

Abstract

A series of 16 new diselenide–acylselenourea conjugates have been designed following the fragment-based drug strategy. Compound in vitro cytotoxic potential was evaluated against six human cancer cell lines and two nonmalignant derived cell lines with the aim of determining their potency and selectivity. Nine derivatives exhibited GI50 values under 10 μM in at least four cancer cell lines. A clear gap situated phenyl substitution over heterocyclic moieties in terms of selectivity. Among carbocyclic compounds, derivatives 2 and 7 significantly inhibited cell growth of breast adenocarcinoma cells with GI50 values of 1.30 and 0.15 nM, respectively, with selectivity indexes 12 and 121 times higher than those obtained for doxorubicin. Preliminary mechanistic studies indicated that compounds 2 and 7 induce cell cycle arrest and autophagy-dependent cell death evidenced by the blockage of cell death with pretreatment with wortmannin or chloroquine and confirmed by the upregulation of the markers Beclin1 and LC3B in MCF-7 cells.
Keywords: Selenium, acylselenoureas, diselenides, autophagy
Cancer can be categorized as a serious clinical problem as it affects millions of patients worldwide. Only in the United States of America statistics show death rate by cancer will reach the alarming ratio of 1600 deaths per day.1 Even though big progress and investment in the field have been made, selectivity and side effects are still issues for current treatments. Moreover, both acquired and de novo resistances to chemotherapy have been documented.2 In this context, over the past decade, selenium compounds have demonstrated important effects on cancer progression, distressing cell growth and proliferation; this activity is both form and dose dependent.3,4 Many mechanisms of action have been identified, including the induction of apoptosis either caspase dependent or independent.5,6 Furthermore, modulation of some kinases activity, antioxidant effects through selenoproteins involved in the modulation of oxidative stress, mitotic catastrophe, autophagy, or a combination of the previously described mechanisms have been proven for selenium-containing molecules.7,8
Autophagy has been deeply studied as a factor of cancer regulation.9 Drug-induced autophagy in breast cancer cell lines has already been described in the past and has been correlated with apoptosis promotion.10 Beyond the interest of the oncosuppressive effect, autophagy assures anticancer inmunosurveillance.11 Some autophagy activators have been approved by FDA for cancer treatment such as everolimus.12
Among the chemical entities containing selenium that have been identified, the chemical form of diselenide stands out as an appealing skeleton for development of new anticancer agents.13 In particular, diphenyl diselenide (PhSe)2 has been widely studied and has proven activity as oxidative stress protector,14,15 presenting also cytotoxic activity.16 In this line of investigation, our research group has explored this chemical entity and the most potent antitumoral structures synthesized in our group containing the diphenyldiselenide moiety corresponds to bis(4-aminophenyl)diselenide structure.17 Therefore, we selected this fragment as a starting point for our structure design with the aim of enhancing its potency and reducing its toxicity.
However, selenourea moiety is emerging as a privileged structural fragment in some antiproliferative studies. Those resulting molecules behaved as reactive oxygen species (ROS) scavengers as well as glutathione peroxidase mimetics and proved to have potent cytotoxic effects.18N,N′-disubstitued selenourea derivatives have been reported as nontoxic polyfunctional antioxidants with potency over sulfur or oxygen analogs.19 We have recently confirmed that selenourea derivatives have more potent in vitro activities against several cancer cell lines than their sulfur analogs.20 Since combination of two or more active structural moieties can possibly enhance bioactivity and reduce adverse effects, we combined (PhSe)2 nucleus and selenourea fragment with the intention of achieving this synergic effect. In addition, and considering that a variety of drugs are activated by biotransformation, the selenoureas were functionalized with different acyl substituents in order to facilitate or to modulate the release of the active groups. These modifications enclose:
-
a)
Systematic variations of substituents at different diversification points in phenyl rings with electron-withdrawing or electron-donor groups in order to obtain the maximal 3D structural enrichment as well as information about the effects of electronic properties in the antitumoral activity (compounds 1–7). In addition, compounds 7 and 8 were included with the aim of evaluating the effect of polymethoxy substitution and biomimetics of combretastatin. This agent has proven ability to induce cell death through various mechanisms including mitotic catastrophe and autophagy induction in various adenocarcinoma-derived cell lines.21
-
b)
Replacing the lateral phenyl group by heterocyclic system (compounds 9–16) possessing a functional group able to form hydrogen bonding as well as antitumor activity against different tumor cell lines as furan,22 thiophene, benzothiophene,23 isoxazole,24 benzodioxole,25 quinoline,26 and phenylquinoline27 derivatives.
