

Abstract
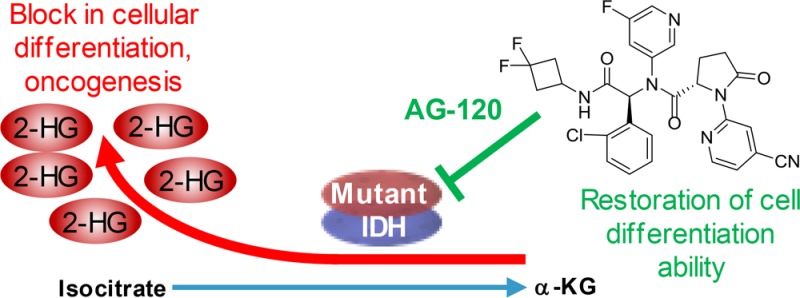
Somatic point mutations at a key arginine residue (R132) within the active site of the metabolic enzyme isocitrate dehydrogenase 1 (IDH1) confer a novel gain of function in cancer cells, resulting in the production of d-2-hydroxyglutarate (2-HG), an oncometabolite. Elevated 2-HG levels are implicated in epigenetic alterations and impaired cellular differentiation. IDH1 mutations have been described in an array of hematologic malignancies and solid tumors. Here, we report the discovery of AG-120 (ivosidenib), an inhibitor of the IDH1 mutant enzyme that exhibits profound 2-HG lowering in tumor models and the ability to effect differentiation of primary patient AML samples ex vivo. Preliminary data from phase 1 clinical trials enrolling patients with cancers harboring an IDH1 mutation indicate that AG-120 has an acceptable safety profile and clinical activity.
Keywords: isocitrate dehydrogenase, mutant IDH1, AG-120, ivosidenib, differentiation therapy, 2-hydroxyglutarate
Point mutations in isocitrate dehydrogenase (IDH) 1 and 2 are found in multiple tumors, including glioma, cholangiocarcinoma, chondrosarcoma, and acute myeloid leukemia (AML).1 Mutant IDH (mIDH) enzymes have a gain-of-function activity that results in accumulation of the oncometabolite d-2-hydroxyglutatrate (2-HG),2 which is structurally similar to α-ketoglutarate (α-KG). 2-HG competitively inhibits α-KG-dependent dioxygenases, which participate in many cellular processes such as histone and DNA demethylation, and adaption to hypoxia, and their inhibition leads to a block in normal cellular differentiation and oncogenic transformation.1,3−5
mIDH inhibitors represent a novel class of targeted cancer metabolism therapy that induces differentiation of proliferating cancer cells. The mIDH2 inhibitor enasidenib, recently approved by the FDA for relapsed/refractory AML, as well as all-trans retinoic acid for the treatment for acute promyelocytic leukemia, support the potential of such differentiation therapy.6−8 We previously reported that the prototype mIDH1 inhibitor AGI-5198 inhibited both biochemical and cellular production of 2-HG.9 AGI-5198 showed robust tumor 2-HG inhibition in an in vivo mIDH1 xenograft model, impaired growth of mIDH1 glioma cells in vivo, and induced epigenetic alterations leading to the expression of genes associated with gliogenic differentiation.5 However, the poor pharmaceutical properties of AGI-5198 precluded its use in clinical studies. Although several additional mIDH1 inhibitors have been disclosed,1,10,11 AG-120 is the first inhibitor of the mIDH1 enzyme to achieve clinical proof of concept in human trials. Lead optimization of AGI-5198 leading to the discovery of AG-120 is described here. The mIDH1-R132H enzyme was utilized for primary biochemical evaluation. Routine profiling in cells was done in the HT1080 chondrosarcoma cell line, which endogenously expresses mIDH1-R132C, and in our experience the potency for mIDH1-R132C is very similar to mIDH1-R132H, as previously reported.9
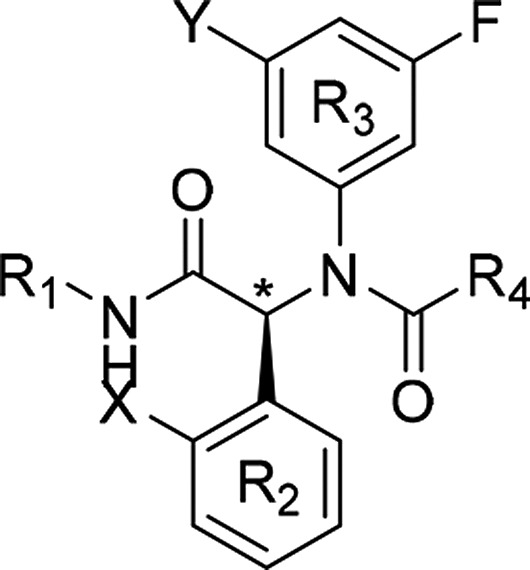
In vitro profiling of AGI-5198 in kinetic solubility and liver microsomal assays pointed to reasonable physicochemical properties but poor metabolic stability across species. Metabolite identification studies conducted in human liver microsomal S9 fraction revealed extensive NADPH-dependent oxidation of the cyclohexyl (R1) and imidazole ring (R4). The following strategies were therefore employed to decrease metabolic clearance (Table 1). At R4, the imidazole ring was replaced with moieties that emerged from broad structure–activity relationship profiling and had similar potency to AGI-5198, as previously described.9 R1 modifications focused on blocking metabolism using fluorinated cycloalkyl groups, and to mitigate any potential oxidative metabolism at R2, the o-Me (X) group was replaced by an o-Cl group.
