

Abstract
Grc3 is an essential well-conserved eukaryotic polynucleotide kinase (PNK) that cooperates with the endoribonuclease Las1 to process the preribosomal RNA (rRNA). Aside from being dependent upon Las1 for coordinated kinase and nuclease function, little is known about Grc3 substrate specificity and the molecular mechanisms governing kinase activity. Here we characterize the kinase activity of Grc3 and identify key similarities and differences between Grc3 and other polynucleotide kinase family members. In contrast to other PNK family members, Grc3 has distinct substrate preference for RNA substrates in vitro. By disrupting conserved residues found at the Grc3 kinase active site, we identified specific residues required to support Grc3-directed Las1-mediated pre-rRNA cleavage in vitro and in vivo. The crosstalk between Grc3 and Las1 ensures the direct coupling of cleavage and phosphorylation during pre-rRNA processing. Taken together, our studies provide key insight into the polynucleotide kinase activity of the essential enzyme Grc3 and its molecular crosstalk with the endoribonuclease Las1.
Keywords: polynucleotide kinase, Grc3, Las1, RNA processing, crosstalk
INTRODUCTION
Polynucleotide kinases (PNKs) are enzymes that play fundamental roles in numerous cellular processes including the repair of DNA, as well as the processing, maturation, repair, and degradation of RNA (Weinfeld et al. 2011; Das et al. 2014; Dikfidan et al. 2014; Remus et al. 2016). PNKs target the 5′-terminus of DNA and RNA substrates and catalyze the transfer of the γ-phosphate from nucleoside triphosphate (NTP) to the 5′-hydroxyl terminus of the polynucleotide substrate. The 5′-phosphorylation status of DNA and RNA is critically important in the cell, thus underscoring the need for the PNK family of enzymes (Peach et al. 2015). For example, phosphorylation of 5′-hydroxyl termini following DNA damage is required for subsequent DNA repair (Weinfeld et al. 2011); whereas RNA turnover by the eukaryotic Xrn 5′-exonuclease family requires a 5′-phosphate for RNA degradation (Heindl and Martinez 2010; Jinek et al. 2011).
All known PNKs are members of the P-loop NTPase superfamily, yet each member has distinct substrate specificity. Bacteriophage T4 PNK is the founding member of the PNK family and is a bifunctional enzyme that harbors both PNK and phosphatase domains for repairing the 5′ and 3′-ends of damaged tRNA (Richardson 1965; Cameron and Uhlenbeck 1977). Furthermore, T4 PNK can efficiently phosphorylate both DNA and RNA substrates in vitro; a feature that has turned T4 PNK into a molecular biology workhorse for labeling DNA/RNA substrates (Richardson 1965; Wang and Shuman 2001; Galburt et al. 2002; Wang et al. 2002). Mammalian polynucleotide kinase/phosphatase (mPNKP) is another well-studied PNK family member which is required for DNA repair and is specific for damaged DNA substrates (Bernstein et al. 2005, 2009). There are also several PNK family members which target RNA in vivo including the bacterial Pnkp/Hen1 complex, which is required for bacterial RNA repair (Wang et al. 2012, 2015); Trl1, which is involved in tRNA maturation (Sawaya et al. 2003); and the Clp1/Grc3 subfamily, which is involved in multiple RNA processing pathways (Dikfidan et al. 2014; Weitzer et al. 2015).
The Clp1/Grc3 subfamily includes homologs of two distinct PNK enzymes, Clp1 and Grc3. In contrast to other well-characterized PNK enzymes, Grc3 and Clp1 are not bifunctional and they lack a phosphatase domain (Galburt et al. 2002). Clp1 is evolutionarily well-conserved across eukaryotes and homologs have been identified in archaea (Shuman and Hurwitz 1979; Jain and Shuman 2009; Weitzer et al. 2015). Clp1 has been implicated in at least three RNA pathways including mRNA, tRNA, and siRNA processing (Weitzer and Martinez 2007; Holbein et al. 2011; Haddad et al. 2012; Hanada et al. 2013; Karaca et al. 2014; Schaffer et al. 2014; Weitzer et al. 2015; Salzman et al. 2016). In contrast to Clp1, much less is known about Grc3 (Nol9 in mammals), which is required for marking preribosomal RNA (rRNA) through 5′-phosphorylation for degradation by the Rat1 (Xrn2 in mammals) 5′-exonuclease (Braglia et al. 2010; Heindl and Martinez 2010; Castle et al. 2012, 2013; Gasse et al. 2015; Pillon et al. 2017).
One major distinguishing feature between Clp1 and Grc3 is that Grc3 is reliant on its binding partner, the endoribonuclease Las1, for kinase activity (Pillon et al. 2017). Las1 is the endoribonuclease responsible for cleavage at the C2 site during pre-rRNA processing (Gasse et al. 2015). C2 cleavage is a critical step during the middle stages of ribosome assembly that triggers the removal of the internal transcribed spacer 2 (ITS2) that lies between the 5.8S and 25S rRNA (Konikkat and Woolford 2017; Konikkat et al. 2017). Failure to remove the ITS2 results in the generation of aberrant 60S particles with translational defects (Sarkar et al. 2017). Las1 cleavage generates the 26S pre-rRNA with a hydroxyl group at the 5′ end that is subsequently phosphorylated by Grc3 (Gasse et al. 2015). Furthermore, we recently established that there is functional crosstalk between the nuclease and kinase active sites within the Grc3/Las1 complex (Pillon et al. 2017). Specifically, Las1-mediated C2 cleavage depends on the functional integrity of the associated Grc3 phosphotransferase, while phosphorylation of the 26S pre-rRNA by Grc3 relies on a functionally competent Las1 nuclease. While this cooperativity likely allows for accurate and efficient processing of the ITS2, our understanding of this molecular regulation is limited (Pillon et al. 2017). To gain further mechanistic insight into kinase activity of the Grc3/Las1 complex, we generated a series of Grc3 PNK variants to determine the requirements for kinase activity. Through a combination of in vitro activity assays and in vivo studies, we establish the roles of four PNK motifs in cell viability, kinase activity, and Grc3-directed Las1 nuclease activity.
RESULTS
Catalytic requirements for recombinant Grc3 phosphotransferase activity
To assess the requirements for Grc3 phosphotransferase activity in vitro, we purified recombinant Saccharomyces cerevisiae Grc3/Las1, as described previously (Pillon et al. 2017) and determined the requirements for metal ions, nucleotide, and substrate. When bound to the minimal Las1 C terminus (residues 469–502) for stability, S. cerevisiae Grc3 harbors weak phosphotransferase activity toward single-stranded RNA, however upon binding full-length Las1, Grc3 assembles into a super-dimer of 2:2 stoichiometry that exhibits robust PNK activity in vitro (Pillon et al. 2017). Therefore, all of the in vitro PNK RNA phosphorylation assays in this study were carried out in the presence of the Grc3/Las1 complex and not Grc3 alone.
Members of the P-loop NTPase superfamily rely on a metal-dependent mechanism to transfer the γ-phosphate from an NTP donor to the 5′-hydroxyl polynucleotide (Wang et al. 2012; Das et al. 2014; Dikfidan et al. 2014). To assess the requirements for metal ions, we set up RNA phosphorylation reactions with 2 μM Grc3/Las1, 15 μM of a fluorescently labeled single-stranded RNA substrate and 1 mM ATP in the presence of 15 mM divalent metal or 5 mM EDTA. RNA phosphorylation was measured with a labeled 5′-hydroxyl 21-mer single-stranded RNA substrate that we previously determined was refractory to Las1 cleavage and has been previously used to study human Clp1 (Weitzer and Martinez 2007; Pillon et al. 2017). These reactions were incubated for 60 min at 37°C, quenched with loading dye and urea, and resolved on sequencing gels (Fig. 1A). Grc3/Las1-mediated RNA phosphorylation was supported in the presence of magnesium, but was significantly hindered with excess EDTA (Fig. 1A). We explored additional divalent metals to determine the metal selectivity of Grc3/Las1. Metals with typical octahedral coordination such as Mg2+, Mn2+, Ca2+, and Ni2+ supported Grc3/Las1 PNK activity in vitro, whereas Zn2+, which is often bound through tetrahedral geometry, could not support catalysis under the experimental conditions examined (Fig. 1A).
FIGURE 1.
