

Abstract
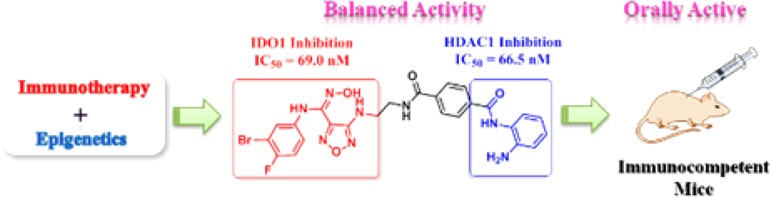
In order to take advantage of both immunotherapeutic and epigenetic antitumor agents, the first generation of dual indoleamine 2,3-dioxygenase 1 (IDO1) and histone deacetylase (HDAC) inhibitors were designed. The highly active dual inhibitor 10 showed excellent and balanced activity against both IDO1 (IC50 = 69.0 nM) and HDAC1 (IC50 = 66.5 nM), whose dual targeting mechanisms were validated in cancer cells. Compound 10 had good pharmacokinetic profiles as an orally active antitumor agent and significantly reduced the l-kynurenine level in plasma. In particular, it showed excellent in vivo antitumor efficacy in the murine LLC tumor model with low toxicity. This proof-of-concept study provided a novel strategy for cancer treatment. Compound 10 represents a promising lead compound for the development of novel antitumor agents and can also be used as a valuable probe to clarify the relationships and mechanisms between cancer immunotherapy and epigenetics.
Keywords: IDO1, HDAC, dual inhibitors, cancer immunotherapy, epigenetics, antitumor efficacy
In the past two decades, immune checkpoint therapy has achieved important clinical advances for the treatment of cancer.1 Rather than targeting the tumor cells directly, cancer immunotherapy acts by activating T cells to enhance patients’ native immune response. The immune checkpoint cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody ipilimumab and programmed death (PD-1) antibodies pembrolizumab and nivolumab were approved by U.S. Food and Drug Administration (FDA).1,2 As compared to antibody drugs, small-molecules for cancer immunotherapy have remarkable advantages, such as oral administration, access to intracellular targets, and greater drug exposure within the tumor microenvironment. Therefore, there is an urgent need to develop small molecules to modulate the immune system and fight against cancer.3
Indoleamine 2,3-dioxygenase 1 (IDO1), an extrahepatic heme-containing dioxygenase, is capable of catalyzing the conversion of l-tryptophan (Trp) to N-formylkynurenine (NFK) in the first rate-limiting step of the kynurenine pathway (KP).4,5 NFK is then metabolized to l-kynurenine (Kyn) and subsequent bioactive metabolites.4 The tryptophan depletion results in inhibiting the proliferation of T lymphocytes, which are sensitive to low Trp levels. The production of KP metabolites can enhance immune tolerance by activating the aryl hydrocarbon receptor (AhR). Both of them contribute to the immunosuppressed state of the tumor microenvironment.6,7 In addition, numerous evidence indicated that elevated levels of IDO1 expression in both tumor cells and antigen-presenting cells were correlated with poor prognosis and reduced survival.8,9 Given the important role in tumor immune escape, IDO1 represents a valuable therapeutic target in cancer immunotherapy. A number of potential small-molecule inhibitors of IDO1 have been disclosed, among which D-1-MT (1), INCB024360 (2), and GDC-0919 (3) have entered clinical trials (Figure 1).10−12
Figure 1.

Chemical structures of IDO1 inhibitors and HDAC inhibitors and design of dual IDO1 and HDAC inhibitors.
