

Abstract
Diversity in the structure and expression of microRNAs, important regulators of gene expression, arises from SNPs, duplications followed by divergence, production of isomiRs, and RNA editing. Inbred mouse strains and crosses using them are important reference populations for genetic mapping, and as models of human disease. We determined the nature and extent of interstrain miRNA variation by (i) identifying miRNA SNPs in whole-genome sequence data from 36 strains, and (ii) examining miRNA editing and expression in hippocampus (Hpc) and frontal cortex (FCx) of six strains, to facilitate the study of miRNAs in neurobehavioral phenotypes. miRNA loci were strongly conserved among the 36 strains, but even the highly conserved seed region contained 16 SNPs. In contrast, we identified RNA editing in 58.9% of miRNAs, including 11 consistent editing events in the seed region. We confirmed the functional significance of three conserved edits in the miR-379/410 cluster, demonstrating that edited miRNAs gained novel target mRNAs not recognized by the unedited miRNAs. We found significant interstrain differences in miRNA and isomiR expression: Of 779 miRNAs expressed in Hpc and 719 in FCx, 262 were differentially expressed (190 in Hpc, 126 in FCx, 54 in both). We also identified 32 novel miRNA candidates using miRNA prediction tools. Our studies provide the first comprehensive analysis of SNP, isomiR, and RNA editing variation in miRNA loci across inbred mouse strains, and a detailed catalog of expressed miRNAs in Hpc and FCx in six commonly used strains. These findings will facilitate the molecular analysis of neurological and behavioral phenotypes in this model organism.
Keywords: inbred mouse strain, genome, single-nucleotide polymorphism, miRNA, RNA editing, isomiR, small RNA-sequencing
INTRODUCTION
MicroRNAs (miRNAs) are small noncoding RNAs that participate in the post-transcriptional regulation of their target mRNAs (Lee et al. 1993; Wightman et al. 1993). They are found in all tissues of all higher organisms where they regulate numerous biological processes that include cell growth, tissue differentiation, embryonic development, apoptosis, and neurobiological functions such as development of the nervous system, synaptic plasticity, and neurodegeneration (Zhao and Srivastava 2007; Kawahara et al. 2012). One miRNA typically targets multiple transcripts, and a single mRNA can be targeted by multiple miRNAs.
Many miRNAs are coexpressed from polycistronic miRNA clusters or belong to families comprised of paralogous miRNA genes that often target several mRNAs within the same biological pathway (Altuvia et al. 2005; Baskerville and Bartel 2005; Juhila et al. 2011; Wolter et al. 2017). miRNAs are initially transcribed as primary miRNAs (pri-miRNAs). Pri-miRNAs are subsequently processed in the nucleus by the RNase III nuclease Drosha to produce hairpin-shaped precursor miRNAs (pre-miRNAs) (Lee et al. 2003). Some miRNAs, called miRtrons, are spliced from introns of protein-coding genes and processed in a Drosha-independent manner. Pre-miRNA hairpins are exported to the cytoplasm where the RNase III nuclease Dicer further cleaves the double-stranded RNA to form the ∼22 bp product. The two strands, called the 5p and 3p arms, are separated to produce mature single-stranded miRNAs (Hutvágner et al. 2001). miRNA biogenesis is usually a relatively stable process but the cut sites of Drosha or Dicer may vary. This leads to the production of multiple forms of a miRNA, called isomiRs, with 5′ or 3′ cut sites either upstream or downstream from the canonical cleavage site. Specific RNA editing enzymes may also change the nucleotide sequence of the mature miRNA post-transcriptionally or add nucleotides to the 3′ end of the mature miRNA. Finally, the mature miRNA is loaded into the miRNA-induced silencing complex (miRISC) with an argonaut protein (AGO). The miRNA component of the complex determines target specificity. Important component of the target recognition is complementary binding of bases 2–8 of the mature miRNA (the seed region) to the 3′ UTR of the target mRNA, which either blocks its translation or activates mRNA degradation (Baek et al. 2008; Guo et al. 2010), resulting in gene silencing.
Inbred mouse strains and crosses using them are important reference populations for genetic mapping, and are widely used genetic and pharmacological models of human disease phenotypes. Catalogs of mouse miRNAs have been previously published (Landgraf et al. 2007; Chiang et al. 2010) but the genetic variation of miRNA loci and expression across different inbred mouse strains has not been systematically investigated. Therefore, commercial miRNA tools are generally only available for the miRBase canonical miRNA sequence and the mouse reference strain C57BL/6J, hindering the use of other mouse strains in biomedical research on miRNAs. The first inbred mouse strains were generated over 100 years ago and now several hundred are commercially available. Their genealogy has been extensively investigated at the DNA (Ideraabdullah et al. 2004; Yalcin et al. 2004; Cervino et al. 2005) and gene expression (Hovatta et al. 2007) levels. Recently the whole-genome sequence (WGS) of 36 classical laboratory and wild-derived strains was determined as a part of the Mouse Genomes Project (Keane et al. 2011; Wong et al. 2012; Adams et al. 2015). Genetic variation in miRNA genes between mouse strains is expected to influence miRNA function, and consequently contribute to the phenotypic differences between the strains.
DNA polymorphisms in miRNA loci, post-transcriptional RNA editing, and production isomiRs can affect miRNA stability, target specificity, and silencing efficiency (Kawahara et al. 2007; Kume et al. 2014). To identify the nature and extent of interstrain miRNA variation, we performed a comprehensive genome-wide characterization of mouse miRNA loci and expression across inbred strains. We investigated DNA polymorphisms and genetic conservation within miRNA loci of 36 strains. Furthermore, we carried out miRNA and isomiR expression profiling in two brain regions of six strains, the frontal cortex (FCx) and hippocampus (Hpc), central regulators of many neurobehavioral traits.
RESULTS
miRNA loci are highly conserved
To identify genetic variation within miRNA loci in the mouse genome, we downloaded SNP information based on the WGS data of 36 inbred mouse strains (Supplemental Table S1) produced by the Mouse Genomes Project (Keane et al. 2011; Wong et al. 2012; Adams et al. 2015). These strains include commonly used inbred laboratory strains and wild-derived inbred strains, and the data are provided in reference to the C57BL/6J strain (Adams et al. 2015). We analyzed variants in the 405 high-confidence pre-miRNAs (34% of the 1193 mouse pre-miRNAs) curated by miRBase (v21) and meeting criteria for functionality (Kozomara and Griffiths-Jones 2014). Of these pre-miRNAs, 51% were located within protein-coding genes and 49% elsewhere in the genome (Fig. 1A). Five miRNAs in coding sequence were located in antisense direction within the same host gene, Rtl1 that they also regulate (Davis et al. 2005), and the remaining three in sense direction within Chpf2, Dgcr8, and Rcan3 genes. The four miRNAs annotated to 3′-UTRs resided within Dvl2, Vmp1, Ppp2ca (sense direction), and Glyctk (antisense direction to the host gene).
FIGURE 1.

