

This review by Shan et al. discusses necroptosis, a form of regulated necrotic cell death mediated by RIPK1 kinase activity, RIPK3, and MLKL, which can be activated under apoptosis-deficient conditions. Both necroptosis and apoptosis can be activated in response to various mutations that result in the abortion of defective embryos and during human inflammatory and neurodegenerative pathologies.
Keywords: apoptosis, MLKL, necroptosis, RIPK1, RIPK3
Abstract
Necroptosis, a form of regulated necrotic cell death mediated by RIPK1 (receptor-interacting protein kinase 1) kinase activity, RIPK3, and MLKL (mixed-lineage kinase domain-like pseudokinase), can be activated under apoptosis-deficient conditions. Modulating the activation of RIPK1 by ubiquitination and phosphorylation is critical to control both necroptosis and apoptosis. Mutant mice with kinase-dead RIPK1 or RIPK3 and MLKL deficiency show no detrimental phenotype in regard to development and adult homeostasis. However, necroptosis and apoptosis can be activated in response to various mutations that result in the abortion of the defective embryos and human inflammatory and neurodegenerative pathologies. RIPK1 inhibition represents a key therapeutic strategy for treatment of diseases where blocking both necroptosis and apoptosis can be beneficial.
Necroptosis is a form of regulated necrotic cell death mediated by the kinase activity of RIPK1 (receptor-interacting protein [RIP] kinase 1), RIPK3, and MLKL (mixed-lineage kinase domain-like pseudokinase). Necroptosis was first defined by its inhibition with specific chemical inhibitors of RIPK1, such as necrostatin-1 (Nec-1) and its improved analog, Nec-1s (Degterev et al. 2005, 2008). The availability of Nec-1 facilitated the research investigating the mechanism of necroptosis and its relevance to human diseases and ignited tremendous interest in this area over the past decade. Necroptosis has been established as an important cell death mechanism (Christofferson and Yuan 2010; Weinlich et al. 2017). While the activation of caspases mediates the signal transduction and execution of apoptotic cell death (Degterev et al. 2003), necroptosis can be activated under apoptosis-deficient conditions, leading to cell death and embryonic lethality. While multiple signals may potentially activate necroptosis, tumor necrosis factor α (TNFα) remains the best-understood and possibly most important trigger of necroptosis under various pathological conditions in humans. In addition, the activation of necroptosis can also robustly promote inflammation by mediating the production of inflammatory cytokines and the release of damage-associated molecular patterns (DAMPs) from disrupted cell membrane, which occurs as a result of necrotic cellular lysis. We focus this review on the mechanisms of necroptosis and a closely related RIPK1-dependent apoptosis (RDA) pathway induced by TNFα and discuss the relevance of necroptosis in development and human diseases.
Necroptosis activated by TNFα signaling
TNFα is an important proinflammatory cytokine involved in mediating a myriad of human inflammatory and degenerative diseases (Vassalli 1992; Aggarwal 2003; Varfolomeev and Vucic 2018). Activation of TNFR1 (TNF receptor 1) by TNFα sets off a rapid chain of intracellular signaling events, including dynamic post-translational modifications such as multiple types of ubiquitination, deubiquitination, and phosphorylation, leading to the formation of distinct protein complexes, which in turn dictate cellular responses. RIPK1 functions as a key integrator of these dynamic regulatory events by acting as a critical cellular signaling hub to modulate the cellular responses and determine cell fate (Ofengeim and Yuan 2013; Christofferson et al. 2014; Peltzer et al. 2016; Weinlich et al. 2017). RIPK1 contains an N-terminal kinase domain, an intermediate domain, and a C-terminal death domain (DD) (Fig. 1). In TNFα-stimulated cells, activation of RIPK1 kinase activity plays an important role in promoting regulated cell death through either necroptosis or apoptosis. Activated RIPK1 interacts with FADD and caspase-8 to form complex IIa to mediate RDA or with RIPK3 to form complex IIb to mediate necroptosis (Fig. 2). The kinase activity of RIPK1 provides a key pharmacological target for inhibiting both necroptosis and RDA. Here we review the signaling mechanisms that regulate the activation of RIPK1 downstream from TNFR1 to control cell death responses.