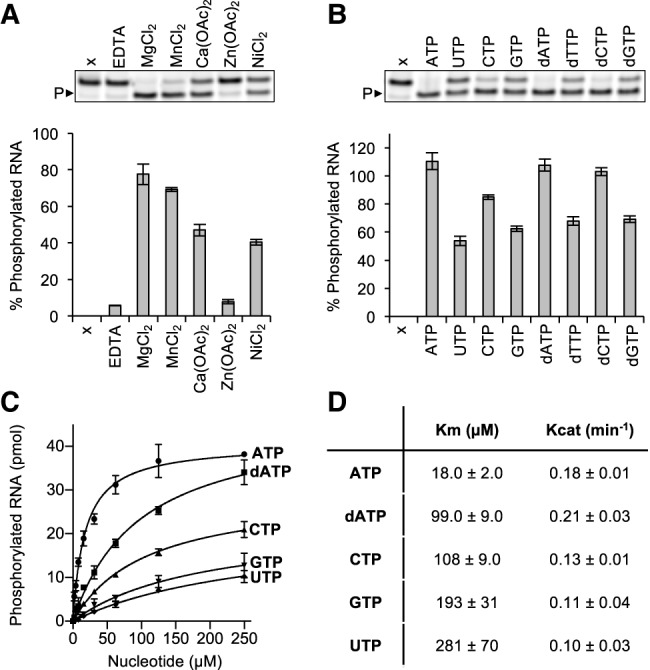
Characterization of recombinant Grc3/Las1 PNK activity. (A) RNA phosphorylation activity of Grc3/Las1 (2 µM) with ssRNA (15 µM) in the presence of 5 mM EDTA or 15 mM divalent metal [MgCl2, MnCl2, Ca(OAc)2, Zn(OAc)2, NiCl2]. A representative gel is shown (above) where P► marks phosphorylated RNA and sample “x” is an RNA alone control. (B) RNA phosphorylation activity of Grc3/Las1 (2 µM) with ssRNA (15 µM) in the presence of an assortment of nucleotides (ATP, UTP, CTP, GTP, dATP, dTTP, dCTP, and dGTP at 1 mM). A representative gel is shown (above) where P► marks phosphorylated RNA and sample “x” is a no nucleotide control. (C) Nucleotide concentration dependence of RNA phosphorylation. Grc3/Las1 (2 µM) was incubated with 15 µM fluorescently labeled ssRNA in the presence of 0–250 µM ATP, dATP, CTP, GTP, and UTP. (D) Data from C was fit using a Michaelis–Menten linear regression to calculate the Michaelis constant (Km) and catalytic constant (Kcat). The mean of three independent replicates were plotted with the standard deviation.
Next, we determined the NTP/dNTP specificity of Grc3. P-loop motifs are a hallmark for NTP binding, often responsible for engaging ATP or GTP (Saraste et al. 1990), and PNK family members have a wide variety of NTP donor specificities (Munir and Shuman 2017). To determine the NTP specificity of the Grc3/Las1 complex, we carried out RNA phosphorylation reactions in the presence of 1 mM ATP, GTP, CTP, UTP, dATP, dGTP, dCTP, and dTTP. Reactions were incubated for 60 min at 37°C with 2 μM Grc3/Las1 and 15 μM of a fluorescently labeled single-stranded RNA substrate. The reactions were quenched with loading dye supplemented with urea and resolved on sequencing gels (Fig. 1B). We did not detect phosphorylation in the absence of nucleotide and observed that Grc3/Las1 can use any NTP or dNTP as the phosphate donor (Fig. 1B). Next, we analyzed the nucleotide specificity of Grc3/Las1 by repeating our phosphorylation reactions with titrations of ATP, CTP, UTP, GTP, and dATP. By varying the NTP/dNTP concentration we observed that Grc3/Las1 shows a clear preference for ATP over dATP, CTP, GTP, and UTP (Fig. 1C). The trend for nucleotide preference corresponds to the variability in binding affinity as measured by the Michaelis constant (Km) while the catalytic constant (Kcat) remained largely unchanged (Fig. 1D).
To confirm that Grc3/Las1 uses the γ-phosphate of ATP for its PNK activity, we asked whether ADP or the nonhydrolyzable ATP analog, ADPnP, could support RNA phosphorylation in the absence of ATP. Similar to other members of the P-loop NTPase superfamily (Wang et al. 2012; Das et al. 2014; Dikfidan et al. 2014), Grc3/Las1 was deficient in RNA phosphorylation in the presence of ADP and ADPnP suggesting Grc3/Las1 transfers the γ-phosphate from ATP to the 5′-hydroxyl RNA (Fig. 2A). To confirm the requirement of a γ-phopshate for RNA phosphorylation, we performed competition assays with ATP and ADP or ADPnP. Each Grc3/Las1 reaction contained 30 μM ATP to which we titrated in an excess of ADP or ADPnP. Increasing amounts of ADP and ADPnP inhibited Grc3/Las1 kinase activity (up to twofold), indicating that neither ADP or ADPnP can support kinase activity (Fig. 2B,C). These results verify that Grc3/Las1 uses the γ-phosphate of ATP for its phosphotransferase activity.
FIGURE 2.

Recombinant Grc3/Las1 requires a γ-phosphate for RNA phosphorylation. (A) RNA phosphorylation activity of Grc3/Las1 (4 µM) with ssRNA (5 µM) in the absence (x) and presence of ATP, ADP, and ADPnP at 1 mM. (B) ATP/ADP competition assay. Grc3/Las1 (4 µM) was incubated with 10 µM fluorescently labeled ssRNA with constant ATP (30 µM) and a concentration series of ADP (0–5 mM). (C) ATP/ADPnP competition assay. Grc3/Las1 (4 µM) was incubated with 10 µM fluorescently labeled ssRNA with constant ATP (30 µM) and a concentration series of ADPnP (0–5 mM). The average and standard deviation were measured from three independent replicates. (*) P < 0.02; (**) P < 0.005; (***) P < 2 × 10−4; (****) P < 2 × 10−5 were calculated from two-tailed Student's t-tests.
Effects of oligonucleotide substrate on PNK activity
We investigated RNA phosphorylation activity of Grc3/Las1 on a mimic of the in vivo substrate. The pre-rRNA sequence harboring the C2 site forms a predicted stem–loop structure (van Nues et al. 1995a,b; Pillon et al. 2017). Las1 cleaves the C2 site following a bulged adenine residue, therefore we created a substrate with the local sequence and structure to mimic the Las1 cleavage product (Fig. 3A; Grc3 primed). To avoid multiple phosphorylation sites, we engineered a loop at the top of the stem to remove the free 5′ end. In the presence of the cleaved C2 RNA mimic, Grc3/Las1 displayed RNA phosphorylation at the C2 site with a stronger binding affinity (Km) than with the nonspecific single-stranded RNA substrate (Fig. 1C,D versus Fig. 3B). Finally, we mixed equimolar amounts of the nonspecific 21-mer with the cleaved C2 mimic and measured RNA phosphorylation over a titration of Grc3/Las1. Grc3/Las1 shows a clear preference for the C2 mimic over the nonspecific 21-mer. The specific activity of Grc3/Las1 was calculated from the linear range of the protein titration to show a 3.5-fold higher specific activity in phosphotransferase activity toward the C2 mimic (Grc3 primed RNA: 1.1 min−1) relative to the nonspecific 21-mer (ssRNA: 0.31 min−1), suggesting that Grc3/Las1 has selectivity for the cleaved C2 substrate over the nonspecific substrate (Fig. 3C).
FIGURE 3.

Effects of oligonucleotide substrate on PNK activity. (A) Cartoon diagram of the C2 cleaved pre-rRNA mimic. The black dot marks the 3′-end labeled fluorophore. (B) ATP concentration dependence of Grc3/Las1 phosphotransferase activity toward Grc3 primed RNA. Grc3/Las1 (2 µM) was incubated with 15 µM fluorescently labeled Grc3 primed RNA in the presence of 0–125 µM ATP. The mean was fit using a Michaelis–Menten linear regression and error bars represent the standard deviation from three independent experiments. (C) RNA substrate competition assay. Grc3/Las1 (0–2 µM) was incubated with equimolar single-stranded 21-mer RNA (15 µM) and Grc3 primed RNA (15 µM) in the presence of 1 mM ATP. The mean and standard deviation were calculated from three independent replicates. (*) P < 0.005; (**) P < 2 × 10−5 were calculated from two-tailed Student's t-tests.
Identification of conserved motifs within the PNK domains from the Clp1/Grc3 family
After establishing the requirements for Grc3 phosphotransferase activity, we next sought to understand the importance of conserved features within the PNK domain of Grc3, which shares ∼20% sequence identity with Clp1 and is flanked by distinct N- and C-terminal domains (Fig. 4A). Recent high-resolution structures of Clp1 from Caenorhabditis elegans in complex with ATP analogs and dinucleotide RNA substrates provide snapshots of this enzyme in several steps along the kinase reaction pathway revealing the presence of four key structural motifs important for catalysis and RNA specificity (Dikfidan et al. 2014). Multiple sequence alignments across Grc3 and Clp1 homologs revealed that these four motifs, including the P-loop, Walker B, Clasp, and Lid, are mostly well-conserved within the Grc3/Nol9 PNK domain (Fig. 4B; Dikfidan 2013). The P (phosphate binding)-loop motif (also known as the Walker A motif), contains the consensus sequence, GxxxxGK[T/S] (where x is any residue), and is characteristic of proteins that bind and hydrolyze ATP or GTP (Saraste et al. 1990). The lysine residue within this motif is traditionally thought to be important for nucleotide binding, however crystal structures of Clp1 suggest that within Clp1 this lysine residue (K127 in C. elegans Clp1) serves as a molecular switch that regulates ATP turnover (Dikfidan et al. 2014). The Walker B motif ([D/E]hhQ, where h is a hydrophobic residue) follows the P-loop and contains a well-conserved D/E residue essential for hydrolysis that is often referred to as the catalytic base (Koonin 1993). Within the Walker B motif from the Clp1/Grc3 family there is also a conserved glutamine residue which interacts with the 5′–3′ bridging phosphate group of the ultimate base and is important for hydrolysis (Dikfidan et al. 2014). Downstream lies the Clasp motif [T/S/L]xGW important for RNA specificity and unique to the Clp1/Grc3 family (Fig. 4B; Dikfidan et al. 2014). The final feature of the PNK domain is the Lid module (Fig. 4B), encoding arginine residues that in Clp1 stabilize the transition state (Dikfidan et al. 2014). Unlike the P-loop, Walker B, and Clasp motifs; the Lid module is not as well-conserved across Clp1 and Grc3 homologs and its functional significance within Grc3 is unknown. To visualize the S. cerevisiae Grc3 PNK active site, we constructed a homology model using the PNK domain from the S. cerevisiae Clp1 crystal structure (PDB ID 2NPI) (Noble et al. 2007). All four conserved Grc3 PNK motifs cluster in space to form a continuous and distinct active site (Fig. 4C) suggesting that these residues are important for Grc3's PNK activity.