Genomic location of miRNAs and SNP variation within high-confidence miRNA loci. (A) Number of miRNAs located within or outside of protein-coding genes. (B) Distribution of SNPs and average nucleotide divergence (π) across miRNA loci. (C) Number of miRNAs with SNPs between 36 inbred mouse strains in the seed, mature miRNA sequence excluding the seed, and pre-miRNA excluding the mature miRNA sequence. (D) Nucleotide divergence (π) in high-confidence miRNA loci, 1 kb of 5′ and 3′ flanking sequences, random protein-coding exons, and noncoding regions. Sixteen SNPs (in 16 miRNAs) were located in the seed region (nucleotides 2–8 from the 5′ start of the canonical mature miRNA), 65 (in 52 miRNAs) in the mature miRNA sequence excluding the seed region, and 159 (in 116 miRNAs) in the pre-miRNA sequence excluding the mature miRNA and the seed sequences. Mean and standard error of the mean is calculated over all miRNA loci. All groups differ significantly from each other, except 5′ and 3′ flanking sequences (Kruskal–Wallis test, π P < 2.2 × 10−16). (CDS) Coding sequence.
We detected 242 SNPs between the strains within the pre-miRNA coordinates (Supplemental Table S2). Of the mature miRNAs (n = 809) derived from the high-confidence pre-miRNAs, 2.1% had SNP variation in the seed and 7.0% elsewhere in the mature sequence (Fig. 1B). Thirteen of the 16 seed-SNPs were detected exclusively among wild-derived strains (CAST/EiJ, MOLF/EiJ, PWK/PhJ, and SPRET/EiJ) and nine were specific to a single strain (PWK/PhJ: n = 2, Spret/EiJ: n = 4, I/LnJ: n = 1, MOLF/EiJ: n = 1, and 129S5SvEvBrd: n = 1). Nine miRNAs with a seed-SNP belonged to a ∼48 kb long, recently expanded highly repetitive miRNA cluster of 82 high-confidence miRNAs (Wang et al. 2011; Zheng et al. 2011), located in the intron of the Sfmbt2 gene. Of all polymorphic miRNAs, only four had SNPs in all three investigated regions of the pre-miRNA, suggesting that genetic variation is not concentrated only within a few miRNA loci with less selective constraint (Fig. 1C). To investigate the extent of sequence variation in different regions of the pre-miRNA loci, we calculated π, the sum of base-wise nucleotide divergence of the region, taking into account the varying lengths of the regions. The seed region π was significantly lower than the mature or precursor region, corroborating the functional importance of the seed in miRNA function (Fig. 1D). Overall, we observed less variation in miRNA loci compared to protein coding or noncoding genomic regions, suggesting high evolutionary constraint on miRNA genes.
Significant miRNA expression level differences among mouse strains
To investigate brain miRNA expression level differences across inbred mouse strains, we performed miRNA sequencing (miRNA-seq) of Hpc and FCx of six commonly used strains (129S1/SvImJ, A/J, C3H/HeJ, C57BL/6J, DBA/2J, and FVB/NJ) that differ in many behavioral phenotypes (Fig. 2; Hovatta et al. 2005; https://phenome.jax.org/). We found 779 (518 high-confidence) miRNAs expressed in the Hpc and 719 (510 high-confidence) in the FCx (Fig. 2C; Supplemental Table S3). We established that 262 miRNAs (176 high-confidence) were significantly differentially expressed between the strains (190 in Hpc, 126 in FCx, and 54 in both; adjusted P < 0.05; Fig. 2D; Supplemental Table S3) by using an unsupervised fuzzy pattern algorithm (Glez-Peña et al. 2009) for data reduction and classification, followed by analysis of variance (ANOVA). Of all expressed miRNAs, 32.8% were differentially expressed. Although most miRNAs were expressed in both brain regions (Fig. 2C), only a minority were differentially expressed in both (Fig. 2D). Expression levels of miRNAs located within a host gene did not correlate with the host gene expression levels (average R2 in Hpc = 0.12 and in FCx = 0.09 when miRNA and mRNA are located on the same strand, and Hpc = 0.07 and FCx = 0.03 when located on the different strands), indicating that miRNA expression is not dependent on host gene expression.
FIGURE 2.

miRNA expression in hippocampus (Hpc) and frontal cortex (FCx) of six inbred mouse strains. Heat map showing the expression levels of the most significantly (adjusted P ≤ 0.001) differentially expressed miRNAs in (A) Hpc (n = 74) and (B) in FCx (n = 43). N = 3 biological replicates per strain and brain region. (C) Overlap of expressed miRNAs in Hpc (n = 779) and in FCx (n = 719). (D) Overlap of differentially expressed miRNAs (adjusted P ≤ 0.05) in Hpc (n = 190) and FCx (n = 126). (DBA) DBA/2J, (AJ) A/J, (129S) 129S1/SvImJ, (C3H) C3H/HeJ, (C57) C57BL/6J, (FVB) FVB/NJ.
miRNA loci produce a broad variety of isomiRs
We used the Hpc and FCx miRNA-seq data to identify expressed isomiRs, defining them as any alternative form of the canonical miRNA (alternative 5′ start site, RNA editing, SNPs compared to the reference miRNA, and 3′ variation). We aligned the Hpc and FCx miRNA-seq reads from the six strains to mature high confidence miRNA sequences in miRBase using Miraligner (Pantano et al. 2010) and calculated the relative proportions of isomiRs for each miRNA. We included 543 Hpc and 544 FCx miRNAs in the isomiR analysis. In both brain regions >90% of miRNAs expressed noncanonical sequences. We found some rare isomiRs, but >90% of the reads aligned to the five most common isomiR (Fig. 3A). The miRBase canonical miRNA was the most highly expressed isomiR of only 53.0% and 53.6% of miRNAs in FCx and Hpc, respectively (Fig. 3B).
FIGURE 3.

Each miRNA gene produces many isomiRs with varying expression levels. (A) Contribution of the five most expressed isomiRs to the total miRNA expression levels in frontal cortex (FCx, accounting for 96.4% of all reads) and in hippocampus (Hpc, accounting for 96.0% of all reads). (B) The proportion of canonical sequences represented within the top five expressed isomiRs in Hpc (87.7%) and FCx (87.2%). (C) Distribution of the five most common isomiRs of miR-22 in the FCx. Top miR-22-3p isomiRs are expressed at a 1640-fold higher level than the 5p arm top isomiRs. The canonical isomiR is the most common, constituting 93.9% of the 3p arm expression. The canonical miRNA sequence is depicted by vertical lines. RNA-edited nucleotides are shown in red and 3′ addition in blue. (D) Distribution of the five most common isomiRs of miR-411 in Hpc. Top miR-411-5p isomiRs are more commonly expressed than miR-411-3p (50:1). Although the canonical isomiR is the most prevalent isomiR, it only constitutes 37.7% of the 5p expression. The second most common isomiR has an alternative 5′ start site and accounts for 36.2% of the overall miR-411-5p expression. (E) The expression levels of the second most common miR-411-5p isomiR vary across the six mouse strains (adjusted P = 0.036).
We detected considerable differences in the isomiR expression patterns between individual miRNAs (Fig. 3C,D). Since the location of the 5′ start site of a miRNA determines the seed position, 5′ start site variation (such as seen in miR-411-5p, Fig. 3D) may have functional importance. Twenty-eight percent of miRNAs in FCx and 27% in Hpc had at least one seed position-altering isomiR with at least 10% frequency. To determine whether Drosha or Dicer differ in the alternative start site production, we compared the number of 5′ variable 5p (determined by Drosha) and 3p (determined by Dicer) miRNAs (406 in Hpc, 408 in FCx). Dicer produced significantly more variation than Drosha both in the Hpc (Wilcoxon rank sum test, W = 91,9310, P < 2.2 × 10−16) and FCx (W = 93,5340, P < 2.2 × 10−16).