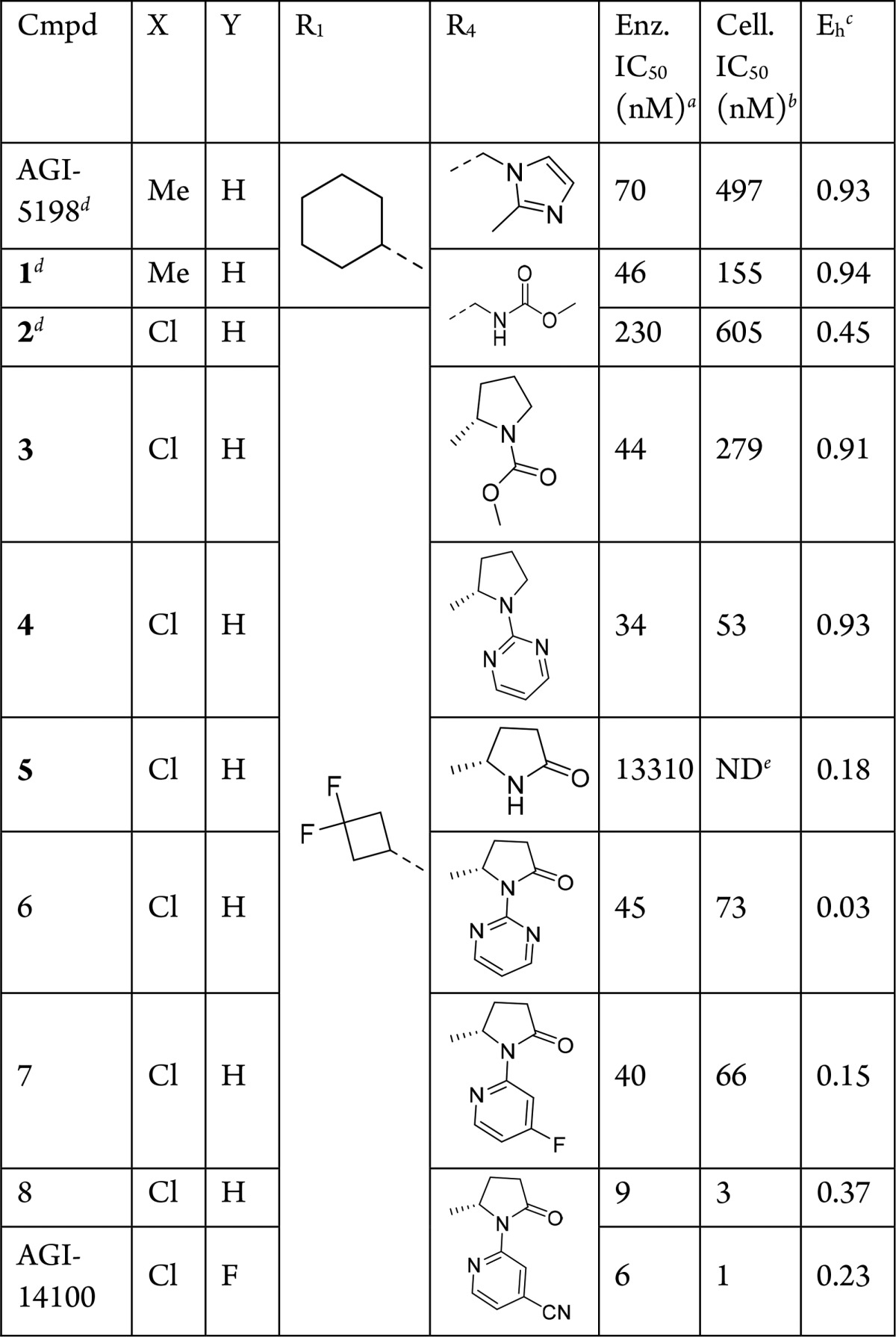
Table 1. Optimization of Microsome Stability and Potency Leading to AGI-14100.