FIGURE 4.

Conserved Grc3 PNK active site motifs. (A) Cartoon diagram of S. cerevisiae (Sc) Grc3 and C. elegans (Ce) Clp1. The homologous central polynucleotide kinase (PNK) domain is colored in yellow, and the variable N- and C-terminal domains (NTD and CTD) are shaded in light and dark gray, respectively. Numbering above the bar diagrams marks the amino acid residue domain boundaries. Arrowheads identify the relative position of the P-loop (red), Walker B (blue), Clasp (purple), and Lid (orange) motifs. (B) Conserved sequence motifs of the Clp1/Grc3 PNK family from Homo sapiens (Hs), Mus musculus (Mm), C. elegans (Ce), Chaetomium thermophilum (Ct), and S. cerevisiae (Sc). Fully conserved and semi-conserved residues are highlighted in dark and light colors, respectively. Sc Grc3 residues targeted for mutagenesis in this study are marked by a black dot. (C) S. cerevisiae Grc3 PNK active site modeled using the SWISS-MODEL server (Arnold et al. 2006) and derived from the crystal structure of S. cerevisiae Clp1 (PDB ID: 2NPI) (Noble et al. 2007). Grc3 P-loop (red), Walker B (blue), Clasp (purple), and Lid (orange) motifs are colored and residues targeted for mutagenesis are labeled.
Grc3 PNK motifs are essential in S. cerevisiae
To investigate the roles of the P-loop, Walker B, Clasp, and Lid motifs for cell viability in S. cerevisiae, we generated a series of single and double Grc3 PNK domain mutants to well-conserved residues within the: P-loop (K252, S253), Walker B (D283, Q286), Clasp (L361, W364), and Lid (R433, R440) (Table 1; Fig. 4C). We then performed genetic complementation assays using a S. cerevisiae strain encoding the tetracycline-inducible promoter (tetO7) upstream of endogenous GRC3. Prior to carrying out complementation assays, we modified the Grc3 tetO7 strain by the addition of a 5X-Flag tag to the C terminus of endogenous Las1 for downstream detection purposes. Addition of doxycycline (DOX) represses expression of endogenous Grc3 leading to the suppression of cell growth (Fig. 5A; Castle et al. 2013). We first tested the complementation of Grc3 WT, with and without an N-terminal 3X-Myc tag in the host strains by repressing endogenous GRC3 expression with DOX at 30°C. Yeast expressing Grc3 WT from the ARS1-CEN4 YCplac vector (Gietz and Sugino 1988) grew well in the presence of doxycycline while the empty YCplac vector could not restore growth in any of the conditions tested (Fig. 5B). Addition of a 3X-Myc tag to WT Grc3 (Grc3 versus Myc-Grc3) or a 5X Flag tag to endogenous Las1 (tet-Grc3 versus tet-Grc3/Las1-Flag strains) did not alter growth (Fig. 5B). Thus, all subsequent variants of Grc3 included a 3X-Myc tag (Table 1), unless specified otherwise.
TABLE 1.
Yeast plasmids used in this study
FIGURE 5.

Grc3 PNK active site motifs are critical in S. cerevisiae. (A) Schematic of tetracycline-off system. In the presence of doxycycline (DOX), the tetracycline-transcription activator (tTA) cannot bind the tetracycline response element (TRE); thus repressing transcription of endogenous GRC3. (B) S. cerevisiae tetO7-GRC3 and S. cerevisiae tetO7-GRC3–Las1 5X Flag strains were transformed with plasmids encoding wild-type Grc3 with and without an N-terminal Myc-tag and the ARS1-CEN4 YCplac vector. Serial dilutions were spotted on YPD agar plates in the absence and presence of DOX (20 µg/mL) and incubated at 30°C for 2–3 d. (C) S. cerevisiae tetO7-GRC3–Las1 5X Flag were transformed with plasmids encoding variants of the Grc3 PNK active site motifs (P-loop [red], Walker B [blue], Clasp [purple], Lid [orange]). Serial dilutions were spotted on YPD agar plates in the absence and presence of doxycycline (20 µg/mL) and incubated at 30°C for 2–3 d. (D) Growth curves from selected Grc3 PNK variants grown in the absence or presence of DOX (20 µg/mL) at 30°C. The absorbance at 595 nm was recorded over a 25-h time period. Each curve is the average of three independent replicates and errors bars mark the standard deviation.
Variants of GRC3 harboring missense mutations at conserved residues within the P-loop (K252A/S253A, K252A, K252R, S253A), Walker B (D283A/Q286A, D283A, D283N, D283E, Q286A), Clasp (L361A/W364A, L361A, L361I, W364A), and Lid (R433A/R440A, R433A, R433K, R440A, R440K) were expressed from the yeast ARS1-CEN4 YCplac vector transformed into tetO7-GRC3-Las1-5XFlag (Table 2). We tested the complementation of these Grc3 PNK variants in the host strain by repressing endogenous GRC3 expression with DOX followed by serial dilution plating and growth at 30°C (Fig. 5C). We further tested the complementation of a subset of these Grc3 PNK variants by recording growth curves. Absorbance was measured at 595 nm over a 25 h time period at 30°C (Fig. 5D).
TABLE 2.
Yeast strains used and constructed in this study

Disruption of the conserved K[T/S] residues within the P-loop had a severe growth defect that could not be restored even with the conservative single K252R mutation (Fig. 5C), highlighting the importance of these residues within the P-loop. Mutation of the aspartic acid residue (D283) within the Walker B motif to either an alanine, glutamic acid, or asparagine residue had a significant growth defect, while mutation of the Walker B glutamine residue (Q286) to alanine, only resulted in a minor growth defect (Fig. 5C,D). Disruption of the Clasp motif also resulted in a severe growth defect, as yeast expressing the Clasp mutants (L361A/W364A, L361A) could not restore growth in the presence of DOX (Fig. 5C,D). Replacement of L361 with the conservative isoleucine mutation could restore growth to almost wild-type levels, while a single mutation of W364A had a moderate effect on cell growth (Fig. 5C,D). In contrast to the P-loop, Walker B, and Clasp motifs, expression of Grc3 with mutations to the Lid motif restored cell proliferation under the experimental conditions tested by serial dilution plating, suggesting that the Lid motif plays a less significant role in Grc3 function in vivo (Fig. 5C). To further assess the role of the Lid motif, we measured growth curves on the single arginine mutants (Fig. 5D). Mutation of the first arginine residue (R433A) within in the Lid motif had no effect on cell growth while mutation of the second arginine residue within the Lid motif (R440A) had a mild effect on rates of cell proliferation (Fig. 5D). This corresponds well with sequence alignment of the Lid motif highlighting the lack of conservation for R433 and semi-conservation of R440 (Fig. 4B). The doubling time for the Lid double mutant (R433A, R440A: 160 min) and Lid R440A mutant (170 min) in YPD supplemented with DOX was slightly longer than the WT strain (140 min) emphasizing the subtlety of this growth defect.
The mild growth phenotype seen by mutagenesis of the Walker B motif at conserved residue Q286 and arginine residues of the Lid module (R433 and R440) was further investigated to dertermine whether these strains exhibited a temperature-sensitive growth defect. We assessed the complementation of the Walker B mutant (Q286A) and Lid variants (R433A, R440A; R433A/K; R440A/K) to the host strain by repressing endogenous GRC3 expression with DOX at 16°C, 22°C, 34°C, and 37°C (Fig. 6). Changes to the conserved glutamine of the Walker B motif and Lid motif arginine residues had no effect on cell growth and proliferation, confirming these residues are not critical for survival.
FIGURE 6.

Complementation of S. cerevisiae GRC3 over a temperature range. S. cerevisiae tetO7-GRC3-Las1 5X Flag were transformed with plasmids encoding the Grc3 Walker B Q286A mutant and Lid motif variants (R433A, R440A; R433A/K; R440A/K). Serial dilutions were spotted on YPD agar plates in the absence and presence of doxycycline (20 µg/mL) and incubated at 16°C, 22°C, 34°C, and 37°C for 2–6 d.
