nearly four times longer than 1. The increased half-life is derived from the nearly 7-fold greater steady-state volume of distribution (Vd = 0.57 L) that we attribute to the enhanced permeability of the cinnolinone. The pKa value of 5 ensures approximately 10% will exist in the neutral form at physiological pH, compared to less than 0.01% for 1. Oral administration (p.o.) of 5 (25 mg/kg) permitted calculation of the oral bioavailability (F), which remained low at 2%, but was more than twice that of 1. Collectively, these results are encouraging and show that bioisosteric replacement of the salicyl-sulfamoyl in 1 by a cinnolone provides an improved PK profile.
Table 2. In Vivo Pharmakokinetic Parameters of Inhibitor 5 in Female Sprague–Dawley Rats (n = 3, Mean ± SD).
| pharmacokinetic indices | analogue 5 | Sal-AMS 1 |
|---|---|---|
| dose i.v., p.o. (mg/kg) | 2.5, 25 | 2.5, 25 |
| AUC0–8h (p.o., μg·min·mL–1) | 58 ± 11 | 66 ± 41 |
| AUC0–8h (i.v., μg·min·mL–1) | 278 ± 27 | 519 ± 115 |
| Vd (i.v., L·kg–1) | 0.57 ± 0.17 | 0.079 ± 0.023 |
| CL (i.v., mL·min–1·kg–1) | 8.9 ± 1.0 | 4.9 ± 0.9 |
| t1/2 (i.v., min) | 40 ± 5 | 11 ± 2 |
| F (%) | 2.1 | <1 |
Figure 4.

Mean plasma concentration versus time curves after single p.o. (25 mg·kg–1) and i.v. (2.5 mg·kg–1) administration of compound 5 to female Sprague–Dawley rats. Error bars represent standard deviation of the mean (n = 3).
In conclusion, we successfully achieved our objective to replace the salicyl-sulfamate moiety of 1 by a bioactive isostere with more favorable physicochemical properties. Conformationally constrained cinnolinone-3-sulfonamides were efficiently synthesized in only two steps from commercially available anthranilates then ligated to adenosine by a Mitsunobu coupling to afford 4–7. Analogue 5 was identified as the most active compound possessing low nanomolar enzyme inhibition of MbtA and a respectable MIC of 2.3 μM against Mtb in iron-deficient GAST medium. We verified 5 inhibited biosynthesis of the lipophilic mycobactin-T and the water-soluble carboxymycobactins using a new LC–MS/MS assay. The ability of salicylate to antagonize 5 provided additional support for the mechanism of action. The improved performance of the cinnolone bioisostere was manifested in a 7-fold enhanced volume of distribution and 2-fold greater oral bioavailability.
Acknowledgments
We thank Dr. Bruce Witthuhn of College of Biological Sciences at the University of Minnesota for technical assistant with LC–MS/MS method development, Carl Nathan at Weil Cornell Medical College for providing Mtb Rv0560c-OE, and Peter Larson in Prof. David Ferguson’s lab for calculating the pKas. We thank College of Pharmacy at the University of Minnesota for providing the software Phoenix WinNonlin 7.0 used for noncompartmental PK analysis.
Glossary
ABBREVIATIONS
- %F
bioavailability
- HPLC
high performance liquid chromatography
- i.v.
intravenous
- MDR-TB
multidrug-resistant tuberculosis
- XDR-TB
extensively drug-resistant tuberculosis
- LDA
lithium diisopropylamide
- MIC
minimum inhibitory concentration
- PK
pharmacokinetic
- p.o.
oral
- SAR
structure–activity relationship
- t1/2
terminal elimination half-life following an i.v. dose
- TB
tuberculosis
- TFA
trifluoroacetic acid
- tPSA
total polar surface area
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.8b00090.
Experimental procedure, biochemical methods, LC–MS/MS method, and 1H and 13C NMR spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by a grant from the NIH (AI070219 to C.C.A.) and the Intramural Research Program of the NIAID, NIH (to C.E.B.).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization. Global tuberculosis report 2017. Executive Summary. http://www.who.int/tb/publications/global_report/en/.