-
c)
Carbonyl group was included as a cleavable linker largely used in targeted drug strategies with intention of modulating metabolism.
Founded on these facts, the experience, and the results of our research team in the field of diselenide and selenourea synthesis,17,20,28,29 a general structure was designed, included in Figure 1. In this entity, the fragments incorporated are bis(4-aminophenyl)diselenide colored in green, selenourea moiety colored in red, and lateral cyclic fragment colored in blue.
Figure 1.
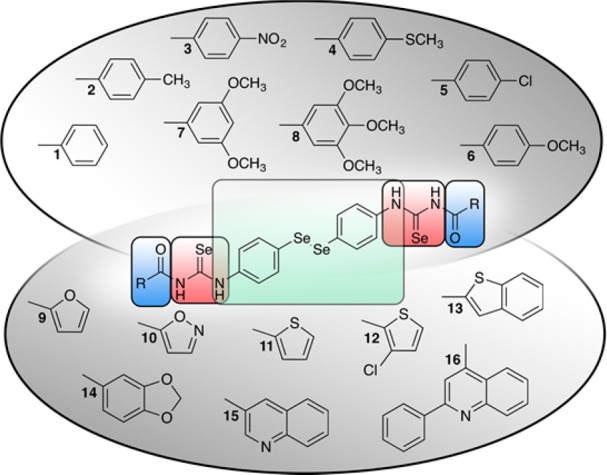
Structures of novel selenium containing compounds.
All the synthesized compounds were screened for their cytotoxic activity against a panel of cancer cell lines, and a preliminary approach to the undergoing mechanism of action was performed.
Sixteen novel compounds matching the designed scaffold described in the introduction were synthesized. They can be classified according to their lateral rings, carbocyclic (1–8) and heterocyclic (9–16), and their structures are summarized in Figure 1. The structures were confirmed by nuclear magnetic resonance (1H NMR and 13C NMR), infrared spectroscopy (IR), and mass spectrometry (MS), and purity was confirmed by elemental microanalysis.
The novel organoseleno derivatives were synthesized starting from bis(4-aminophenyl)diselenide, which has been synthesized according to a previously published procedure.29 The first step of the proposed mechanism (Scheme 1) is an electrophilic aromatic substitution that curses through an intermediate stabilized by the four possible resonance structures leading to 4-aminophenylselenocyante by deprotonation. Second, treatment with NaBH4 causes dimerization of the 4-aminophenylselenocyanate to yield bis(4-aminophenyl)diselenide.
Scheme 1. Mechanism Proposed for the Synthesis of Bis(4-aminophenyl)diselenide.

The reaction of benzoyl chlorides (1–8) or hetero aroyl chlorides (9–16) with potassium selenocyanate in a molar ratio 1:1, respectively, in dry acetone yielded the corresponding acyl isoselenocyanates. These intermediates were used without purification, and the reaction in situ with bis(4-aminophenyl)diselenide in acetone at room temperature generated the final compounds.
Reaction times range from 1 h up to 12 h and was then quenched with cold water where potassium chloride from the formation of isoselenocyanate is dissolved. At this point, some compounds precipitate along with some starting reagents. Purification is performed by various methodologies such as washing with ethyl ether or extraction with dichloromethane. After purification yields range from 16% to 95%, this variability in yield values may be due to acyl chloride reactivity or to other physical properties of the reagents such as solubility in the solvent used.
Given the absence of commercially available acyl chlorides such as those needed for the synthesis of compounds 12, 13, 14, and 15, they were obtained by chlorination of their corresponding carboxylic acids with thionyl chloride under reflux conditions according to the literature.