Enzymatic IC50 values for the mIDH1-R132H homodimer are the mean of at least two determinations performed as described in the Supporting Information.
Cellular IC50 from HT1080 chondrosarcoma cell line.
Microsome stability recorded as the hepatic extraction ratio in human liver microsomes.
Racemic.
Not determined.
Replacing the R4 imidazole group with glycine carbamate in 1 slightly improved the enzymatic potency but maintained the same high metabolic clearance. Simultaneously switching the o-Me group on R2 to o-Cl and the cyclohexyl in R1 to difluoro cyclobutyl in 2 incurred only a 5-fold potency loss, but brought the metabolic stability into the medium clearance range. Next, replacement of the glycine carbamate group at R4 with proline carbamate (3) restored the biochemical potency but lost the improvement in the hepatic extraction ratio (Eh). A metabolite identification study of 3 revealed that mono- and dioxidation of the proline carbamate moiety were the major metabolic pathways, allowing us to stabilize the R1 site of oxidative metabolism. Eliminating oxidative liabilities at R4 was the next focus. Replacing the methyl carbamate with a heterocyclic “mimic” gave the pyrimidine analog 4, which maintained biochemical potency but did not improve metabolic stability. Removal of the pyrimidine ring in 4 in concert with oxidation of the proline ring at the 2-position eliminated nearly all biochemical potency but resulted in much improved metabolic stability for 5, giving another important insight into stabilization of oxidative metabolism at R4. Addition of the pyrimidine ring on the oxidized proline moiety at R4 provided 6, which maintained low metabolic clearance and restored enzyme potency. Optimization then focused on improving the biochemical/cellular potency while maintaining low metabolic clearance.
A scan of heterocycles at R4 revealed that pyridines substituted with electron-withdrawing groups at the 4-position could achieve the desired potency and metabolic stability profile as shown for 7 and 8. Finally, additional fluorine substitution at the 5-position of the R3 aromatic group provided the compound AGI-14100, with a good balance of single-digit nM potency in enzyme and cell-based assays and desirable metabolic stability.
To further assess the suitability of AGI-14100 as a potential development candidate, additional pharmacokinetic (PK) properties were evaluated. Low clearance in liver microsomal incubations was observed across species, which was also observed in the rat, dog, and cynomolgus monkey in vivo (Table S1). However, assessment in the human pregnane X receptor (hPXR) screen indicated that AGI-14100 was potentially a cytochrome P450 (CYP) 3A4 inducer. hPXR activation by AGI-14100 was approximately 70% that of rifampicin, a known strong CYP 3A4 inducer. CYP induction studies using human hepatocytes confirmed the results (data not shown).
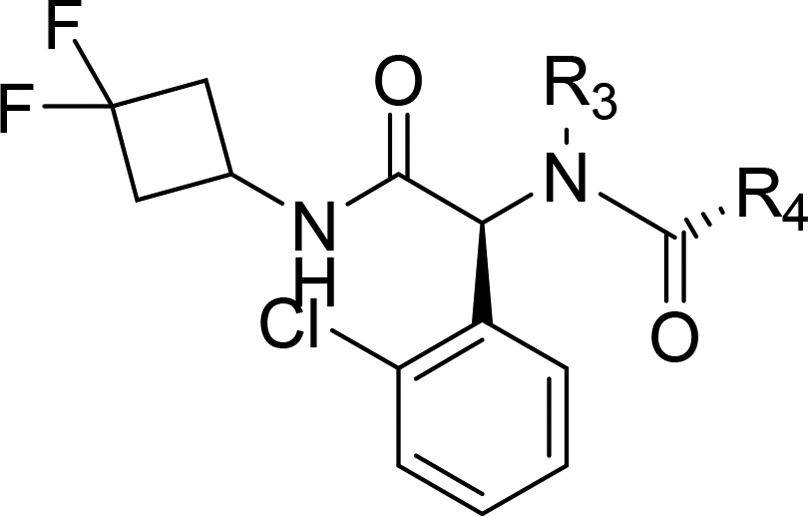
To mitigate the CYP induction liabilities, further medicinal chemistry optimization was conducted to eliminate hPXR activation (Table 2). Since the R1 and R2 substituents require hydrophobic character to maintain potency, our strategy focused on introducing polarity at R3 and R4 to decrease hPXR activation12 while maintaining enzymatic and cellular potency, good metabolic stability, and avoiding efflux that may affect in vivo clearance.