IDO1 inhibitors control and eradicate the growth of tumor cells by enhancing antitumor immune responses. However, a number of preclinical studies revealed that IDO1 inhibitors only exhibited moderate antitumor activity when used as single agents.8,13 Indeed, preclinical and clinical data indicated that IDO1 inhibitors are generally developed as combination therapies with cytotoxic antitumor agents, radiotherapy, therapeutic vaccination, and PD-1 antibodies.8,13,14 For example, compound 2, an orally active and competitive IDO1 inhibitor, is currently evaluated in phase III clinical trials for the treatment of multiple tumor types in combination with immune checkpoint inhibitors.15 Despite the synergistic effects observed by the combination of IDO1 inhibitors with other therapies, drug combination strategies are always limited by complex pharmacokinetics and drug–drug interactions.16 To overcome the problem, designing a single agent that simultaneously targets two or more synergistic mechanisms has attracted great interests.17,18
Histone deacetylases (HDACs) are a family of enzymes, which catalyze the deacetylation of the lysine residues at the amino terminal of histones.19 HDAC inhibitors (HDACIs) can induce cell cycle arrest, differentiation, and apoptosis by blocking abnormal HDAC deacetylation.20 Several HDACIs, such as SAHA (4) and Mocetinostat (5), have been approved for the treatment of various hematological malignancies.21 Importantly, recent studies indicated that HDACIs could also improve tumor recognition and reverse immune suppression via various mechanisms.22,23 Therefore, the discovery of dual IDO1 and HDAC inhibitors may provide a novel strategy for cancer treatment by taking advantages of both immunotherapeutic and epigenetic drugs. Herein, the first dual IDO1 and HDAC inhibitors were designed and evaluated. Interestingly, a highly potent inhibitor with balanced activity against IDO1/HDAC was successfully identified, which was orally active and showed excellent in vivo antitumor potency.
The dual IDO1 and HDAC inhibitors were designed by a pharmacophore fusion strategy. IDO1 inhibitor 2 and HDACIs 4(24) and 5(25) were used as the templates for drug design. Binding mode analysis of the docked conformation of compound 2 (Figure S1 in Supporting Information) revealed that the oxygen of the hydroxyamidine bound to heme iron in the active site (pocket A) of IDO1, which was identified as a crucial functional group for IDO1 inhibition.26 The aminoethyl-sulfamide substituent projected out of the active site toward solvent (pocket B),26 which could be modified without impairing IDO1 binding affinity. Thus, the zinc binding functional group (hydroxamic acid or benzamide) that is essential for HDAC inhibition can be introduced on the IDO heme binding scaffold directly or via a proper spacer (Figure 1). As a result, a series of novel IDO1 and HDAC dual targeting molecules were designed, synthesized, and assayed.
The preparation of target compounds 10–23 are shown in Scheme 1. Amidation of protected and substituted carboxylic acids 7a–d with hydroiodide salt intermediate 6, using HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate) as the coupling reagent, led to intermediates 8a–d. After alkaline cleavage with NaOH and removal of Boc protecting group with trifluoroacetic acid, benzamide analogues 10–13 were obtained. Amide condensation of intermediate 6 with 4-(methoxycarbonyl)benzoic acid 7e, 2-methoxycarbonylvinyl benzoic acids 7f–g, 4-phenyl-1H-1,2,3-triazol carboxylic acids 7h–j, or aliphatic carboxylic acids 7k–n gave esters 8e–n, which were converted to corresponding hydroxamic acids 14–23 using hydroxylamine hydrochloride in the presence of KOH. Intermediates 6 and 7a–n were prepared as outlined in Schemes S1 and S2 (Supporting Information).
Scheme 1. Synthesis of Compounds 10–23.

Reagents and conditions: (a) HATU, DIPEA, DMF, rt, overnight, yield 49–59%; (b) NaOH, MeOH, rt, 2 h, yield 71–85%; (c) CF3COOH, DCM, 40 °C, 3 h, yield 62–77%; (d) HATU, DIPEA, DMF, rt, overnight, yield 53–85%; (e) NH2OH-HCl, KOH, MeOH, rt, 2 h, yield 43–83%.
