

Abstract
Patient: Female, 34
Final Diagnosis: Sjogren’s syndrome • osteomalacia • Fanconi syndrome
Symptoms: Bone fractures • osteomuscular pain • xerophtalmia • xerostomia
Medication: —
Clinical Procedure: —
Specialty: Rheumatology
Objective:
Rare co-existance of disease or pathology
Background:
Sjögren’s syndrome is a chronic inflammatory autoimmune disease, which is also known as sicca syndrome, due to the symptoms of dry eyes and dry mouth, and is associated with other connective tissue diseases and autoimmune diseases. Sjögren’s syndrome can also be associated with renal involvement. Fanconi’s syndrome is associated with impaired reabsorption in the proximal renal tubule associated with tubulointerstitial nephritis and is associated with renal tubular acidosis and hypophosphatemia. Osteomalacia is a rare association with Sjögren’s syndrome, which may result from renal disease.
Case Report:
We report the case of a 34-year-old woman who presented with xerostomia, xerophthalmia, bone fractures, and osteomuscular pain. A Schirmer test showed reduced tear production, and a biopsy of a minor salivary gland of the lip, with high titers of antinuclear antibodies (ANA), and positive anti-SSA/Ro and anti-SSB/La antibodies confirmed the diagnosis of Sjögren’s syndrome. Serum and urinary laboratories tests and clinical manifestations confirmed Fanconi’s syndrome associated with osteomalacia. The patient was treated with potassium supplements, 25-hydroxyvitamin D (25(OH)D), hydroxychloroquine, mycophenolate mofetil, and prednisone, with a favorable response.
Conclusions:
This case is of a rare association between Sjögren’s syndrome, Fanconi’s syndrome, and osteomalacia. Even though these are rare clinical associations, early detection can improve the quality of life and prevent further complications.
MeSH Keywords: Fanconi Syndrome, Osteomalacia, Sjogren’s Syndrome
Background
Sjögren’s syndrome is a chronic inflammatory autoimmune disease, which is also known as sicca syndrome, due to the symptoms of dry eyes and dry mouth, and is associated with other connective tissue diseases and autoimmune diseases [1–3]. Sjögren’s syndrome has a prevalence between 1–3% in the general population, and can present in any age group but is more common between the fifth and eighth decades of life, and it is nine times more frequent among women [1,2]. Sicca symptoms, which include dryness of the mouth and eyes, are the most common clinical findings [3].
Tubulointerstitial nephritis is a recognized systemic manifestation in patients with Sjögren’s syndrome [4]. Because the symptoms are unspecific, the diagnosis is usually delayed, and the complications can be lethal [1]. Renal tubular acidosis and tubulointerstitial nephritis can be presenting features of patients with Sjögren’s syndrome, and are characterized by metabolic acidosis due to defects in the renal tubules, which can be proximal or distal [5]. Fanconi’s syndrome is a generalized disorder in the proximal renal tubule that leads to defects in the transport of amino acids, glucose, phosphate, uric acid, bicarbonate, and other substances leading to conditions that include osteomalacia [6]. Osteomalacia is a metabolic disease that presents with bone fractures [7], or bone deformity and weakness [4]. Osteomalacia rarely occurs in patients with Sjögren’s syndrome, but when it does occur, it is associated with renal involvement [4].
To best of our knowledge, few cases have been reported on the association between Sjögren’s syndrome, Fanconi’s syndrome, and osteomalacia.
Case Report
A 34-year-old woman attended the Ophthalmology Department due to keratoconjunctivitis sicca, but approximately four years previously she had presented with symptoms of xerostomia and xerophthalmia, treated with artificial tears as her initial treatment. Sicca symptoms progressed with the development of osteomuscular pain. During this time, electromyography (EMG) was performed, which showed normal electrical activity with no neurologic or muscular abnormalities.
Three years previously, before this presentation, the patient was admitted to the emergency department complaining of intense osteomuscular pain. A computed tomography (CT) scan (Somatom Sensation 64) (Siemens, Erlangen, Germany) showed a right femoral neck fracture (Figure 1). The orthopedic team decided to perform osteosynthesis surgery, which did not result in improvement of the patient’s pain or functional capacity. The following year, a new linear fracture was detected in the left femoral neck, and another osteosynthesis was performed. The pain continued to increase, and the patient was treated with nonsteroidal anti-inflammatory drugs (NSAIDs), which only provided temporary relief. The difficulty with walking worsened as the muscle mass decreased, and the patient began to use crutches.
Figure 1.