Grc3 PNK motifs do not disrupt Las1 interaction
To confirm that our Grc3 PNK variants did not compromise Grc3 stability or disrupt Las1 binding, we carried out a series of coimmunoprecipitation experiments. Grc3 PNK variants were grown in the tetO7-GRC3-Las1-5XFlag strain (Table 2) to mid-log phase in the absence and presence of DOX at 30°C. Cells were harvested, lysed, run on SDS-PAGE gels, and analyzed by western blot. Grc3 was detected using an anti-Myc antibody, while endogenous Las1 was detected with an anti-Flag antibody and tubulin was used as a loading control. The addition of DOX to the medium to repress endogenous Grc3 did not significantly impact the expression of endogenous Las1 with the exception of the vector only control (Fig. 7A versus 7B). This confirms earlier reports that Grc3 and Las1 are dependent upon one another for protein stability (Castle et al. 2013). Moreover, the presence or absence of DOX did not affect the protein levels of the Grc3 PNK variants, indicating that the PNK mutations do not severely impact the stability of Grc3 in S. cerevisiae. Finally, cells grown in the presence of DOX were coimmunoprecipitated with Myc-tagged Grc3. All Grc3 PNK variants were able to pull down endogenous Las1, indicating that these mutations do not hinder Las1 binding in S. cerevisiae (Fig. 7B). Taken together, these data suggest the growth defects observed upon disrupting the Grc3 P-loop, Walker B, Clasp, and Lid motifs are not due to instability or Las1 dissociation, but a defect in Grc3 function.
FIGURE 7.

Grc3 PNK active site motifs do not disrupt interaction with Las1 in S. cerevisiae. (A,B) S. cerevisiae tetO7-GRC3–Las1 5X Flag strains transformed with plasmids encoding Grc3 PNK variants were grown to mid-log phase at 30°C in the absence (A) and presence (B) of DOX (20 µg/mL). Cells were harvested, then lysed, and equal amounts of whole-cell lysate (WCL) were separated by SDS-PAGE and analyzed by western blot with an anti-Myc antibody (Grc3), an anti-Flag antibody (Las1), and an antitubulin antibody (loading control). Cells grown in the presence of DOX were further analyzed by coimmunoprecipitation with anti-Myc resin. Immunoprecipitated proteins were separated by SDS-PAGE and analyzed by western blot using an anti-Myc antibody (Grc3) and an anti-Flag antibody (Las1).
Grc3 PNK motifs are essential for RNA phosphorylation in vitro
After establishing the significance for the Grc3 P-loop, Walker B, Clasp, and Lid motifs for cell viability in S. cerevisiae, we wanted to determine their role in RNA phosphorylation in vitro. First, we generated a series of double mutations to the PNK motifs in an Escherichia coli expression vector (Table 3). The Grc3 PNK variants were coexpressed with full-length poly-histidine tagged Las1 in E. coli and purified using Ni2+-affinity chromatography followed by gel filtration (Fig. 8A). All four double Grc3 PNK variants bound by Las1 were stably expressed and eluted similarly by gel filtration when compared to wild-type (WT) Grc3/Las1, confirming our in vivo results which showed that these mutations do not disrupt or destabilize the interaction between Grc3 and Las1 (Fig. 7B). Moreover, we measured the stability of the Grc3/Las1 PNK variants by thermal shift, and did not observed any significant changes to the stability of the variants in comparison to the Grc3/Las1 WT complex (Fig. 8B). We performed a protein titration of Grc3 variants bound by Las1 with constant 5′-hydroxyl single-stranded RNA in the presence of ATP (Fig. 8C). The Grc3/Las1 WT complex displayed a 17-fold higher specific activity in phosphotransferase activity (0.87 min−1) when compared to the Lid variant (0.05 min−1) and 40-fold higher specific activity relative to the Clasp mutant (0.02 min−1). Intriguingly, despite causing marked defects in phosphotransferase activity in vitro, the Lid double mutant only caused a mild growth phenotype while the Clasp double mutant was inviable (Fig. 5C,D). These data suggest that the Lid mutant presents a lower limit in PNK activity which can still support survival. The P-loop and Walker B variants had almost no detectable RNA phosphorylation activity under the experimental conditions examined preventing the measurement of their specific activity.This is in agreement with our cell viability assays, which revealed that the P-loop and Walker B variants could not support growth.
TABLE 3.
E. coli expression plasmids used and constructed in this study
FIGURE 8.

Grc3 PNK active site motifs are critical for kinase activity in vitro. (A) SDS-PAGE analysis of recombinant Sc Las1 bound to Grc3 WT, P-loop, Walker B, Clasp, and Lid variants. (B) Melting temperature of Grc3/Las1 PNK variants determined by thermal shift experiments. The mean and standard deviation were calculated from three independent replicates. (C) RNA phosphorylation of 0–2 µM Grc3/Las1 variants incubated with 21-nt ssRNA (15 µM). Specific activity was calculated from the moles of phosphorylated RNA in the 60 min PNK reaction per mole of input Grc3/Las1 in the linear range of the protein titration. (D) SDS-PAGE analysis of recombinant Sc Las1 bound to Grc3 WT, and single P-loop, Walker B, Clasp, and Lid variants. (E) RNA phosphorylation of 16 µM Grc3/Las1 variants incubated with 21-nt ssRNA (10 µM). The mean and standard deviation were calculated from three independent replicates. (*) P < 5 × 10−4; (**) P < 2 × 10−4; n.s., not significant were calculated from two-tailed Student's t-tests.
To determine the individual roles of specific residues within the four PNK motifs in RNA phosphotransferase activity, we generated 13 single mutants (Table 3). The single PNK mutants were expressed and purified as described above for the double Grc3 PNK variants. All 13 single mutants were stably expressed, copurified with Las1, and eluted similarly by gel filtration as seen with the WT and double PNK variants (Fig. 8D). We measured RNA phosphorylation at a fixed protein concentration of the single point mutants (Fig. 8E). Individual point mutants to the P-loop (K252A, K252R, and S253A), Walker B (D283A, D283N, D283E), and Clasp (W364A) motifs had almost no detectable kinase activity, while other single mutants from the Walker B (Q286A) and Clasp (L361A) motifs had a four- to fivefold reduction in total phosphorylated RNA compared to WT (Fig. 8E). Within the Lid motif, we could not detect a significant defect in activity with the R433A/K mutants, but observed a decrease in activity for the R440A/K mutants (approximately four- to fivefold reduction). With the exception of L361A, the in vitro kinase assays nicely complement the in vivo growth assays. Thus, mutations that abolish kinase activity cause severe growth defects, while mutants that only impair kinase activity have a more modest effect on cell growth. Residue L361 presents an exception to this trend since mutation of L361 to an alanine shows a moderate reduction in PNK activity, but a severe growth defect. These results suggest that aside from supporting kinase activity L361 may have an additional role in vivo.
Grc3 PNK motifs are critical for assembly of the large ribosomal subunit
Grc3 and its mammalian counterpart, Nol9, are essential for production of the large ribosomal subunit (60S) (Heindl and Martinez 2010; Castle et al. 2013; Pillon et al. 2017). To assess whether the reduction in cell proliferation and the defects in RNA phosphorylation in vitro cause deficiencies in ribosome production, we performed sucrose gradient fractionation from tetO7-GRC3-Las1-5XFlag strains expressing WT Grc3 and select Grc3 PNK variants (K252A, S253A, D283A, L361A/W364A, R433A/R440A) in the presence of DOX. Ribosomes purified from yeast expressing WT Grc3 produced three distinct peaks representing the small subunit (40S), large subunit (60S), and intact 80S ribosomes, as well as a series of peaks indicating the presence of polyribosomes (polysomes) (Fig. 9; WT). In comparison to WT Grc3, the five Grc3 PNK mutants (K252A, S253A, D283A, L361A/W364A, R433A/R440A) that we analyzed marked a reduction in the 60S ribosomal subunit and a corresponding decrease in the 80S monosome and polysomes (Fig. 9). Moreover, the accumulation of ribosome halfmers further emphasize the importance for Grc3 phosphotransferase activity in ribosome assembly. Interestingly, mutations to the Lid motif caused a mild-phenotype in 60S production, in agreement with our growth assays which revealed that mutations to this motif had a mild effect on cell proliferation (Fig. 9; LidR433A/R440A). We interpret this to mean the Grc3 Lid motif plays an important role in enhancing the efficiency of ribosome production, but is not strictly required for cell viability like the P-loop, Walker B and Clasp motifs. To determine whether Grc3 variants and endogenous Las1 still associate with preribosome particles, we analyzed the sedimentation pattern of Grc3 variants and Las1 in our sucrose gradient fractions by western blot. Myc-Grc3 variants and Las1-Flag co-sediment with fractions corresponding to the 60S ribosomal particle (Fig. 9). This confirms that mutations to the Grc3 PNK motifs do not hinder Grc3/Las1 binding to pre-60S particles.
FIGURE 9.