Figure 1.

A schematic diagram of RIPK1 domains, interacting proteins, post-translational modifications, and the catalytic enzymes that read or write these modifications. RIPK1 contains an N-terminal kinase domain, an intermediate domain with a RIP homotypic interaction motif (RHIM), and a C-terminal DD. The phosphorylation and ubiquitination sites as well as types of ubiquitin linkages on RIPK1 are indicated together with their respective catalytic enzymes. K63 ubiquitination chains on Lys377 mediate the recruitment of TAB2/3 and the activation of transforming growth factor-β-activated kinase 1 (TAK1), which in turn phosphorylates IKKs for activating the NF-κB pathway. M1 ubiquitination of RIPK1 is regulated by the linear ubiquitination assembly complex (LUBAC) and the deubiqutinating complex CYLD/SPATA2. The ubiquitin-binding proteins ABIN-1, Optineurin (OPTN), and NEMO, each carrying a UBAN motif that can bind with the linear ubiquitination chains, play important roles in regulating the activation of RIPK1. The RHIM motif of RIPK1 is required for binding with RIPK3 to mediate necroptosis and may also interact with TRIF, DAI, and Toll-like receptors to promote inflammation. Autophosphorylation of Ser166 is a biomarker for RIPK1 activation. The small molecule Nec-1s is caged in a hydrophobic pocket between the N and C lobes of the kinase domain and forms an H bond between its nitrogen atom and the hydroxyl oxygen of Ser161 on the activation loop to inhibit the activation of RIPK1. The cleavage of RIPK1 after Asp324 by caspase-8 or the phosphorylation on Ser320/331/333/335 in human RIPK1 and Ser321/332/334/336 in murine RIPK1 by TAK1 or MK2 leads to the suppression of RIPK1 activation. E3 ligase Pellino 1 (PELI1) mediates K63 ubiquitination on Lys115 of activated RIPK1 to promote complex IIb formation and necroptosis. The DD is not only crucial for the initiation of TNFR1 signaling but also indispensable for RIPK1 activation by mediating RIPK1 dimerization during the transition from complex I to complex II.
Figure 2.
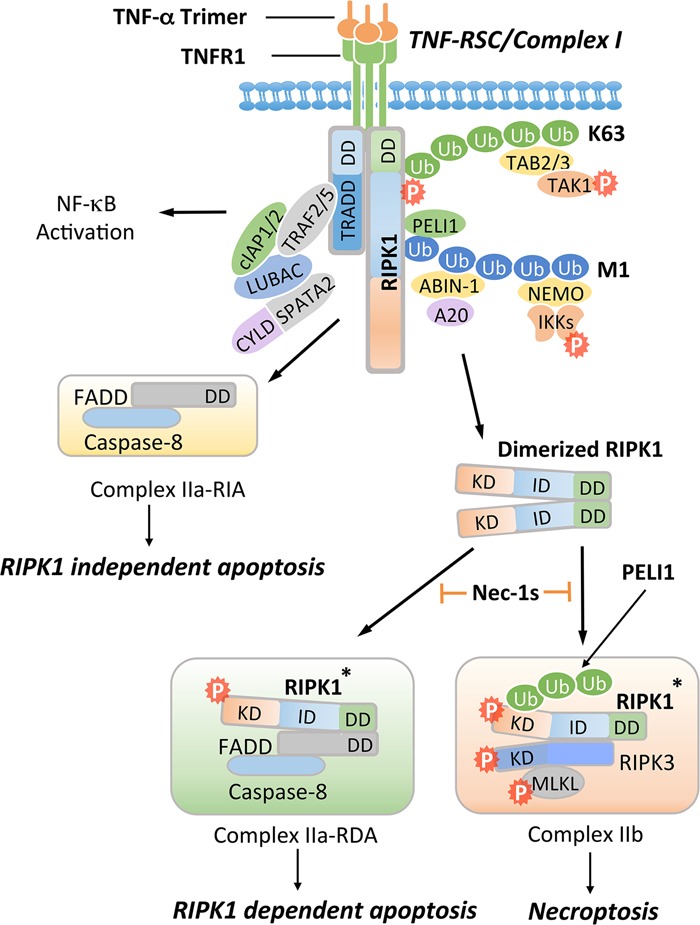
Activation of TNFR1 may promote multiple alternative signaling pathways, including the activation of NF-κB, RIPK1-independent apoptosis (RIA), RDA, and necroptosis. Activation of TNFR1 by trimerized TNFα induces the formation of a membrane-associated transient complex (named complex I or TNF-RSC), which includes RIPK1, TRADD, TRAF2/5, cIAP1/2, LUBAC, ABIN-1, A20, NEMO, PELI1, CYLD, and SPATA2. In complex I, RIPK1 is rapidly polyubiquitinated by K63-linked and M1-linked ubiquitin chains mediated by cIAP1/2 and LUBAC, respectively. K63 ubiquitination chains