We next identified differentially expressed isomiRs (adjusted P < 0.05) between the six strains by using the fuzzy pattern algorithm followed by ANOVA. Of the 17,257 isomiRs in Hpc, 2268 (13.1%) were differentially expressed between the strains. Of the 14063 FCx isomiRs, 1051 (7.4%) were differentially expressed. Of these isomiRs, 190 were differentially expressed in both brain regions (Supplemental Table S4). An example of a differentially expressed isomiR of miR-411-5p is shown in Figure 3E.
Several miRNAs are consistently edited
MiRNAs are commonly post-transcriptionally modified by RNA editing enzymes that deaminate adenine and cytosine, or trim or insert nucleotides in the mature miRNA by nucleotidyl transferases and 3′–5′ exonucleases (Neilsen et al. 2012). To establish the frequency and variation in post-transcriptional modification of miRNAs by RNA editing enzymes, we assessed mismatches between the miRNA-seq and the WGS data. We detected 572 mismatch positions in 466 miRNAs (Fig. 4A), corresponding to 58.9% of miRNAs expressed in Hpc or FCx. Of note, 106 miRNAs (13.3%) were edited within their canonical sequence (Supplemental Table S5), the rest within ±3 bases up- or downstream from the canonical sequence, frequently expressed by isomiRs.
FIGURE 4.

Mature miRNAs are frequently edited. (A) Distribution and frequency of RNA–DNA mismatch positions in canonical mature miRNA and ±3 bases up- and downstream. Editing frequency and Shannon entropy, a measure of DNA diversity, were averaged over miRNAs with editing in the corresponding position. Editing frequencies correlated well with entropy (R2 = 0.82), indicating that higher editing frequency is associated with greater diversity in the edited position. (B) Editing frequency of miRNAs. (C) Distribution of RNA–DNA mismatches separately for the seed region, anchor sequence, and 3′-tail.
The edited reads constituted the minority of all reads in most miRNAs (Fig. 4B), as previously observed in the mouse cerebellum (Wyman et al. 2011). Overall, there was more editing within the 3′ than the 5′ end of the canonical mature miRNA (Fig. 4C). Of all RNA–DNA mismatches, 37.4% coincided with RNA-specific adenosine deaminase (ADAR) A-to-I deamination activity (detected as an A-to-G mismatch), which dominated editing of the seed (bases 2–7) and anchor (bases 10–16) regions that interact with the AGO complex and the target mRNA (Fig. 4C; Filipowicz et al. 2008). We also detected 12 RNA–DNA mismatches within bases 1–16 not compatible with ADAR activity (Supplemental Table S5). One of these, a U-to-G editing (60% of reads) of let-7a-5p at the position 9 is known to stabilize the miRNA:mRNA duplex (Reid et al. 2008). The various RNA–DNA mismatches in the 3′ end of the miRNAs are likely accountable for nucleotidyl transferase activity.
We detected nine miRNAs with editing in the seed, with editing frequencies varying from 14.8% to 46.0% on average across strains and brain regions (Supplemental Table S5). We ruled out the possibility of a DNA sequencing error in the WGS data set by capillary sequencing of all seed edited miRNAs using independent genomic DNA from the six strains. Eight of the nine seed editing events were consistent with ADAR activity. These miRNAs (miR-99b-3p, miR-411-5p, miR-379-5p, miR-376b-3p, miR-376c-3p, miR-467d-5p) were all located in clusters that are often expressed as polycistronic units, or were in an intron of a gene (miR-1251-5p and miR-3099-3p), in line with ADAR preferring longer targets than a single pre-miRNA (Pinto et al. 2014).
Editing frequencies of six A-to-I events varied significantly between brain regions, strains, or both (Fig. 5). They were found in miR-99b-3p, intronic miR-3099-3p (within gene Usp29), miR-467d-5p from the Sfmbt2 miRNA cluster, and miR-411-5p, miR-376b-3p, and miR-376c-3p from the miR-379/miR-410 cluster located in the Dlk1-Dio3 locus (Davis et al. 2005; Sekita et al. 2008; Marty et al. 2016). As RNA editing can affect miRNA target recognition, stability, and expression, it may provide significant spatiotemporal plasticity even when the DNA sequence of the miRNA is not polymorphic (Rosenthal 2015). Many of the differentially A-to-I seed edited miRNAs were also differentially expressed (miR-3099-3p, miR-467d-5p, miR-411-5p, miR-376b-3p, and miR-376c-3p) (Supplemental Table S3).
FIGURE 5.

Differences in ADAR seed editing levels between strains and brain regions. ADAR editing frequency is shown for miRNAs with significant differences between strains or brain regions in two-way ANOVA. Table shows the main effect of strain and brain region, and their interaction. Error bars denote standard error. (Hpc) Hippocampus, (FCx) frontal cortex.
Seed-edited miRNAs gain novel targets
SNPs and RNA editing within the miRNA seed region may affect its target specificity. To predict mRNA targets for the canonical and alternative forms of miRNAs, we used TargetScanMouse (Agarwal et al. 2015). On average 46.7% (range 14.6%–100.0%) of target genes of a specific miRNA were predicted both with the canonical and alternative seeds but we found large variation depending on the miRNA and position of the SNP or editing event (Supplemental Table S6). If the variation was at the end of the seed, many 8 or 7-mer target sites in the canonical sequence were instead predicted as 6-mer sites in the alternative form. When we excluded 6-mer sites from the analysis, the average overlap between the canonical and alternative predictions was 30.2% (range 7.3%–66.7%).
To validate the functional consequences of seed editing, we carried out luciferase reporter gene assays with three miRNAs (miR-376b-3p, miR-376c-3p, and miR-411-5p), all located within the miR-379/410 cluster and having an A-to-G RNA–DNA mismatch within the seed, indicative of ADAR activity. These miRNAs were highly expressed and differentially edited both between the mouse strains and brain regions. The functional consequence of these conserved editing events has not been established. For each miRNA, we selected two or three mRNA targets predicted to be silenced by the edited but not the nonedited form of the miRNA. We verified that all target genes were expressed in the corresponding brain region using RNA sequencing from the same total RNA. Selected edited miRNAs significantly silenced five of the seven predicted acquired targets and none of the targets of the canonical miRNA (Fig. 6). Thus, RNA editing changes the target repertoire of the miR-379/410 cluster miRNAs and may thereby have significant functional consequences.
FIGURE 6.
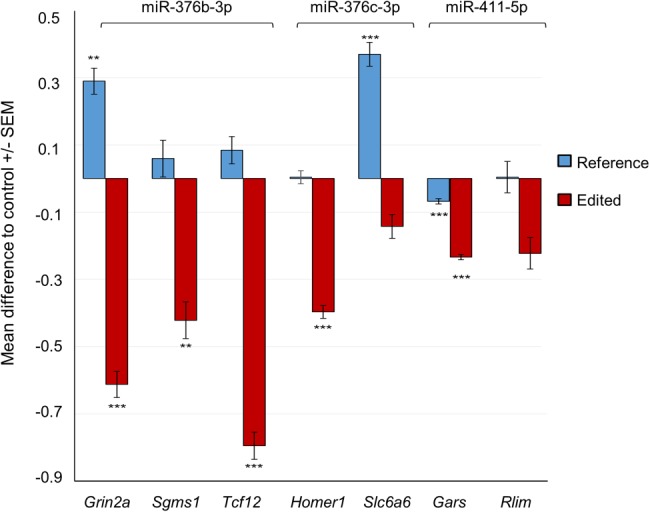
RNA-edited miRNAs acquire novel target mRNAs not recognized by the unedited form. Relative levels of luciferase activity (firefly/Renilla luciferase ratio) after cotransfecting HEK293FT cells with either the edited form of the miRNA (mimic containing A) or the nonedited reference form (mimic containing G) and the target site sequence predicted for the edited miRNA. For all the analyses, the miRNA effect was compared with the negative control miRNA effect. We performed two independent experiments for each miRNA and carried out two-way ANOVA adjusting for the assay batch. Pairwise comparisons were calculated with Tukey HSD, adjusting for multiple testing. Five of seven tested alternative targets were significantly silenced by the edited form of the miRNA, but not the reference form. (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. Grin2a, NMDA2A ionotropic glutamate receptor; Sgms1, sphingomyelin synthase 1; Tcf12, transcription factor 12; Homer1, homer scaffolding protein 1; Slc6a6, solute carrier family 6 (neurotransmitter transporter, taurine), member 6; Gars, glycyl-tRNA synthetase; and Rlim, ring finger protein, LIM domain interacting.