Grc3 PNK active site motifs are critical for ribosome production in S. cerevisiae. Polysome profile analysis of transformed tetO7-GRC3–Las1 5X Flag expressing WT and PNK variants (P-loopK252A, P-loopS253A, Walker BD283A, ClaspL361A/W364A, LidR433A/R440A) of Grc3. Black arrows mark ribosome halfmers and brackets highlight the polysomes. Sucrose gradient fractions were precipitated, run on SDS-PAGE gels, and analyzed by western blot with anti-Myc (Grc3), anti-Flag (Las1), and anti-RPL8 (60S ribosomal protein) antibodies.
Las1 crosstalk is dependent on the integrity of the Grc3 PNK domain
The higher-order assembly of the Grc3/Las1 super-dimer is critical for supporting crosstalk between the Las1 nuclease and the Grc3 polynucleotide kinase (Pillon et al. 2017). Disrupting the Grc3 Walker B motif causes a deficiency in crosstalk leading to a reduction in Las1-mediated C2 cleavage of the S. cerevisiae ITS2 in vitro (Pillon et al. 2017). To assay whether the Grc3 P-loop, Clasp, and Lid motifs are also required for Grc3 to direct efficient Las1-mediated C2 cleavage, we performed in vitro C2 cleavage assays using an ITS2 RNA mimic (Fig. 10A) and Grc3/Las1 complexes with the double PNK variants. As shown previously, mutations to the Walker B motif cause a significant reduction in C2 cleavage. Changes to the Lid motif resulted in a similar RNA cleavage deficiency while mutations to the Clasp motif significantly impaired C2 cleavage in vitro (Fig. 10B). Conversely, the Grc3 P-loop was dispensable for Grc3/Las1 crosstalk, since C2 cleavage was similar to WT (Fig. 10B). To assess the individual contribution of the residues within the PNK motifs, we also carried out nuclease assays at a fixed protein concentration with single point mutants (Fig. 10C). From these individual mutants, we identified three residues (D283, L361, R440) which are important for Grc3-directed C2 cleavage activity. These cleavage assays highlight the contributions of the Grc3 PNK motifs in supporting the coordinated crosstalk between these two essential RNA processing enzymes.
FIGURE 10.

PNK active site motifs facilitate Grc3/Las1 crosstalk in vitro and in vivo. (A) S. cerevisiae ITS2 model RNA substrate. C2 cleavage site is marked by an arrowhead. Black dot marks the 3′-end labeled fluorophore. (B) C2 RNA cleavage by Las1 bound to Grc3 PNK variants. A protein concentration series (0–0.8 µM) of Grc3/Las1 WT and PNK variants (P-loop [K252A/S253A], Walker B [D283A/Q286A], Clasp [L361A/W364A], and Lid [R433A, R440A]) were incubated with 0.1 µM fluorescently labeled Las1 primed RNA. (C) Quantification of C2 RNA cleavage by Grc3/Las1 variants (0.8 µM) incubated with 0.1 µM Las1 primed RNA. (*) P < 0.01; (**) P < 0.005 were calculated by a two-tailed Student's t-test. (D) Simplified S. cerevisiae pre-rRNA processing pathway. The C2 site (red) and probes are marked on the S. cerevisiae pre-rRNA processing pathway. (E) Northern blot analysis of tetO7-GRC3 transformed with plasmids encoding Grc3 PNK active site motif mutants (P-loop [K252A/S253A], Walker B [D283A/Q286A], Clasp [L361A/W364A], and Lid [R433A, R440A]). RNA loading was monitored by methylene blue staining. (F) The integrative density of rRNA was normalized to the mature 18S rRNA. Relative abundance (RA) of pre-rRNA intermediates was calculated by dividing the integrative density of rRNA to the equivalent rRNA from the wild-type strain. Quantification of rRNA intermediates was calculated from a single representative northern blot presented in panel E.
To assess whether the observed Grc3/Las1 crosstalk also occurs in S. cerevisiae, we looked at the accumulation of pre-rRNA intermediates by northern blot analysis. We extracted RNA from tetO7-GRC3 strains expressing Grc3 PNK variants grown in the presence of DOX. Defects to the Grc3 P-loop, Walker B, and Clasp motifs, lead to a reduction in the mature 25S rRNA and an accumulation in RNA intermediates, including the 35S, 27S, and 7S precursor rRNA, highlighting their importance in pre-rRNA processing (Fig. 10D–F). Disruption of the Lid motif also lead to a pronounced abundance of the 27S, but unlike the P-loop, Walker B, or Clasp motifs, there was no stark reduction in the mature 25S or a severe accumulation of either the 35S or 7S (Fig. 10F). This is in good agreement with our in vitro biochemical assays and in vivo yeast viability and sucrose gradient assays suggesting the Lid motif plays a minor role in pre-rRNA processing. Interestingly, the accumulation of the 27S intermediate indicates a defect in C2 cleavage (Fig. 10D). The most dramatic C2 cleavage defect is observed with the Clasp variant (L361A/W364A); whereas, the P-loop mutant (K252A/S253A) has comparable levels to WT (Fig. 10E,F). This observation corresponds well with our in vitro C2 cleavage assay suggesting the P-loop is dispensable for Las1 nuclease activity. Moreover, yeast expressing the double Grc3 P-loop variant struggled to process the 7S intermediate suggesting a functional consequence in a critical downstream processing step involving the RNA exosome (Castle et al. 2013; Fromm et al. 2017). Las1 has been previously linked to 7S processing, however its role is currently unknown (Schillewaert et al. 2012). We also observed an accumulation in the 35S pre-rRNA upon disrupting the P-loop, Walker B, and Clasp motifs (Fig. 10F). This may either reflect a regulatory feedback loop that is activated due to the inability to phosphorylate the 26S or it may highlight an unknown role for Grc3/Las1 in earlier steps of pre-rRNA processing. Together, these data show for the first time that Grc3/Las1 crosstalk occurs in vivo and that the Grc3 PNK motifs support varying roles for pre-rRNA processing at the C2 site and 7S pre-rRNA maturation in S. cerevisiae.
DISCUSSION
Prior to this study little was known about the activity of the PNK domain of the essential polynucleotide kinase Grc3, therefore we characterized the Grc3 PNK domain through a series of in vivo and in vitro assays. Here we establish that the PNK domain has a requirement for a divalent metal ion for catalysis and has a nucleotide preference for ATP. Similar to the Trl1 and Pnkp/Hen1 phosphotransferases, the NTP donor selectivity of Grc3/Las1 includes all four nucleotides, but like Clp1, Grc3/Las1 shows a preference for ATP (Sawaya et al. 2003; Noble et al. 2007; Das et al. 2013). We also determined that Grc3/Las1 has enhanced activity toward the Las1 cleaved C2 substrate. Similarly, mammalian PNKP has a substrate preference for DNA oligonucleotides with recessed 5′-OH groups, which is consistent with its role in certain DNA repair pathways (Bernstein et al. 2005). Taken together, our results show that the Grc3 polynucleotide kinase shares similar metal- and nucleotide-selectivity to known members of the P-loop NTPase superfamily, but exhibits critical differences in its substrate selection in comparison with other PNK enzymes. These differences in substrate selection likely influence Grc3's biological target(s) in vivo.
We further enhanced our understanding of the Grc3 PNK domain by generating missense mutations within four key motifs that are shared with the ancestral Clp1, including the P-loop, Walker B, Clasp, and Lid (Fig. 11). The P-loop, Walker B, and Clasp motifs are all essential for cell viability, kinase activity, and ribosome production (Fig. 11). In contrast, the Lid motif is not essential for cell viability, but disruption of the Lid motif results in minor impairments to cell growth, ribosome production and a 17-fold reduction in specific activity of the phosphotransferase (Fig. 11). We interpret this to mean the Lid motif is important for efficient rRNA processing, but is not essential for survival. This is in agreement with the report that pre-rRNA transcription is the rate limiting step in ribosome assembly (Nomura et al. 1984), and not pre-rRNA processing. Similar to the Lid mutant, mutagenesis of other ribosome assembly factors, such as Fap7 and Has1, displayed defects in enzymatic function in vitro, while maintaining wild-type cell growth and proliferation (Granneman et al. 2005; Rocak et al. 2005; Dembowski et al. 2013).
FIGURE 11.

Model of Grc3/Las1 molecular crosstalk. Grc3/Las1 assembles into a super dimer driven by homodimerization of the Las1 nuclease domain and Grc3 PNK domain (Pillon et al. 2017). Each Grc3 PNK domain encodes four motifs, including the P-loop (red), Walker B (blue), Clasp (purple), and Lid (orange), important for kinase activity, nuclease activity, and cell viability. Approximate positions of the motifs relative to one another are based upon the homology model from Figure 4C. PNK residues identified to be critical for molecular crosstalk are encircled (Grc3 D283 and L361). Yellow asterisks highlight the Grc3 C-terminal tail and the Las1 HEPN active site, which were previously established to also mediate molecular crosstalk (Pillon et al. 2017). The X marks residues that are dispensable, whereas the checkmark identifies critical residues.