We propose a two-phase reaction to obtain the novel diselenides. With regard to the proposed mechanism (Scheme 2), the first reaction could be a nucleophilic attack of the described negative charge over the nitrogen atom over the carbonylic carbon following an acyl substitution mechanism to yield the isoselenocyanate. Second, the lone pair of electrons of the nitrogen atom attached to the phenyl ring could perform a nucleophilic attack over the isoselenocyanate carbon atom leading to the formation of the final structure.
Scheme 2. Proposed Mechanisms for the Two-Phase Reaction To Form the Novel Diselenides.

The 16 novel compounds were screened for their effect on cancer cell viability against a panel of six human cancer cell lines: breast adenocarcinoma (MCF-7), colon carcinoma (HT-29), lymphocytic leukemia (K-562), prostate cancer (PC-3), lymphoblastic leukemia (CCRF-CEM), and lung carcinoma (HTB-54). The cytotoxic potency was evaluated by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay according to the methodology previously described.28 Initially, each compound was tested at five different concentrations from 0.01 to 100 μM after 72 h treatment; for some compounds, further dilutions were needed due to their potency. Doxorubicin, etoposide, and cisplatin were used as reference drugs. Methylseleninic acid (MSA) was introduced as a selenium containing molecule with promising cytotoxic profile.30
Compound 6 was not screened due to its lack of solubility under the assay conditions. As an approximation, compound 4 will be considered as an analogue in terms of electronic and steric modulations.
As Table 1 illustrates, CCRF-CEM, PC-3, and MCF-7 were generally the most sensitive cell lines. However, K-562 cell line was less sensitive in the general picture. TGI and LC50 values for every cell line are also provided in the Supporting Information (Tables S1 and S2).
Table 1. Average GI50 Values (μM) and Selectivity Indexes.

Calculated as GI50 184B5)/GI50(MCF-7).
Calculated as GI50(BEAS-2B)/GI50(HTB-54).
Interestingly, compounds with carbocyclic lateral rings (1–5, 7, and 8) and those with heterocyclic rings (9–16) displayed different cytotoxic profiles. Whereas compounds with carbocyclic substituted endings exhibited GI50 values in the nanomolar range in MCF-7, PC-3, and CCRF-CEM, no heterocyclic derivatives matched this condition.
The assay was also performed on two cell lines derived from nonmalignant cells, one mammary gland derived (184B5) and the other derived from bronchial epithelium (BEAS-2B). Selectivity index (SI) for breast and cancer cells was calculated as the ratio of the GI50 values obtained for the nonmalignant derived and homologue cancer cell line.
Moreover, there are also clear differences in SI values for breast cell lines. Table 1 shows a clear range gap correlated with ending core nature, with the exception of compound 8 that did not pair the level of selectivity shown by the rest of the carbocyclic lateral ring compounds (1–5, 7, and 8).
In spite of this, GI50 values of compounds 9–15 are under the 10 μM threshold for HT-29 treatment and in the same order of magnitude as cisplatin. Furthermore, among the different heterocyclic substituents, furan and isoxazole (9–10) fulfill the 10 μM GI50 value condition in at least five out of six tested cell lines. Besides, GI50 values for compound 10 are in the same order of magnitude as those exhibited by cisplatin and MSA in those cell lines where data is available. However, although compound 10 was cytotoxic showing LD50 values under 10 μM in CCRF-CEM and MCF-7, this activity was not accompanied by the required selectivity for breast adenocarcinoma cells. If we draw the focus to MCF-7, PC-3, and CCRF-CEM cell lines, all seven carbocyclic derivative compounds presented GI50 values under 10 μM in all three cell lines.
In CCRF-CEM cells, compounds 4, 5, and 7 present GI50 values at least 10-fold lower than etoposide, cisplatin, and MSA. Comparison of those reference drugs in PC-3 cells reveals that compounds 1, 2, and 3 satisfy these same criteria.
When antiproliferative activities for structures with carbocyclic core ending were analyzed in MCF-7 cells, compounds 1–5 and 7 showed GI50 values at least two orders of magnitude lower than those for cisplatin, etoposide, and MSA. Due to this data, a more potent reference drug is needed for comparison such as doxorubicin. Besides, doxorubicin was highly selective for the breast adenocarcinoma cell line (SI184B5/MCF-7 = 574), which makes it an ideal reference for compounds 1–5 and 7. Taking into account this benchmark, five out of the six compounds (1, 2, 4, 5, and 7) equal the nanomolar range for GI50 values. Moreover, selectivity index for mammary adenocarcinoma exceeded the one calculated for doxorubicin in up to 120-fold (compound 7).