Identification of 32 putative novel miRNAs
MiRNA expression profiling using next-generation sequencing reveals putative novel expressed miRNA genes, which can be truly novel or are orthologous to miRNAs described in other species (Desvignes et al. 2015). We detected 32 miRNA candidates within our deep miRNA-seq data predicted by both miRDeep2 (Friedländer et al. 2008) and sRNAbench (Rueda et al. 2015) programs (Fig. 7; Supplemental Table S7). Twenty-eight (87.5%) were located in an intron of a protein-coding gene, and five of them intersected an exon, suggesting that they may be 5′ or 3′ tailed miRtrons. Twenty-four (75%) novel candidates met the miRBase high-confidence criteria (http://www.mirbase.org/; Kozomara and Griffiths-Jones 2014), the remaining eight failing due to excessive 3′ overhang. We investigated whether the novel miRNA candidates were orthologs of known miRNAs in other species. Most of the novel miRNAs were expressed at a low level in the bulk tissue but some seem to be differentially expressed between the strains (Fig. 7). None of the 5p and 3p pairs aligned together to any animal pre-miRNAs found in miRBase, but two pre-miRNA sequences aligned to the rat genome with 93.1%–96.9% homology (Supplemental Table S6). None aligned to the human genome. Thus, these miRNAs are novel and they may be rodent-specific. Since our data set only contains sequence reads of mature miRNAs, we selected four candidate pre-miRNAs for validation using PCR amplification of cDNA using independent RNA samples from FCx (Supplemental Table S7). All full pre-miRNAs were expressed corroborating that they are derived from active miRNA loci.
FIGURE 7.
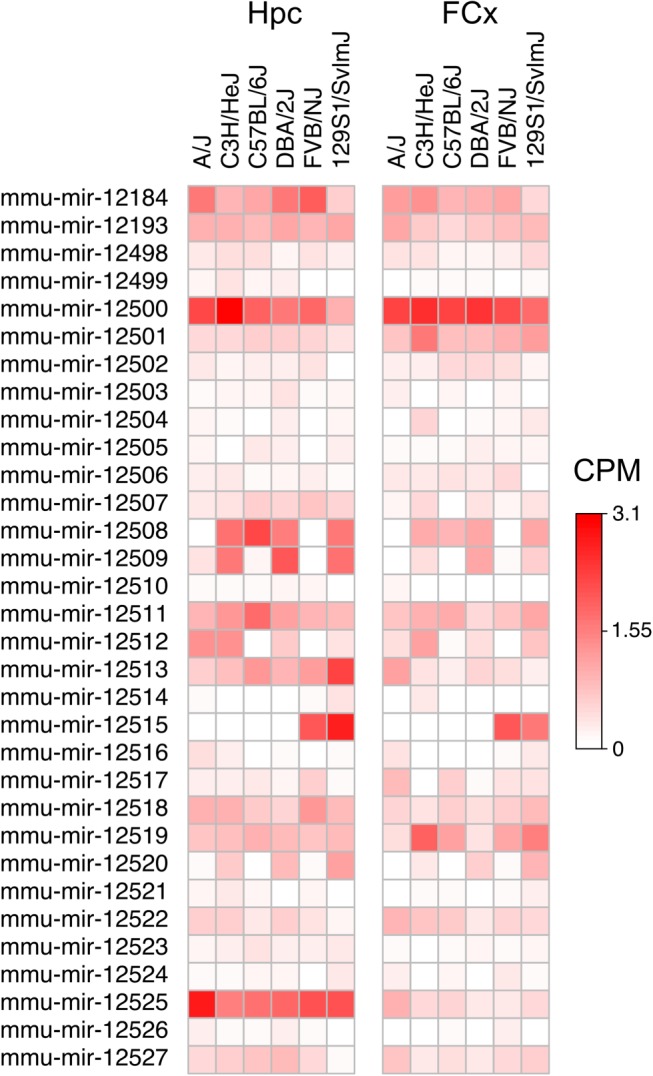
Predicted novel miRNAs and their expression levels in hippocampus (Hpc) and frontal cortex (FCx). (CPM) Counts per million.
DISCUSSION
We have performed a comprehensive analysis of genetic variation within mouse miRNA loci, and determined the diversity of miRNAs expressed in the mouse brain. By using WGS data from 36 inbred laboratory and wild-derived strains, we established that although the functionally important regions of the miRNA genes were conserved, 7.0% of mature miRNAs have SNP variation. Using small RNA sequencing of Hpc and FCx we demonstrated that 58.9% of miRNAs are consistently edited, mostly in a manner compatible with known RNA editing enzymes. A significant proportion of variation in the expressed mature miRNAs was also due to variation in Drosha and Dicer processing leading to 5′ and 3′ heterogeneity, with Dicer causing significantly more variation than Drosha, as reported before (Hu et al. 2009).
We found miRNA loci to be highly conserved, implying that mutations in these genes will likely be deleterious. The entire pre-miRNA region was on average more conserved than protein-coding genes, as described before in Cichlid fish species (Franchini et al. 2016). The strong conservation of the whole locus reflects the importance of the entire pre-miRNA sequence in miRNA function, including maintenance of the proper hairpin structure for Drosha and Dicer processing, mediation of target specificity, and miRNA stability. The seed was the most conserved region, as previously observed in humans (Chen and Rajewsky 2006; Saunders et al. 2007; Quach et al. 2009), and only 2.1% of miRNAs had SNP variation within the seed. Not surprisingly, most of this variation occurred in wild-derived strains because of higher genetic diversity compared to the laboratory strains.
Of the 16 SNPs we observed within the seed, nine were located within the repetitive Sfmbt2 miRNA cluster that has expanded relatively recently among rodents through duplications (Zheng et al. 2011). This situation has likely provided relaxed selection on miRNA copies allowing mutations to produce variable seeds, leading to the opportunity for the miRNAs to gain new functions. We did not observe comparable occurrence of seed SNPs in a much older large miR-379/miR-410 cluster (also known as C14MC in humans) on chromosome 12, with evolution dating back to the common ancestor of placental mammals (Marty et al. 2016). However, we observed considerable RNA level variation within miRNAs of both clusters. miRNAs of the miR-379/miR-410 cluster were often edited (see below), and four of the 82 high confidence miRNAs in the Sfmtb2 cluster and 21 of the 86 miRNAs in the miR-379/410 cluster were differentially expressed between the strains. Both Sfmbt2 and miR-379/miR-410 clusters are imprinted in mouse providing means for miRNA clusters to convey epigenetic information to offspring (Wang et al. 2011; Zheng et al. 2011). Both clusters are involved in the regulation of metabolism and emotional behavior in a parent-of-origin-dependent manner (Marty et al. 2016; Wu et al. 2016).