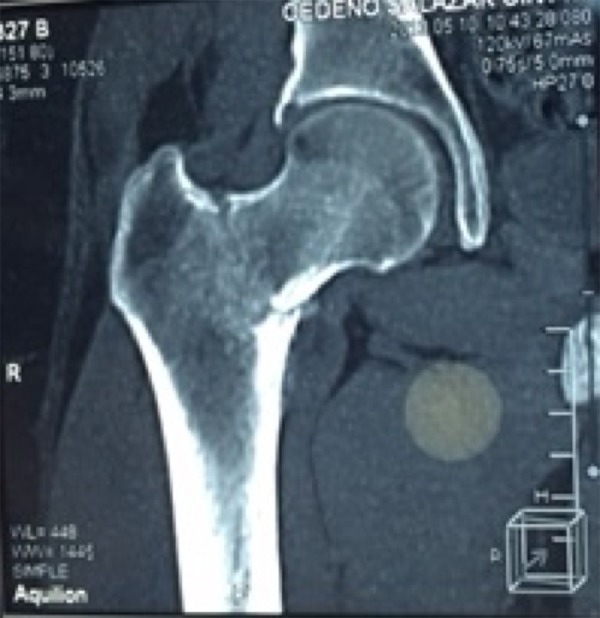
Computed tomography (CT) image of the right femur CT image in the coronal plane shows the fracture of the neck of the femur.
There were no laboratory metabolic studies performed. The bone pain was described as localized in the long bones, with no radiation and was severe, while the muscle pain was localized to the pelvis and shoulder girdle. The patient developed increasing muscle weakness and difficulty with walking.
On physical examination on hospital admission, the patient appeared well, alert, and oriented. Her height, body weight and body mass index were 145 cm, 45.5 kg, and 21.64 kg/m2, respectively. Her vital signs were within normal limits, with a temperature of 36.5°C, blood pressure of 100/65 mmHg, heart rate of 90 beats/min, and a respiration rate of 14/min. There was no lymphadenopathy. Cardiac, respiratory, and abdominal examinations were unremarkable. The tongue was dry, red, and glossy. The patient had tenderness of the long bones and pelvic bones with a conserved articular range of motion, and no evidence of synovitis. Further neurologic examination showed mild muscular atrophy in the deltoid muscles and severe muscular atrophy in quadriceps and gastrocnemius muscles of both lower limbs. Motor examination showed muscle strength of 4/5 in the scapular and pelvic musculature. Deep tendon reflexes were 1+, and sensation was normal throughout. Laboratory studies (shown in Table 1) showed reduced serum sodium, potassium, phosphorus, and uric acid, a normal complete blood count (CBC), normal total calcium, glucose, albumin, globulin, and high levels of chloride. Urinalysis showed glucosuria, proteinuria, and increased pyrilinks-D test. However, urinary calcium and phosphate excretion were within the normal range, but urinary pH was borderline. Arterial blood gases showed metabolic acidosis with a pH of 7.11, bicarbonate of 17.6 mEq/L, and a normal anion gap (9 mEq/L). Increased levels of serum alkaline phosphatase (ALP), parathyroid hormone (PTH), and thyroid stimulating hormone (TSH) were present, but 25-hydroxyvitamin D (25(OH)D) levels were reduced. The erythrocyte sedimentation rate (ESR) was at mildly raised at 21 mm/hr (N: <20 mm/hr) and C reactive protein (CRP) was low at 2.81 mg/L (N: 0–20 mg/L). Serum protein electrophoresis showed a polyclonal hypergammaglobulinemia.
Table 1.
Laboratory data on hospital admission and after six weeks.