Through the generation of individual missense mutations we were able to discern differences in activity within individual motifs. One of the most significant differences was observed for two key residues within the Clasp motif ([T/S/L]xGW). The L361A mutation resulted in a lethal phenotype while the W364A mutation only impaired growth. However when we carried out in vitro kinase assays we found that the W364A mutation had a more significant impact on phosphotransferase activity than the L361 mutation. These results suggested that aside from supporting kinase activity, L361 must have another critical role in the cell and could be important for supporting Las1 nuclease activity.
To delineate the role of the PNK motifs in molecular crosstalk between the Las1 nuclease and the Grc3 kinase active sites, we looked at Las1 nuclease activity in vitro and in vivo. We previously established that Grc3 and Las1 are dependent upon one another for higher ordered assembly and enzyme activation (Pillon et al. 2017). Mutation of the key active site residues within Las1 (R129/H134) disrupts Grc3 kinase activity (Fig. 11). Likewise, mutation of the Walker B motif within the Grc3 PNK active site and truncation of the Grc3 C terminus (residues 623–632) disrupts Las1 nuclease function (Fig. 11; Pillon et al. 2017). Here we establish that in addition to the Walker B motif and C-terminal tail, the Clasp motif and, to a lesser extent, the Lid motif are important for Grc3-directed Las1 nuclease activity in vivo and in vitro. Through the generation of single point mutants we were able to identify two residues critical for directing crosstalk including the Walker B catalytic base, D283, and L361 from the Clasp motif, which lie in close proximity to one another in our homology model (Fig. 4C). Thus, molecular crosstalk between Grc3 and Las1 relies on a series of motifs within the nuclease active site (R129/H134 from Las1), the kinase active site (D283/L361 from Grc3), and the C-terminal tail of Grc3 (residues 623–632) (Fig. 11). Together, Grc3/Las1 generate cleavage and phosphorylation products with RNA ends that cannot be resealed by any known ligase. Phosphorylation of the 26S pre-rRNA by Grc3/Las1 also primes the 26S pre-rRNA for processing by Rat1/Rai1 (Gasse et al. 2015). Therefore, the unique combination of enzymatic activities of the Grc3/Las1 complex commits the cell to processing the ITS2 following C2 cleavage.
In contrast to the Walker B, Lid, and Clasp motifs, the P-loop motif is dispensable for Las1 nuclease activity. This marks a striking difference between Grc3/Las1 and the related endoribonucleases Ire1 and RNase L, which are both dependent upon their kinase domains for high-ordered assembly and activation of their nuclease domains (Pillon and Stanley 2017). In contrast to Grc3/Las1, both Ire1 and RNase L require ATP binding to their respective kinase/pseudo kinase domains for nuclease activation (Wreschner et al. 1982; Dong et al. 1994; Sidrauski and Walter 1997; Dong and Silverman 1999; Huang et al. 2014). We previously established that ATP binding is not a requirement for Las1 nuclease function and this is further supported by mutation of the P-loop residues, critical for ATP binding (Castle et al. 2013; Pillon et al. 2017). One of the major differences between Grc3/Las1 and Ire1 and RNase L, is that Grc3 phosphotransferase activity is vital in vivo as Grc3 directly phosphorylates the Las1 cleavage product. From a functional standpoint, coupling C2 cleavage with RNA phosphotransferase activity would be beneficial as this ensures the Grc3/Las1 RNA processing machinery strictly commits to C2 cleavage if both catalytic sites of the Grc3/Las1 complex are functionally competent. This is achieved through a molecular crosstalk network supported by the catalytic base from the Walker B motif as opposed to the P-loop. Thus, the alternative wiring of the crosstalk network seen for Grc3/Las1 compared to its related RNA processing machinery, Ire1 and RNase L, may reflect a structural feature or unknown function of the distinct Grc3 N-terminal and C-terminal domains.
Together, this work reveals the checks and balances necessary for cells to commit to ITS2 processing through the Grc3/Las1 RNA processing machinery. We hypothesize that the Grc3/Las1 active sites share key residues, including Grc3 Walker B and Clasp motif residues, to form a composite active site to properly position the RNA substrate for efficient and accurate cleavage and phosphorylation. While this hypothesis awaits high resolution structural determination for validation, our data provides additional insight into the intricate molecular crosstalk between the essential enzymes Grc3 and Las1 that ensures C2 cleavage and phosphorylation of the 26S pre-rRNA is directly coupled.
MATERIALS AND METHODS
Cloning of Grc3/Las1 variants
Generation of bacterial expression plasmids of S. cerevisiae (Sc) Grc3/Las1 (pMP 001; Grc3 residues 1–632 and Las1 residues 1–502) and the Walker B variant Grc3D283A/Q286A/Las1 (pMP 335; Grc3 residues 1–632 and Las1 residues 1–502) were described previously (Pillon et al. 2017). Kinase-deficient Grc3/Las1 variants were generated by Q5 site-directed mutagenesis (NEB) using pMP 001 as a template (Table 3). The Grc3 (pMP 003; residues 1–632) plasmid used for proliferation assays was modified from the original by the inclusion of three additional Myc tags (pMP 484) onto the N terminus for detection purposes. Kinase-deficient Grc3 variants were generated by Q5 site-directed mutagenesis (NEB) using pMP 484 as a template and are listed in Table 1. All plasmids were verified by DNA sequencing (GeneWiz).
Expression and purification of Grc3 and Las1 constructs
Grc3/Las1 variant proteins were produced as described previously (Pillon et al. 2017) with minor modifications. Briefly, Grc3/Las1 variants were overexpressed in E. coli LOBTSR (Kerafast) cells. Cells were harvested by centrifugation and resuspended in 50 mM Tris pH 8.0, 500 mM NaCl, 5 mM MgCl2, 1%(v/v) Triton X-100, 10% (v/v) glycerol and disrupted by sonication. Lysate was clarified at 26,916g for 50 min at 4°C and the supernatant was applied to a gravity flow column loaded with His60 Ni Superflow Resin (Clontech). The column was washed with 200 mL of wash buffer (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM MgCl2, 15 mM imidazole, 10% glycerol) and eluted with imidazole (50 mM Tris pH 8.0, 500 mM NaCl, 5 mM MgCl2, 200 mM imidazole, 10% glycerol). Samples were resolved by a HiLoad 16/600 Superdex-200 Prep Grade (GE Healthcare) gel filtration column equilibrated with storage buffer (20 mM Tris pH 8.0, 200 mM NaCl, 5 mM MgCl2, 5% glycerol).
Thermal shift assay
The melting temperature (Tm) was measured for purified Sc Grc3/Las1 variants (1.0 μM) in storage buffer supplemented with 5× SYPRO Orange Protein Gel Stain (Sigma-Aldrich). Mixtures were heated from 25°C–95°C at a rate of 0.05°C/sec. Relative fluorescence was measured using excitation and emission wavelengths 490 nm and 580 nm, respectively, using a QuantStudio 7 Flex (Applied Biosystems). The mean and standard deviation were calculated from three independent replicates.
Phosphorylation assay
RNA phosphorylation was measured with 2 μM Grc3/Las1 variants, unless specified otherwise, in kinase reaction buffer (50 mM Tris pH 8.0, 100 mM NaCl, 15 mM MgCl2, and 1 mM ATP). Fluorescently labeled RNA (10–15 μM) encoding a 5′-hydroxyl included: single-stranded 21-mer RNA (5′-ACGUACGCGGAAUACUUCGAA-TAMRA-3′) and Grc3 primed RNA (5′-GGUUUUACCAACUGCGGCUAAUCGUACGGUCGUUUUA-TAMRA-3′). To ascertain NTP and dNTP concentration dependence, ATP, UTP, CTP, GTP, and dATP titrations (0–250 μM) were used where indicated. To test metal selectivity, we purified Grc3/Las1 variants in storage buffer lacking metal and supplemented reactions with 5 mM EDTA or 15 mM divalent metal ion, where indicated. Nucleotide selectivity was measured using modified kinase reaction buffer with 1 mM nucleotide, as indicated. ATP/ADP and ATP/ADPnP competition mixtures included constant ATP (30 µM) with a titration of ADP or ADPnP (0–5 mM). Phosphorylation reactions were incubated for 60 min at 37°C and quenched by adding equal volume urea loading dye (20 mM Tris pH 8.0, 8 M urea, 0.05% bromophenol blue, 1 mM EDTA). Reaction mixtures were boiled and loaded onto a 15% polyacrylamide (8 M urea) gel in 0.5× tris–borate–EDTA buffer. Gels were visualized on a Typhoon FLA 9500 (GE Healthcare). All phosphorylation reactions were done in triplicate and representative gels are shown in the figures. Gels were analyzed using ImageJ software (Abramoff et al. 2004).