Although many miRNAs are conserved between species, new miRNAs emerge in the genome through gene duplication and from introns of protein-coding genes (Berezikov et al. 2011; Meunier et al. 2013). We identified 32 putative novel miRNAs expressed in the mouse brain, and most of them were coded from introns of protein-coding genes in line with this notion. Seventeen were expressed (>0.75 CPM) in more than one studied laboratory strain and nine in all strains, suggesting that they have emerged before the separation of these strains but after evolution of rodents, since only two predicted miRNAs aligned against the rat genome and the rest were mouse-specific.
We detected considerable brain miRNA expression differences between the six studied laboratory strains as 262 of the 800 expressed miRNAs were significantly differentially expressed in one (26.0%) or both (6.8%) brain regions. Furthermore, we observed substantial additional variation in the expressed mature miRNAs created by consistent RNA editing and production of multiple isomiRs from a single miRNA locus. We observed 58.9% of the expressed miRNAs to be edited in the brain. The majority of the edits were located in the 3′ end of the miRNAs likely affecting miRNA stability as polyadenylation generally promotes stability and polyuridylation degradation (Chung et al. 2017). The most common pattern in the 5′ and seed regions was consistent with ADAR activity, which was responsible for 37.4% of all events. ADAR compatible editing is the most common editing form in the whole-brain mRNAs of inbred mouse strains, with more than 80% of edited reads compatible with ADAR activity (Danecek et al. 2012). In humans, ADAR activity is estimated to affect 16% of pri-miRNAs (Kawahara et al. 2008). We found nine editing events within the miRNA seed, all previously described in the mouse or human with similar editing frequencies (Kawahara et al. 2008; Chiang et al. 2010; Ekdahl et al. 2012; Vesely et al. 2012, 2014). Four seed-edited miRNAs belong to the miR-379/miR-410 cluster and are highly enriched in neurons compared to other brain cell types (Jovičić et al. 2013). We established that the editing frequencies of miR-376b-3p, miR-376c-3p, and miR-411-5p differ significantly between the six strains and brain regions (Fig. 5). They are all known targets of ADAR and the editing sites are conserved between mouse and human (Warnefors et al. 2014), suggesting they have functional importance in the brain that dates back to the common ancestor of these taxa. Although editing frequencies can differ significantly between tissues, many editing sites are conserved across mammals (Vesely et al. 2014), supporting the relevance of mouse as a model to investigate consequences of brain RNA editing for traits shared with humans.
Another layer of diversity is provided by the expression of isomiRs, mature miRNAs produced from the same hairpin but differing in their 5′ and 3′ termini or having RNA editing. They can be due to variation in Drosha and Dicer processing and nontemplated 3′ nucleotide additions (for review, see Berezikov et al. 2011). While we detected a large number of isomiRs, most sequence reads typically mapped to a few highly expressed isomiRs. miRBase canonical miRNA sequence is defined as the most highly expressed isomiR of the first experiment in which the miRNA was described. In our brain miRNA expression data, the miRBase canonical sequence was the most highly expressed isomiR of 60% of miRNAs. For 12% of brain expressed miRNAs the canonical sequence was not among the top five most expressed isomiRs. Overall, our finding on the production of numerous isomiRs from a single locus agrees with recent work on retinal miRNAs (Karali et al. 2016) and supports the notion that instead of focusing on canonical miRNAs, the full diversity of expressed isomiRs should be considered (Desvignes et al. 2015).
What are the functional consequences of miRNA seed SNPs, isomiRs, and seed editing? While the 3′ end of the mature miRNA is considered to contribute relatively little to the target specificity, variation in the 5′ end that changes the seed position, and SNPs or RNA editing in the seed sequence are expected to have a larger effect. In the case of 5′ isomiRs, seed shifting may be a mechanism for miRNAs to acquire divergent function, such as in the case of Nematostella and bilaterian miR-100 (Grimson et al. 2008). We observed several seed SNPs between the mouse strains and seed editing that were predicted to affect target specificity. In the cases where the SNP or editing event was observed at the end of the seed, the predicted target often remained the same, but having a 1 nt shorter seed, possibly leading to weaker targeting. If the SNP or editing event was located in the middle of the seed, the overlap of the predicted targets for the two alternative alleles was small.
To demonstrate the functional effect of seed editing, we examined three miRNAs from the miR-379/miR-410 cluster and their editing sites that are conserved between mouse and human. Previously, RNA editing of miR-376a within the cluster was shown to change its target gene repertoire (Kawahara et al. 2007). The deletion of the entire cluster affects anxiety-like behavior in mice (Marty et al. 2016), a phenotype regulated by FCx and Hpc and segregating in the six mouse strains we studied (Hovatta et al. 2005). We demonstrated that edited forms of 376b-3p, miR-376c-3p, and miR-411-5p that were all differentially edited between the mouse strains and brain regions, gained the ability to inhibit the expression of novel targets, also expressed within the same brain regions. Thus, RNA editing may have considerable functional significance to the gene regulatory networks of the miR-379/miR-410 cluster members and thereby modulate the phenotypes these miRNAs regulate. Demonstrating the RNA editing effect on a network level remains challenging due to technical limitations, but editing can have functional importance also when occurring outside the seed region, and it may alter miRNA binding properties, stability, processing, and expression levels of mature miRNAs (Kawahara et al. 2008; Zhang and Zeng 2010).
To conclude, we demonstrate that miRNA loci were strongly conserved between 36 inbred mouse strains, but even the highly conserved seed region contained 16 SNPs, although 13 were found only in wild-derived strains. In contrast, strain variation had a substantial impact on miRNA expression and most miRNA loci produced several isomiRs and/or were post-transcriptionally edited in a strain or brain region-dependent manner. miRNAs may therefore provide an example of contrasting selective regimes within the mouse genome, with purifying selection at the level of genes but diversifying selection at the level of miRNA expression. The laboratory mouse strains have been created by selective breeding but the mechanistic processes underpinning miRNA function are the same as in natural populations. Our findings provide the first comprehensive analysis of SNP, isomiR, and RNA editing variation in miRNA loci across inbred mouse strains, and a detailed catalog of expressed miRNAs in Hpc and FCx in six commonly used strains. These findings will facilitate the molecular analysis of neurological and behavioral phenotypes in inbred mouse strains and the crosses created using them, such as the Diversity Outbred or Collaborative Cross strains (Churchill et al. 2004, 2012).
MATERIALS AND METHODS
SNP variation in miRNA genes
We downloaded the publicly available variant call data generated as part of the Mouse Genomes Project from 36 inbred mouse strains (http://www.sanger.ac.uk/science/data/mouse-genomes-project; Supplemental Table S1). These data contain sequence variation relative to the C57BL/6J mouse reference genome.
We analyzed genomic variation in high-confidence mouse miRNA loci of miRBase release 21 (www.mirbase.org), including 405 pre-miRNA loci giving rise to 809 mature miRNAs. To estimate sequence variability of genetic regions of interest with length L, we calculated length-normalized per mille sum of position-wise nucleotide divergence π over positions i:
using VCFtools v0.1.13 (Nei and Li 1979; Danecek et al. 2011). From variation estimates of the 5′ and 3′ flanking regions, we excluded clustered miRNAs and miRtrons due to overlap with other miRNAs or exons (remaining n = 177). We also analyzed the degree of DNA variation from a random subset of 60-bp intergenic noncoding fragments (n = 388) and protein-coding exons (n = 500).