Taking into account that one of the goals of fragment-based design strategy is to improve potency and reduce the toxicity, we compared those results with the ones obtained for bis(4-aminophenyl)diselenide itself. GI50 values (μM) for each cell line were 0.75 (MCF-7), 0.80 (PC-3), 0.21 (HTB-54), <0.01 (HT-29), 0.95 (K-562), and 5.28 (CCRF-CEM). Selectivity-wise, the GI50 value obtained when the assay is performed in 184B5 cell cultures was of 6.29 μM leading to a SI of 8.39. Our target when SI was calculated as GI50(BEAS-2B)/GI50(HTB-54) was a value of 3.44 as the GI50 for BEAS-2B was 0.73 μM. In general terms, this compound could be considered potent as it induces 50% cell growth inhibition under 10 μM concentrations in all six cell cancer cell lines. Although HT-29 was the most sensitive cell line while CCRF-CEM was the most resistant, when we have a look at SI values we can state that the selectivity for this compound is, in general, scarce. However, when we compare these values with the results obtained for the novel diselenide derivatives we can observe that compounds 1–5 and 7 have a striking selectivity for MCF-7 cell line over the rest of the cancer cell lines and over 184B5 cell line. From the data, we can conclude that the introduction of the acylselenourea group plays a determinant role in terms of selectivity when compared to the core structure bis(4-aminophenyl)diselenide alone.
Though the number of synthesized compounds is too small to make solid structure–activity relationship statements, some conclusions can be withdrawn from the results. When structure ending core is replaced by heterocyclic rings, a loss of antiproliferative activity and selectivity was observed. As for the compounds bearing carbocyclic rings, a trend might be considered as electron-donating substituents enhance activity in MCF-7 cells, peaking this effect with the presence of two methoxy groups (compound 7). However, the presence of three electron-donating groups seems to revert this effect, presumably due to steric hindrance.
At this point and considering all the previous results, compounds 2 and 7 were selected for further biological studies due to their activity and selectivity, especially against breast adenocarcinoma cells (MCF-7).
Apoptosis and cell cycle arrest are targets for many anticancer drugs such as doxorubicin. Influence of some selenium containing compounds over those processes has been proven.31,32 As a preliminary approach to the mechanism of action, the cell cycle distribution and cell death status was studied in MCF-7 cultures treated with either compounds 2 or 7. These tests were performed by flow cytometry using Apo-Direct Kit (BD Pharmigen) based on the TUNEL technique under the conditions described by the manufacturer. Treatment with 6 μM camptothecin was used as a positive control.
As shown in Figure 2, both compounds 2 and 7 increased the percentages of subdiploid cells in the cultures in a dose- and time-dependent manner. In fact, both derivatives induced a significant increase in cell death when added at 5 μM concentration (Figure 2A). At 10 μM concentration, an increase of cell death was detected as soon as 6 h for both compounds (Figure 2B).
Figure 2.

Compounds 2 and 7 induced cell death in a dose- and time-dependent manner in MCF-7 cell cultures. Cells were treated with increasing concentrations of compounds 2 and 7 for 24 h (A) or with 10 μM concentration for different periods of time (B).
As expected for compounds that induce cell death, when cell cycle distribution was studied, the number of the hypodiploid sub-G1 cell population was increased. This result reaches significance at a 1 μM concentration for derivative 7 (Figure 3B) and at 5 μM treatment for compound 2 (Figure 3A). Concomitant with this, a 10 μM treatment for 6 h with either compound 2 or 7 augmented the number of cells in sub-G1 phase (Figure 3C,D). Furthermore, both compounds 2 and 7 induced arrest in G2/M in a time- and dose-dependent manner. A very remarkable increment of G2/M phase was evidenced for the 24 h treatment even at the lowest concentration tested (1 μM) peaking at 10 and 20 μM with over 40% of cells in this phase (Figure 3A,B). Similar results were found for time course analysis; after 10 μM treatment for 6 h, the growth of G2/M phase population was meaningful (Figure 3C,D).
