

Abstract
Farber disease (FD) is a debilitating lysosomal storage disorder (LSD) caused by a deficiency of acid ceramidase (ACDase) activity due to mutations in the gene ASAH1. Patients with ACDase deficiency may develop a spectrum of clinical phenotypes. Severe cases of FD are frequently associated with neurological involvement, failure to thrive, and respiratory complications. Mice homozygous (Asah1P361R/P361R) for an orthologous patient mutation in Asah1 recapitulate human FD. In this study, we show significant impairment in lung function, including low compliance and increased airway resistance in a mouse model of ACDase deficiency. Impaired lung mechanics in Farber mice resulted in decreased blood oxygenation and increased red blood cell production. Inflammatory cells were recruited to both perivascular and peribronchial areas of the lung. We observed large vacuolated foamy histiocytes that were full of storage material. An increase in vascular permeability led to protein leakage, edema, and impacted surfactant homeostasis in the lungs of Asah1P361R/P361R mice. Bronchial alveolar lavage fluid (BALF) extraction and analysis revealed accumulation of a highly turbid lipoprotein-like substance that was composed in part of surfactants, phospholipids, and ceramides. The phospholipid composition of BALF from Asah1P361R/P361R mice was severely altered, with an increase in both phosphatidylethanolamine (PE) and sphingomyelin (SM). Ceramides were also found at significantly higher levels in both BALF and lung tissue from Asah1P361R/P361R mice when compared with levels from wild-type animals. We demonstrate that a deficiency in ACDase leads to sphingolipid and phospholipid imbalance, chronic lung injury caused by significant inflammation, and increased vascular permeability, leading to impaired lung function.
INTRODUCTION
Farber disease (FD; OMIM no. 228000) is a rare lysosomal storage disorder (LSD) caused by an inherited deficiency in acid ceramidase (ACDase) activity, the enzyme that hydrolyzes the bioactive sphingolipid ceramide into sphingosine and a fatty acid. Case reports catalog ACDase deficiency as a spectrum disorder with varying symptoms. Patients with the classical form of FD die in early childhood (2, 11, 32). To date, there is no known cure for FD.
Manifestations of ACDase deficiency include growth of subcutaneous nodules that cause pain in the joints, development of a hoarse cry that progresses to aphonia, and severe neurological deficits. Other characteristics of this debilitating disorder may include hepatosplenomegaly and respiratory difficulties (32). Decreased lung function and increased respiratory infections are commonly described in patients with FD. Although there are no detailed studies addressing lung involvement per se, case reports have mentioned impairments such as atelectasis (44), lung consolidation, and calcification of nodules in lungs (59). The most reported cause of death is respiratory complications from bronchial pneumonia (6, 56, 68). Despite this, aside from superficial descriptions of pulmonary manifestations within these occasional case reports, there has not been a systemic analysis of the pathophysiology of lungs in FD.
Lipoproteinosis causes severe lung pathology in several other LSDs, such as Gaucher disease and Niemann-Pick disease types B, C1, and C2 (19, 46, 51–53). Corresponding mouse models have been instrumental in characterizing disease progression (10, 12, 26). We previously reported the first viable murine model of ACDase deficiency, wherein a human ASAH1 mutation [proline (P) 362 to arginine (R)] was “knocked in” to a conserved region of the murine Asah1 locus (P361R) (33). Mice homozygous (Asah1P361R/P361R) for this mutation develop a phenotype that mirrors symptoms reported in FD patients, including systemic accumulation of ceramides, shortened life spans, decreased weight, hepatosplenomegaly, accumulation of foamy macrophages in organs, elevation of inflammatory cytokines, and failure to thrive (3).
Ceramide accumulation is a hallmark of FD. Ceramides and their metabolites are vital constituents of membranes that regulate a diverse array of cellular responses and functions ranging from inflammation to apoptosis (22). Recently, several important studies have linked sphingolipid signaling with lung health and pathologies (58, 60, 65).
Here, we present a systematic examination of the lung pathology in the Asah1P361R/P361R mouse with the objective of obtaining better insights into the consequences of ACDase deficiency. Specifically, we present the signature of the various phospholipids and sphingolipids that accumulate in the lung due to ACDase deficiency. We also performed a combination of functional, biochemical, histological, ultrastructural, and lipidomic approaches within the lungs and bronchial alveolar lavage fluid (BALF). We report that ACDase deficiency leads to chronic lung injury characterized by inflammation, increased vascular permeability, and perturbed surfactant and phospholipid homeostasis, all culminating in impaired lung function.
MATERIALS AND METHODS
Animal breeding.
To generate homozygous Asah1P361R/P361R mice, we crossed heterozygotes (3). All animal procedures were approved by and carried out in strict accordance with the policies of the University Health Network Animal Care Committee and the Medical College of Wisconsin Biomedical Resource Center Advisory Committee.
Pulmonary function test.
Mice were anesthetized with an intraperitoneal injection of ketamine-xylazine (50 and 10 mg/kg) and underwent tracheostomy, followed by intubation with an 18-gauge cannula (BD Biosciences Canada, Mississauga, ON, Canada), which was then connected to the FlexiVent system (Scireq, Montreal, QC, Canada) for lung function measurements. Before measurement, the FlexiVent system was calibrated for mouse weight and sex. Mice were ventilated at 150 breaths/min, with a tidal volume of 10 ml/kg. The total lung capacity and snapshop (single frequency forced oscillation) maneuvers were applied. For the snapshot maneuver, measurements for resistance, elastance, and compliance were recorded with the forced oscillation technique that applied a stepwise incremental pressure from 2 to 30 cmH2O and an oscillatory waveform set at 150 breaths/min.
Blood gas, oximetry, and blood counts.
For blood gas measurements, mice were anesthetized with an intraperitoneal injection of ketamine-xylazine (50 and 10 mg/kg, respectively). A 20-gauge catheter was inserted into the trachea. Each mouse was then connected to the Servo-I mechanical ventilator (Maquet, Germany) for respiratory support with a peak inspiratory pressure of 10 cmH2O, positive end-expiratory pressure of 2 cmH2O, and a respiratory rate of 120 breaths/min. Following a 5-min acclimatization on the ventilator, the chest cavity was exposed and the left ventricle of the heart identified. Oxygenated blood was collected using a 25-gauge heparinized needle/syringe and immediately transferred to a Radio Medical ABL800 Flex (Radio Medical, Copenhagen, Denmark) blood gas analyzer. Partial pressure of O2 (), partial pressure of CO2 (), and blood pH were measured. Fraction of inspired O2 () of 21% was used to calculate the / ratio. The MouseOX Plus apparatus (Star Life Sciences, Oakmont, PA) was used to measure pulse, respiration rate, and arterial oxygen saturation (). Blood samples were taken from the saphenous vein of animals into EDTA-coated tubes. Complete blood counts were performed on a Hemavet 950 FS machine (Drew Scientific Group, Miami, FL).
Lung histopathology and immunohistochemistry.
Harvested mouse lungs were inflated with 10% phosphate-buffered formalin and then immersed in the same fixative solution for 24–48 h. The lungs were trimmed, and the lobes were separated and embedded in paraffin and then sectioned at 4 μm. Sections were stained with hematoxylin-eosin and Masson’s trichrome. For immunohistochemistry, the following primary and secondary antibodies and reagents were used: rat anti-mouse neutrophil (clone 7/4; Cedarlane, Burlington, ON, Canada), rat anti-mouse Mac-2 (Galectin-3 clone M3/38; Cedarlane), rabbit anti-Pro-SP-C (Sevenhills Bioreagents, Cincinnati, OH), biotinylated rabbit anti-rat (Vector, Burlingame, CA), biotinylated goat anti-rabbit IgG (Vector), avidin-biotin/horseradish peroxidase (HRP) (Vector), diaminobenzidine kit (Vector), and Vectastain ABC Elite Standard kit (Vector). For the primary antibodies, we relied on the testing of the specificity of staining as performed by the respective companies from whom those reagents were purchased. Our own assays included a control wherein secondary antibody was added alone; this measured background signal from the staining of nonspecific epitopes. Slides were assessed by a pathologist, and no signs of atelectasis or tissue deformation were observed. All histology slides were scanned on the Aperio AT2 apparatus (Leica Biosystems, Buffalo Grove, IL) and analyzed using Aperio ImageScope analysis software (Leica Biosystems).
Electron microscopy.
Posteuthanasia and lung inflation with 4% paraformaldehyde, lungs were immersed in the same fixative solution for 24–48 h. Lungs of each animal were sampled (4–6 individual samples) from the central (hilar) and peripheral (subpleural) areas. Each sample was postfixed in 3% glutaraldehyde. Samples were further washed and postfixed with 2% OsO4 in phosphate buffer. After dehydration, samples were then embedded in Durcupan Epon (Fluka, Hatfield, PA). Ultrathin sections (60 nm) were cut from selected tissue blocks using an Ultracut ultramicrotome (Reichert-Jung, Munich, Germany) and placed on copper grids (regular Cu 200 Mesh; SPI, West Chester, PA). Ultrathin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1400+ (JEOL, Tokyo, Japan) transmission electron microscope (TEM) equipped with Olympus Veleta CCD camera and Radius software.
Alveolar type II cell quantitation.