Initially, HDAC1 and IDO1 were used as the primary assays with compounds 2 and 4 as the reference drugs. Then, pan-HDAC activity assay was performed for a representative compound to investigate whether it is a pan-HDAC inhibitor or a selective inhibitor. As shown in Table 1, the designed compounds generally showed potent inhibitory activity against both targets. The linkers between HDAC1 Zn binding group and IDO1 heme binding scaffold played an important role in enzyme inhibition. Compound 10 containing the benzamide-N-phenylamine group showed excellent activity toward both IDO1 (IC50 = 69.0 nM) and HDAC1 (IC50 = 66.5 nM). Replacement of the phenyl group in compound 10 to pyridine (compound 11) and thiophene (compound 12) or changing the position of amide (compound 13) resulted in the decreased activity against both targets. When the phenylamine group was replaced by hydroxamic acid, compound 14 showed decreased activity against both targets. Interestingly, the insertion of a double bond between the hydroxamic acid and phenyl group in compound 15 led to the improved activity against both targets. Compounds 15 (IC50 = 46.2 nM) and 16 (IC50 = 70.5 nM) were highly active against HDAC1. In particular, compound 16 was the most active IDO1 inhibitor (IC50 = 27.0 nM), which was about 3-fold more potent than compound 2. Triazole is widely used as the linker in HDAC-based multitargeting antitumor agents.27,28 Our results also showed that the 1,2,3-triazol derivatives 17–19 with the increasing of the carbon chain length exhibited better inhibitory activities against HDAC1, which were consistent with the docking results (Table S1 and Figure S2 in Supporting Information). The order of influence of carbon chain length on HDAC1 inhibition was five carbons (22) > six carbons (23) > three carbons (20) > four carbons (21). Notably, compound 22 was the most potent HDAC1 inhibitor (IC50 = 9 nM). However, no improvement of the IDO1 inhibitory was observed for these compounds with a long linker.
Table 1. HDAC1 and IDO1 Inhibitory and Antiproliferative Activities of the Target Compounds.
| compds | HDAC1 IC50 (nM)a | IDO-1 IC50 (nM)a or % inhibition at 1 μM | LLC (IC50, μM) | CT-26 (IC50, μM) | A549 (IC50, μM) | HCT-116 (IC50, μM) | HT-29 (IC50, μM) |
|---|---|---|---|---|---|---|---|
| 2 | NTb | 77.8 ± 6.4 | >100 | >100 | >100 | >100 | >100 |
| 4 | 14.4 ± 3.8 | NTb | 9.68 ± 0.15 | 5.97 ± 0.85 | 2.63 ± 0.54 | 3.07 ± 0.55 | 1.78 ± 0.89 |
| 10 | 66.5 ± 3.3 | 69.0 ± 7.1 | 17.62 ± 1.06 | 59.84 ± 8.51 | 16.73 ± 2.12 | 5.12 ± 0.43 | 11.71 ± 1.54 |
| 11 | 604.4 ± 44.2 | 260.3 ± 13.5 | 15.13 ± 1.85 | 23.3 ± 4.35 | 20.65 ± 5.66 | 6.36 ± 1.25 | 12.24 ± 3.11 |
| 12 | 1429.5 ± 325.6 | 76% | 18.34 ± 4.21 | 38.82 ± 7.71 | 14.52 ± 3.27 | 7.12 ± 1.72 | 20.26 ± 6.07 |
| 13 | 632.7 ± 53.8 | 79% | 31.38 ± 5.67 | 25.51 ± 4.35 | 27.76 ± 7.73 | 16.18 ± 3.12 | 46.42 ± 9.43 |
| 14 | 262.4 ± 16.7 | 95% | >100 | >100 | 89.79 ± 6.99 | 28.08 ± 6.44 | 68.01 ± 7.19 |
| 15 | 46.2 ± 5.9 | 167.9 ± 8.7 | 21.64 ± 2.16 | 12.79 ± 3.13 | 25.56 ± 6.84 | 5.89 ± 0.54 | 14.15 ± 2.01 |
| 16 | 70.5 ± 3.8 | 27.0 ± 3.5 | 53.30 ± 8.97 | 38.94 ± 7.56 | 41.66 ± 8.46 | 12.44 ± 2.53 | 23.31 ± 4.12 |
| 17 | 894.8 ± 93.6 | 88% | >100 | >100 | >100 | 37.53 ± 7.59 | >100 |
| 18 | 66.5 ± 4.9 | 87% | >100 | >100 | >100 | >100 | >100 |
| 19 | 23.5 ± 1.5 | 209.6 ± 18.7 | 35.95 ± 7.66 | 90.18 ± 5.85 | 45.48 ± 10.55 | 29.44 ± 8.12 | 23.88 ± 2.73 |
| 20 | 121.1 ± 9.1 | 133.0 ± 14.1 | >100 | >100 | >100 | 80.24 ± 8.46 | >100 |
| 21 | 308.1 ± 10.5 | 122.5 ± 10.5 | >100 | >100 | >100 | 85.37 ± 7.91 | >100 |
| 22 | 9.2 ± 0.08 | 113.4 ± 11.2 | 90.11 ± 13.05 | 95.46 ± 20.28 | 40.66 ± 9.56 | 17.46 ± 5.61 | 28.74 ± 4.84 |
| 23 | 47.7 ± 5.3 | 139.8 ± 13.3 | 56.58 ± 5.68 | 97.45 ± 15.34 | 36.03 ± 8.95 | 4.70 ± 0.38 | 14.88 ± 3.09 |
IC50 values are the mean of at least three independent assays, presented as mean ± SD.