Figure 3.

Cell cycle analysis after treatment with compounds 2 (A,C) and 7 (B,D). Cells were treated with increasing concentrations of compounds 2 (A) and 7 (B) for 24 h. Panels C and D correspond to treatments with 10 μM concentration for different periods of time for compounds 2 (C) and 7 (D).
Some general considerations can be extracted from these results for derivative 2. The arrest of cells in G2/M phase becomes significant at the lowest times and doses tested. For this population, building up seems to plateau over the 5 μM concentration for 24 h treatment (Figure 3A). If we analyze the 10 μM treatment over the time, population of cells in G2/M phase peak for the 24 h treatment, while in the 48 h experiment this population seems to have reached its limit value as cells start to then accumulate in the hypodiploid sub-G1 phase.
This same phenomenon can be observed when compound 7’s behavior is studied; in this case, arrest in G2/M phase peaks at 10 μM treatment for 24 h. Higher doses or longer times of exposure cause cells to accumulate in sub-G1 phase and the number of cells in S phase to drop below the 10% value. A slight revision of G2/M arrest can be perceived for 20 μM treatment for 24 h or 10 μM for 48 h.
With the aim of narrowing down the pathways implicated in this cell death processes, MCF-7 cells were incubated with compounds 2 and 7 in the presence or absence of the pan-caspase inhibitor Z-VAD-FMK, the PI3K inhibitor wortmannin, and chloroquine. As observed in Figure 4A, preincubation with both wortmannin and chloroquine blocked cell death caused by both compounds 2 and 7, reverting apoptotic population levels to those present in the culture treated only with wortmannin or chloroquine on their own. Rapamycin was used as a reference autophagy inductor. This suggests that autophagy is involved in the cell death induced by these compounds. However, preincubation with Z-VAD-FMK could not prevent compounds 2 and 7 induced cell death (Figure 4B), which rules out caspase-dependent cell death.
Figure 4.

Cell death induced by compounds 2 and 7 is blocked by wortmannin and chloroquine but not by caspase inhibitor Z-VAD-FMK. MCF-7 cells were preincubated for 1 h with 50 mM of pan-caspase inhibitor Z-VAD-FMK (A), 100 nM of the autophagy inhibitor wortmannin, and 10 μM of chloroquine (B) or in absence of inhibitor prior the addition of 10 μM 2 and 7 or DMSO (control) for 24 h. Thirty micromolar rapamycin was used as a reference autophagy control. Cells were then processed with the Apo-Direct kit and analyzed by flow cytometry.
As suggested by the inhibitor assay results and with the aim of confirming that autophagy is involved in the mechanism of action by which compounds 2 and 7 cause cell death, we proceeded to the evaluation by Western blot of autophagy markers (Beclin1 and LC3B).33 MCF-7 cells were treated with 10 μM of compounds 2 and 7 for 24 h. Formation of autophagosomes starts with an initial nucleation step that requires the ULK1 complex that includes Beclin1, which, as shown in Figure 5, is upregulated by treatment with both selected compounds. Consistent effects are observed when LC3B-I and LC3B-II were studied.
Figure 5.

Significant upregulation of autophagy markers studied was confirmed by densitometry as shown: Beclin1 (A), LC3B-I (B), and LC3B-II (C).
In conclusion, a total of 16 novel acylselenourea-diselenide derivatives were designed following the fragment-based strategy and synthesized: eight with carbocyclic endings (1–8) and eight with heterocyclic endings (9–16). The evaluation of their in vitro cytotoxic activity revealed that, in general, MCF-7, PC-3, and CCRF-CEM cell lines are the most sensitive among our panel of six cancer cell lines. Among the different substituents, carbocyclic endings proved in general to be more potent and selective than their heterocyclic homologues. Many of the derivatives synthesized were found to be more effective and selective as agents compared to bis(4-aminophenyl)diselenide on the cancer cell lines tested, hence validating our approach. Compounds 2 and 7, with 4-methylphenyl and 3,5-dimethoxyphenyl moieties, respectively, were selected for further biological studies due to their high potency and selectivity against breast adenocarcinoma cells. Cell cycle analysis through flow cytometry revealed that cell death and G2/M phase arrest are both time- and dose-dependent. Moreover, in the presence of wortmannin or chloroquine, cell death was reverted to negative control levels, indicating that autophagy might be implicated in cell death mechanism. This hypothesis is confirmed when autophagy marker analysis is performed by Western blot for LC3B and Beclin1.