| Variables | Reference range | On admission | 6 weeks after treatment |
|---|---|---|---|
| Serum Levels | |||
| Hemoglobin (g/dL) | 12.3–15.3 | 13.1 | |
| Leukocytes (per mm3) | 4,400–11,300 | 7,100 | |
| Platelets (per mm3) | 150,000–450,000 | 278,000 | |
| ESR (mm/h) | <20 | 21 | |
| C-reactive protein (mg/L) | 0–20 | 2.81 | |
| Glucose (mg/dL) | 70–99 | 85 | 88 |
| Uric acid (mg/dL) | 2.6–6 | 1.28 | |
| Albumin (g/dL) | 3.5–5.2 | 4.95 | |
| Globulin (g/dL) | 2.5–3.5 | 3.55 | |
| Calcium (mg/dL) | 8.4–10.2 | 9.2 | 8.8 |
| Phosphorus (mg/dL) | 2.3–4.7 | 1.46 | 2.29 |
| PTH (pg/mL) | 12–72 | 114 | |
| 25(OH)D (ng/mL) | 32–70 | 22.78 | |
| Alkaline Phosphatase (UI/L) | 40–150 | 322 | |
| Potassium (mEq/L) | 3.5–5.1 | 2.85 | 3.8 |
| Sodium (mEq/L) | 136–145 | 134.5 | 136 |
| Chloride (mg/dL) | 98–107 | 111.47 | 108 |
| Bicarbonate (mEq/L) | 23–32 | 17.6 | 22.8 |
| pH | 7.35–7.45 | 7.11 | |
| Anion Gap (mEq/L) | 8–12 | 9 | |
| AAN | <1:40 | 1:2560 | |
| Anti-Ro (U/mL) | 0–16 | 145 | |
| Anti-La (U/mL) | 0–16 | 89.88 | |
| TSH (mIU/L) | 0.4–4 | 5.4 | 3 |
| fT4 (ng/dL) | 0.76–1.39 | 1.25 | 1.25 |
| T3 (nm/L) | 1.30–2,60 | 1.07 | 1.29 |
| Anti-TPO | – | Negative | |
| ATG | – | Negative | |
| Urinalysis | |||
| pH | 5–7.5 | 7.5 | 7 |
| Gravity | 1.015–1.020 | 1.008 | |
| Glucose (mg/dL) | – | 500 | 200 |
| Proteins (mg/dL) | – | 150 | |
| Pyrilinks-D (nMDPD/mM) | 3–7.4 | 14.04 | |
| Calcium (mg/24 h) | 100–300 | 112 | |
| Phosphorus (mg/24 h) | 400–1300 | 525.3 | |
| Ophtalmological tests | |||
| Schirmer (5 min) | 0 mm | ||
| Tear break-up time | Unstable | ||
| Fluorescein eye stain | Positive |
A lip salivary gland biopsy showed Chisholm–Mason stage IV, and Greenspan focus score of 2 lesions (Figure 2), while an ophthalmological examination showed reduced tear production by the Schirmer test (0 mm for both eyes). Antinuclear antibodies (ANA) were present in a titer of 1: 2560 (N<1: 40) with positive anti-SSA and anti-SSB autoantibodies.
Figure 2.
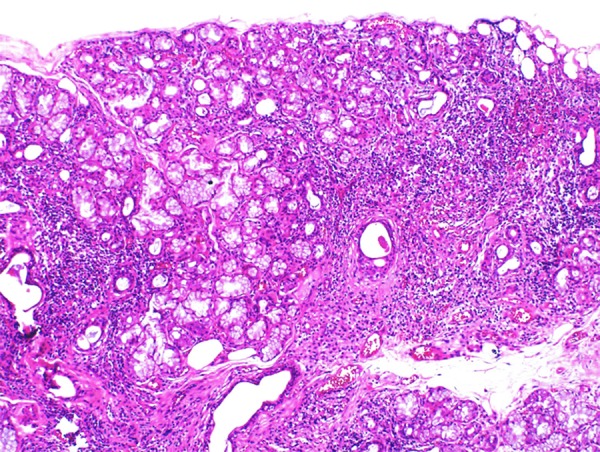
Photomicrograph of the histology of the biopsy of the minor salivary gland. Histology of the biopsy of the minor salivary gland shows a diffuse mononuclear inflammatory lymphoplasmacytic infiltrate with the formation of groups of >50 lymphocytes equivalent to Chisholm-Mason stage IV (based on assessing a 4 mm2 area of salivary gland tissue) and Greenspan focus score (FS) of 2.
According to the presence of dry mouth and eyes, an abnormal ophthalmic test, the histology of the lip biopsy, high titers of antinuclear antibodies (ANA), and positive anti-SSA/Ro and anti-SSB/La antibodies, the patient was diagnosed with Sjögren’s syndrome. The presence of hyperchloremic metabolic acidosis with a normal anion gap, hypophosphatemia, hypouricemia, glycosuria (normal glycemic levels), aminoaciduria, and borderline levels of urinary pH supported a diagnosis Fanconi’s syndrome. The presence of tubulointerstitial nephritis was believed to be related to Sjögren’s syndrome, which also contributed to the development of osteomalacia, characterized by osteomuscular pain, recurrent fractures, muscle weakness, hypophosphatemia, and high levels of ALP. The patient also had associated features of secondary hyperparathyroidism presenting with a raised PTH, and low 25(OH)D levels, and subclinical hypothyroidism, with decreased T3, normal T4, and increased levels of TSH.