- McIlleron H.; Meintjes G.; Burman W. J.; Maartens G. Complications of antiretroviral therapy in patients with tuberculosis: drug interactions, toxicity, and immune reconstitution inflammatory syndrome. J. Infect. Dis. 2007, 196 (Suppl 1), S63–75. 10.1086/518655. [DOI] [PubMed] [Google Scholar]
- Miethke M.; Marahiel M. A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. 10.1128/MMBR.00012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratledge C.; Dover L. G. Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 2000, 54, 881–941. 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]
- Schaible U. E.; Kaufmann S. H. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953. 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- Gobin J.; Moore C. H.; Reeve J. R.; Wong D. K.; Gibson B. W.; Horwitz M. A. Iron acquisition by Mycobacterium tuberculosis: Isolation and characterization of a family of iron-binding exochelins. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 5189–5193. 10.1073/pnas.92.11.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan C. A.; Martinot A. J.; Wei J.-R.; Madduri A.; Cheng T.-Y.; Young D. C.; Layre E.; Murry J. P.; Rubin E. J.; Moody D. B. Lipidomic analysis links mycobactin synthase K to iron uptake and virulence in M. tuberculosis. PLoS Pathog. 2015, 11, e1004792. 10.1371/journal.ppat.1004792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Voss J. J.; Rutter K.; Schroeder B. G.; Su H.; Zhu Y.; Barry C. E. 3rd The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 1252–1257. 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P. V.; Puri R. V.; Chauhan P.; Kar R.; Rohilla A.; Khera A.; Tyagi A. K. Disruption of mycobactin biosynthesis leads to attenuation of Mycobacterium tuberculosis for growth and virulence. J. Infect. Dis. 2013, 208, 1255–1265. 10.1093/infdis/jit250. [DOI] [PubMed] [Google Scholar]
- Tufariello J. M.; Kerantzas C. A.; Vilchèze C.; Calder R. B.; Nordberg E. K.; Fischer J. A.; Hartman T. E.; Yang E.; Driscoll T.; Cole L. E.; Sebra R.; Maqbool S. B.; Wattam A. R.; Jacobs W. R. Jr. Separable roles for Mycobacterium tuberculosis ESX-3 effectors in iron acquisition and virulence. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 348–357. 10.1073/pnas.1523321113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreras J. A.; Ryu J. S.; Di Lello F.; Tan D. S.; Quadri L. E. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005, 1, 29–32. 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- Miethke M.; Bisseret P.; Beckering C. L.; Vignard D.; Eustache J.; Marahiel M. A. Inhibition of aryl acid adenylation domains involved in bacterial siderophore synthesis. FEBS J. 2006, 273, 409–419. 10.1111/j.1742-4658.2005.05077.x. [DOI] [PubMed] [Google Scholar]
- Somu R. V.; Boshoff H.; Qiao C.; Bennett E. M.; Barry C. E. 3rd; Aldrich C. C. Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 31–34. 10.1021/jm051060o. [DOI] [PubMed] [Google Scholar]
- Lun S.; Guo H.; Adamson J.; Cisar J. S.; Davis T. D.; Chavadi S. S.; Warren J. D.; Quadri L. E.; Tan D. S.; Bishai W. R. Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother. 2013, 57, 5138–5140. 10.1128/AAC.00918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao C. H.; Gupte A.; Boshoff H. I.; Wilson D. J.; Bennett E. M.; Somu R. V.; Barry C. E. 3rd; Aldrich C. C. 5′-O-[(N-cyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem. 2007, 50, 6080–6094. 10.1021/jm070905o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannada J.; Bennett E. M.; Wilson D. J.; Boshoff H. I.; Barry C. E. 3rd; Aldrich C. C. Design, synthesis, and biological evaluation of β-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org. Lett. 2006, 8, 4707–4710. 10.1021/ol0617289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somu R. V.; Wilson D. J.; Bennett E. M.; Boshoff H. I.; Celia L.; Beck B. J.; Barry C. E. 3rd; Aldrich C. C. Antitubercular nucleosides that inhibit siderophore biosynthesis: SAR of the glycosyl domain. J. Med. Chem. 2006, 49, 7623–7635. 10.1021/jm061068d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neres J.; Labello N. P.; Somu R. V.; Boshoff H. I.; Wilson D. J.; Vannada J.; Chen L.; Barry C. E. 3rd; Bennett E. M.; Aldrich C. C. Inhibition of siderophore biosynthesis in Mycobacterium tuberculosis with nucleoside bisubstrate analogues: structure-activity relationships of the nucleobase domain of 5′-O-[N-(salicyl)sulfamoyl]adenosine. J. Med. Chem. 2008, 51, 5349–5370. 