We manually inspected all seed SNPs and omitted two as likely false positives due to alignment issues. We observed two different alleles of the multicopy miR-669a in the PWK/PhJ and SPRET/EiJ strains (chr2:10479332, chr2:10501360; Supplemental Table S2). At the same time, there was a deletion in this locus compared to the reference C57BL/6J strain, suggesting complex genetic rearrangements that are challenging to resolve with short read data (http://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1505).
We classified miRNAs based on whether their pre-miRNA coordinates overlapped completely an exon or intron of a protein-coding gene using GRCm38/mm10 genome annotation and BEDtools v2.17.0 (Quinlan and Hall 2010). In case of annotation to multiple features, annotation to CDS was selected over UTR, and UTR over intron. We defined miRtrons as described in Ladewig et al. (2012).
RNA sequence data
We conducted miRNA-seq of FCx and Hpc from 7-wk-old male mice (The Jackson Laboratory) of six strains: A/J, 129S1/SvImJ, C57BL/6J, C3H/HeJ, DBA/2J, and FVB/NJ, three animals per strain. Mice were singly housed for one week before dissections and killed by cervical dislocation between 8 a.m. and 11 a.m. Dissections were performed on a Petri dish filled with ice. FCx included 2 mm of the anterior part of the cortex. The Hpc was dissected whole. Samples were directly frozen in liquid nitrogen and stored in −80oC. We extracted total RNA with TriReagent according to the manufacturer's instructions and verified its quality by the Agilent Bioanalyzer Small RNA Analysis and RNA 6000 Nano Kits. We prepared the sequencing libraries with the TruSeq Small RNA Library Preparation Kit (Illumina), enriched the libraries for miRNA inserts by size selection using the S2 ultrasonicator (Covaris), and sequenced (single-end, 101 bp) them on Illumina HiSeq 2000 (Illumina) by the Finnish Institute for Molecular Medicine (FIMM) Technology Centre. Data distribution is presented in Supplemental Table S8.
We pre-processed miRNA-seq reads keeping adapter clipped reads of 17–32 bp and containing no unknown nucleotides. We aligned them to the mouse reference miRNAs (miRBase v21) using miRDeep2 (Friedländer et al. 2008) and SeqBuster (Pantano et al. 2010), and to the mouse genome (GRCm38) using bowtie v1.1.1 (--all --strata --best) (Langmead et al. 2009), allowing three alignment mismatches due to miRNA and strain variability.
miRNA and isomiR expression
To quantify expression levels, miRDeep2 read counts were normalized with the limma package and voom (Smyth 2004; Law et al. 2014), and Hpc samples were batch-adjusted using the ComBat function of the package SVA (Johnson et al. 2007; Leek and Storey 2008) in R. Differences in normalized expression levels were determined by the DFP package (Glez-Peña et al. 2009), followed by one-way ANOVA, and P-values were adjusted for multiple testing using the Benjamini–Hochberg method (Benjamini and Hochberg 1995). Heat map was drawn from the statistically most significant differentially expressed miRNAs (adjusted P ≤ 0.001) using z-scores as it scales better for plotting than more variable fold change.
To determine 5′ and 3′ isomiR variation, we analyzed SeqBuster alignments for reads mapping uniquely to high-confidence miRNAs, and having a frequency of at least 10 reads combined across all samples. By limiting the analysis to high-confidence miRNAs, we wanted to focus on miRNAs with real functional importance and to limit the chance of unreliable calls to skew results.
To analyze 5′ variation due to Drosha and Dicer processing, we examined miRNA 5′ start site differences in 5p and 3p arms of 406 (in Hpc) and 408 (in FCx) high-confidence miRNAs with both arms listed in miRBase. Based on the miraligner results, reads were separated to canonical 5′ cut sites and alternative 5′ sites, and a ratio between their frequencies was calculated per miRNA. P-values for the differences between the 5p and 3p arms were calculated in R using the Wilcoxon rank sum test.
miRNA editing
We called RNA–DNA mismatch positions from unique genome alignments using FreeBayes v0.9.21 (Garrison and Marth 2012) in pooled-continuous mode. To call edited positions we required at minimum 10 high-quality bases (Phred ≥ 20) supporting the reference base and alternative calls with at least 5% frequency for both, and including only variant calls of Phred ≥ 30. The RNA–DNA mismatches originating from the correct strand in each miRNA locus were annotated to mature miRNAs and to the three bases from the canonical miRNA start and end positions. We omitted 19 RNA–DNA mismatches from the analysis as they coincided with known SNPs between the strains and one that was called due to an indel in the C3H/HeJ strain. In addition, manual curation of RNA–DNA mismatches in functionally important regions overlapping the seed and anchor sequences led us to remove four editing events, which were likely called due to misalignments between miRNAs with nearly identical sequence.
To further validate the seed-located RNA editing events, we carried out Sanger sequencing of genomic DNA (Jackson Laboratory) from all six strains. We PCR-amplified the genomic locus 50–150 bp around the mature miRNA gene using Phusion High-Fidelity PCR mix (Thermo Scientific) and sequenced the amplicons using ABI3700 capillary sequencer (Applied Biosystems).
To calculate the magnitude of miRNA editing, we calculated for each variable position the average editing frequency and average Shannon entropy H:
where pi is the frequency of each of four nucleotides i at the miRNA position and n is the total number of miRNAs with variation at the position.
Target prediction
We performed target predictions using TargetScanMouse v7.1 for all miRNAs that had seed SNPs (n = 16) or editing events (n = 9). No conservation or context++ scores were used. To limit the predicted targets to those expressed in the corresponding brain region, we built a custom set of expressed UTRs based on mRNA sequence alignment files from the same total RNA samples as used in miRNA-seq. We computed transcript predictions with Cufflinks v2.2.1 using alignment files produced with TopHat 2.0.0 (Trapnell et al. 2009) to the mouse genome (NCBIM37). We subtracted protein-coding exons from these predictions and selected UTR-matching expressed sequences from the M1 encode gene map with BEDtools v2.17.0 and custom R-scripts, assigning each predicted transcript to the transcript UTR with which it had the longest overlap of coordinates. This resulted in 21059 UTR-overlapping sequences on the same strand as the coding gene region.
Reporter gene assays
We carried out target prediction using TargetScan and miRDB v5.0 (Wong and Wang 2015; Wang 2016) for edited and canonical forms of miR-411-5p, miR-376c-3p, and miR-376b-3p. We selected targets for reporter gene assays using the following criteria: (i) predicted by both algorithms; (ii) predicted to be targeted by the seed-edited, but not the canonical miRNA; and (iii) reported importance for brain function. This resulted in the following targets: miR-411-5p (fourth seed base A-to-G; targets Gars, Rlim), miR-376c-3p (fifth seed base A-to-G, targets Homer1, Slc6a6), and miR-376b-3p (fifth seed base A-to-G; targets Grin2a, Sgms1, and Tcf12). Target sequence together with approximately ±30 bp flanking sequence were cloned into pmirGLO dual-luciferase expression vector (Promega). Vector constructs (100 ng) and miRNA or negative control mimics (1 µmol, Dharmacon) were reverse co-transfected using DharmaFECT Duo (0.12 µL/well; Dharmacon) to HEK293FT cells cultured in 70 µL of DMEM medium on 96-well plates. Each assay was done in six replicates and repeated twice. Cells were grown for 40 h, and the signal of primary reporter including miRNA target sequence in 3′-UTR (luc2) and control reporter (hRluc-neo) were quantified with Dual-Luciferase Reporter Assay System (Promega) using EnSpire2300 plate reader (PerkinElmer). Data were analyzed with two-way ANOVA and Tukey HSD pairwise comparisons in R. We used DISC1 UTR construct and siRNA (Rossi et al. 2014) as a positive control on each reaction plate, and it silenced the Renilla luciferase calibrated firefly luciferase signal on average by 67.3% (P < 2 × 10−16).