on RIPK1 mediate the recruitment of TAB2/3 to facilitate the activation of TAK1, leading to phosphorylation of the IKK complex and RIPK1 on S320/S331/S333/S335. M1 ubiquitination chains on RIPK1 mediate the recruitment of PELI1, ABIN-1, and NEMO. ABIN-1 in turn recruits phosphorylated A20 to promote K63 deubiquitination of RIPK1. CYLD/SPATA2 promote the M1 deubiquitination of RIPK1. The activated IKK/NEMO complex promotes the activation of NF-κB. When the output of the NF-κB pathway is inhibited by cycloheximide, which blocks the expression of cFLIPL, FADD interacts with caspase-8 to form complex IIa-RIA to promote RIA. Alternatively, dimerization of RIPK1 can mediate the activation of RIPK1 during the transition from complex I to complex II. Activated RIPK1* can interact with FADD and caspase-8 to form complex IIa-RDA to promote RDA. When caspase-8 is inhibited, activated RIPK1* is ubiquitinated by PELI1 on K115 and binds with RIPK3 to form complex IIb, leading to MLKL activation and necroptosis. Nec-1s inhibits the activation of RIPK1 kinase and in turn prevents the formation of complex IIa to block RDA and complex IIb to block necroptosis.
The activation of RIPK1 kinase is controlled by ubiquitination
Activation of TNFR1 by TNFα induces the formation of a transient complex, named complex I or TNF-RSC, associated with the intracellular DD of trimerized TNFR1, which recruits RIPK1 and TRADD by binding with their DDs (Micheau and Tschopp 2003). TRADD in turn recruits the adaptor proteins TRAF2/5 and the E3 ubiquitin ligases cIAP1/2 into complex I to catalyze the K63 ubiquitin modifications on RIPK1 (Bertrand et al. 2008). K63 ubiquitin chains on RIPK1 mediate the recruitment of TAB2/3 to facilitate the activation of transforming growth factor-β-activated kinase 1 (TAK1; also called MAP3K7). K63 ubiquitination of complex I also mediates the recruitment of the linear ubiquitination (M1-Ubi) assembly complex (LUBAC), composed of heme-oxidized IRP2 ubiquitin ligase 1 (HOIL-1), the catalytic subunit HOIL-1-interacting protein (HOIP), and shank-associated RH domain-interacting protein (SHARPIN). Interestingly, the complex of deubiqitinating enzyme CYLD and its adaptor, spermatogenesis-associated 2 (SPATA2), which can disassemble M1-Ubi, are recruited together with the LUBAC into complex I (Kovalenko et al. 2003; Trompouki et al. 2003; Draber et al. 2015; Elliott et al. 2016; Kupka et al. 2016; Schlicher et al. 2016; Wei et al. 2017). Simultaneous recruitment of M1-Ubi and deubiquitinating complexes suggests that the M1-Ubi of complex I components, such as TNFR1 and RIPK1, is modulated by both addition and trimming.