Table 2. Reduction of hPXR Activation Leading to AG-120.

Human pregnane X receptor activation was determined as the fold activation relative to reference compound (rifampicin).
The cell-permeability coefficient (Papp) was determined in both directions (apical to basolateral [A–B] and basolateral to apical [B–A]) across the Caco2 cell monolayer. The efflux ratio was estimated as Papp[B–A]/Papp[A–B].
Total polar surface area.
Starting from AGI-14100, the introduction of additional polarity (hydroxyl group) on the pyrrolidinone ring of R4 in 9 maintained similar potency, somewhat decreased hPXR activation at 1 μM, but also decreased overall permeability and increased the efflux ratio. Further increasing the polarity at R4 by transitioning from cyanopyridine to cyanopyrimidine heterocycle in 10 abolished the hPXR activation but led to poor cellular potency and decreased metabolic stability.
Next, functional group changes at R3 (replacing one of the F atoms with a sulfonamide group in 11) dramatically increased the polarity of the molecule, leading to low hPXR activation values, but coupled with high microsomal clearance and efflux ratio. Lastly, changing one of the C–F bonds at R3 with an N atom embedded in the ring led to AG-120, with a balance of desirable properties: good enzyme and cellular potency, good stability in human liver microsomes, reduced hPXR activation, good permeability, and low efflux ratio. Synthesis of AG-120 and all related analogues was accomplished as described in Scheme S1 and the Supporting Information.
Biochemical and cell biology profiling revealed that AG-120 inhibited several IDH1-R132 mutants with potency similar to that seen for R132H (Table 3) and was highly selective for IDH1 isoforms, showing no inhibition of IDH2 (WT or mutant) isoforms at micromolar concentrations (Table S2). AG-120 at 100 μM did not inhibit multiple dehydrogenases tested (Table S3).
Table 3. Biochemical and Cell Biology Profiling of AG-120.
| assay type | mutationa | IC50b (nM) |
|---|---|---|
| enzyme | IDH1-R132H | 12 |
| IDH1-R132C | 13 | |
| IDH1-R132G | 8 | |
| IDH1-R132L | 13 | |
| IDH1-R132S | 12 | |
| IDH1-R132H/IDH1-WT heterodimer + NADP+/NADPH @ 1 h | 12 | |
| IDH1-R132H/IDH1-WT heterodimer + NADP+/NADPH @ 16 h | 5 | |
| IDH1-WT + NADP+ @ 1h | 71 | |
| IDH1-WT + NADP+ @ 16h | 24 | |
| cell-based | U87 MG (R132H) | 19 |
| neurospheres (R132H) | 3 | |
| HT1080 (R132C) | 8 | |
| COR-L105 (R132C) | 15 | |
| HCCC-9810 (R132S) | 12 |
All cell lines described here express mIDH1 endogenously, except U87 MG, which is an overexpression system.
For activity against enzyme, the enzyme and compound were preincubated for 1 or 16 h either in the presence or absence of cofactor as described in the Supporting Information.
In vitro, AG-120 exhibited rapid-equilibrium inhibition against the mIDH1-R132 homodimer. Kinetic studies of binding to demonstrate mode of action were inconclusive due to persistent prebound NADP(H) in all soluble mIDH1 enzyme preparations (Supporting Information, Figures S1 and S2). Surprisingly, AG-120 demonstrated slow-tight binding inhibition against the IDH1-WT homodimer (Figure S3 and S4).
AG-120 also showed good cellular potency across multiple mIDH1-R132 endogenous and overexpressing cell lines (Table 3), indicating its potential for use across all mIDH1-R132 cancers. AG-120 has a low turnover rate in liver microsomes derived from multiple species, including humans. PK studies performed in Sprague–Dawley rats, beagle dogs, and cynomolgus monkeys showed rapid oral absorption, low total body plasma clearance (CLp) and moderate to long half-life (t1/2) (Table S4). Although moderate exposure reduction was observed in a repeat-dose study in rodents (data not shown), no exposure reduction occurred in cynomolgus monkeys, and in patients with cancer a long t1/2 and accumulation of AG-120 following multiple doses were observed.13,14
Following a single oral dose of 50 mg/kg to rats with an intact blood–brain barrier, AG-120 exhibited brain penetration of 4.1% (AUC0–8h [brain]/AUC0–8h [plasma]). However, brain penetration is likely to be higher in glioma patients who have a compromised blood–brain barrier. Given that AG-120 is very potent and well tolerated, it has the potential to achieve therapeutic concentration in the brain, and its therapeutic benefit in glioma is being evaluated in clinical trials.