The patient was treated with alkalinization therapy (500 mg of sodium bicarbonate twice a day), potassium supplements (2 gm daily), vitamin D (50.000 units weekly), hydroxychloroquine (200 mg daily), levothyroxine (50 mcg daily, increasing to 75 mcg daily), mycophenolate mofetil (500 mg twice a day), and prednisone at 30 mg daily for six weeks, tapered to 5 mg daily. The electrolyte imbalance was corrected (laboratory results are shown in Table 1), the osteomuscular pain diminished, and ambulation and muscular strength improved gradually over a period of six weeks.
Discussion
A case is presented of a female patient with Sjögren’s syndrome that presented with osteomuscular pain due to the osteomalacia following the development of Fanconi’s syndrome. The renal manifestations of patients with Sjögren’s syndrome have been reported to show that tubulointerstitial involvement is more common than the glomerular disease, but Fanconi’s syndrome is an infrequent association [8]. A study by Ren et al. involving 130 patients with Sjögren’s syndrome with renal tubular dysfunction, reported that 70% (91/130) presented with distal renal tubular acidosis, and only 3.1% (4/130) presented with Fanconi’s syndrome [1].
The mechanisms by which patients with Sjögren’s syndrome develop Fanconi’s syndrome remain unclear. However, two possible mechanisms have been described. Tubular damage by lymphocytic infiltration and hypergammaglobulinemia includes the deposition of immunoglobulin light chains, resulting in an inflammatory response and a direct inhibitory effect on transporters of glucose, amino acids, phosphate, and Na+K+ATP-ase in the proximal tubules [8]. In the patient described in this report, the tubular lymphocytic infiltration might have been the mechanism by which she developed Fanconi’s syndrome, as protein electrophoresis was normal. This diagnosis was confirmed by the presence of metabolic acidosis with a normal anion gap, hypophosphatemia, hyperuricemia, and glycosuria, with normal blood glucose, which supports a diagnosis of Fanconi’s syndrome [9]. Patients with Fanconi’s syndrome may develop hypokalemia due to metabolic acidosis associated with renal potassium loss, and its treatment consists of potassium replacement therapy with correction of the acid-base imbalance [8].
Previously, corticosteroids have been successfully used in patients with interstitial nephritis and Sjögren’s syndrome. However, some patients develop resistance to therapy, relapse when the dose is tapered, or develop intolerance to therapy. In these cases, patients have been treated with alternative immunosuppressant drugs [10]. In a series of 12 patients with tubulointerstitial nephritis and primary Sjögren’s syndrome, 11 patients were treated with mycophenolate mofetil (median dose 1,000 mg/QD) and showed improving serum creatinine and estimated glomerular filtration rate (eGFR) after treatment [11]. Mycophenolate mofetil is an inhibitor of lymphocyte proliferation and showed positive results in these case series; a dose of 500 mg/bd was prescribed in the patient in this case, together with prednisone, which significantly improved her symptoms.
Osteomalacia is a metabolic bone disease characterized by loss of bone density due to abnormalities in vitamin D or phosphates [12]. In patients with connective tissue disorders, osteomalacia may be associated with renal involvement [4]. Osteomalacia can be induced by distal or proximal tubular acidosis, but proximal renal tubular acidosis is a more common association [13]. Osteomalacia in patients with Fanconi’s syndrome is caused by hypophosphatemia and metabolic acidosis [9,14,15]. The phosphate reabsorption defect in patients with Fanconi’s syndrome leads to hypophosphatemia producing impaired bone mineralization [16]. Metabolic acidosis results in a decrease in osteoblast ALP activity [13]. Patients with Fanconi’s syndrome who present with osteomalacia can have a relative decrease in vitamin D [16]. Clinically, these patients have muscle weakness, bone pain, and can present with fractures. Laboratory abnormalities include hypophosphatemia with elevated serum ALP, normal or low calcium, and decreased 25(OH)D [8]. All these parameters were found in this patient, except for low calcium. In this patient, calcium levels remained normal due to an increase in PTH. Osteomalacia is rarely reported as the leading manifestation of patients with Sjögren’s syndrome. However, a previously published study by Yang et al. reported a female patient with Sjögren’s syndrome, Fanconi’s syndrome, and osteomalacia that presented with muscle weakness and bone deformities. The treatment of patients with osteomalacia should include calcium, vitamin D, and alkalinization, which, as shown in this case, can substantially improve musculoskeletal disorders [17].
Conclusions
At this time, less than ten cases of Sjögren’s syndrome associated with Fanconi’s syndrome and osteomalacia have been reported. Although this association is not frequent, it is important to suspect of osteomalacia in Sjögren’s syndrome patients that present bone pain, myalgia, and repeated fractures, because early detection can improve the quality of life and prevent complications.
Footnotes
Conflict of interest
None.