Identification of novel miRNAs
We predicted novel miRNAs with miRDeep2 v0.0.7 (Friedländer et al. 2008) and sRNAbench (sRNAtoolbox v1.0; Rueda et al. 2015) using 633 million miRNA-seq reads from the Hpc and FCx of the six strains. Predictions were done with all read data combined for greatest sensitivity. To consider a reported locus as a candidate novel miRNA, we required overlap of pre-miRNA coordinates by >90% by both software and designation as high-quality prediction by sRNAbench or score >5 in miRDeep2. These candidates were then investigated for their confidence based on miRBase criteria (Kozomara and Griffiths-Jones 2014). To detect origin of the predicted novel miRNAs, we aligned read libraries individually to the predicted miRNAs using bowtie v1.1.1. To detect homology with known miRNAs, we aligned the predicted 5p and 3p miRNAs to the miRBase pre-miRNA sequences of all mammalian species with bowtie, allowing three mismatches after trimming three 3′ bases, and to rat (RGSC 6.0/rn6) and human (GRCh38/hg38) genomes with NCBI BLAST.
We chose four candidates for validation using PCR amplification. One µg of total RNA, extracted from FCx of C57BL/6J strain using TriReagent (MRC Inc.), was treated with 1 U DNase I (Thermo Scientific) and converted to cDNA using the iScript first strand select cDNA synthesis kit (Bio-Rad) with pre-miRNA-specific primers. Unincorporated primers were removed from cDNA reactions with NucleoSpin PCR purification columns (Macherey-Nagel) and cDNA was PCR-amplified using the Phusion enzyme with GC buffer (Thermo Scientific). Amplification products were length-separated on a 2.5% agarose gel by electrophoresis and cloned into the pCR2.1 vector (Invitrogen). Inserts were Sanger sequenced to confirm identity with the predicted pre-miRNA.
DATA DEPOSITION
The miRNA-seq data set and variant call table for miRNA–DNA mismatches are available in the GEO repository (http://www.ncbi.nlm.nih.gov/geo/), accession number GSE84408.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Centre for Scientific Computing (CSC), Espoo, Finland for computing facilities; Pekka Ellonen, Jesus Lopez Marti, and Pirkko Mattila from the Finnish Institute for Molecular Medicine, Technology Centre, University of Helsinki for carrying out RNA-seq and miRNA-seq; Jenni Lahtinen and Suvi Saarnio for assistance with laboratory work; and Mikko Frilander, Katherine Icay, Ari Löytynoja, and Hovatta laboratory members for helpful discussions. This work was funded by the European Research Council Starting Grant (GenAnx 281559), Academy of Finland (grant no. 140747 and 218017), and the Sigrid Jusélius Foundation (to I.H.).
Author contributions: I.H. conceived of the study and led the research. K.T., J.V., T.S., D.G., and I.H. contributed to the design of the study and critical discussion of the results. K.T., J.V., and I.H. performed the laboratory work and data analysis. K.T., J.V., and I.H. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064881.117.
REFERENCES
- Adams DJ, Doran AG, Lilue J, Keane TM. 2015. The Mouse Genomes Project: a repository of inbred laboratory mouse strain genomes. Mamm Genome 26: 403–412. [DOI] [PubMed] [Google Scholar]
- Agarwal V, Bell GW, Nam J, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H. 2005. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 33: 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455: 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baskerville S, Bartel DP. 2005. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA 11: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300. [Google Scholar]
- Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung J, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. 2011. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 21: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervino AC, Li GY, Edwards S, Zhu J, Laurie C, Tokiwa G, Lum PY, Wang S, Castellini LW, Lusis AJ, et al. 2005. Integrating QTL and high-density SNP analyses in mice to identify Insig2 as a susceptibility gene for plasma cholesterol levels. Genomics 86: 505–517. [DOI] [PubMed] [Google Scholar]
- Chen K, Rajewsky N. 2006. Natural selection on human microRNA binding sites inferred from SNP data. Nat Genet 38: 1452–1456. [DOI] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. 2010. Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev 24: 992–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CZ, Seidl LE, Mann MR, Heinemann IU. 2017. Tipping the balance of RNA stability by 3′ editing of the transcriptome. Biochim Biophys Acta 1861: 2971–2979. [DOI] [PubMed] [Google Scholar]
- Churchill G, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, Beavis WD, Belknap JK, Bennett B, Berrettini W, et al. 2004. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet 36: 1133–1137. [DOI] [PubMed] [Google Scholar]
- Churchill GA, Gatti DM, Munger SC, Svenson KL. 2012. The diversity outbred mouse population. Mamm Genome 23: 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Nellåker C, McIntyre RE, Buendia-Buendia JE, Bumpstead S, Ponting CP, Flint J, Durbin R, Keane TM, Adams DJ. 2012. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol 13: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis E, Caiment F, Tordoir X, Cavaillé J, Ferguson-Smith A, Cockett N, Georges M, Charlier C. 2005. RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol 15: 743–749. [DOI] [PubMed] [Google Scholar]
- Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, Singer A, Postlethwait JH. 2015. miRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet 31: 613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekdahl Y, Farahani HS, Behm M, Lagergren J, Öhman M. 2012. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res 22: 1477–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. 2008. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9: 102–114. [DOI] [PubMed] [Google Scholar]
- Franchini P, Xiong P, Fruciano C, Meyer A. 2016. The role of microRNAs in the repeated parallel diversification of lineages of Midas cichlid fish from Nicaragua. Genome Biol Evol 8: 1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedländer MR, Chen W, Adamidi C, Maaskola J, Einspanier R, Knespel S, Rajewsky N. 2008. Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol 26: 407–415. [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv:1207.3907. [Google Scholar]
- Glez-Peña D, Alvarez R, Díaz F, Fdez-Riverola F. 2009. DFP: a Bioconductor package for fuzzy profile identification and gene reduction of microarray data. BMC Bioinformatics 10: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP. 2008. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature 455: 1193–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, Ellison JA, Schadt EE, Verma IM, Lockhart DJ, et al. 2005. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 438: 662–666. [DOI] [PubMed] [Google Scholar]
- Hovatta I, Zapala MA, Broide RS, Schadt EE, Libiger O, Schork NJ, Lockhart DJ, Barlow C. 2007. DNA variation and brain region-specific expression profiles exhibit different relationships between inbred mouse strains: implications for eQTL mapping studies. Genome Biol 8: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu HY, Yan Z, Xu Y, Hu H, Menzel C, Zhou YH, Chen W, Khaitovich P. 2009. Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics 10: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. 2001. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science 293: 834–838. [DOI] [PubMed] [Google Scholar]
- Ideraabdullah FY, de la Casa-Esperón E, Bell TA, Detwiler DA, Magnuson T, Sapienza C, de Villena FP. 2004. Genetic and haplotype diversity among wild-derived mouse inbred strains. Genome Res 14: 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8: 118–127. [DOI] [PubMed] [Google Scholar]