Morphometric analyses were performed on the right upper lobes. Lungs were prepared and inflated as previously mentioned; no signs of macroscopic atelectasis were observed. Lung sections were taken from randomly oriented blocks and then stained with an anti-Pro-SP-C antibody for immunohistochemistry detection. Scoring was performed by two independent observers who were blinded from group allocation. The dimension of each field scored was 250 × 450 µm. Quantification was performed on three tissue sections per animal; 10 random nonoverlapping images per section were scored. The individual section average was calculated from the cell counts of the 10 images, and then an average of the three sections was used to produce a count for each individual animal.
Western blots.
Lung samples were collected, lysed in RIPA lysis buffer (Thermo Scientific, Waltham, MA), and homogenized with a Dounce tissue homogenizer. Forty micrograms of protein was separated by 12% SDS-PAGE, transferred to PVDF membranes by wet transfer, and blocked overnight at 4°C with 5% nonfat dry milk. The following primary and secondary antibodies were used: anti-β-actin (1:1,000 dilution; Millipore, Billerica, MA), anti-surfactant protein (SP)-A (Santa Cruz Biotechnology, Dallas, TX; 1:300 dilution), anti-SP-B (1:500 dilution; Millipore), anti-SP-C (1:500; Santa Cruz Biotechnology), anti-SP-D (1:1,000 dilution; Abcam, Cambridge, MA), anti-rabbit IgG-HRP (Thermo Scientific), anti-mouse IgG-HRP (BD Biosciences Canada, Mississauga, ON, Canada), anti-goat-IgG-HRP (Santa Cruz Biotechnology), and anti-mouse IgG-HRP (GE Healthcare, Waukesha, WI). The secondary antibody was detected with ECL (Thermo Scientific Pierce, Waltham, MA) and autoradiography film.
BALF cytospins and differentials.
Mice were euthanized by CO2 inhalation. An 18-gauge catheter (BD Biosciences Canada) was inserted and tied into the trachea. The lungs were washed three times by gentle flushing with 0.9 ml of cold phosphate-buffered saline (PBS). BALF was collected into 1.5-ml tubes and centrifuged at 300 g for 10 min. The supernatant was removed and stored at −80°C. The cell pellet was resuspended in 300–500 µl of PBS and used for total cell counts, which were performed on trypan blue-stained samples on a Countess II FL (Life Technologies, Carlsbad, CA). The remainder of the resuspended cells were diluted to a total volume of 150 µl with PBS, and a cytospin was performed with the Shandon CytoSpin III Cytocentrifuge (Thermo Shandon, Waltham, MA) for 5 min at 800 rpm. For cell type quantification, cytospins of 2 × 105 cells/slide were prepared and stained with the Kwik-Diff kit (Thermo Scientific Pierce, Waltham, MA). Differential cell counts were calculated by averaging the cell counts in 10 nonoverlapping zones, where a total of 200 cells were scored in each zone. Cell counts were scored by a “blinded” observer.
BALF turbidity, cytokine analysis, and ELISA.
Turbidity was determined by measuring BALF supernatant on the NanoDrop One (Thermo Scientific, Wilmington, DE) apparatus at 600 nm. Cytokine levels in mouse BALF were analyzed with the Cytokine 20-Plex Mouse Panel (Thermo Scientific Pierce) as per the manufacturer’s instructions. Luminescence was quantified on the Luminex 100 instrument (Luminex, Austin, TX). Samples with a low bead count (<45 beads) were omitted. IgG, IgM, albumin (Bethyl Laboratories, Montgomery, TX), and surfactant proteins A, B, C, and D were measured in BALF using commercially-available ELISA kits for mouse samples (Cloud-Clone, Wuhan, China) as per the manufacturer’s instructions.
Vascular permeability and wet-to-dry ratio.
To test for vascular permeability, 150 µl of Evans Blue dye (EBD) solution (Thermo Scientific Pierce) in PBS (50 mg/kg) was injected into the tail vein of mice. Thirty minutes after injection, animals were euthanized by CO2 inhalation and perfused with PBS. Organs were extracted, weighed, and dried overnight at 55°C. Dried organs were incubated in 2 ml of formamide (Thermo Scientific Pierce) at 60°C for 24 h, followed by centrifugation at 5,000 g for 30 min. The supernatant was collected and the absorbance measured at 620 and 740 nm (Biotek ELx800, Winooski, VT). The following formula was used to adjust for the contaminating hemoglobin pigment: E620(EBD) = E620 − (1.426 × E740 + 0.030) (55). To calculate the wet-to-dry lung weight ratio, age-matched mice were used. Posteuthanization by CO2 inhalation, lungs were rinsed in PBS, weighed immediately, and then transferred to an oven at 60°C. Lung samples were reweighed after 4 days to obtain dry weights.
Phospholipid and sphingolipid analyses.
Lipids from BALF and postlavage lungs were extracted in CH2Cl2/MeOH (2% AA)/water (2.5:2.5:2, vol/vol/vol) in the presence of internal standards: ceramide d18:1/15:0 (16 ng), sphingomyelin (SM) d18:1/12:0 (16 ng), phosphatidylethanolamine (PE) 12:0/12:0 (180 ng), phosphatidylcholine (PC) 13:0/13:0 (16 ng), phosphatidylinositol (PI) 14:1/17:0 (30 ng), and phosphatidylserine (PS) 12:0/12:0 (156.25 ng) (Avanti Polar Lipids, Alabaster, AL) according to a procedure adapted from Bligh and Dyer (9). The relative quantifications of the phospholipid (PC, PE, PI, PS) and sphingolipid (ceramide and SM) species were obtained in the multiple reaction monitoring mode on a triple quadrupole liquid chromatrography mass spectrometer (Agilent 6460, Santa Clara, CA) equipped with a Kinetex HILIC column (50 × 4.6 mm, 2.6 μm; Phenomenex, Torrance, CA), as previously described (34, 54).
Statistical analyses.
Data are expressed as means ± SE. Statistical significance between Asah1+/+ and Asah1P361R/P361R groups was determined by Student’s t-test. Significant differences are expressed in the figures as P < 0.05, P < 0.01, and P < 0.001. No corrections for multiple comparisons were performed for this exploratory study.
RESULTS
Impaired lung mechanics and decreased blood oxygenation in Asah1P361R/P361R mice.
To assess respiratory function in Asah1P361R/P361R mice, lung mechanics were measured with the FlexiVent system. Lung volume measurement revealed a decrease in total lung capacity (Fig. 1A) in 8- to 9-wk-old Asah1P361R/P361R mice compared with age-matched Asah1+/+ mice, which is in line with the decreased animal size we reported previously (3). Within the single-frequency force oscillation program, lungs from Asah1P361R/P361R mice displayed increased airway resistance and increased elastance as well as decreased compliance (Fig. 1, B–D). Lung mechanics were also investigated in younger ACDase-deficient and control mice at 5 wk of age. No difference was seen in lung volume, possibly because the mice are relatively similar in size at this age, but an analogous impairment was observed in airway resistance, elastance, and compliance (Fig. 1, A–D).
Fig. 1.
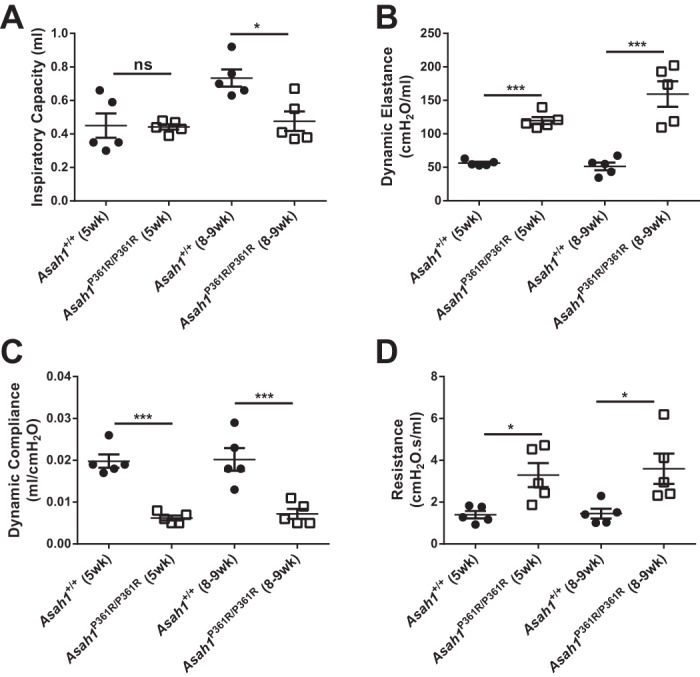
Impaired lung mechanics in the Asah1P361R/P361R lungs. Total inspiratory capacity (A), dynamic elastance (B), dynamic compliance (C), and resistance (D) were measured as described in materials and methods. All comparisons were made between Asah1+/+ and Asah1P361R/P361R mice (n = 5 mice for each genotype) at 5 and 8–9 wk of age. *P < 0.05; ***P < 0.001.