References:
- 1.Ren H, Wang W-M, Chen X-N, et al. Renal involvement and followup of 130 patients with primary Sjogren’s syndrome. J Rheumatol. 2008;35(2):278–84. [PubMed] [Google Scholar]
- 2.Bell M, Askari A, Bookman A, et al. Sjogren’s syndrome: A critical review of clinical management. J Rheumatol. 1999;26(9):2051–61. [PubMed] [Google Scholar]
- 3.Arman F, Shakeri H, Nobakht N, Rastogi A, Kamgar M. A case of kidney involvement in primary Sjögren’s syndrome. Am J Case Rep. 2017;18:622–26. doi: 10.12659/AJCR.903476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang YS, Peng CH, Sia SK, Huang CN. Acquired hypophosphatemia osteomalacia associated with Fanconi’s syndrome in Sjögren’s syndrome. Rheumatol Int. 2007;27(6):593–97. doi: 10.1007/s00296-006-0257-6. [DOI] [PubMed] [Google Scholar]
- 5.Ataoglu EH, Demir B, Tuna M, et al. Sjögren syndrome presenting with hypopotassemic periodic paralysis due to renal tubular acidosis. Am J Case Rep. 2012;13:187–90. doi: 10.12659/AJCR.883326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roth KS, Foreman JW, Segal S. The Fanconi syndrome and mechanisms of tubular transport dysfunction. Kidney Int. 1981;20(6):705–16. doi: 10.1038/ki.1981.200. [DOI] [PubMed] [Google Scholar]
- 7.Shi M, Chen L. Sjögren’s syndrome complicated with Fanconi syndrome and Hashimoto’s thyroiditis: Case report and literature review. J Int Med Res. 2016;44(3):753–59. doi: 10.1177/0300060515593767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang C-C, Shiang J-C, Huang W-T, Lin S-H. Hypokalemic paralysis as primary presentation of Fanconi syndrome associated with Sjogren syndrome. J Clin Rheumatol. 2010;16(4):178–80. doi: 10.1097/RHU.0b013e3181df903f. [DOI] [PubMed] [Google Scholar]
- 9.François H, Mariette X. Renal involvement in primary Sjögren syndrome. Nat Rev Nephrol. 2016;12(2):82–93. doi: 10.1038/nrneph.2015.174. [DOI] [PubMed] [Google Scholar]
- 10.Preddie DC, Markowitz GS, Radhakrishnan J, et al. Mycophenolate mofetil for the treatment of interstitial nephritis. Clin J Am Soc Nephrol. 2006;1(4):718–22. doi: 10.2215/CJN.01711105. [DOI] [PubMed] [Google Scholar]
- 11.Evans RDR, Laing CM, Ciurtin C, Walsh SB. Tubulointerstitial nephritis in primary Sjögren syndrome: clinical manifestations and response to treatment. BMC Musculoskelet Disord. 2016;17(1):2. doi: 10.1186/s12891-015-0858-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wintermeyer E, Ihle C, Ehnert S, et al. Crucial role of vitamin D in the musculoskeletal system. Nutrients. 2016;8(6) doi: 10.3390/nu8060319. pii: E319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saoud B, Bahiri R, Benbouazza K, et al. Osteomalacia revealing Sjogren’s syndrome. Joint Bone Spine. 2005;72(6):594–95. doi: 10.1016/j.jbspin.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 14.Nakamura H, Kita J, Kawakami A, et al. Multiple bone fracture due to Fanconi’s syndrome in primary Sjögren’s syndrome complicated with organizing pneumonia. Rheumatol Int. 2009;30(2):265–67. doi: 10.1007/s00296-009-0924-5. [DOI] [PubMed] [Google Scholar]
- 15.Khandelwal D, Bhattacharya S, Gadodia A, et al. Metabolic bone disease as a presenting manifestation of primary Sjögren’s syndrome: Three cases and review of literature. Indian J Endocrinol Metab. 2011;15(4):341–45. doi: 10.4103/2230-8210.85599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke B, Wynne A, Wilson D, Fitzpatrick L. Osteomalacia associated with adult Fanconi’s syndrome: clinical and diagnostic features. Clin Endocrinol (Oxf) 1995;43:479–90. doi: 10.1111/j.1365-2265.1995.tb02621.x. [DOI] [PubMed] [Google Scholar]
- 17.Cherif E, Ben Hassine L, Kaoueche Z, Khalfallah N. Osteomalacia as inaugural manifestation of Sjögren syndrome. BMJ Case Rep. 2013;2013 doi: 10.1136/bcr-2013-201052. pii: bcr2013201052. [DOI] [PMC free article] [PubMed] [Google Scholar]