NT = not tested.
Given the potent enzyme inhibitory activities, we further evaluated the antiproliferative activities of the IDO1/HDAC dual inhibitors against LLC (lewis lung cancer), CT-26 (mouse colon cancer), A549 (human lung cancer), HCT-116 (human colon cancer), and HT-29 (human colon cancer) cell lines by the CCK8 (Cell Counting Kit-8) assay. Compounds 2 and 4 were used as the positive controls. As shown in Table 1, HDAC inhibitor 4 was active in the low micromolar range, whereas compound 2 was inactive against the five solid tumor cell lines because IDO1 inhibitors do not destroy tumor cells directly. Generally, the target compounds showed modest to good antitumor activities. Among the tested cell lines, the dual inhibitors were more active against the HCT-116 cell line. Particularly, compounds 10 (IC50 = 5.12 μM), 15 (IC50 = 5.89 μM), and 23 (IC50 = 4.70 μM) exhibited comparable antitumor activity to compound 4 (IC50 = 3.07 μM) in the HCT-116 cell line.
To clarify whether the inhibition in cell growth is associated with apoptosis, HCT-116 cells were treated with DMSO or various concentrations of compounds 10, 15, and 23 for 48 h. The cells were stained with Annexin-V and propidium iodide (PI), and the apoptotic ratio was determined by flow cytometry. As shown in Figure 2A,B, the percentage of apoptotic cells for compound 15 was 26.7% (5 μM), 45.4% (10 μM), and 61.3% (20 μM), respectively. The ability of compound 15 to induce apoptosis was stronger than that of compounds 10 (18.8%, 29.9%, and 48.9%) and 23 (25.5%, 31.1%, and 44.5%). These results demonstrated that dual IDO1 and HDAC inhibitors induced HCT116 cells apoptosis in a dose-dependent manner.
Figure 2.
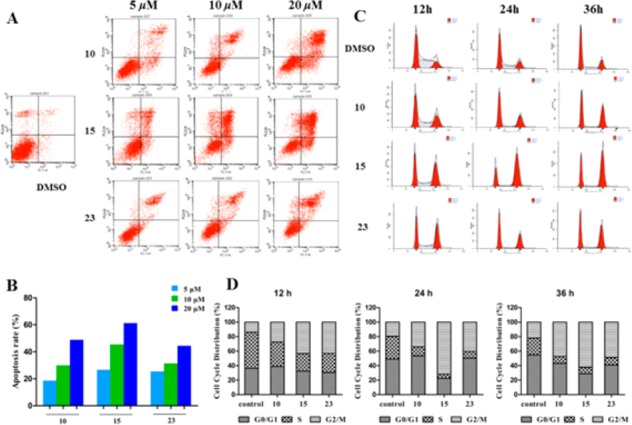
Compounds 10, 15, and 23 induce cell apoptosis and cycle arrest. (A,B) Apoptotic index analysis at different concentrations in HCT116 cells. (C,D) Time-dependent effects on cell cycle progression. The cell cycle and the proportions are shown in each phase of HCT116 cells treated with or without compounds 10, 15, and 23.