We then assessed whether the impairment in lung mechanics had any effect on blood oxygenation. Arterial blood gas measurements from 8- to 9-wk-old Asah1P361R/P361R mice revealed a lower , an increased , and noticeable decreases in the Po2/ ratio and were compared with Asah1+/+ mice (Fig. 2, A, B, and D). Arterial blood analysis also revealed a decrease in blood pH, suggesting possible respiratory or combined respiratory/metabolic acidosis (Fig. 2F). Mouse pulse oximetry measurements confirmed that Asah1P361R/P361R mice had significantly lower levels (Fig. 2C). In addition, Asah1P361R/P361R mice displayed both increased respiration and increased heart rates (Fig. 2, E and G). Previously, we reported that Asah1P361R/P361R mice had increased levels of red blood cells (RBCs) from complete blood counts (3). Here, we further characterized the RBCs and show that, in addition to developing an elevated cell count, RBCs from Asah1P361R/P361Rmice also demonstrated an increase in both hemoglobin and hematocrit values, a lower mean corpuscular volume (MCV), and no change in mean corpuscular hemoglobin concentration (MCHC) (Fig. 2, H–L). Taken together, these vital readings suggest that Asah1P361R/P361R mice demonstrate signs of chronic hypoxemia/hypercapnia and polycythemia.
Fig. 2.

Impaired blood oxygenation and polycythemia in the Asah1P361R/P361R mice. Arterial blood gas measurements: arterial blood oxygen partial pressure (; A), arterial partial pressure of carbon dioxide (; B), the ratio of arterial oxygen partial pressure to fractional inspired oxygen (D), and blood pH measurements (F); n = 5 mice for each genotype. Pulse oximeter measurements: arterial oxygen saturation () percentage (C), respiratory rate (E), and heart rate (G); n = 5 each. Erythrocyte indices from hematology analysis: red blood cell (RBC) counts (H), hemoglobin (Hb) levels (I), hematocrit (J), mean corpuscular volume (MCV; K), and mean corpuscular hemoglobin concentration (MCHC; L); n = 10 mice for each genotype. All comparisons were made between 8- and 9-wk-old Asah1+/+ and Asah1P361R/P361R mice. *P < 0.05; **P < 0.01; ***P < 0.001.
Cellular infiltration and storage pathology in the ACDase-deficient lungs.
Hematoxylin-eosin staining of 8- to 9-wk-old Asah1P361R/P361R inflated mouse lungs revealed signs of inflammation. There was an increased presence of both alveolar (Fig. 3B) and interstitially localized (Fig. 3C) macrophages with foamy cytoplasm, the latter being abundant in perivascular and peribronchial tissue as well as in the terminal alveolar septa (Fig. 3, B and C). Importantly, these cells were found both singly and in aggregated clusters. Lungs from 8- to 9-wk-old Asah1P361R/P361R mice also contained substantial amounts of eosinophilic cellular debris scattered throughout the alveolar space. To further characterize the extent of inflammation, immunohistochemistry (IHC) with antibodies specific for Mac-2 (galectin-3) and a mouse neutrophil marker was performed. The accumulations of foamy macrophages were greatly highlighted (Fig. 3, E, G, and H). Macrophages were variable in size, and on occasion we could detect large foamy multinucleated cells. Similarly, IHC staining for a neutrophil marker showed that recruitment of neutrophils is widely abundant through the lung parenchyma of Asah1P361R/P361R mice (Fig. 3J).
Fig. 3.

Acid ceramidase (ACDase)-deficient lungs show immune infiltration. Light micrographs of 8- to 9-wk-old Asah1+/+ and Asah1P361R/P361R mouse lung sections stained with hematoxylin-eosin (A–C); large numbers of large foamy storage laden macrophages (alveolar and interstitial) are found in the Asah1P361R/P361R mouse. Immunohistochemistry (IHC) for MAC-2 (D–H) and a neutrophil marker (I and J) show significant infiltration by the foamy macrophages and increased presence of neutrophils in the mutants. The interstitial foamy macrophages can be found individually or clustered in the alveolar septa (C, E, G, H, and J) and peribronchial/perivascular tissue (E). Black arrowheads, foamy interstitial macrophages; open arrowheads, foamy alveolar macrophages; black arrows, cells with segmented nuclei/neutrophil granulocytes; open arrows, abnormal alveolar debris. *Bronchial lumen; **lumen of the hilar vessel. Scale bars, 100 (A, D, and E); 25 (B, C, G, and H), and 50 μm (F, I, and J).
Ultrastructure analyses were performed on both 8- to 9-wk-old Asah1+/+ and Asah1P361R/P361R mouse lungs (Fig. 4). Extensive storage products were found in interstitial as well as within resident alveolar macrophages in 8- to 9-wk-old Asah1P361R/P361R mice lungs. Ultrastructural pathology of these cells was hallmarked by extensive cytoplasmic vacuolization (Fig. 4B). The limiting membranes of many of these vacuoles were fusing. Importantly, curved-linear tubular profiles (Farber bodies; Fig. 4C) represented the almost exclusive contents of these storage vacuoles. On the contrary, storage vacuoles observed in alveolar macrophages were structurally more pleiomorphic, and many of them adopted the lamellar “zebra body” appearance (Fig. 4E). On occasion, the alveolar storage-laden macrophages displayed degenerative cytoplasmic changes and loss of cytoplasmic membrane integrity (Fig. 4F). Extracellular alveolar space contained excessive amounts of biological debris that was formed by membranous lamellar whorls and condensed Farber body-like/fingerprint-like profiles (Fig. 4, H and I). In addition to macrophages, subcellular storage pathology could be found in pulmonary endothelial cells (Fig. 5, B and C) and ciliary cells of the respiratory epithelium (Fig. 5, E and F). Finally, we observed subtle storage abnormalities in the microvillous epithelial and Clara cells (data not shown).
Fig. 4.

Ultrastructural pathology in macrophages and contents of alveolar space in Asah1P361R/P361R mice. Electron micrographs of 8- to 9-wk-old Asah1+/+ (A, D, and G) and Asah1P361R/P361R mouse lungs. B: features include macrophages in the alveolar septa (nucleus marked by a white asterisk). The macrophages that contain excess cytoplasmic vacuolar storage bodies are labeled with open arrowheads. Black arrows demark alveolar type II cells. C: storage bodies of the interstitial macrophages contain curved linear tubular profiles (Farber bodies in C; detail corresponding to the box in B). E: an alveolar macrophage laden with pleomorphic storage vacuoles [white asterisk, nucleus; black arrowheads, Zebra bodies; open arrowheads, vacuoles containing Farber bodies; compare with Asah1+/+ alveolar macrophages (white asterisks) in A and D]. White arrows highlight erythrocytes in the alveolar septal capillary. F: discontinuity and rupture of the cytoplasmic membrane of the alveolar macrophage (black arrows in F; detail corresponding to the box in E). H: the abnormal extracellular alveolar content that is formed partly by concentric membranous whorls (black arrowheads) and Farber/finger print-like profiles (open arrowheads). The latter pattern is shown in detail in I (and corresponds to the box in H). Compare with limited amounts of concentric lamellar structures (black arrowhead in D) or completely clear alveolar spaces in Asah1+/+ mice (G). Scale bars, 5 (A, B, D, and E) and 3 μm (G and H).
Fig. 5.

Ultrastructural pathology of pulmonary endothelial cells and respiratory epithelial cells in the Asah1P361R/P361R mice. Electron micrographs of 8- to 9-wk-old Asah1P361R/P361R mouse lungs featuring endothelial cells in pulmonary capillaries containing storage vacuoles (open arrowheads) with curved linear tubular profiles (Farber bodies; B and C; detail corresponding to the box in B). Lamellar “zebra” storage bodies can also be identified within ciliated cells of the respiratory epithelium (white arrowheads; E and F; detail corresponding to the rectangle in E). Compare the findings in the Asah1P361R/P361R mice to Asah1+/+ mice (A and D). Scale bars, 1.2 (A and B) and 2 μm (D and E).
Absence of lung fibrosis but presence of lung edema in Asah1P361R/P361R mice.
Slides of hematoxylin-eosin-stained lung tissue from Asah1P361R/P361R mice did not show significant perturbation (e.g., large areas of atelectasis, excessive emphysema, and/or widespread alveolar edema) in tissue architecture (Fig. 3, B and C). Trichrome staining was performed on lung sections to rule our fibrosis. Although Asah1P361R/P361R mice lungs did not appear to show fibrosis and collagen accumulation, we did notice that the cellular debris within the parenchyma stained positive for collagen (Fig. 6B). This nonspecific staining maybe caused by the released products of the large macrophages. Because the lung contains increased cell infiltration and cellular debris, lung tissue weights were performed. Measurement of both the normalized wet and dry lung weights from Asah1P361R/P361R mice revealed an increased tissue weight. This may indicate pulmonary remodeling or increased cellularity (Fig. 6, C and D). Interestingly the wet-to-dry ratio as normalized to body weight was higher in lungs from Asah1P361R/P361R mice, indicating the likely presence of lung edema (Fig. 6E).
Fig. 6.

Asah1P361R/P361R mice lungs do not express fibrosis but develop interstitial edema. Light micrographs of 8- to 9-wk-old Asah1+/+ and Asah1P361R/P361R mouse lung sections were stained with trichrome [A and B; alveolar storage macrophages (white arrowheads), interstitial storage macrophages (black arrowheads), and alveolar debris (black arrows)]; scale bars, 100 μm. Lung wet and dry weights were normalized to body weight. Wet lung/body weight percentage (C), dry lung/body weight percentage (D), and wet-to-dry lung ratio were normalized to body weight (E); n = 10 mice at 8–9 wk of age for each genotype. ***P < 0.001.