In complex I, RIPK1 is extensively modulated by different types of ubiquitin modifications, including M1 and K63 (Fig. 1). The ratio of M1/K63 ubiquitination on RIPK1 is critical for regulating its activation (Wei et al. 2017; Dziedzic et al. 2018). The activation of RIPK1 is regulated by both the M1-Ubi enzyme complex LUBAC and the deubiquitinating complex CYLD/SPATA2 (Wei et al. 2017). The lack of M1-Ubi, such as in HOIP-deficient cells, highly sensitizes cells to the activation of RIPK1 kinase and cell death in response to TNFα. On the other hand, CYLD and SPATA2 deficiencies partially protect cells against TNFα-induced apoptosis and necroptosis (Hitomi et al. 2008; Wei et al. 2017). CYLD promotes the activation of RIPK1 by predominantly removing M1-Ubi and, less so, K63 ubiquitin chains on RIPK1 after TNFα stimulation. Loss of SPATA2 impairs the recruitment of CYLD to complex I and preserves the M1-Ubi of RIPK1; as a consequence, RIPK1 activation is impaired (Hitomi et al. 2008; Moquin et al. 2013; Draber et al. 2015; Wei et al. 2017).
A20 (TNFAIP3) is a ubiquitin-editing enzyme and a negative regulator of NF-κB activation that is also recruited into complex I following TNFα stimulation (Wertz et al. 2004; Vucic et al. 2011; Harhaj and Dixit 2012). A20 contains an OTU domain that functions as a K63 deubiquitinase to remove K63 ubiquitination of RIPK1, and its ZnF4 motif promotes K48 ubiquitin linkage on RIPK1 to direct proteasome degradation (Wertz et al. 2004, 2015; Bosanac et al. 2010; Shembade et al. 2010). A20 exerts an opposing activity to CYLD, as A20 deficiency reduces the M1-Ubi levels of RIPK1 and promotes the activation of RIPK1 and its pronecroptotic partner, RIPK3 (Draber et al. 2015; Onizawa et al. 2015; Dziedzic et al. 2018). As a result, A20-deficient cells are highly sensitized to TNFα-induced RDA and necroptosis.
The recruitment of A20 to the TNFR1 is controlled by ABIN-1, which, along with A20, binds to M1 ubiquitin chains (Draber et al. 2015; Wertz et al. 2015; Dziedzic et al. 2018). Lack of ABIN-1 leads to the loss of phosphorylated A20 from complex I, elevated K63 ubiquitination of RIPK1, and hypersensitivity to necroptosis (Dziedzic et al. 2018), highlighting the function of the ABIN-1/A20 axis in restricting RIPK1 kinase activation by modulating its K63 ubiquitination.
Overall, K63 and M1 ubiquitin modifications in complex I may provide the code to dictate the activation of RIPK1 (Ofengeim and Yuan 2013). While it is still not clear how the cell decodes the various ubiquitination modifications on RIPK1, we now know that there are three ubiquitin-binding proteins (NEMO, ABIN-1, and Optineurin [OPTN]) that help to transmit and regulate the signals provided by various ubiquitination events on RIPK1. These three ubiquitin-binding proteins all carry UBAN domains and can bind to predominantly M1 ubiquitin chains (Rahighi et al. 2009; Hadian et al. 2011; Nanda et al. 2011; Ito et al. 2016; Nakazawa et al. 2016; Dziedzic et al. 2018). Strikingly, the recruitment of NEMO into complex I not only enables the formation of the IKK complex and phosphorylation of IKKα/β by TAK1 to promote the activation of NF-κB and MAPK signaling pathways but also suppresses the activation of RIPK1-mediated necroptosis and apoptosis (Wang et al. 2001; Kanayama et al. 2004; Haas et al. 2009; Legarda-Addison et al. 2009). As mentioned above, M1-dependent recruitment of ABIN-1 provides a scaffold to recruit A20 into complex I to mediate K63 deubiquitination of RIPK1 and inhibition of cell death (Dziedzic et al. 2018). On the other hand, OPTN, which is mutated in both familial and sporadic amyotrophic lateral sclerosis (ALS) patients, promotes K48 ubiquitination and degradation of RIPK1 and prevents neuroinflammation and axonal degeneration (Ito et al. 2016).