To investigate the effect of compounds on the various phases of cell cycle, HCT116 cells were treated with 10 μM of compounds 10, 15, and 23 for the indicated time interval and analyzed by flow cytometry (Figure 2C,D). Compared with the DMSO control, the cell cycle showed all the compounds arrested HCT116 mainly in G2/M phase. Compound 10 also induced the increase of G2/M-phase cells (27.2% to 47.4%) in a time-dependent manner. Compound 10, with the balanced inhibitory activity against HDAC1 (IC50 = 66.5 nM, Figure 3A) and IDO1 (IC50 = 69.0 nM, Figure 3B) and excellent apoptosis inducing activity, was selected for cellular mechanism and in vivo studies.
Figure 3.

Compound 10 inhibits IDO1 and HDAC1 in vitro. (A,B) Concentration–effect curves for IDO1 and HDAC1 enzyme inhibitory activity of compound 10. (C) Compound 10 effectively inhibits Kyn production in IFN-γ treated human HeLa cells. (D) Cytotoxic effect of compound 10 on HeLa cells. (E) Western blot analysis of acetylated histone H3 after 12 h treatment with compound 10 in HCT116 cells. (F) Quantification of HDAC inhibitory responses by densitometry. Data points represent a mean ± SD of biological triplicates from a representative experiment.
In order to evaluate the IDO1 inhibitory activity of compound 10 under the cellular environment (EC50), a HeLa cell-based assay29 measuring the Kyn was performed (Figure 3C). Moreover, its cytotoxic activity (LC50) against HeLa cells was also assayed (Figure 3D). Compound 10 had an EC50 and LC50 value of 0.41 and 24.77 μM, respectively, with a LC50/EC50 ratio of 60.4. These results suggested that compound 10 effectively inhibited IDO1 activity in HeLa cells and that the cell-based IDO1 activity was not caused by the cytotoxicity. In order to investigate whether compound 10 inhibits HDAC in cancer cells, we evaluated its effect on the acetylation of histone H3 using the Western blot assay. After the incubation with compound 10 or compound 4 in HCT116 cells for 12 h, the acetylation level of histone 3 was elevated in a dose-dependent manner (Figure 3E,F), indicating that it could inhibit HDAC in cells. To explore the inhibitory activity toward other HDAC isoforms, compound 10 was tested for its inhibitory activity of HDAC2, HDAC3, HDAC4, HDAC6, and HDAC8 (Table S2 in Supporting Information). Compound 10 displayed nanomolar activity against HDAC2 (IC50 = 179 nM), HDAC3 (IC50 = 45 nM), and HDAC6 (IC50 = 70 nM). The results indicated that compound 10 was a pan-HDAC inhibitor at the molecular level. In male mouse liver microsome, compound 10 had a half-life of 48.37 min with apparent clearance of 0.0287 mL/min/mg (Figure S2 in Supporting Information).
Inspired by the excellent IDO1/HDAC inhibition of compound 10, it was subjected for in vivo pharmacokinetic evaluation in male Sprague–Dawley (SD) rats. Compound 10 was administered in a single intravenous (iv) dose of 2 mg/kg or an oral dose of 100 mg/kg, and the main pharmacokinetic parameters are listed in Table 2. It demonstrated good oral drug exposure (AUC(0-inf) = 33534 ± 225 ng/mL × h) with oral bioavailability of 18%, suggesting that oral administration would be a suitable dosing route for further pharmacodynamic study (Table 2). Inhibition of IDO1 activity in vivo could reduce the plasma Kyn levels. We observed that treatment of wild-type female C57BL/6 mice with compound 10 (100 mg/kg) significantly decreased the plasma Kyn levels between 3–8 h (Figure 4). Similarly, compound 2 decreased plasma Kyn levels within 1 h and the levels remained at least 50% of suppression at 8 h. Taken together, the results indicated that compound 10 was an orally active agent and able to effectively suppress tryptophan catabolism in mice. Moreover, compound 10 was stable in the PBS buffer after 24 h at room temperature, but its water solubility (<0.01 g/L) remains to be improved.