- Jovičić A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, Luthi-Carter R. 2013. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci 33: 5127–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhila J, Sipilä T, Icay K, Nicorici D, Ellonen P, Kallio A, Korpelainen E, Greco D, Hovatta I. 2011. MicroRNA expression profiling reveals miRNA families regulating specific biological pathways in mouse frontal cortex and hippocampus. PLoS One 6: e21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karali M, Persico M, Mutarelli M, Carissimo A, Pizzo M, Marwah VS, Ambrosio C, Pinelli M, Carrella D, Ferrari S, et al. 2016. High-resolution analysis of the human retina miRNome reveals isomiR variations and novel microRNAs. Nucleic Acids Res 44: 1525–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. 2007. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 315: 1137–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Megraw M, Kreider E, Iizasa H, Valente L, Hatzigeorgiou AG, Nishikura K. 2008. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res 36: 5270–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara H, Imai T, Okano H. 2012. MicroRNAs in neural stem cells and neurogenesis. Front Neurosci 6: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. 2011. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477: 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S. 2014. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42: D68–D73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume H, Hino K, Galipon J, Ui-Tei K. 2014. A-to-I editing in the miRNA seed region regulates target mRNA selection and silencing efficiency. Nucleic Acids Res 42: 10050–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. 2012. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 22: 1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, Pfeffer S, Rice A, Kamphorst AO, Landthaler M, et al. 2007. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129: 1401–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. 2014. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15: r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. 1993. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han JJ, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, et al. 2003. The nuclear RNase III Drosha initiates microRNA processing. Nature 425: 415–419. [DOI] [PubMed] [Google Scholar]
- Leek JT, Storey JD. 2008. A general framework for multiple testing dependence. Proc Natl Acad Sci 105: 18718–18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty V, Labialle S, Bortolin-Cavaillé M, De Medeiros GF, Moisan M, Florian C, Cavaillé J. 2016. Deletion of the miR-379/miR-410 gene cluster at the imprinted Dlk1-Dio3 locus enhances anxiety-related behaviour. Hum Mol Genet 25: 728–739. [DOI] [PubMed] [Google Scholar]
- Meunier J, Lemoine F, Soumillon M, Liechti A, Weier M, Guschanski K, Hu H, Khaitovich P, Kaessmann H. 2013. Birth and expression evolution of mammalian microRNA genes. Genome Res 23: 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Li WH. 1979. Mathematical model for studying genetic-variation in terms of restriction endonucleases. Proc Natl Acad Sci 76: 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen CT, Goodall GJ, Bracken CP. 2012. IsomiRs—the overlooked repertoire in the dynamic microRNAome. Trends Genet 28: 544–549. [DOI] [PubMed] [Google Scholar]
- Pantano L, Estivill X, Martí E. 2010. SeqBuster, a bioinformatic tool for the processing and analysis of small RNAs datasets, reveals ubiquitous miRNA modifications in human embryonic cells. Nucleic Acids Res 38: e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto Y, Cohen HY, Levanon EY. 2014. Mammalian conserved ADAR targets comprise only a small fragment of the human editosome. Genome Biol 15: R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach H, Barreiro LB, Laval G, Zidane N, Patin E, Kidd KK, Kidd JR, Bouchier C, Veuille M, Antoniewski C, et al. 2009. Signatures of purifying and local positive selection in human miRNAs. Am J Hum Genet 84: 316–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JG, Nagaraja AK, Lynn FC, Drabek RB, Muzny DM, Shaw CA, Weiss MK, Naghavi AO, Khan M, Zhu H, et al. 2008. Mouse let-7 miRNA populations exhibit RNA editing that is constrained in the 5′-seed/cleavage/anchor regions and stabilize predicted mmu-let-7a:mRNA duplexes. Genome Res 18: 1571–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal JJ. 2015. The emerging role of RNA editing in plasticity. J Exp Biol 218: 1812–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Kilpinen H, Muona M, Surakka I, Ingle C, Lahtinen J, Hennah W, Ripatti S, Hovatta I. 2014. Allele-specific regulation of DISC1 expression by miR-135b-5p. Eur J Hum Genet 22: 840–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueda A, Barturen G, Lebrón R, Gómez-Martin C, Alganza Á, Oliver JL, Hackenberg M. 2015. sRNAtoolbox: an integrated collection of small RNA research tools. Nucleic Acids Res 43: W467–W473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders MA, Liang H, Li W. 2007. Human polymorphism at microRNAs and microRNA target sites. Proc Natl Acad Sci 104: 3300–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekita Y, Wagatsuma H, Nakamura K, Ono R, Kagami M, Wakisaka N, Hino T, Suzuki-Migishima R, Kohda T, Ogura A, et al. 2008. Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet 40: 243–248. [DOI] [PubMed] [Google Scholar]
- Smyth GK. 2004. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, von Haeseler A, Jantsch MF. 2012. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res 22: 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesely C, Tauber S, Sedlazeck FJ, Tajaddod M, von Haeseler A, Jantsch MF. 2014. ADAR2 induces reproducible changes in sequence and abundance of mature microRNAs in the mouse brain. Nucleic Acids Res 42: 12155–12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. 2016. Improving microRNA target prediction by modeling with unambiguously identified microRNA-target pairs from CLIP-ligation studies. Bioinformatics 32: 1316–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Chow J, Hong J, Smith AF, Moreno C, Seaby P, Vrana P, Miri K, Tak J, Chung ED, et al. 2011. Recent acquisition of imprinting at the rodent Sfmbt2 locus correlates with insertion of a large block of miRNAs. BMC Genomics 12: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnefors M, Liechti A, Halbert J, Valloton D, Kaessmann H. 2014. Conserved microRNA editing in mammalian evolution, development and disease. Genome Biol 15: r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruckun G. 1993. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern-formation in C. elegans. Cell 75: 855–862. [DOI] [PubMed] [Google Scholar]
- Wolter JM, Le HH, Linse A, Godlove VA, Nguyen TD, Kotagama K, Lynch A, Rawls A, Mangone M. 2017. Evolutionary patterns of metazoan microRNAs reveal targeting principles in the let-7 and miR-10 families. Genome Res 27: 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, Wang X. 2015. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res 43: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, Bumpstead S, Van Der Weyden L, Reinholdt LG, Wilming LG, Adams DJ, Keane TM. 2012. Sequencing and characterization of the FVB/NJ mouse genome. Genome Biol 13: R72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Lu Y, Jiao Y, Liu B, Li S, Li Y, Xing F, Chen D, Liu X, Zhao J, et al. 2016. Paternal psychological stress reprograms hepatic gluconeogenesis in offspring. Cell Metab 23: 735–743. [DOI] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, Tewari M. 2011. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res 21: 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalcin B, Fullerton J, Miller S, Keays DA, Brady S, Bhomra A, Jefferson A, Volpi E, Copley RR, Flint J, et al. 2004. Unexpected complexity in the haplotypes of commonly used inbred strains of laboratory mice. Proc Natl Acad Sci 101: 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zeng Y. 2010. The terminal loop region controls microRNA processing by Drosha and Dicer. Nucleic Acids Res 38: 7689–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Srivastava D. 2007. A developmental view of microRNA function. Trends Biochem Sci 32: 189–197. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Ravi A, Gould GM, Burge CB, Sharp PA. 2011. Genome-wide impact of a recently expanded microRNA cluster in mouse. Proc Natl Acad Sci 108: 15804–15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.