Acknowledgments
The authors wish to express their gratitude to University of Calgary Chemistry Department and to Thomas G. Back for the opportunity to perform the 77Se NMR experiments.
Glossary
ABBREVIATIONS
- GI50
concentration that reduces growth by 50% compared to control
- TGI
concentration that completely inhibits cell growth
- LC50
concentration that kills 50% of cells
- SI
selectivity index
- n.d.
not determined
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00482.
Chemical synthesis, characterization of target compounds, protocols of biological assays, cytotoxicity complete data and cell cycle, and cell death and protein analysis exemplification (PDF)
This research was founded in part by “la Caixa” Banking Foundation and Asociación de Amigos de la Universidad de Navarra (a grant to P. Garnica), by plan de Investigación de la Universidad de Navarra, PIUNA (ref 2014–26), and Fundación Caja Navarra–UNED-“La Caixa”.
The authors declare no competing financial interest.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2016. Ca-Cancer J. Clin. 2016, 66, 7–30. 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Mummaneni P.; Shord S. S. Epigenetics and oncology. Pharmacotherapy 2014, 34, 495–505. 10.1002/phar.1408. [DOI] [PubMed] [Google Scholar]
- Allen N. E.; Travis R. C.; Appleby P. N.; Albanes D.; Barnett M. J.; Black A.; Bueno-de-Mesquita H. B.; Deschasaux M.; Galan P.; Goodman G. E.; Goodman P. J.; Gunter M. J.; Heliovaara M.; Helzlsouer K. J.; Henderson B. E.; Hercberg S.; Knekt P.; Kolonel L. N.; Lasheras C.; Linseisen J.; Metter E. J.; Neuhouser M. L.; Olsen A.; Pala V.; Platz E. A.; Rissanen H.; Reid M. E.; Schenk J. M.; Stampfer M. J.; Stattin P.; Tangen C. M.; Touvier M.; Trichopoulou A.; van den Brandt P. A.; Key T. J. Selenium and Prostate Cancer: Analysis of Individual Participant Data From Fifteen Prospective Studies. J. Natl. Cancer Inst 2016, 108, djw153. 10.1093/jnci/djw153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes A. P.; Gandin V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 1642–1660. 10.1016/j.bbagen.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Singletary K.; Milner J. Diet, autophagy, and cancer: a review. Cancer Epidemiol., Biomarkers Prev. 2008, 17, 1596–1610. 10.1158/1055-9965.EPI-07-2917. [DOI] [PubMed] [Google Scholar]
- Wallenberg M.; Misra S.; Wasik A. M.; Marzano C.; Bjornstedt M.; Gandin V.; Fernandes A. P. Selenium induces a multi-targeted cell death process in addition to ROS formation. J. Cell Mol. Med. 2014, 18, 671–684. 10.1111/jcmm.12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad G. K.; Tomar R. S. Ebselen, a promising antioxidant drug: mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. 10.1007/s11033-014-3417-x. [DOI] [PubMed] [Google Scholar]
- Wallenberg M.; Misra S.; Bjornstedt M. Selenium cytotoxicity in cancer. Basic Clin. Pharmacol. Toxicol. 2014, 114, 377–386. 10.1111/bcpt.12207. [DOI] [PubMed] [Google Scholar]
- Chen N.; Debnath J. Autophagy and tumorigenesis. FEBS Lett. 2010, 584, 1427–1435. 10.1016/j.febslet.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z. Z.; Li M. M.; Deng P. F.; Wang S.; Wang L.; Lu X. P.; Hu L. B.; Chen Z.; Jie H. Y.; Wang Y. F.; Liu X. X.; Liu Z. Paris saponin-induced autophagy promotes breast cancer cell apoptosis via the Akt/mTOR signaling pathway. Chem.-Biol. Interact. 2017, 264, 1–9. 10.1016/j.cbi.2017.01.004. [DOI] [PubMed] [Google Scholar]