Table 2. Pharmacokinetic Profiles of Compound 10 in Sprague–Dawley Ratsa.
| compds | Cmax (ng/mL) | Tmax (h) | T1/2 (h) | AUC(0-inf) (ng/mL·h) | F (%) |
|---|---|---|---|---|---|
| 2 (Po) | 5560 ± 967 | 0.8 | 4.5 ± 0.8 | 30747 ± 785 | |
| 10 (Po) | 5765 ± 1276 | 2.0 | 3.4 ± 0.1 | 33534 ± 225 | 18 |
| 10 (Iv) | 12039 ± 1762 | 0.083 | 1.4 ± 0.05 | 3646 ± 201 |
Data are presented as mean ± SD.
Figure 4.
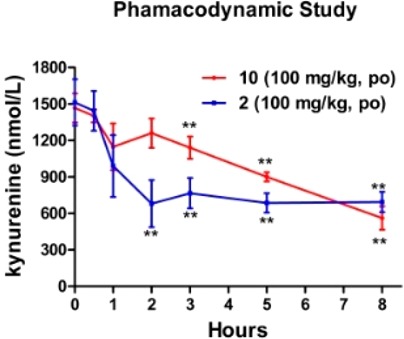
Compound 10 efficiently suppresses Kyn in wild-type mice. Wild-type female C57BL/6 mice were orally administrated compound 10 or 2 at 100 mg/kg, and blood was harvested at the indicated times. Average values from the plasma for three mice (±SD), analyzed for Kyn levels by LC/MS/MS, are shown. All data between 0.5 and 8 h for effects in wild-type mice are statistically significant (**, P < 0.01).
Compound 10 was further investigated for its in vivo antitumor efficacy in a LLC tumor growth model in immunocompetent mice. Compound 10 (100 mg/kg, bid), compound 2 (100 mg/kg, bid), and compound 4 (100 mg/kg, qd) were dosed orally by gavage for 14 consecutive days. As shown in Figure 5, treatment with compound 10 showed good tumor growth control (TGI = 56.0%) without significant changes in body, which was comparable to compound 2 (TGI = 54.3%) and superior to compound 4 (TGI = 41.4%). From the pharmacokinetic data, compound 2 achieved higher exposure (AUC 70.2 μM·h vs 60.0 μM·h, calculated from ng/mL·h) and a longer half-life (4.5 h vs 3.4 h) than compound 10, which may explain why compound 10 did not exhibit higher in vivo antitumor effect than compound 2. Thus, further structural optimization of dual IDO1/HDAC inhibitor 10 is required to improve the in vivo antitumor efficacy.
Figure 5.
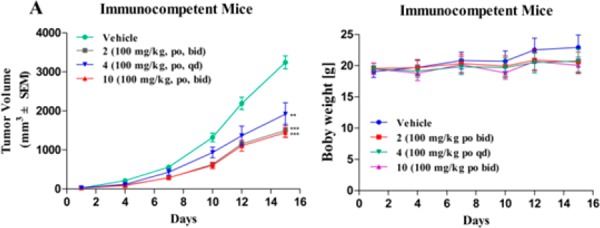
Antitumor efficacy of compound 10 in murine LLC tumor model. (A) Mean tumor volumes (mm3) ± SEM (n = 6 mice/group) are shown from the initiation of dosing (∼100 mm3). The statistical difference was determined by Student’s t test. **P < 0.01, ***P < 0.001 compared with the control group. (B) Body weights were measured three times per week, and data are presented as the mean (g) ± SD.