Elevation of leukocytes in lungs of Asah1P361R/P361R mice.
To quantitate and characterize the immune cell infiltration in the Asah1P361R/P361R mouse lungs, cells collected from bronchoalveolar lavage fluids (BALF) were stained with the Kwik-Diff reagent for cytology. Analysis of the slides revealed significantly more inflammatory cells in the lungs of Asah1P361R/P361R mice (Fig. 7, A and B). Proteinaceous material was also widely present throughout the background of the slide. Evaluation of blood cell types revealed that Asah1P361R/P361R mice had significantly higher quantities of all infiltrating immune cells in BALF samples (Fig. 7, C–F). In addition, the percentage of multinucleated macrophages within the total macrophage pool in BALF from Asah1P361R/P361R mice was severalfold higher than that in their Asah1+/+ counterparts (Fig. 7G).
Fig. 7.

Immune cell infiltration in bronchoalveolar lavage fluid (BALF) of Asah1P361R/P361R mice. Light micrograph of BALF cells; scale bars, 100 µm at equal 1:2 dilutions (A and B). Cell counts and differentials from Kwik-Diff stained cytospin slides in BALF samples show BALF cell count (C), macrophages (D), neutrophils (E), lymphocytes (F), and percentage of multinucleated macrophages of total macrophages count (G). All comparisons were made between 8- and 9-wk-old Asah1+/+ and Asah1P361R/P361R mice; n = 5 mice for each genotype. ***P < 0.001.
Vascular permeability in multiple organs.
The consistency of the BALF collected from the 8–9-wk-old Asah1P361R/P361R mice lungs was milky and opaque (Fig. 8A). Analysis of that BALF revealed markedly increased turbidity (Fig. 8B), accumulation of albumin, and the presence of IgG and IgM antibodies (Fig. 8C–E), which are indicative of protein leakage. Compared with age-matched Asah1+/+ mice, the lungs, heart, thymus, liver, and spleen of Asah1P361R/P361R mice demonstrated increased vascular permeability as measured by parenchymal dye accumulation following Evans Blue injections (Fig. 8F, G).
Fig. 8.

Vascular permeability leads to protein leakage in Asah1P361R/P361R mouse lungs. BALF was characterized showing opaque samples from Asah1P361R/P361R vs. Asah1+/+ (A) and turbidity as measured by absorbance at 600 nm (B); n = 8 from 8- to 9-wk-old mice for each genotype. Levels of albumin (C), IgG antibodies (D), and IgM antibodies (E) collected from BALF were determined by ELISA from 8- to 9-wk-old mice; n = 5 mice for each genotype. Evans Blue (EB) dye injected via tail vein revealed dye accumulation (F and G) in the lungs and other organs in 8- to 9-wk-old Asah1+/+ and Asah1P361R/P361R mice; n = 5 mice for each genotype. *P < 0.05; **P < 0.01; ***P < 0.001.
Increased cytokine production in BALF.
Systemic inflammation is a characteristic of FD. A multiplex cytokine panel assay was performed on BALF from 8- to 9-wk-old mice. An increase in monocyte chemoattractant protein-1 (MCP-1) concentration was seen in BALF from Asah1P361R/P361R mouse lungs (Fig. 9A), similar to the MCP-1 elevation we reported previously in the plasma of Asah1P361R/P361R mice and FD patients (13). IL-4, VEGF, and TNFα concentrations were also increased (Fig. 9, B–D). No significant change was found in macrophage inflammatory protein-1α (MIP-1α) or GM-CSF levels (Fig. 9, E and F). No changes were observed for bFGF, IL-1β, IL-10, IL-13, IL-6, IL-17, IL-5, IGNγ, IL-2, IP-10, MIG, or KC (data not shown). No data were obtained for the IL-20 and IL-1α samples due to low bead counts (data not shown).
Fig. 9.
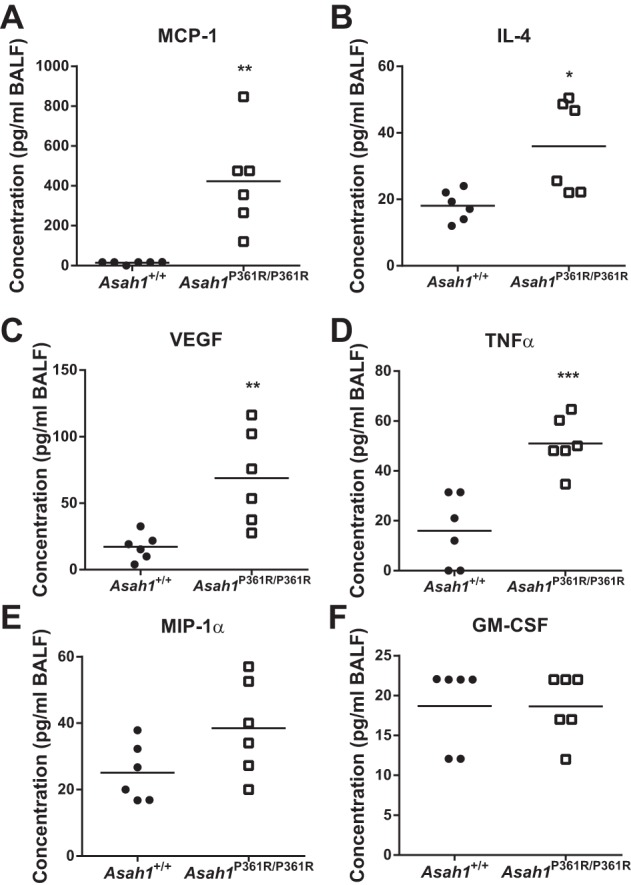
Cytokine elevation in the BALF of Asah1P361R/P361R mice. Cytokines were measured in BALF samples of 8- to 9-wk-old animals using a multiplex assay. Levels of CCL2 [monocyte chemoattractant protein-1 (MCP-1); A], interleukin 4 (IL-4; B), vascular endothelial growth factor (VEGF; C), tumor necrosis factor-α (TNFα; D), CCL3 [macrophage inflammatory protein-1α (MIP-1α); E], and granulocyte macrophage colony-stimulating factor (GM-CSF; F) were measured; n = 6 mice for each genotype. *P < 0.05; **P < 0.01; ***P < 0.001.
Abnormal lamellar body formation and increased expression of surfactant proteins.
In contrast to cell types listed earlier, ultrastructural analyses of alveolar type II cells in Asah1P361R/P361R mice did not show any apparent storage bodies within the cytoplasm (Fig. 10B). Unexpectedly though, we observed that the surfactant-containing lamellar bodies that were readily detected in Asah1+/+ mice (Fig. 10A) showed partial condensed contents in Asah1P361R/P361R mice (Fig. 10B). To access expression of surfactants, we performed IHC staining for prosurfactant C (pro-SP-C) on lung sections from 8- to 9-wk-old Asah1P361R/P361R mice compared with Asah1+/+ mice (Fig. 10, D and E). The staining which marks alveolar type II cells also showed positive staining in patches of alveolar cellular debris and within the foamy alveolar macrophages (Fig. 10E). Quantitation of alveolar type II cells from 8- to 9-wk-old pro-SP-C stained slides of Asah1+/+ and Asah1P361R/P361R mice showed no differences (Fig. 10C). Western blot analyses of lung tissue lysates confirmed the increase in SP-A levels; of note is that a second unknown band of lower molecular weight was also demarcated (Fig. 10F). SP-C and SP-D levels were increased, whereas SP-B was present at normal levels when compared with samples from Asah1+/+ lungs (Fig. 10F). Because surfactants are also secreted into the lung airspaces, we further measured surfactant proteins in BALF via ELISA. In BALF, the content of all surfactant proteins was higher in samples from Asah1P361R/P361R mice than from Asah1+/+ controls (Fig. 10, G–J). This observed increase in surfactant protein may be a compensatory mechanism caused by increased production and/or decreased degradation in the Asah1P361R/P361R mice as a result of their chronic lung injury.
Fig. 10.

Lamellar body formation and accumulation of surfactant proteins in Asah1P361R/P361R mice. A: electron micrograph of an alveolar type II cell in Asah1+/+ lungs containing normal, formed lamellar bodies (black arrowheads). B: electron micrograph of an alveolar type II cell in Asah1P361R/P361R lung suggesting an altered phenotype of the surfactant-containing granules. Contents are largely condensed into electron-dense clumps (black arrowheads). C: quantitation of alveolar type II cells from IHC staining for pro-surfactant protein-C (pro-SP-C) in Asah1+/+ and Asah1P361R/P361R mice; n = 6 mice for each genotype. D and E: micrographs of IHC for pro-SP-C in Asah1+/+ [alveolar type II cells (black arrowheads); D] and Asah1P361R/P361R mice [alveolar type II cells (black arrowheads) and alveolar storage macrophages (white arrowheads); E] at 8–9 wk of age. F: Western blots from lung tissue lysate show levels of SP-A, SP-B, SP-C, SP-D, and β-actin in samples from Asah1+/+ and Asah1P361R/P361R mice. G–J: ELISA was performed on BALF samples from 8- to 9-wk-old mice for SP-A (G), SP-B (H), SP-C (I), and SP-D (J) from Asah1+/+ and Asah1P361R/P361R samples; n = 6–8 mice for each genotype. *P < 0.05; ***P < 0.001. Scale bars, 2 (A), 1.2 (B), and 50 μm (D and E).