AG-120 showed robust tumor 2-HG reduction in female nude BALB/c mice inoculated with HT1080 cells. Each mouse received a single oral dose of vehicle or AG-120 at 50 or 150 mg/kg by gavage. Tumor 2-HG concentration declined rapidly, with maximum inhibition (92.0% and 95.2% at the 50 mg/kg and 150 mg/kg doses, respectively) achieved at ∼12 h post dose. Tumor 2-HG concentrations approached baseline levels 48–72 h following a single dose of AG-120 (Figure 1), consistent with the reversible nature of AG-120 inhibition.
Figure 1.
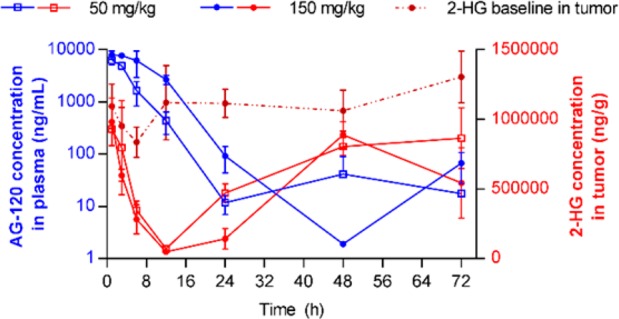
Mean ± SD concentrations of AG-120 in plasma and 2-HG in tumor after single oral administration of AG-120 at 50 or 150 mg/kg in a mouse HT1080 xenograft tumor model (n = 3 at each time point).
IDH mutations have been shown to block normal cellular differentiation via epigenetic and metabolic rewiring.1,3−5 To determine the effect of mIDH1 inhibition in primary human AML blast cells, mIDH1-R132H, mIDH1-R132C, and IDH1-WT, bone marrow or peripheral blood samples from patients (Table S5) were treated with AG-120 in an ex vivo assay. Living blast cells were sorted and cultured in medium containing cytokines (at a density of 0.5 × 106 cells/mL) in the presence or absence of AG-120. In mIDH1 samples, AG-120 reduced the level of intracellular 2-HG by 96% at the lowest tested dose (0.5 μM) and by 98.6% and 99.7%, respectively, at 1 and 5 μM (Figure 2). 2-HG was not measurable in multiple IDH1-WT patient samples assessed. AG-120 induced differentiation of primary mIDH1-R132H and mIDH1-R132C (but not IDH1-WT) blast cells from patients with AML treated ex vivo, as shown by enhanced ability to form differentiated colonies in methylcellulose assays, increased levels of cell-surface markers of differentiation, and increases in the proportion of mature myeloid cells (Figure S5).
Figure 2.
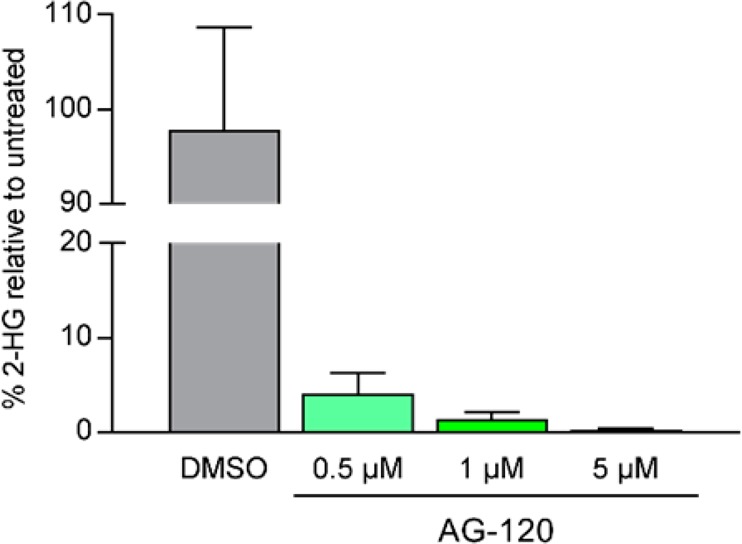
Percent intracellular 2-HG remaining relative to DMSO control after 6 days’ treatment with AG-120 in mIDH1-R132H or mIDH1-R132C patient samples (mean ± SEM from cells from four patients with mIDH1 AML).
Together, these compelling preclinical data provided the rationale to advance AG-120 into clinical development.