In summary, the first dual IDO1/HDAC inhibitors were designed and evaluated. Compound 10, a highly active dual inhibitor, was successfully identified, which showed balanced activity against both IDO1 and HDAC1. It significantly induced the apoptosis in the HCT116 cells with a G2/M cell cycle arrest. Compound 10 had acceptable pharmacokinetic profiles as an orally active antitumor agent and significantly reduced Kyn levels in plasma over an 8 h period. It showed good in vivo antitumor efficacy in the murine LLC tumor model with low toxicity. This proof-of-concept study provided a novel strategy for the development of novel antitumor agents. Compound 10 represents a promising lead compound for drug development. It can also be used as a valuable probe to clarify the relationships and mechanisms between cancer immunotherapy and epigenetics. Also, it should be noted that structural optimization of compound 10 and the synergistic effects of dual IDO1/HDAC inhibitors with immune checkpoint inhibitors (e.g., PD-1 antibodies) remain to be further investigated. Such studies are in progress in our lab.
Glossary
ABBREVIATIONS
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- PD-1
programmed death
- IDO1
indoleamine 2,3-dioxygenase 1
- Kyn
l-kynurenine
- Trp
l-tryptophan
- NFK
N-formylkynurenine
- AhR
aryl hydrocarbon recepto
- HDACs
histone deacetylases
- TGI
tumor growth inhibition
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- DIPEA
N,N-diisopropylethylamine
- PBS
phosphate buffer saline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00487.
Chemical synthesis and structural characterization of the target compounds; protocols of biological assays (PDF)
Author Contributions
∥ K.F., G.D., and Y.L. contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Key R&D Program of China (2017YFA0506000 to C.S.), National Natural Science Foundation of China (21738002 to W.W. and 81725020 to C.S.), and Science and Technology Commission of Shanghai Municipality (Grant 17XD1404700).
The authors declare no competing financial interest.
Supplementary Material
References
- Sharma P.; Allison J. P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Markham A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1–6. [DOI] [PubMed] [Google Scholar]
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Dounay A. B.; Tuttle J. B.; Verhoest P. R. Challenges and Opportunities in the Discovery of New Therapeutics Targeting the Kynurenine Pathway. J. Med. Chem. 2015, 58, 8762–8782. 10.1021/acs.jmedchem.5b00461. [DOI] [PubMed] [Google Scholar]
- Macchiarulo A.; Camaioni E.; Nuti R.; Pellicciari R. Highlights at the gate of tryptophan catabolism: a review on the mechanisms of activation and regulation of indoleamine 2,3-dioxygenase (IDO), a novel target in cancer disease. Amino Acids 2009, 37, 219–229. 10.1007/s00726-008-0137-3. [DOI] [PubMed] [Google Scholar]
- Curti A.; Trabanelli S.; Salvestrini V.; Baccarani M.; Lemoli R. M. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood 2009, 113, 2394. 10.1182/blood-2008-07-144485. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Sharma M. D.; Baban B.; Harding H. P.; Zhang Y.; Ron D.; Mellor A. L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Uyttenhove C.; Pilotte L.; Théate I.; Stroobant V.; Colau D.; Parmentier N.; Boon T.; Van den Eynde B. J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- Godin-Ethier J.; Hanafi L. A.; Piccirillo C. A.; Lapointe R. Indoleamine 2,3-dioxygenase expression in human cancers: clinical and immunologic perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. 10.1158/1078-0432.CCR-11-1331. [DOI] [PubMed] [Google Scholar]
- Röhrig U. F.; Majjigapu S. R.; Vogel P.; Zoete V.; Michielin O. Challenges in the Discovery of Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors. J. Med. Chem. 2015, 58, 9421–9437. 10.1021/acs.jmedchem.5b00326. [DOI] [PubMed] [Google Scholar]