- Ladoire S.; Enot D.; Senovilla L.; Chaix M.; Zitvogel L.; Kroemer G. Positive impact of autophagy in human breast cancer cells on local immunosurveillance. Oncoimmunology 2016, 5, e1174801. 10.1080/2162402X.2016.1174801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petibone D. M.; Majeed W.; Casciano D. A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. 10.1002/jat.3393. [DOI] [PubMed] [Google Scholar]
- Kim C.; Lee J.; Park M. S. Synthesis of new diorganodiselenides from organic halides: their antiproliferative effects against human breast cancer MCF-7 cells. Arch. Pharmacal Res. 2015, 38, 659–665. 10.1007/s12272-014-0407-4. [DOI] [PubMed] [Google Scholar]
- Sartori G.; Jardim N. S.; Sari M. H.; Flores E. F.; Prigol M.; Nogueira C. W. Diphenyl Diselenide Reduces Oxidative Stress and Toxicity Caused by HSV-2 Infection in Mice. J. Cell. Biochem. 2017, 118, 1028–1037. 10.1002/jcb.25667. [DOI] [PubMed] [Google Scholar]
- Shaaban S.; Negm A.; Sobh M. A.; Wessjohann L. A. Organoselenocyanates and symmetrical diselenides redox modulators: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2015, 97, 190–201. 10.1016/j.ejmech.2015.05.002. [DOI] [PubMed] [Google Scholar]
- Caeran Bueno D.; Meinerz D. F.; Allebrandt J.; Waczuk E. P.; dos Santos D. B.; Mariano D. O.; Rocha J. B. Cytotoxicity and genotoxicity evaluation of organochalcogens in human leucocytes: a comparative study between ebselen, diphenyl diselenide, and diphenyl ditelluride. BioMed Res. Int. 2013, 2013, 537279. 10.1155/2013/537279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plano D.; Baquedano Y.; Ibanez E.; Jimenez I.; Palop J. A.; Spallholz J. E.; Sanmartin C. Antioxidant-prooxidant properties of a new organoselenium compound library. Molecules 2010, 15, 7292–7312. 10.3390/molecules15107292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Hernandez L. L.; Merino-Montiel P.; Montiel-Smith S.; Meza-Reyes S.; Vega-Baez J. L.; Abasolo I.; Schwartz S. Jr.; Lopez O.; Fernandez-Bolanos J. G. Diosgenin-based thio(seleno)ureas and triazolyl glycoconjugates as hybrid drugs. Antioxidant and antiproliferative profile. Eur. J. Med. Chem. 2015, 99, 67–81. 10.1016/j.ejmech.2015.05.018. [DOI] [PubMed] [Google Scholar]
- Neganova M. E.; Proshin A. N.; Redkozubova O. M.; Serkov I. V.; Serkova T. P.; Dubova L. G.; Shevtsova E. F. N,N’-Substituted Selenoureas as Polyfunctional Antioxidants. Bull. Exp. Biol. Med. 2016, 160, 340–342. 10.1007/s10517-016-3165-0. [DOI] [PubMed] [Google Scholar]
- Alcolea V.; Plano D.; Karelia D. N.; Palop J. A.; Amin S.; Sanmartin C.; Sharma A. K. Novel seleno- and thio-urea derivatives with potent in vitro activities against several cancer cell lines. Eur. J. Med. Chem. 2016, 113, 134–144. 10.1016/j.ejmech.2016.02.042. [DOI] [PubMed] [Google Scholar]
- Greene L. M.; O’Boyle N. M.; Nolan D. P.; Meegan M. J.; Zisterer D. M. The vascular targeting agent Combretastatin-A4 directly induces autophagy in adenocarcinoma-derived colon cancer cells. Biochem. Pharmacol. 2012, 84, 612–624. 10.1016/j.bcp.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Thapa P.; Karki R.; Thapa U.; Jahng Y.; Jung M. J.; Nam J. M.; Na Y.; Kwon Y.; Lee E. S. 2-Thienyl-4-furyl-6-aryl pyridine derivatives: synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure-activity relationship study. Bioorg. Med. Chem. 2010, 18, 377–386. 10.1016/j.bmc.2009.10.049. [DOI] [PubMed] [Google Scholar]