The discovery of enasidenib, which is active against mIDH2, and now AG-120 (ivosidenib) against mIDH1 as described here, presents a novel class of cancer therapy based on cellular differentiation. AG-120 is a potent mIDH1 inhibitor with favorable nonclinical and clinical safety profiles that has shown promising clinical activity in phase 1 clinical trials for both solid and hematologic malignancies. In patients with relapsed/refractory mIDH1 AML, interim results from the ongoing phase 1 trial have demonstrated an overall response rate of 42% and a complete response rate of 22% (median duration of complete response 9.3 months).15 Long-term stable disease has been observed in patients with previously treated nonenhancing mIDH1 gliomas,16 and in heavily pretreated patients with mIDH1 cholangiocarcinoma, where the median progression-free survival was 3.8 months and the 6-month progression-free survival rate was 40%.17 In these two single arm, phase 1 studies, AG-120 has demonstrated an acceptable safety profile to date.15−18 AG-120 is currently in late-stage clinical development in adults with mIDH1 AML (ClinicalTrials.gov NCT03173248), and with previously treated advanced mIDH1 cholangiocarcinoma (NCT02989857).
Acknowledgments
Writing assistance was provided by Christine Ingleby, Ph.D., of Excel Scientific Solutions, Horsham, U.K., and supported by Agios Pharmaceuticals, Inc. We thank Jean-Baptiste Micol and Christophe Willekens for clinical specimens, Nathalie Auger for cytogenetic analyses, and Zenon Konteatis for insightful discussions during the optimization of hPXR activation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00421.
Synthesis and profiling of AG-120, experimental procedures, synthetic details and characterization of compounds, and abbreviations (PDF)
Author Present Address
∇ (J.P.-M., S.-S.M.S.) Decibel Therapeutics, Cambridge, MA.
Author Present Address
⊥ (R.L.M., E.A., E.T.) KSQ Therapeutics, Cambridge, MA.
Author Present Address
# (J.O.S.) JOSC LLC Consulting, Lincoln, MA.
Author Present Address
▲ (F.G.S.) Sage Therapeutics, Cambridge, MA.
Author Present Address
□ (J.T.) Shire, Lexington, MA.
Author Present Address
⊗ (D.Z.) GSK R&D Shanghai, Shanghai, China.
Author Present Address
∞ (K.S.) Vertex Pharmaceuticals, Boston, MA.
Author Present Address
@ (L.U.) Spero Therapeutics, Cambridge, MA.
Author Present Address
+ (A.O.) Akebia Therapeutics, Cambridge, MA.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
These studies were funded by Agios Pharmaceuticals Inc., the French National Institute of Health (INSERM-AVIESAN; to M.D. David and V. Penard-Lacronique), French National Cancer League (LNCC), the Institut National du Cancer (INCa-DGOS-Inserm_6043 and INCa 2012–1-RT- 09), and the Fondation Association pour la Recherche sur le Cancer (ARC, SL220130607089 Programme Labellisé to V. Penard-Lacronique and S. de Botton). M.D. David was funded by a fellowship from the Institut National du Cancer (INCa-DGOS_5733).
The authors declare the following competing financial interest(s): S.d.B. serves on advisory boards for Agios and Celgene. S.S.M.S is a consultant for Agios.
Supplementary Material
References