Modulation of RIPK1 activation and downstream outputs by phosphorylation
In complex I, K63 ubiquitination of RIPK1 by cIAP1/2 leads to the recruitment of TAB2/3 to promote the activation of TAK1. TAK1 performs multiple important prosurvival functions in complex I, including phosphorylation of IKKα/β to promote the activation of NF-κB as well as transient phosphorylation of RIPK1 on multiple sites (including S321, S332, and S334) to control activation of its kinase activity (Figs 1, 2; Ea et al. 2006; Geng et al. 2017). In particular, transient S321 phosphorylation on RIPK1 inhibits its binding to FADD and activation of RDA. On the other hand, sustained TAK1-mediated phosphorylation of RIPK1 on multiple sites of the intermediate domain, including S321, promotes its interaction with RIPK3 to mediate necroptosis. In addition, the IKK complex, including NEMO and IKKα/β, can also mediate inhibitory phosphorylation on RIPK1. NEMO deficiency, IKKα/IKKβ double deficiency, or treatment with IKK inhibitors such as TPCA-1 highly sensitizes cells to RDA independent of the NF-κB pathway (Dondelinger et al. 2015; Koppe et al. 2016).
TAK1 is also important for mediating the activation of MAP kinase p38 and its downstream kinase, MK2. MK2 can inhibit the activation of cytosolic RIPK1 by directly phosphorylating S321 and S336 (Figs 1,2; Dondelinger et al. 2017; Jaco et al. 2017; Menon et al. 2017). Inhibition of MK2 or S321/336A mutation in murine RIPK1 promotes the activation of RIPK1 kinase in the cytosol and complex II formation (Dondelinger et al. 2017; Jaco et al. 2017; Menon et al. 2017). Thus, TAK1 orchestrates multiple phosphorylation events on RIPK1 both directly and indirectly via promoting the activation of IKK and MK2 and in both complex I and cytosol to control the direction of RIPK1 signaling.
Activation of RIPK1-independent apoptosis (RIA) and RDA
Necroptosis can occur when apoptosis-deficient cells are stimulated by TNFα. In apoptosis-competent cells, TNFα stimulation can induce apoptosis in either a RIPK1-independent or a RIPK1-dependent manner. When protein synthesis is inhibited by cycloheximide, which blocks the expression of caspase-8 inhibitor cFLIPL, mediated by the NF-κB pathway, TNFα stimulation activates caspase-8 to promote RIA (Kreuz et al. 2001; Micheau et al. 2001). In RIA, RIPK1 may serve as a substrate of caspase-8 (Lin et al. 1999). The cleavage of RIPK1 at the C terminus of its kinase domain (after residue D324) provides an important mechanism by which apoptosis may preempt the activation of RIPK1 kinase and necroptosis. Inactivation of caspase-8 by chemical inhibitors, such as zVAD.fmk, as well as FADD or caspase-8 deficiency prevents the cleavage of RIPK1 to permit the activation of necroptosis in cells stimulated by TNFα.
On the other hand, in the absence of caspase inhibition, disrupting the ubiquitination and phosphorylation status of complex I by either cIAP1/2, LUBAC, TAK1, or NEMO/IKK deficiency promotes the activation of RIPK1 kinase to lead to RDA (Dondelinger et al. 2015; Geng et al. 2017; Wei et al. 2017). In addition, inactivation of MK2, a downstream target of TAK1, also sensitizes cells to RDA (Dondelinger et al. 2017; Jaco et al. 2017; Menon et al. 2017; Mohideen et al. 2017). Phosphorylation of S321 in RIPK1 is important for suppressing the activation of RIPK1, as S321A mutation in murine RIPK1 switches cells from RIA to RDA even though S321A mutation has no effect on NF-κB activation (Geng et al. 2017; Jaco et al. 2017). Activation of RDA is also affected by cFLIP isoforms present in the cells. While long isoform FLIPL forms a heterodimeric complex with caspase-8 in which the latter is partially active and inhibits RIPK1-dependent cell death (Oberst et al. 2011), short isoform FLIPS forms caspase-8 heterodimers completely devoid of catalytic activity, promoting RIPK1 kinase activation and necroptosis (Feoktistova et al. 2011).