Accumulation and disruption of phospholipid composition.
Total phospholipids were analyzed by mass spectrometry in BALF samples obtained from 8- to 9-wk-old mice. There were shifts in the phospholipid composition of BALF from Asah1P361R/P361R mice compared with Asah1+/+ mice. SM and PE were increased in abundance in BALF obtained from Asah1P361R/P361R mice, whereas PC levels were decreased (Fig. 11A). Although PC decreased proportionally as a percent of total phospholipids, the total amount of measurable PC was still significantly increased (Fig. 11, B and D). This includes an increase in saturated PC (Sat-PC), the major phospholipid components in pulmonary surfactant (Fig. 11C). Significant increases were also detected in all lipid species of PI, PE, and PS in Asah1P361R/P361R BALF samples (Fig. 11, E–G). When we analyzed the postlavage lungs themselves, only the PC was increased; there was no change in PE, PS, or PI (Fig. 11, H–K). This observation suggests that much of the accumulation in BALF is from secreted lipids that are released into the air space.
Fig. 11.

Impaired phospholipid homeostasis in BALF from Asah1P361R/P361R mice as measured by mass spectrometry. BALF phospholipid composition (A), total fold change of various phospholipids in BALF (B), total saturated PC (Sat-PC) in BALF (C), phosphatidylcholine (PC) species in BALF (D), phosphatidylinositol (PI) species in in BALF species (E), phosphatidylethanolamine (PE) species in BALF (F), and phosphatidylserine (PS) species in BALF (G). Postlavage lung tissue phospholipid profiles as measured by mass spectrometry. Molecular species of PC (H), PI (I), PE (J), and PS (K) in lung lysates. Lipid concentrations were calculated as ng/mg protein of BALF or lung tissue. All samples were obtained from 8- to 9-wk-old mice; n = 6 mice for each genotype. *P < 0.05; **P < 0.01; ***P < 0.001.
Ceramide and sphingomyelin accumulation in BALF and lungs from Farber mice.
We have previously reported that ceramides are increased in various organs in Asah1P361R/P361R mice (3). Here, we quantified ceramides in both BALF and the postlavage lung tissue. In BALF, all ceramide species were significantly increased in Asah1P361R/P361R mice compared with Asah1+/+. Total ceramides were increased ∼300-fold, with the most prominently elevated species being C24:1, followed by C16:0 (Fig. 12A). In addition, the relative abundance of some individual ceramide species in BALF was altered in that the C24:1 increased, whereas C22:0 and C24:0 decreased (Fig. 12B). The finding of increased ceramide species was the same in lung tissue proper. There, however, total ceramides were increased only 12-fold, and no changes were seen in the relative abundances. The fact that we can detect a significantly higher-fold difference of ceramides within the BALF may suggest that the Asah1P361R/P361R lungs are leaking out ceramides due to increased vascular permeability (Fig. 12, C and D).
Fig. 12.

Significant accumulation of both ceramides and sphingomyelin (SM) in BALF and lungs from Asah1P361R/P361R mice. Ceramide species in BALF (A), relative abundance of ceramide species in BALF (B), ceramide species in postlavage lung tissue (C), relative abundance of ceramide species in postlavage lung tissue (D), SM species in BALF (E), relative abundance of SM species in BALF (F), SM species in postlavage lung tissue (G), and relative abundance of SM species in postlavage lung tissue as measured by mass spectrometry (H). Lipid concentrations were calculated as ng/mg protein of BALF or lung tissue. All samples were obtained from 8- to 9-wk-old mice; n = 6 mice for each genotype. *P < 0.05; **P < 0.01; ***P < 0.001.
When we analyzed the SM content, all species were found to be accumulated in the Asah1P361R/P361R mouse BALF. We detected a 25-fold increase of total SM species in Asah1P361R/P361R mice BALF when compared with Asah1+/+ (Fig. 12E). This contrasted with the results from the postlavage tissue, where only one species, SM 16:1, was significantly increased (Fig. 12G). Changes in the relative SM pattern were similar to those seen in the ceramide pattern, wherein BALF from Asah1P361R/P361R mice had significant changes in composition abundance but postlavage tissue samples did not (Fig. 12, F and H).
DISCUSSION
Deficiencies in ACDase result in systemic accumulation of ceramide. Patient symptoms may manifest along a spectrum with the cardinal phenotypes, including the presence of subcutaneous nodules, joint pain, and a hoarse voice (32). Respiratory complications have been documented in several case reports of Farber disease; nodular opacities in X-rays have been seen, and the terminal respiratory distress has resulted in early morbidity (16, 38). Here, we provide a comprehensive analysis of the lung pathology present in the ACDase-deficient mouse. We demonstrate that a deficiency in ACDase leads to chronic lung injury and impaired pulmonary function. Importantly, we also provide insights that could underlie such modifications. These alterations are likely a result of a several factors. A heightened inflammatory state that led to significant pulmonary inflammation resulted in an increase in vascular permeability, a buildup of storage material, and impaired surfactant homeostasis. Lung mechanic tests on Asah1P361R/P361R mice showed significantly decreased values in lung compliance and an increase in airway resistance.
Of the few Farber case reports that mention lung involvement, most only describe the presence of infiltrates and the development of infections at the end of life (14). Here, we provide the first evidence that ACDase deficiency leads to a wider scope of lung defects, thus expanding the role of respiratory involvement in FD.
Light and electron microscopy analyses showed an inflammatory phenotype similar to other tissues present in Asah1P361R/P361R mice (3, 53). The pulmonary pathology was dominated by the presence of the abnormal storage-laden macrophages. These cells populated individually and in clustered aggregates in the interstitial peribronchial and perivascular tissue as well as in the alveolar septa. This observation mirrors those found in early cases of FD, where patients were reported to have pervasive infiltrating foam cells in the lung and lipid laden macrophages from BALF samples (4, 15, 67). Alveolar macrophages, which are important surfactant scavengers, were also severely affected by the storage pathology. The histopathological, immunohistochemical, and ultrastructural findings we observed suggest that some of these abnormal alveolar macrophages degenerate, and their released contents contribute to the substrate burden for other surrounding cells involved in phagocytosis. This released content may in part contribute to the proteinaceous and highly turbid material that was present throughout the air spaces and in BALF samples. Increased immune cell recruitment and lipoprotein accumulation is a recurring observation in animal models of sphingolipid enzyme deficiencies and LSDs. For example, the ceramide synthase 2-deficient mouse, the sphingosine-1-phosphate lyase knockout mouse, and the acid sphingomyelinase-deficient mouse all have increased alveolar macrophages and/or the presence of protein-like material within the alveolar space or in BALF samples (12, 40, 61). This similarity in phenotype suggests that impairment in sphingolipid-metabolizing enzymes may lead to comparable pathology because their metabolism is tightly interconnected. It also demonstrates that homeostasis of these bioactive lipids is vital for proper respiratory function.
The inflammation and protein accumulation in the lungs of Asah1P361R/P361R mice may be explained in part by increases in the levels of cytokines such as MCP-1. We previously demonstrated that this cytokine is elevated in the plasma of Farber patients and that treatment by hematopoietic stem cell transplantation normalizes MCP-1 levels (13). In addition to its role in the recruitment of monocytes, there is evidence that during lung injury MCP-1 also mediates the recruitment of neutrophils into the lung (5). TNFα and VEGF, which are both elevated in BALF from Asah1P361R/P361R mice, are also implicated in lung inflammation, edema, and promotion of vascular permeability (36, 37, 64). Although the cytokines increased in BALF from Asah1P361R/P361R mice may cause lung complications, this may actually be a secondary consequence of the accumulation of ceramide.
Accumulation of albumin, IgG, and IgM in Asah1P361R/P361R BALF demonstrates impairment of lung vasculature barriers. Sphingolipids have been shown to alter pulmonary vascular permeability (31). For example, sphingosine-1-phosphate (S1P) enhances barrier function by its interaction with the G protein-coupled S1P1 receptor and subsequent downstream activation of a small Rho GTPase (62). Ceramide, however, has been shown to function oppositely by increasing vascular permeability via the regulation of endothelial Ca2+ signaling and the formation of nitric oxide (NO) (66). The relationship between ceramide and edema formation has been demonstrated in an in vivo experiment wherein ceramide was directly instilled in rat lungs (43). Another study demonstrated that lung edema caused by platelet-activating factor (PAF) is mediated by ASMase and ceramide (18). Compared with control mice, lung edema was attenuated in ASMase-deficient mice when they were treated with PAF (18). In the same study, direct perfusion of C2 ceramide into isolated Asah1+/+ rat lungs resulted in interstitial edema formation (18). Furthermore, when control mice were treated with PAF and ceramide-specific antibodies, the resulting lung edema was less severe than when the animals were treated only with PAF (18). These studies allude to sphingomyelin-derived ceramide as the key source of ceramides that affect barrier function. Our results coincide with these observations, as we also report edema from our wet-to-dry ratio measurements along with an increase in all ceramide species from our MS analyses. Taken together with the EB dye accumulation we measured in the lungs, vascular permeability and edema are contributing factors to the impaired lung function we reported.