- Dang L.; Su S. M. Isocitrate Dehydrogenase Mutation and (R)-2-Hydroxyglutarate: From Basic Discovery to Therapeutics Development. Annu. Rev. Biochem. 2017, 86, 305–331. 10.1146/annurev-biochem-061516-044732. [DOI] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. Cancer-associated IDH1 Mutations Produce 2-Hydroxyglutarate. Nature 2009, 462 (7274), 739–744. 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa M. E.; Abdel-Wahab O.; Lu C.; Ward P. S.; Patel J.; Shih A.; Li Y.; Bhagwat N.; Vasanthakumar A.; Fernandez H. F.; Tallman M. S.; Sun Z.; Wolniak K.; Peeters J. K.; Liu W.; Choe S. E.; Fantin V. R.; Paietta E.; Lowenberg B.; Licht J. D.; Godley L. A.; Delwel R.; Valk P. J.; Thompson C. B.; Levine R. L.; Melnick A. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18 (6), 553–567. 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Ward P. S.; Kapoor G. S.; Rohle D.; Turcan S.; Abdel-Wahab O.; Edwards C. R.; Khanin R.; Figueroa M. E.; Melnick A.; Wellen K. E.; O’Rourke D. M.; Berger S. L.; Chan T. A.; Levine R. L.; Mellinghoff I. K.; Thompson C. B. IDH Mutation Impairs Histone Demethylation and Results in a Block to Cell Differentiation. Nature 2012, 483 (7390), 474–478. 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D.; Popovici-Muller J.; Palaskas N.; Turcan S.; Grommes C.; Campos C.; Tsoi J.; Clark O.; Oldrini B.; Komisopoulou E.; Kunii K.; Pedraza A.; Schalm S.; Silverman L.; Miller A.; Wang F.; Yang H.; Chen Y.; Kernytsky A.; Rosenblum M. K.; Liu W.; Biller S. A.; Su S. M.; Brennan C. W.; Chan T. A.; Graeber T. G.; Yen K. E.; Mellinghoff I. K. An Inhibitor of Mutant IDH1 Delays Growth and Promotes Differentiation of Glioma Cells. Science 2013, 340 (6132), 626–630. 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. M.; DiNardo C. D.; Pollyea D. A.; Fathi A. T.; Roboz G. J.; Altman J. K.; Stone R. M.; DeAngelo D. J.; Levine R. L.; Flinn I. W.; Kantarjian H. M.; Collins R.; Patel M. R.; Frankel A. E.; Stein A.; Sekeres M. A.; Swords R. T.; Medeiros B. C.; Willekens C.; Vyas P.; Tosolini A.; Xu Q.; Knight R. D.; Yen K. E.; Agresta S.; de Botton S.; Tallman M. S. Enasidenib in Mutant IDH2 Relapsed or Refractory Acute Myeloid Leukemia. Blood 2017, 130 (6), 722–731. 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein E. M.; Yen K. Targeted Differentiation Therapy with Mutant IDH Inhibitors: Early Experiences and Parallels with Other Differentiation Agents. Annu. Rev. Canc. Biol. 2017, 1, 379–401. 10.1146/annurev-cancerbio-050216-122051. [DOI] [Google Scholar]
- Amatangelo M. D.; Quek L.; Shih A.; Stein E. M.; Roshal M.; David M. D.; Marteyn B.; Farnoud N. R.; de Botton S.; Bernard O. A.; Wu B.; Yen K. E.; Tallman M. S.; Papaemmanuil E.; Penard-Lacronique V.; Thakurta A.; Vyas P.; Levine R. L. Enasidenib Induces Acute Myeloid Leukemia Cell Differentiation to Promote Clinical Response. Blood 2017, 130 (6), 732–741. 10.1182/blood-2017-04-779447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovici-Muller J.; Saunders J. O.; Salituro F. G.; Travins J. M.; Yan S.; Zhao F.; Gross S.; Dang L.; Yen K. E.; Yang H.; Straley K. S.; Jin S.; Kunii K.; Fantin V. R.; Zhang S.; Pan Q.; Shi D.; Biller S. A.; Su S. M. Discovery of the First Potent Inhibitors of Mutant IDH1 That Lower Tumor 2-HG in Vivo. ACS Med. Chem. Lett. 2012, 3 (10), 850–855. 10.1021/ml300225h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch S.; Krausert S.; Fischer V.; Balss J.; Ott M.; Schrimpf D.; Capper D.; Sahm F.; Eisel J.; Beck A. C.; Jugold M.; Eichwald V.; Kaulfuss S.; Panknin O.; Rehwinkel H.; Zimmermann K.; Hillig R. C.; Guenther J.; Toschi L.; Neuhaus R.; Haegebart A.; Hess-Stumpp H.; Bauser M.; Wick W.; Unterberg A.; Herold-Mende C.; Platten M.; von Deimling A. Pan-mutant IDH1 Inhibitor BAY 1436032 for Effective Treatment of IDH1 Mutant Astrocytoma In Vivo. Acta Neuropathol. 2017, 133 (4), 629–644. 10.1007/s00401-017-1677-y. [DOI] [PubMed] [Google Scholar]