- Dolušić E.; Frédérick R. Indoleamine 2,3-dioxygenase inhibitors: a patent review (2008–2012). Expert Opin. Ther. Pat. 2013, 23, 1367–1381. 10.1517/13543776.2013.827662. [DOI] [PubMed] [Google Scholar]
- Austin C. J.; Rendina L. M. Targeting key dioxygenases in tryptophan-kynurenine metabolism for immunomodulation and cancer chemotherapy. Drug Discovery Today 2015, 20, 609–617. 10.1016/j.drudis.2014.11.007. [DOI] [PubMed] [Google Scholar]
- Muller A. J.; Duhadaway J. B.; Donover P. S.; Sutantoward E.; Prendergast G. C. Inhibition of indoleamine 2,3-dioxygenase, an immunoregulatory target of the cancer suppression gene Bin1, potentiates cancer chemotherapy. Nat. Med. 2005, 11, 312–319. 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- Li M.; Bolduc A. R.; Hoda M. N.; Gamble D. N.; Dolisca S. B.; Bolduc A. K.; Hoang K.; Ashley C.; Mccall D.; Rojiani A. M. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J. Immunother. Cancer 2014, 2, 21. 10.1186/2051-1426-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletti A.; Greco F. A.; Dolciami D.; Sardella R.; Camaioni E.; Pallotta M. T.; Volpi C.; Orabona C.; Grohmann U.; Macchiarulo A. Advances in Indolemine 2,3-Dioxygenase 1 Medicinal Chemistry: Structure-Function and Structure-Activity Relationships of the Enzyme and its Inhibitors. MedChemComm 2017, 8, 1378–1392. 10.1039/C7MD00109F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues A. D.Drug–Drug Interactions; CRC Press, 2008. [Google Scholar]
- Peters J. U. Polypharmacology - Foe or Friend?. J. Med. Chem. 2013, 56, 8955–8971. 10.1021/jm400856t. [DOI] [PubMed] [Google Scholar]
- Anighoro A.; Bajorath J.; Rastelli G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. 10.1021/jm5006463. [DOI] [PubMed] [Google Scholar]
- Marks P. A.; Xu W. S. Histone deacetylase inhibitors: Potential in cancer therapy. J. Cell. Biochem. 2009, 107, 600–608. 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfi P. P. Histone deacetylases and transcriptional therapy with their inhibitors. Cancer Chemother. Pharmacol. 2001, 48, S17–S19. 10.1007/s002800100322. [DOI] [PubMed] [Google Scholar]
- Li Z.; Zhu W. G. Targeting Histone Deacetylases for Cancer Therapy: From Molecular Mechanisms to Clinical. Int. J. Biol. Sci. 2014, 10, 757–770. 10.7150/ijbs.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn J.; Rao S. Epigenetics and immunotherapy: The current state of play. Mol. Immunol. 2017, 87, 227–239. 10.1016/j.molimm.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Booth L.; Roberts J. L.; Poklepovic A.; Kirkwood J.; Dent P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. 10.18632/oncotarget.17950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M.; Talpur R.; Hazarika P.; Kelly C.; Chiao J. H.; Reilly J. F.; Ricker J. L.; Richon V. M. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlevi C. L.; Crump M.; Andreadis C.; Rizzieri D.; Copeland A.; Potvin D.; Chao R. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br. J. Haematol. 2017, 178, 431–441. 10.1111/bjh.14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Glenn J.; Zhu W. INCB24360 (Epacadostat), a Highly Potent and Selective Indoleamine-2,3-dioxygenase 1 (IDO1) Inhibitor for Immuno-oncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao L.; Mustafa N.; Tan E. C.; Poulsen A.; Singh P.; Duongthi; Chng W. J.; Yen J. J. Y. Design and Synthesis of Ligand Efficient Dual Inhibitors of Janus Kinase (JAK) and Histone Deacetylase (HDAC) Based on Ruxolitinib and Vorinostat. J. Med. Chem. 2017, 60, 8336–8357. 10.1021/acs.jmedchem.7b00678. [DOI] [PubMed] [Google Scholar]
- Ganesan A. Multitarget Drugs: an Epigenetic Epiphany. ChemMedChem 2016, 11, 1227–1241. 10.1002/cmdc.201500394. [DOI] [PubMed] [Google Scholar]
- Cheng M. F.; Hung M. S.; Song J. S.; Lin S. Y.; Liao F. Y.; Wu J. S.; Chao Y. S. Discovery and structure-activity relationships of phenyl benzenesulfonylhydrazides as novel indoleamine 2,3-dioxygenase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3403–3406. 10.1016/j.bmcl.2014.05.084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.