- Rouf A.; Tanyeli C. Bioactive thiazole and benzothiazole derivatives. Eur. J. Med. Chem. 2015, 97, 911–927. 10.1016/j.ejmech.2014.10.058. [DOI] [PubMed] [Google Scholar]
- Sambasiva Rao P.; Kurumurthy C.; Veeraswamy B.; Poornachandra Y.; Ganesh Kumar C.; Narsaiah B. Synthesis of novel 5-(3-alkylquinolin-2-yl)-3-aryl isoxazole derivatives and their cytotoxic activity. Bioorg. Med. Chem. Lett. 2014, 24, 1349–1351. 10.1016/j.bmcl.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Bajji A.; Tangallapally R.; Markovitz B.; Trovato R.; Shenderovich M.; Baichwal V.; Bartel P.; Cimbora D.; McKinnon R.; Robinson R.; Papac D.; Wettstein D.; Carlson R.; Yager K. M. Discovery of (2S)-1-[4-(2-{6-amino-8-[(6-bromo-1,3-benzodioxol-5-yl)sulfanyl]-9H-purin-9-yl}et hyl)piperidin-1-yl]-2-hydroxypropan-1-one (MPC-3100), a purine-based Hsp90 inhibitor. J. Med. Chem. 2012, 55, 7480–7501. 10.1021/jm3004619. [DOI] [PubMed] [Google Scholar]
- Afzal O.; Kumar S.; Haider M. R.; Ali M. R.; Kumar R.; Jaggi M.; Bawa S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. 10.1016/j.ejmech.2014.07.044. [DOI] [PubMed] [Google Scholar]
- Zhao Y. L.; Chen Y. L.; Chang F. S.; Tzeng C. C. Synthesis and cytotoxic evaluation of certain 4-anilino-2-phenylquinoline derivatives. Eur. J. Med. Chem. 2005, 40, 792–797. 10.1016/j.ejmech.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Diaz M.; Gonzalez R.; Plano D.; Palop J. A.; Sanmartin C.; Encio I. A diphenyldiselenide derivative induces autophagy via JNK in HTB-54 lung cancer cells. J. Cell Mol. Med. 2018, 22, 289–301. 10.1111/jcmm.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plano D.; Baquedano Y.; Moreno-Mateos D.; Font M.; Jimenez-Ruiz A.; Palop J. A.; Sanmartin C. Selenocyanates and diselenides: a new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011, 46, 3315–3323. 10.1016/j.ejmech.2011.04.054. [DOI] [PubMed] [Google Scholar]
- Wang L.; Guo X.; Wang J.; Jiang C.; Bosland M. C.; Lu J.; Deng Y. Methylseleninic Acid Superactivates p53-Senescence Cancer Progression Barrier in Prostate Lesions of Pten-Knockout Mouse. Cancer Prev. Res. 2016, 9, 35–42. 10.1158/1940-6207.CAPR-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi A. R.; Jo M. J.; Jung M. J.; Kim H. S.; Yoon S. Selenate specifically sensitizes drug-resistant cancer cells by increasing apoptosis via G2 phase cell cycle arrest without P-GP inhibition. Eur. J. Pharmacol. 2015, 764, 63–69. 10.1016/j.ejphar.2015.06.046. [DOI] [PubMed] [Google Scholar]
- Suzuki M.; Endo M.; Shinohara F.; Echigo S.; Rikiishi H. Rapamycin suppresses ROS-dependent apoptosis caused by selenomethionine in A549 lung carcinoma cells. Cancer Chemother. Pharmacol. 2011, 67, 1129–1136. 10.1007/s00280-010-1417-7. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Liu H.; Jiang C. C.; Fang L.; Chen C.; Zhang X. D.; Jiang Z. W. Connecting endoplasmic reticulum stress to autophagy through IRE1/JNK/beclin-1 in breast cancer cells. Int. J. Mol. Med. 2014, 34, 772–781. 10.3892/ijmm.2014.1822. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.