- Cho Y. S.; Levell J. R.; Liu G.; Caferro T.; Sutton J.; Shafer C. M.; Costales A.; Manning J. R.; Zhao Q.; Sendzik M.; Shultz M.; Chenail G.; Dooley J.; Villalba B.; Farsidjani A.; Chen J.; Kulathila R.; Xie X.; Dodd S.; Gould T.; Liang G.; Heimbach T.; Slocum K.; Firestone B.; Pu M.; Pagliarini R.; Growney J. D. Discovery and Evaluation of Clinical Candidate IDH305, a Brain Penetrant Mutant IDH1 Inhibitor. ACS Med. Chem. Lett. 2017, 8 (10), 1116–1121. 10.1021/acsmedchemlett.7b00342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. D.; Olson S. H.; Balkovec J. M.; Zhu Y.; Royo I.; Yabut J.; Evers R.; Tan E. Y.; Tang W.; Hartley D. P.; Mosley R. T. Attenuating Pregnane X Receptor (PXR) Activation: A Molecular Modelling Approach. Xenobiotica 2007, 37 (2), 124–138. 10.1080/00498250601050412. [DOI] [PubMed] [Google Scholar]
- Fan B.; Goyal L.; Lowery M. A.; Pandya S. S.; Manyak E.; Le K.; Jiang L.; Auer J.; Dai D.. Pharmacokinetic/pharmacodynamic (PK/PD) Profile of AG-120 in Patients with IDH1-Mutant Cholangiocarcinoma From a Phase 1 Study of Advanced Solid Tumors. J. Clin. Oncol. 2017, 35 ( (15 Suppl), ), Abstract 4082. 10.1200/JCO.2017.35.15_suppl.4082 [DOI] [Google Scholar]
- Fan B.; Le K.; Manyak E.; Liu H.; Prahl M.; Bowden C. J.; Biller S.; Agresta S.; Yang H.. Longitudinal Pharmacokinetic/Pharmacodynamic Profile of AG-120, a Potent Inhibitor of the IDH1 Mutant Protein, in a Phase 1 Study of IDH1-Mutant Advanced Hematologic Malignancies. Blood 2015, 126 ( (23), ), Abstract 1310. [Google Scholar]
- DiNardo C. D.; de Botton S.; Stein E. M.; Roboz G. J.; Mims A. S.; Pollyea D. A.; Swords R. T.; Altman J. K.; Collins R. H.; Mannis G. N.; Uy G. L.; Donnellan W.; Pigneux A.; Fathi A. T.; Stein A. S.; Erba H. P.; Prince G. T.; Foran J. M.; Traer E.; Stuart R. K.; Arellano M. L.; Slack J. L.; Sekeres M. A.; Yen K.; Kapsalis S. M.; Liu H.; Goldwasser M.; Agresta S.; Attar E. C.; Tallman M. S.; Stone R. M.; Kantarjian H. M.. Ivosidenib (AG-120) in Mutant IDH1 AML and Advanced Hematologic Malignancies: Results of a Phase 1 Dose Escalation and Expansion Study. Blood 2017, 130 ( (Suppl), ), Abstract 725. [Google Scholar]
- Mellinghoff I. K.; Touat M.; Maher E.; de la Fuente M.; Cloughesy T. F.; Holdhoff M.; Cote G. M.; Burris H.; Janku F.; Huang R.; Young R. J.; Ellingson B.; Nimkar T.; Jiang L.; Ishii Y.; Choe S.; Fan B.; Steelman L.; Yen K.; Bowden C.; Pandya S.; Wen P. Y. AG-120, a First-In-Class Mutant IDH1 Inhibitor in Patients with Recurrent or Progressive IDH1 Mutant Glioma: Updated Results From the Phase 1 Non-Enhancing Glioma Population. Neuro-Oncology 2017, 19 (Suppl 6), vi10. 10.1093/neuonc/nox168.037. [DOI] [Google Scholar]
- Lowery M. A.; Abou-Alfa G. K.; Burris H. A.; Janku F.; Shroff R. T.; Cleary J. M.; Azad N. S.; Goyal L.; Maher E. A.; Gore L.; Hollebecque A.; Beeram M.; Trent J. C.; Jiang L.; Ishii Y.; Auer J.; Gliser C.; Agresta S. V.; Pandya S. S.; Zhu A. X. Phase I Study of AG-120, an IDH1 Mutant Enzyme Inhibitor: Results From the Cholangiocarcinoma Dose Escalation and Expansion Cohorts. J. Clin. Oncol. 2017, 35 (15 Suppl), 4015. 10.1200/JCO.2017.35.15_suppl.4015. [DOI] [Google Scholar]
- DiNardo C. D.; de Botton S.; Stein E. M.; Roboz G. J.; Swords R. T.; Pollyea D. A.; Fathi A. T.; Collins R.; Altman J. K.; Flinn I. W.; Mannis G. N.; Mims A. S.; Foran J. M.; Pigneux A.; Prince G. T.; Uy G. L.; Tallman M. S.; Kantarjian H. M.; Liu H.; Attar E. C.; Sacolick J.; Yen K.; Hurov J. B.; Choe S.; Wu B.; Stone R. M.. Determination of IDH1 Mutational Burden and Clearance Via Next-Generation Sequencing in Patients with IDH1 Mutation-Positive Hematologic Malignancies Receiving AG-120, a First-in-Class Inhibitor of Mutant IDH1. Blood 2016, 128 ( (22), ), Abstract 1070. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.