The turbid BALF observed in Asah1P361R/P361R mice is likely a result of massive protein extravasation into the air space resulting in a pulmonary alveolar proteinosis (PAP)-like phenotype. PAP is characterized by compromised macrophage function due to a deficiency in GM-CSF that leads to impaired surfactant catabolism (50). Here, both Asah1P361R/P361R and Asah1+/+ mice had similar levels of GM-CSF in their BALF, and macrophages from Asah1P361R/P361R mouse lungs stained positive for pro-surfactant protein-C, suggesting active surfactant catabolism. Interestingly, BALF from Asah1P361R/P361R mice were seen to have elevations in IL-4 and MCP-1, which has been noted in rare cases of PAP (27, 28). Together, this suggests that Asah1P361R/P361R mice have an atypical presentation of PAP.
When we characterized the BALF and tissue lysates in Asah1P361R/P361R mice, we detected increases in all surfactant proteins and Sat-PC. In addition, although IHC staining for pro-SP-C was significantly increased in the Asah1P361R/P361R mice, there was no increase in the number of marked alveolar type II cells. Rather, the increased pro-SP-C signal was due to staining within the foamy macrophages and extracellular debris. Together, this suggests that Asah1P361R/P361R mice have increased surfactant production and release. Unexpectedly, from the TEM observations, the alveolar type II cells in the Asah1P361R/P361R mice exhibited condensed (non-lamellar) contents in the secretory bodies. Although further work is required, this could suggest an abnormality in the formation of surfactant and/or differences in the lipid composition of the lamellar bodies. The secretory lamellar bodies within alveolar type II cells are a subclass of lysosome-related organelles (63). Due to related functions between lysosomes and secretory bodies, it is possible that ACDase deficiency may contribute to the abnormal secretory function of alveolar type II cells, resulting in the irregular lamellar body phenotype. Furthermore, there have been reports demonstrating that surfactant surface tension can be perturbed in the presence of hemoglobin, albumin, and other plasma proteins (24, 25, 29, 48, 49). In addition, ceramides themselves may be able to impede surfactant function (43). This relationship in our Asah1P361R/P361R mice has not been determined and will require future study.
From our lipidomic analyses of sphingolipids and phospholipids, we found that BALF samples contained significantly increased levels of the measured compounds compared with the tissue proper. This finding, added with the observation of degeneration of some alveolar macrophages, suggests that there may be inefficient macrophage scavenging activity, further aggravating the imbalance in surfactant homeostasis in Asah1P361R/P361R mice.
The phospholipid composition analyses of tissues from the Asah1P361R/P361R mice demonstrated a markedly altered relative abundance of each phospholipid type. Asah1P361R/P361R mice were observed to have a reduction in the percentage of PC, coinciding with an increase in SM and PE phospholipids. Interestingly, other LSDs, such as Niemann-Pick types A, B, C1, and C2, as well as Sandhoff disease, display a combination of either dysfunction in surfactant homeostasis and/or changes to phospholipid abundance, indicating the importance of tight sphingolipid regulation in this context (8, 10, 26).
The change in phospholipid profiles and impaired surfactant homeostasis appears to be a generalized characteristic of lung injury. One such example is in patients who develop acute respiratory distress syndrome (ARDS). BALF from patients with this critical illness have been shown to have a decrease in PC and an associated increase in PI and SM (20). Furthermore, the BALF from ARDS patients has been shown to acquire an elevated SM-to-PC ratio (21), and glycolipids such as lactosyl-ceramides accumulate in the airspaces (42).
Ceramide, a central signaling sphingolipid, is essential for maintenance of lung health. Various respiratory diseases have been shown to manifest when the sphingolipid balance is perturbed (17, 39). Increases in ceramide due to various stimuli can lead to downstream effects such as lung cell death in the case of exposure to cigarette smoke and COPD. Also, the use of sphingosine analogs to reduce ceramide levels may provide an antimicrobial effect in models of cystic fibrosis (41). These reports demonstrate that ceramide production in the lung can be triggered acutely by a variety of stimuli. In the case of chronic ceramide accumulation such as in FD, alternative pathways may be activated in response to the sphingolipid buildup. It is clear that more work will be required to fully understand the role of ceramide signaling in FD and lung pathobiology.
In the Asah1P361R/P361R mouse, ceramide accumulation is chronic and likely progressive. The mouse has a drastic reduction in lifespan and recapitulates many features of human FD (3). The lung involvement brought on by significant inflammation and accumulation of lipoprotein and surfactant within the respiratory system suggests that impaired oxygenation and chronic lung injury are potential contributors to mortality. Asah1P361R/P361R mice typically die between 8 and 10 wk of age, so our analyses represent phenotypes seen near the end of life (3). The precise mechanisms of how the various species of ceramide act at the molecular level within the lungs to cause the spectrum of phenotypes observed remain to be uncovered. Within the human context, respiratory involvement is most prevalent in the severe forms of FD (1, 15, 44). The main phenotypes documented in patients are the infiltration of foamy cells and proteinaceous material in the lungs (15, 35, 57). Some cases have also noted the presence of respiratory stridor, autopsy descriptions of poorly expanded lungs, and atelectasis from radiology (7, 15, 38, 45). Although we don’t report the presence of lung atelectasis in the Asah1P361R/P361R mice, the increased airway resistance and low compliance may be consequences of the aforementioned human observations. From our study, several parallels may be drawn from the mouse and human. However, due to the wide clinical spectrum of FD, these results should be taken with caution, as there may also be important species differences. For example, one case revealed that out of six patients who had attenuated forms of FD, only three were reported to have lung involvement (16). Nonetheless, our results do offer clinical insight since respiratory failure and pneumonia are contributors to mortality in classical FD (15, 16, 30). This susceptibility is likely caused for a few reasons: immune dysfunction, poor clinical state, and possibility impaired bronchial clearance. These reasons, along with the expanded lung phenotype seen in the ACDase, mouse warrant the need to perform pulmonary tests in FD patients as part of a natural history study. Furthermore, from a therapeutic perspective, our study indicates that there might be merit in evaluating whether treatment with partial lung lavage, commonly performed on patients with PAP, may help manage the pulmonary aspects of this disorder (50). In addition, with the advent of recombinant enzyme therapy, use of aerosolized ACDase (23, 47) in conjunction with other treatments may help to manage respiratory symptoms and assist in treatment of patients with FD.
GRANTS
The electron microscopic analyses were supported by the Operační Program Praha - Konkurenceschopnost (CZ.2.16/3.1.00/24509) and NCMG LM2015091 projects and by the Charles University Institutional Research and Development schemes PRVOUK P24 and PROGRESS Q26. These studies were also supported by National Institutes of Health Grant 1R21-NS-078191-01A1 (to J. A. Medin) and by the Midwest Athletes Against Childhood Cancer Fund Professorship (to J. A. Medin) from Medical College of Wisconsin.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.P.Y., M.L., H.Z., and J.A.M. conceived and designed research; F.P.Y., D.I., J.S., S.D., J.G., L.L.-V., W.M.K., and T.L. performed experiments; F.P.Y., J.S., J.G., and J.A.M. analyzed data; F.P.Y., D.I., J.S., J.G., M.L., W.M.K., T.L., H.Z., and J.A.M. interpreted results of experiments; F.P.Y., J.S., S.D., and J.G. prepared figures; F.P.Y. and J.S. drafted manuscript; F.P.Y., D.I., J.S., S.D., L.L.-V., M.L., W.M.K., T.L., H.Z., and J.A.M. edited and revised manuscript; F.P.Y. and J.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the staff at the Centre for Phenogenomics, the Centre for Modeling Human Diseases Pathology core in Toronto, and the Children’s Hospital of Wisconsin Children’s Research Institute Histology Core for technical assistance with histology and immunohistochemistry services. We also thank the Advanced Optical Microscopy Facility at the University Health Network for providing whole slide scanning for pathology analysis. We acknowledge Irena Knesplova for technical assistance.