RIPK1 dimerization promotes its activation in the transition from complex I to complex II
Multiple key mediators of the TNFR1 signaling pathway are members of the DD family, including TNFR1, RIPK1, TRADD, and FADD (Park et al. 2007). The six-helical bundle structural folds of DDs mediate the formation of key intracellular signaling complexes by forming homotypic interactions with itself and other members of the family. The interaction of the C-terminal DD of RIPK1 with the DDs of TNFR1 and TRADD results in the recruitment of RIPK1 into complex I. Interestingly, recent evidence suggests that the dimerization of RIPK1 mediated by its C-terminal DD also plays an important role in controlling its activation during the transition from complex I to complex II. A mutation of K584 (murine RIPK1)/K599 (human RIPK1), a lysine located on the surface of the DD, to arginine does not affect the recruitment of RIPK1 into complex I but blocks RIPK1 activation in necroptosis and RDA and the formation of complex II (Meng et al. 2018). Thus, the K584/K599 residue is required for the RIPK1-DD to homodimerize but not heterodimerize with the DDs of TNFR1 or TRADD. The DD homodimerization-mediated activation of RIPK1 is critical in vivo, as K584R knock-in mutant mice are highly resistant to TNFα-induced systematic inflammatory response syndrome (SIRS), similar to what was observed in RIPK1 kinase-dead knock-in D138N and K45A mice as well as ripk3−/− mice (Duprez et al. 2011; Polykratis et al. 2014; Shutinoski et al. 2016). The dimerization-mediated activation of RIPK1 may provide a mechanism to control its activation in a concentration-dependent manner so that pathological conditions or cell types that express higher levels of RIPK1 may be sensitized to the activation of RIPK1 by dimerization.
Regulations for the formation of necroptotic complex IIb
Activation of necroptosis by RIPK1 also requires ubiquitination events downstream from complex I. Pellino 1 (PELI1), a member of the Pellino family, is an E3 ubiquitin ligase known to be involved in mediating the Toll-like receptor 3 (TLR3)/TLR4 signaling (Jiang et al. 2003; Chang et al. 2009). K63 ubiquitination of activated RIPK1 on K115 by PELI1 is critical for the formation of complex IIb (Wang et al. 2017). PELI1 is recruited into complex I in a manner regulated by RIPK1 ubiquitination, as the lack of RIPK1, TRADD, cIAP1/2, or LUBAC blocks the recruitment of PELI1; however, the lack of PELI1 does not affect the ubiquitination status of complex I or the activation of RIPK1. PELI1 is critical specifically for the formation of complex IIb, as peli1−/− mouse embryonic fibroblasts (MEFs) were unable to form complex IIb and therefore were highly resistant to necroptosis. Ubiquitination of RIPK1 by PELI1 is unusual, as it can be inhibited by Nec-1s; thus, ubiquitination of RIPK1 by PELI1 occurs only on activated RIPK1. The phosphorylation of RIPK3 S232 and MLKL S345, the markers for the activation of RIPK3 and MLKL, respectively, was blocked in peli1−/− MEFs stimulated by TNFα/SM-164/zVAD.fmk.
The intermediate domain of RIPK1 contains a 16-amino-acid motif named the RIP homotypic interaction motif (RHIM) that is also found in the C terminus of RIPK3 (Sun et al. 2002). This domain is critical for the binding of RIPK1 to RIPK3 and in mediating necroptosis (Cho et al. 2009). The interaction of activated RIPK1 with RIPK3 leads to the RHIM-dependent formation of complex IIb (or necrosome), a core execution complex of necroptosis that may be present in an amyloid-like conformation (Li et al. 2012). RIPK1mRHIM mutant MEFs and primary fetal liver macrophages carrying a knock-in mutation that changes the core RHIM sequence 529–531 (QIG) to AAA are highly resistant to necroptosis due to the inability of mutated RIPK1 to bind to RIPK3 (Lin et al. 2016; Newton et al. 2016b). On the other hand, the expression of ZBP1, another RHIM-containing protein, is induced in keratinocytes to mediate skin inflammation in mice with epidermis-specific RIPK1 deficiency but not epidermis-specific FADD deficiency. However, since only RIPK1-deficient keratinocytes have been shown to induce the expression of ZBP1, which is known as an innate sensor of viral infection (Kuriakose and Kanneganti 2018), the interaction of RIPK1-deficient keratinocytes with innate immune activators might be involved in inducing ZBP1. Thus, while the RHIM domain of RIPK1 is essential for binding with RIPK3 and mediating TNFα-induced necroptosis, the lack of RIPK1 can induce the expression of ZBP1 to promote RIPK3 activation and necroptosis in the skin.