10.1021/jm800567v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajczyk A.; Zeidler J.; Januszczyk P.; Dawadi S.; Boshoff H. I.; Barry C. E. 3rd; Ostrowski T.; Aldrich C. C. 2-Aryl-8-aza-3-deazaadenosine analogues of 5′-O-[N-(salicyl)sulfamoyl]adenosine: nucleoside antibiotics that block siderophore biosynthesis in Mycobacterium tuberculosis. Bioorg. Med. Chem. 2016, 24, 3133–143. 10.1016/j.bmc.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson K. M.; Viswanathan K.; Dawadi S.; Duckworth B. P.; Boshoff H. I.; Barry C. E.; Aldrich C. C. Synthesis and pharmacokinetic evaluation of siderophore biosynthesis inhibitors for Mycobacterium tuberculosis. J. Med. Chem. 2015, 58, 5459–5475. 10.1021/acs.jmedchem.5b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawadi S.; Viswanathan K.; Boshoff H. I.; Barry C. E. III; Aldrich C. C. Investigation and conformational analysis of fluorinated nucleoside antibiotics targeting siderophore biosynthesis. J. Org. Chem. 2015, 80, 4835–4850. 10.1021/acs.joc.5b00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch A.; Coutsogeorgopoulos C. Inhibition of protein synthesis by 5′-sulfamoyladenosine. Biochemistry 1971, 10, 4394–4398. 10.1021/bi00800a007. [DOI] [PubMed] [Google Scholar]
- Engelhart C. A.; Aldrich C. C. Synthesis of chromone, quinolone, and benzoxazinone sulfonamide nucleosides as conformationally constrained inhibitors of adenylating enzymes required for siderophore biosynthesis. J. Org. Chem. 2013, 78, 7470–7481. 10.1021/jo400976f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad R. A.; White W. A.. 4-(1H)-Oxocinnoline-3-carboxylic acid derivatives. U.S. Patent 4,379,929, April 12, 1983.
- Ikeuchi M.; Meyer M. E.; Ding Y.; Hiratake J.; Richards N. G. A critical electrostatic interaction mediates inhibitor recognition by human asparagine synthetase. Bioorg. Med. Chem. 2009, 17, 6641–6650. 10.1016/j.bmc.2009.07.071. [DOI] [PubMed] [Google Scholar]
- Schaible U. E.; Sturgill-Koszycki S.; Schlesinger P. H.; Russell D. G. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 1998, 160, 1290–1296. [PubMed] [Google Scholar]
- Vandal O. H.; Pierini L. M.; Schnappinger D.; Nathan C. F.; Ehrt S. A membrane protein preserves intrabacterial pH in intraphagosomal Mycobacterium tuberculosis. Nat. Med. 2008, 14, 849–854. 10.1038/nm.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner J. L. Nonheritable resistance to chloramphenicol and other antibiotics induced by salicylates and other chemotactic repellents in Escherichia coli K-12. Proc. Natl. Acad. Sci. U. S. A. 1985, 82, 8771–8774. 10.1073/pnas.82.24.8771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekshun M. N.; Levy S. B. The mar regulon: multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 1999, 7, 410–413. 10.1016/S0966-842X(99)01589-9. [DOI] [PubMed] [Google Scholar]
- Schaller A.; Sun Z.; Yang Y.; Somoskovi A.; Zhang Y. Salicylate reduces susceptibility of Mycobacterium tuberculosis to multiple antituberculosis drugs. Antimicrob. Agents Chemother. 2002, 46, 2636–2639. 10.1128/AAC.46.8.2636-2639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. R.; Li D. F.; Fleming J.; Zhou Y. F.; Liu Y.; Deng J. Y.; Zhou L.; Zhou J.; Zhu G. F.; Zhang X. E.; Wang D. C.; Bi L. J. Structural analysis of the regulatory mechanism of MarR protein Rv2887 in M. tuberculosis. Sci. Rep. 2017, 7, 6471. 10.1038/s41598-017-01705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier T.; Kapilashrami K.; Argyrou A.; Ioerger T. R.; Little D.; Murphy K. C.; Nandakumar M.; Park S.; Gold B.; Mi J.; Zhang T.; Meiler E.; Rees M.; Somersan-Karakaya S.; Porras-De Francisco E.; Martinez-Hoyos M.; Burns-Huang K.; Roberts J.; Ling Y.; Rhee K. Y.; Mendoza-Losana A.; Luo M.; Nathan C. F. N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4523–E4530. 10.1073/pnas.1606590113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler D. L.; Parish T. The promoter of Rv0560c is induced by salicylate and structurally-related compounds in Mycobacterium tuberculosis. PLoS One 2012, 7, e34471. 10.1371/journal.pone.0034471. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.