REFERENCES
- 1.Abul-Haj SK, Martz DG, Douglas WF, Geppert LJ. Farber’s disease. Report of a case with observations on its histogenesis and notes on the nature of the stored material. J Pediatr 61: 221–232, 1962. doi: 10.1016/S0022-3476(62)80257-1. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad A, Mazhar AU, Anwar M. Farber disease: a rare neurodegenerative disorder. J Coll Physicians Surg Pak 19: 67–68, 2009. 01.2009/JCPSP.6768. [PubMed] [Google Scholar]
- 3.Alayoubi AM, Wang JC, Au BC, Carpentier S, Garcia V, Dworski S, El-Ghamrasni S, Kirouac KN, Exertier MJ, Xiong ZJ, Privé GG, Simonaro CM, Casas J, Fabrias G, Schuchman EH, Turner PV, Hakem R, Levade T, Medin JA. Systemic ceramide accumulation leads to severe and varied pathological consequences. EMBO Mol Med 5: 827–842, 2013. doi: 10.1002/emmm.201202301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonarakis SE, Valle D, Moser HW, Moser A, Qualman SJ, Zinkham WH. Phenotypic variability in siblings with Farber disease. J Pediatr 104: 406–409, 1984. doi: 10.1016/S0022-3476(84)81106-3. [DOI] [PubMed] [Google Scholar]
- 5.Balamayooran G, Batra S, Balamayooran T, Cai S, Jeyaseelan S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect Immun 79: 2567–2577, 2011. doi: 10.1128/IAI.00067-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barrière H, Gillot F. [Farber’s lipogranulomatosis]. Nouv Presse Med 2: 767–770, 1972. [PubMed] [Google Scholar]
- 7.Bierman SM, Edgington T, Newcomer VD, Pearson CM. A disorder of mucopolysaccharide metabolism with articular, respiratory, and neurologic manifestations. Arthritis Rheum 9: 620–630, 1966. [Google Scholar]
- 8.Bjurulf B, Spetalen S, Erichsen A, Vanier MT, Strøm EH, Strømme P. Niemann-Pick disease type C2 presenting as fatal pulmonary alveolar lipoproteinosis: morphological findings in lung and nervous tissue. Med Sci Monit 14: CS71–CS75, 2008. [PubMed] [Google Scholar]
- 9.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917, 1959. doi: 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- 10.Buccoliero R, Ginzburg L, Futerman AH. Elevation of lung surfactant phosphatidylcholine in mouse models of Sandhoff and of Niemann-Pick A disease. J Inherit Metab Dis 27: 641–648, 2004. doi: 10.1023/B:BOLI.0000042958.22066.6c. [DOI] [PubMed] [Google Scholar]
- 11.Devi ARR, Gopikrishna M, Ratheesh R, Savithri G, Swarnalata G, Bashyam M. Farber lipogranulomatosis: clinical and molecular genetic analysis reveals a novel mutation in an Indian family. J Hum Genet 51: 811–814, 2006. doi: 10.1007/s10038-006-0019-z. [DOI] [PubMed] [Google Scholar]
- 12.Dhami R, He X, Gordon RE, Schuchman EH. Analysis of the lung pathology and alveolar macrophage function in the acid sphingomyelinase—deficient mouse model of Niemann-Pick disease. Lab Invest 81: 987–999, 2001. doi: 10.1038/labinvest.3780311. [DOI] [PubMed] [Google Scholar]
- 13.Dworski S, Lu P, Khan A, Maranda B, Mitchell JJ, Parini R, Di Rocco M, Hugle B, Yoshimitsu M, Magnusson B, Makay B, Arslan N, Guelbert N, Ehlert K, Jarisch A, Gardner-Medwin J, Dagher R, Terreri MT, Marques Lorenco C, Barillas-Arias L, Tanpaiboon P, Solyom A, Norris JS, He X, Schuchman EH, Levade T Medin JA. Acid Ceramidase Deficiency is characterized by a unique plasma cytokine and ceramide profile that is altered by therapy. BBA Mol Basis Dis 186: 386–394, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ehlert K, Frosch M, Fehse N, Zander A, Roth J, Vormoor J. Farber disease: clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol Online J 5: 15, 2007. doi: 10.1186/1546-0096-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farber S, Cohen J, Uzman LL. Lipogranulomatosis; a new lipo-glycoprotein storage disease. J Mt Sinai Hosp N Y 24: 816–837, 1957. [PubMed] [Google Scholar]
- 16.Fiumara A, Nigro F, Pavone L, Moser HW. Farber disease with prolonged survival. J Inherit Metab Dis 16: 915–916, 1993. doi: 10.1007/BF00714300. [DOI] [PubMed] [Google Scholar]
- 17.Ghidoni R, Caretti A, Signorelli P. Role of sphingolipids in the pathobiology of lung inflammation. Mediators Inflamm 2015: 487508, 2015. doi: 10.1155/2015/487508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Göggel R, Winoto-Morbach S, Vielhaber G, Imai Y, Lindner K, Brade L, Brade H, Ehlers S, Slutsky AS, Schütze S, Gulbins E, Uhlig S. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med 10: 155–160, 2004. doi: 10.1038/nm977. [DOI] [PubMed] [Google Scholar]
- 19.Griese M, Brasch F, Aldana VR, Cabrera MM, Goelnitz U, Ikonen E, Karam BJ, Liebisch G, Linder MD, Lohse P, Meyer W, Schmitz G, Pamir A, Ripper J, Rolfs A, Schams A, Lezana FJ. Respiratory disease in Niemann-Pick type C2 is caused by pulmonary alveolar proteinosis. Clin Genet 77: 119–130, 2010. doi: 10.1111/j.1399-0004.2009.01325.x. [DOI] [PubMed] [Google Scholar]
- 20.Günther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med 153: 176–184, 1996. doi: 10.1164/ajrccm.153.1.8542113. [DOI] [PubMed] [Google Scholar]
- 21.Hallman M, Spragg R, Harrell JH, Moser KM, Gluck L. Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J Clin Invest 70: 673–683, 1982. doi: 10.1172/JCI110662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139–150, 2008. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 23.He X, Dworski S, Zhu C, DeAngelis V, Solyom A, Medin JA, Simonaro CM, Schuchman EH. Enzyme replacement therapy for Farber disease: Proof-of-concept studies in cells and mice. BBA Clinc 7: 85–96, 2017. doi: 10.1016/j.bbacli.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holm BA, Notter RH, Finkelstein JN. Surface property changes from interactions of albumin with natural lung surfactant and extracted lung lipids. Chem Phys Lipids 38: 287–298, 1985. doi: 10.1016/0009-3084(85)90022-2. [DOI] [PubMed] [Google Scholar]
- 25.Holm BA, Notter RH. Effects of hemoglobin and cell membrane lipids on pulmonary surfactant activity. J Appl Physiol (1985) 63: 1434–1442, 1987. doi: 10.1152/jappl.1987.63.4.1434. [DOI] [PubMed] [Google Scholar]
- 26.Ikegami M, Dhami R, Schuchman EH. Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am J Physiol Lung Cell Mol Physiol 284: L518–L525, 2003. doi: 10.1152/ajplung.00258.2002. [DOI] [PubMed] [Google Scholar]
- 27.Ikegami M, Whitsett JA, Chroneos ZC, Ross GF, Reed JA, Bachurski CJ, Jobe AH. IL-4 increases surfactant and regulates metabolism in vivo. Am J Physiol Lung Cell Mol Physiol 278: L75–L80, 2000. doi: 10.1152/ajplung.2000.278.1.L75. [DOI] [PubMed] [Google Scholar]
- 28.Iyonaga K, Suga M, Yamamoto T, Ichiyasu H, Miyakawa H, Ando M. Elevated bronchoalveolar concentrations of MCP-1 in patients with pulmonary alveolar proteinosis. Eur Respir J 14: 383–389, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Keough KM, Parsons CS, Phang PT, Tweeddale MG. Interactions between plasma proteins and pulmonary surfactant: surface balance studies. Can J Physiol Pharmacol 66: 1166–1173, 1988. doi: 10.1139/y88-192. [DOI] [PubMed] [Google Scholar]
- 30.Kim YJ, Park SJ, Park CK, Kim SH, Lee CW. A case of Farber lipogranulomatosis. J Korean Med Sci 13: 95–98, 1998. doi: 10.3346/jkms.1998.13.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuebler WM, Yang Y, Samapati R, Uhlig S. Vascular barrier regulation by PAF, ceramide, caveolae, and NO - an intricate signaling network with discrepant effects in the pulmonary and systemic vasculature. Cell Physiol Biochem 26: 29–40, 2010. doi: 10.1159/000315103. [DOI] [PubMed] [Google Scholar]
- 32.Levade T, Sandhoff K, Schulze H, Medin J. Acid ceramidase deficiency: farber lipogranulomatosis. In: Scriver’s OMMBID (Online Metabolic and Molecular Bases of Inherited Diseases), edited by Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, and Ballabio A). New York: McGraw-Hill, 2014, Ch. 143. [Google Scholar]
- 33.Li CM, Park JH, He X, Levy B, Chen F, Arai K, Adler DA, Disteche CM, Koch J, Sandhoff K, Schuchman EH. The human acid ceramidase gene (ASAH): structure, chromosomal location, mutation analysis, and expression. Genomics 62: 223–231, 1999. doi: 10.1006/geno.1999.5940. [DOI] [PubMed] [Google Scholar]
- 34.Loiseau N, Polizzi A, Dupuy A, Therville N, Rakotonirainy M, Loy J, Viadere JL, Cossalter AM, Bailly JD, Puel O, Kolf-Clauw M, Bertrand-Michel J, Levade T, Guillou H, Oswald IP. New insights into the organ-specific adverse effects of fumonisin B1: comparison between lung and liver. Arch Toxicol 89: 1619–1629, 2015. doi: 10.1007/s00204-014-1323-6. [DOI] [PubMed] [Google Scholar]