Yeast growth conditions and proliferation assays
The S. cerevisiae strain encoding a tetracycline-titratable promoter (tetO7) upstream of the endogenous GRC3 gene was obtained from Open Biosystems (GE Dharmacon). A 5XFlag tag was added onto the C terminus of endogenous Las1 in the tetO7-GRC3 strain for detection purposes with the pFA6a-5FLAG-natMX6 vector (Noguchi et al. 2008). The tetO7-GRC3-Las1-5XFlag strain was transformed with plasmids encoding Grc3 variants (see Table 1) and expression of endogenous GRC3 was repressed by supplementing YPD media with doxycycline (20 µg/mL). Proliferation assays spotted transformed tetO7-GRC3-Las1-5XFlag strains (Table 2) on YPD in the absence and presence of doxycycline (20 µg/mL) and incubated at 30°C for 2–3 d, unless specified otherwise. Growth curves were generated from cultures (100 µL) with and without doxycycline (20 µg/mL) inoculated at an OD600 of 0.05 and incubated at 30°C for 25 h. Saturation of the cultures were monitored by measuring the absorbance at 595 nm every 15 min using an Infinite 200 Pro (Tecan). The mean and standard deviation were calculated from three independent replicates.
Western blot and coimmunoprecipitation
TetO7-GRC3-Las1-5XFlag strain was transformed with plasmids listed in Table 1. Cultures were grown in YPD in the absence and presence of doxycycline (20 µg/mL) and harvested at mid-log phase. Whole-cell lysate was prepared by lysing the cells using glass beads followed by protein precipitation using trichloroacetic acid. Coimmunoprecipitation was carried out by resuspending cells in 50 mM Tris pH 8.0, 300 mM NaCl, 5 mM MgCl2, 0.5% NP-40 prior to cell lysis using glass beads. Whole-cell lysate was incubated with anti-myc resin (Pierce) for 30 min at 4°C and washed using 50 mM Tris pH 8.0, 300 mM NaCl, 5 mM MgCl2, 5% Glycerol. Samples were resolved by SDS-PAGE and analyzed by western blot using anti-Myc (Grc3; EMD Millipore), anti-Flag (Las1; Sigma-Aldrich), and anti-α-tubulin (Abcam) antibodies.
RNA extraction and northern blot analysis
RNA isolation and northern blot analysis were performed as described previously (Pillon et al. 2017). Briefly, transformed tetO7-GRC3 strains (Table 2) were maintained in log phase in the presence of doxycycline (20 µg/mL). Extracted RNA (3 µg) was resolved over a 1% formaldehyde agarose gel and transferred onto Hybond XL membrane (GE Healthcare). Ribosomal RNA was detected using Digoxigenin (DIG)-conjugated DNA probes described previously (Castle et al. 2013; Pillon et al. 2017), and blots were visualized with anti-DIG-alkaline phosphatase (Sigma-Aldrich) as per manufacturer's instructions. RNA loading was monitored by methylene blue staining to visualize the mature 25S and 18S rRNA. The integrated density of pre-rRNA intermediates were measured with Image J software (Abramoff et al. 2004) and normalized to the mature 18S rRNA. The relative abundance of rRNA was determined by dividing the density of rRNA by the equivalent rRNA from the wild-type strain. Quantification was performed on a single representative northern blot presented in Figure 10E.
Sucrose gradient and polysome profiling
Sucrose gradients and polysome profiling were performed as described in Masek et al. (2011) with minor modifications. Cultures of transformed tetO7-GRC3-Las1-5XFlag (Table 2) were grown at 30°C in the presence of doxycycline (20 μg/mL) to OD600 ∼0.5. Cycloheximide (0.1 mg/mL) was added to the cultures and the cultures were incubated for 5 min on ice before harvesting cells by centrifugation. Cells were resuspended in extraction buffer (20 mM Tris pH 7.4, 60 mM KCl, 10 mM MgCl2, 1 mM DTT, 1% [v/v] Triton X-100, 0.1 mg/mL cycloheximide, 0.2 mg/mL heparin [ammonium salt]) and disrupted using glass beads for 5 min at 4°C. Lysate was clarified at 8000g for 5 min at 4°C and 0.8 mg RNA was loaded onto a 7%–47% sucrose gradient. Gradients were subjected to 260,110g for 2.5 h at 4°C and immediately loaded onto a gradient fractionator (Brandel). Fractions from the gradient were collected and then precipitated by trichloroacetic acid. The precipitants were resupspended in 2× SDS-PAGE loading dye and analyzed by western blots with anti-Myc (Grc3: EMD Millipore), anti-Flag (Las1; Sigma-Aldrich), and anti-RPL8 (Thermo Fisher Scientific) antibodies.
C2 RNA cleavage assays
Las1 endoribonuclease activity was measured as previously described (Pillon et al. 2017) with minor modifications. Briefly, Grc3/Las1 variants (0–0.8 μM) were incubated with fluorescently labeled S. cerevisiae rRNA mimic (0.1 μM) encoding the C2 site (5′-GUCGUUUUAGGUUUUACCAACUGCGGC-FAM-3′) for 60 min at 37°C. Samples were resolved on 15% polyacrylamide (8 M urea) gels in 1× tris–borate–EDTA buffer and visualized using the Typhoon FLA 9500. The mean and standard deviations were calculated from three independent replicates.
ACKNOWLEDGMENTS
We thank Dr. Traci Hall and Dr. Jessica Williams for their critical reading of this manuscript. We would also like to thank Dr. Mellissa Wells for help with creating yeast strains. This work was supported by the US National Institutes of Health Intramural Research Program; US National Institute of Environmental Health Sciences (NIEHS) (ZIA ES103247 to R.E.S.). Additional support for this work comes from the Canadian Institutes of Health Research (CIHR, 146626 to M.C.P.).
Author contributions: M.C.P. and R.E.S. designed the experiments. M.C.P., M.S., and R.E.S. carried out experiments, analyzed data, and prepared figures. M.C.P. and R.E.S. wrote the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.065037.117.
REFERENCES
- Abramoff MD, Magalhaes PJ, Ram SJ. 2004. Image processing with ImageJ. Biophotonics Int 11: 36–42. [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T. 2006. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Bernstein NK, Williams RS, Rakovszky ML, Cui D, Green R, Karimi-Busheri F, Mani RS, Galicia S, Koch CA, Cass CE, et al. 2005. The molecular architecture of the mammalian DNA repair enzyme, polynucleotide kinase. Mol Cell 17: 657–670. [DOI] [PubMed] [Google Scholar]
- Bernstein NK, Hammel M, Mani RS, Weinfeld M, Pelikan M, Tainer JA, Glover JN. 2009. Mechanism of DNA substrate recognition by the mammalian DNA repair enzyme, polynucleotide kinase. Nucleic Acids Res 37: 6161–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braglia P, Heindl K, Schleiffer A, Martinez J, Proudfoot NJ. 2010. Role of the RNA/DNA kinase Grc3 in transcription termination by RNA polymerase I. EMBO Rep 11: 758–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron V, Uhlenbeck OC. 1977. 3′-Phosphatase activity in T4 polynucleotide kinase. Biochemistry 16: 5120–5126. [DOI] [PubMed] [Google Scholar]
- Castle CD, Cassimere EK, Denicourt C. 2012. LAS1L interacts with the mammalian Rix1 complex to regulate ribosome biogenesis. Mol Biol Cell 23: 716–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle CD, Sardana R, Dandekar V, Borgianini V, Johnson AW, Denicourt C. 2013. Las1 interacts with Grc3 polynucleotide kinase and is required for ribosome synthesis in Saccharomyces cerevisiae. Nucleic Acids Res 41: 1135–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang LK, Smith P, Shuman S. 2013. Structural and biochemical analysis of the phosphate donor specificity of the polynucleotide kinase component of the bacterial Pnkp•Hen1 RNA repair system. Biochemistry 52: 4734–4743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Wang LK, Smith P, Jacewicz A, Shuman S. 2014. Structures of bacterial polynucleotide kinase in a Michaelis complex with GTP*Mg2+ and 5′-OH oligonucleotide and a product complex with GDP*Mg2+ and 5′-PO4 oligonucleotide reveal a mechanism of general acid-base catalysis and the determinants of phosphoacceptor recognition. Nucleic Acids Res 42: 1152–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowski JA, Kuo B, Woolford JL Jr. 2013. Has1 regulates consecutive maturation and processing steps for assembly of 60S ribosomal subunits. Nucleic Acids Res 41: 7889–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikfidan A. 2013. “Structural and functional characterization of Clp1, a eukaryotic RNA-specific polynucleotide kinase.” PhD thesis, University of Heidelberg, Heidelberg. [Google Scholar]
- Dikfidan A, Loll B, Zeymer C, Magler I, Clausen T, Meinhart A. 2014. RNA specificity and regulation of catalysis in the eukaryotic polynucleotide kinase Clp1. Mol Cell 54: 975–986. [DOI] [PubMed] [Google Scholar]
- Dong B, Silverman RH. 1999. Alternative function of a protein kinase homology domain in 2′, 5′-oligoadenylate dependent RNase L. Nucleic Acids Res 27: 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong B, Xu L, Zhou A, Hassel BA, Lee X, Torrence PF, Silverman RH. 1994. Intrinsic molecular activities of the interferon-induced 2–5A-dependent RNase. J Biol Chem 269: 14153–14158. [PubMed] [Google Scholar]
- Fromm L, Falk S, Flemming D, Schuller JM, Thoms M, Conti E, Hurt E. 2017. Reconstitution of the complete pathway of ITS2 processing at the pre-ribosome. Nat Commun 8: 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galburt EA, Pelletier J, Wilson G, Stoddard BL. 2002. Structure of a tRNA repair enzyme and molecular biology workhorse: T4 polynucleotide kinase. Structure 10: 1249–1260. [DOI] [PubMed] [Google Scholar]
- Gasse L, Flemming D, Hurt E. 2015. Coordinated ribosomal ITS2 RNA processing by the Las1 complex integrating endonuclease, polynucleotide kinase, and exonuclease activities. Mol Cell 60: 808–815. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Sugino A. 1988. New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74: 527–534. [DOI] [PubMed] [Google Scholar]
- Granneman S, Nandineni MR, Baserga SJ. 2005. The putative NTPase Fap7 mediates cytoplasmic 20S pre-rRNA processing through a direct interaction with Rps14. Mol Cell Biol 25: 10352–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad R, Maurice F, Viphakone N, Voisinet-Hakil F, Fribourg S, Minvielle-Sebastia L. 2012. An essential role for Clp1 in assembly of polyadenylation complex CF IA and Pol II transcription termination. Nucleic Acids Res 40: 1226–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada T, Weitzer S, Mair B, Bernreuther C, Wainger BJ, Ichida J, Hanada R, Orthofer M, Cronin SJ, Komnenovic V, et al. 2013. CLP1 links tRNA metabolism to progressive motor-neuron loss. Nature 495: 474–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heindl K, Martinez J. 2010. Nol9 is a novel polynucleotide 5′-kinase involved in ribosomal RNA processing. EMBO J 29: 4161–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holbein S, Scola S, Loll B, Dichtl BS, Hübner W, Meinhart A, Dichtl B. 2011. The P-loop domain of yeast Clp1 mediates interactions between CF IA and CPF factors in pre-mRNA 3′ end formation. PLoS One 6: e29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Zeqiraj E, Dong B, Jha BK, Duffy NM, Orlicky S, Thevakumaran N, Talukdar M, Pillon MC, Ceccarelli DF, et al. 2014. Dimeric structure of pseudokinase RNase L bound to 2–5A reveals a basis for interferon-induced antiviral activity. Mol Cell 53: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Shuman S. 2009. Characterization of a thermostable archaeal polynucleotide kinase homologous to human Clp1. RNA 15: 923–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Coyle SM, Doudna JA. 2011. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol Cell 41: 600–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaca E, Weitzer S, Pehlivan D, Shiraishi H, Gogakos T, Hanada T, Jhangiani SN, Wiszniewski W, Withers M, Campbell IM, et al. 2014. Human CLP1 mutations alter tRNA biogenesis, affecting both peripheral and central nervous system function. Cell 157: 636–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konikkat S, Woolford JL Jr. 2017. Principles of 60S ribosomal subunit assembly emerging from recent studies in yeast. Biochem J 474: 195–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konikkat S, Biedka S, Woolford JL Jr. 2017. The assembly factor Erb1 functions in multiple remodeling events during 60S ribosomal subunit assembly in S. cerevisiae. Nucleic Acids Res 45: 4853–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 1993. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res 21: 2541–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masek T, Valasek L, Pospisek M. 2011. Polysome analysis and RNA purification from sucrose gradients. Methods Mol Biol 703: 293–309. [DOI] [PubMed] [Google Scholar]
- Munir A, Shuman S. 2017. Characterization of Runella slithyformis HD-Pnk, a bifunctional DNA/RNA end-healing enzyme composed of an N-terminal 2′,3′ -phosphoesterase HD domain and a C-terminal 5′-OH polynucleotide kinase domain. J Bacteriol 199: pe00739-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble CG, Beuth B, Taylor IA. 2007. Structure of a nucleotide-bound Clp1-Pcf11 polyadenylation factor. Nucleic Acids Res 35: 87–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi C, Garabedian MV, Malik M, Noguchi E. 2008. A vector system for genomic FLAG epitope-tagging in Schizosaccharomyces pombe. Biotechnol J 3: 1280–1285. [DOI] [PubMed] [Google Scholar]
- Nomura M, Gourse R, Baughman G. 1984. Regulation of the synthesis of ribosomes and ribosomal components. Annu Rev Biochem 53: 75–117. [DOI] [PubMed] [Google Scholar]
- Peach SE, York K, Hesselberth JR. 2015. Global analysis of RNA cleavage by 5′-hydroxyl RNA sequencing. Nucleic Acids Res 43: e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillon MC, Stanley RE. 2017. Nuclease integrated kinase super assemblies (NiKs) and their role in RNA processing. Curr Genet 64: 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillon MC, Sobhany M, Borgnia MJ, Williams JG, Stanley RE. 2017. Grc3 programs the essential endoribonuclease Las1 for specific RNA cleavage. Proc Natl Acad Sci 114: E5530–E5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus BS, Schwer B, Shuman S. 2016. Characterization of the tRNA ligases of pathogenic fungi Aspergillus fumigatus and Coccidioides immitis. RNA 22: 1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson CC. 1965. Phosphorylation of nucleic acid by an enzyme from T4 bacteriophage-infected Escherichia coli. Proc Natl Acad Sci 54: 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocak S, Emery B, Tanner NK, Linder P. 2005. Characterization of the ATPase and unwinding activities of the yeast DEAD-box protein Has1p and the analysis of the roles of the conserved motifs. Nucleic Acids Res 33: 999–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman DW, Nakamura K, Nallur S, Dookwah MT, Metheetrairut C, Slack FJ, Weidhaas JB. 2016. miR-34 activity is modulated through 5′-end phosphorylation in response to DNA damage. Nat Commun 7: 10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A. 1990. The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci 15: 430–434. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Thoms M, Barrio-Garcia C, Thomson E, Flemming D, Beckmann R, Hurt E. 2017. Preribosomes escaping from the nucleus are caught during translation by cytoplasmic quality control. Nat Struct Mol Biol 24: 1107–1115. [DOI] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. 2003. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem 278: 43928–43938. [DOI] [PubMed] [Google Scholar]
- Schaffer AE, Eggens VR, Caglayan AO, Reuter MS, Scott E, Coufal NG, Silhavy JL, Xue Y, Kayserili H, Yasuno K, et al. 2014. CLP1 founder mutation links tRNA splicing and maturation to cerebellar development and neurodegeneration. Cell 157: 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schillewaert S, Wacheul L, Lhomme F, Lafontaine DL. 2012. The evolutionarily conserved protein Las1 is required for pre-rRNA processing at both ends of ITS2. Mol Cell Biol 32: 430–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S, Hurwitz J. 1979. 5′-Hydroxyl polyribonucleotide kinase from HeLa cell nuclei. Purification and properties. J Biol Chem 254: 10396–10404. [PubMed] [Google Scholar]
- Sidrauski C, Walter P. 1997. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90: 1031–1039. [DOI] [PubMed] [Google Scholar]
- van Nues RW, Rientjes JM, Morre SA, Mollee E, Planta RJ, Venema J, Raue HA. 1995a. Evolutionarily conserved structural elements are critical for processing of Internal Transcribed Spacer 2 from Saccharomyces cerevisiae precursor ribosomal RNA. J Mol Biol 250: 24–36. [DOI] [PubMed] [Google Scholar]
- van Nues RW, Venema J, Rientjes JM, Dirks-Mulder A, Raue HA. 1995b. Processing of eukaryotic pre-rRNA: the role of the transcribed spacers. Biochem Cell Biol 73: 789–801. [DOI] [PubMed] [Google Scholar]
- Wang LK, Shuman S. 2001. Domain structure and mutational analysis of T4 polynucleotide kinase. J Biol Chem 276: 26868–26874. [DOI] [PubMed] [Google Scholar]
- Wang LK, Lima CD, Shuman S. 2002. Structure and mechanism of T4 polynucleotide kinase: an RNA repair enzyme. EMBO J 21: 3873–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Das U, Smith P, Shuman S. 2012. Structure and mechanism of the polynucleotide kinase component of the bacterial Pnkp-Hen1 RNA repair system. RNA 18: 2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Selvadurai K, Huang RH. 2015. Reconstitution and structure of a bacterial Pnkp1-Rnl-Hen1 RNA repair complex. Nat Commun 6: 6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinfeld M, Mani RS, Abdou I, Aceytuno RD, Glover JN. 2011. Tidying up loose ends: the role of polynucleotide kinase/phosphatase in DNA strand break repair. Trends Biochem Sci 36: 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzer S, Martinez J. 2007. The human RNA kinase hClp1 is active on 3′ transfer RNA exons and short interfering RNAs. Nature 447: 222–226. [DOI] [PubMed] [Google Scholar]
- Weitzer S, Hanada T, Penninger JM, Martinez J. 2015. CLP1 as a novel player in linking tRNA splicing to neurodegenerative disorders. Wiley Interdiscip Rev RNA 6: 47–63. [DOI] [PubMed] [Google Scholar]
- Wreschner DH, Silverman RH, James TC, Gilbert CS, Kerr IM. 1982. Affinity labelling and characterization of the ppp(A2′p)nA-dependent endoribonuclease from different mammalian sources. Eur J Biochem 124: 261–268. [DOI] [PubMed] [Google Scholar]