- 35.Moser HW, Prensky AL, Wolfe HJ, Rosman NP. Farber’s lipogranulomatosis. Report of a case and demonstration of an excess of free ceramide and ganglioside. Am J Med 47: 869–890, 1969. doi: 10.1016/0002-9343(69)90202-2. [DOI] [PubMed] [Google Scholar]
- 36.Mura M, Binnie M, Han B, Li C, Andrade CF, Shiozaki A, Zhang Y, Ferrara N, Hwang D, Waddell TK, Keshavjee S, Liu M. Functions of type II pneumocyte-derived vascular endothelial growth factor in alveolar structure, acute inflammation, and vascular permeability. Am J Pathol 176: 1725–1734, 2010. doi: 10.2353/ajpath.2010.090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel BV, Wilson MR, O’Dea KP, Takata M. TNF-induced death signaling triggers alveolar epithelial dysfunction in acute lung injury. J Immunol 190: 4274–4282, 2013. doi: 10.4049/jimmunol.1202437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pellissier JF, Berard-Badier M, Pinsard N. Farber’s disease in two siblings, sural nerve and subcutaneous biopsies by light and electron microscopy. Acta Neuropathol 72: 178–188, 1986. doi: 10.1007/BF00685981. [DOI] [PubMed] [Google Scholar]
- 39.Petrache I, Berdyshev EV. Ceramide signaling and metabolism in pathophysiological states of the lung. Annu Rev Physiol 78: 463–480, 2016. doi: 10.1146/annurev-physiol-021115-105221. [DOI] [PubMed] [Google Scholar]
- 40.Petrache I, Kamocki K, Poirier C, Pewzner-Jung Y, Laviad EL, Schweitzer KS, Van Demark M, Justice MJ, Hubbard WC, Futerman AH. Ceramide synthases expression and role of ceramide synthase-2 in the lung: insight from human lung cells and mouse models. PLoS One 8: e62968, 2013. doi: 10.1371/journal.pone.0062968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pewzner-Jung Y, Tavakoli Tabazavareh S, Grassmé H, Becker KA, Japtok L, Steinmann J, Joseph T, Lang S, Tuemmler B, Schuchman EH, Lentsch AB, Kleuser B, Edwards MJ, Futerman AH, Gulbins E. Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol Med 6: 1205–1214, 2014. doi: 10.15252/emmm.201404075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rauvala H, Hallman M. Glycolipid accumulation in bronchoalveolar space in adult respiratory distress syndrome. J Lipid Res 25: 1257–1262, 1984. [PubMed] [Google Scholar]
- 43.Ryan AJ, McCoy DM, McGowan SE, Salome RG, Mallampalli RK. Alveolar sphingolipids generated in response to TNF-alpha modifies surfactant biophysical activity. J Appl Physiol (1985) 94: 253–258, 2003. doi: 10.1152/japplphysiol.00184.2002. [DOI] [PubMed] [Google Scholar]
- 44.Samuelsson K, Zetterström R, Ivemark B.. Studies on a case of lipogranulomatosis (Farber’s disease) with protracted course. In: Sphingolipids, Sphingolipidoses and Allied Disorders, edited by Volk B and Aronson S. New York: Springer, 1972, p. 533–548. [Google Scholar]
- 45.Samuelsson K, Zetterström R. Ceramides in a patient with lipogranulomatosis (Farber’s disease) with chronic course. Scand J Clin Lab Invest 27: 393–405, 1971. doi: 10.3109/00365517109080235. [DOI] [PubMed] [Google Scholar]
- 46.Schneider EL, Epstein CJ, Kaback MJ, Brandes D. Severe pulmonary involvement in adult Gaucher’s disease. Report of three cases and review of the literature. Am J Med 63: 475–480, 1977. doi: 10.1016/0002-9343(77)90288-1. [DOI] [PubMed] [Google Scholar]
- 47.Schuchman EH. Acid ceramidase and the treatment of ceramide diseases: The expanding role of enzyme replacement therapy. Biochim Biophys Acta 1862: 1459–1471, 2016. doi: 10.1016/j.bbadis.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 48.Seeger W, Grube C, Günther A, Schmidt R. Surfactant inhibition by plasma proteins: differential sensitivity of various surfactant preparations. Eur Respir J 6: 971–977, 1993. [PubMed] [Google Scholar]
- 49.Seeger W, Stöhr G, Wolf HR, Neuhof H. Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J Appl Physiol (1985) 58: 326–338, 1985. doi: 10.1152/jappl.1985.58.2.326. [DOI] [PubMed] [Google Scholar]
- 50.Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med 166: 215–235, 2002. doi: 10.1164/rccm.2109105. [DOI] [PubMed] [Google Scholar]
- 51.Sheth J, Joseph JJ, Shah K, Muranjan M, Mistri M, Sheth F. Pulmonary manifestations in Niemann-Pick type C disease with mutations in NPC2 gene: case report and review of literature. BMC Med Genet 18: 5, 2017. doi: 10.1186/s12881-017-0367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sideris GA, Josephson M. Pulmonary alveolar proteinosis and Niemann Pick disease type B: An unexpected combination. Respir Med Case Rep 19: 37–39, 2016. doi: 10.1016/j.rmcr.2016.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sikora J, Dworski S, Jones EE, Kamani MA, Micsenyi MC, Sawada T, Le Faouder P, Bertrand-Michel J, Dupuy A, Dunn CK, Yang Xuan Ingrid C, Casas J, Fabrias G, Hampson DR, Levade T, Drake Richard R, Medin JA, Walkley SU. Acid ceramidase deficiency in mice results in a broad range of central nervous system abnormalities. Am J Pathol 187: 864–883, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sommer U, Herscovitz H, Welty FK, Costello CE. LC-MS-based method for the qualitative and quantitative analysis of complex lipid mixtures. J Lipid Res 47: 804–814, 2006. doi: 10.1194/jlr.M500506-JLR200. [DOI] [PubMed] [Google Scholar]
- 55.Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol 155: 1515–1524, 1995. [PubMed] [Google Scholar]
- 56.Sugita M, Dulaney JT, Moser HW. Ceramidase deficiency in Farber’s disease (lipogranulomatosis). Science 178: 1100–1102, 1972. doi: 10.1126/science.178.4065.1100. [DOI] [PubMed] [Google Scholar]
- 57.Tanaka T, Takahashi K, Hakozaki H, Kimoto H, Suzuki Y. Farber’s disease (disseminated lipogranulomatosis)—a pathological, histochemical and ultrastructural study. Acta Pathol Jpn 29: 135–155, 1979. [DOI] [PubMed] [Google Scholar]
- 58.Tibboel J, Reiss I, de Jongste JC, Post M. Sphingolipids in lung growth and repair. Chest 145: 120–128, 2014. doi: 10.1378/chest.13-0967. [DOI] [PubMed] [Google Scholar]
- 59.Toppet M, Vamos-Hurwitz E, Jonniaux G, Cremer N, Tondeur M, Pelc S. Farber’s disease as a ceramidosis: clinical, radiological and biochemical aspects. Acta Paediatr Scand 67: 113–119, 1978. doi: 10.1111/j.1651-2227.1978.tb16287.x. [DOI] [PubMed] [Google Scholar]
- 60.Uhlig S, Gulbins E. Sphingolipids in the lungs. Am J Respir Crit Care Med 178: 1100–1114, 2008. doi: 10.1164/rccm.200804-595SO. [DOI] [PubMed] [Google Scholar]
- 61.Vogel P, Donoviel MS, Read R, Hansen GM, Hazlewood J, Anderson SJ, Sun W, Swaffield J, Oravecz T. Incomplete inhibition of sphingosine 1-phosphate lyase modulates immune system function yet prevents early lethality and non-lymphoid lesions. PLoS One 4: e4112, 2009. doi: 10.1371/journal.pone.0004112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang L, Dudek SM. Regulation of vascular permeability by sphingosine 1-phosphate. Microvasc Res 77: 39–45, 2009. doi: 10.1016/j.mvr.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weaver TE, Na CL, Stahlman M. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin Cell Dev Biol 13: 263–270, 2002. doi: 10.1016/S1084952102000551. [DOI] [PubMed] [Google Scholar]
- 64.Yang G, Hamacher J, Gorshkov B, White R, Sridhar S, Verin A, Chakraborty T, Lucas R. The dual role of TNF in pulmonary edema. J Cardiovasc Dis Res 1: 29–36, 2010. doi: 10.4103/0975-3583.59983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang Y, Uhlig S. The role of sphingolipids in respiratory disease. Ther Adv Respir Dis 5: 325–344, 2011. doi: 10.1177/1753465811406772. [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Yin J, Baumgartner W, Samapati R, Solymosi EA, Reppien E, Kuebler WM, Uhlig S. Platelet-activating factor reduces endothelial nitric oxide production: role of acid sphingomyelinase. Eur Respir J 36: 417–427, 2010. doi: 10.1183/09031936.00095609. [DOI] [PubMed] [Google Scholar]
- 67.Yeager AM, Armfield Uhas K, Coles CD, Davis PC, Krause WL, Moser HW. Bone marrow transplantation for infantile ceramidase deficiency (Farber disease). Bone Marrow Transplant 26: 357–363, 2000. doi: 10.1038/sj.bmt.1702489. [DOI] [PubMed] [Google Scholar]
- 68.Zetterström R. Disseminated lipogranulomatosis (Farber’s disease). Acta Paediatr 47: 501–510, 1958. doi: 10.1111/j.1651-2227.1958.tb07665.x. [DOI] [PubMed] [Google Scholar]