The intermediate domain of RIPK1, which contains the RHIM, is regulated by both ubiquitination and phosphorylation. In addition to phosphorylation of S321/S332/S334/S336 as discussed above, LUBAC has been shown to mediate M1-Ubi on RIPK1 in the intermediate domain, and thus M1-Ubi on RIPK1 may directly interfere with its binding with RIPK3 to inhibit the formation of complex IIb (Wei et al. 2017).
Execution of necroptosis by MLKL polymerization
Activation and phosphorylation of MLKL—a pseudokinase composed of an N-terminal four-helical bundle domain (4HBD) and a C-terminal pseudokinase domain—by RIPK3 marks an important execution step in necroptosis (Cho et al. 2009; He et al. 2009; Zhang et al. 2009; Sun et al. 2012; Zhao et al. 2012). RIPK3-mediated phosphorylation of MLKL induces a conformational change in MLKL that transitions into an open active conformation to expose the 4HBD (Murphy et al. 2013; Hildebrand et al. 2014). The phosphorylated MLKL has been reported to lead to changes in its conformation to form disulfide bond-dependent amyloid-like polymers and disrupt the plasma membrane to execute necroptosis (Cai et al. 2014; Chen et al. 2014; Wang et al. 2014; Liu et al. 2017a; Reynoso et al. 2017). The T357/S358 phosphorylation of MLKL and the four cysteines responsible for the disulfide bonds on the N-terminal domain (NTD) of MLKL are essential for the tetramer and subsequent polymer formation of MLKL. Neither T357A/S358A MLKL nor the mutant in which the four cysteines are mutated can form a functional tetramer or polymers and consequently cannot induce necroptosis (Wang et al. 2014; Liu et al. 2017a). Necrosulfonamide (NSA), which can form a covalent adduct with Cys86 of human MLKL and inhibit necroptosis of human cells, modestly blocks MLKL tetramer formation but completely blocks MLKL polymerization downstream from MLKL phosphorylation in complex IIb. Furthermore, forced polymerization of the NTD of MLKL directly triggers necroptosis even in the absence of RIPK3. This is still blocked by NSA through the inhibition of high-molecular-weight polymerization but not the formation of tetramers (Sun et al. 2012; Liu et al. 2017a), suggesting that MLKL polymerization is indispensable for necroptosis.
Thioredoxin-1 (Trx1), an oxidoreductase, is reported to bind with MLKL and maintain MLKL at a reduced state under basal conditions, thus blocking necroptosis by preventing MLKL disulfide bond formation and polymerization (Reynoso et al. 2017). Both Trx1 knockdown and the addition of Trx1 inhibitors promoted MLKL polymerization and necroptosis (Reynoso et al. 2017). These data suggest that Trx1 may play an important role in negative regulation of MLKL activation. However, necroptosis is often not sensitive to the addition of reducing agents in many cell lines and conditions (Degterev et al. 2005), a critical feature of necroptosis that distinguishes it from other forms of necrosis, such as ferroptosis; thus, MLKL polymerization during necroptosis is likely not regulated through nonspecific oxidative mechanisms.
Recombinant MLKL can bind to phosphatidylinositol phosphates and cardiolipin and disrupt liposomes (Dondelinger et al. 2014; Wang et al. 2014). Na+ or Ca++ influx may also occur upon necroptosis induction to induce the osmotic disruption of the plasma membrane (Cai et al. 2014; Chen et al. 2014). In the latter case, activated MLKL was proposed to bind to the TRPM7 cation channel to promote necroptosis (Cai et al. 2014). In addition, recent studies show that activation of the endosomal sorting complexes required for transport-III (ESCRT-III) machinery downstream from the MLKL step leads to the release of damaged plasma membrane vesicles with an exposed phagocytic phosphatidylserine signal. Such mechanisms may work together with the release of cytokines and DAMPs during necroptosis to allow dying cells to signal the surrounding cells (Gong et al. 2017; Yoon et al. 2017).
Necroptosis in development
Apoptosis is involved in mediating normal development. Mutant mice that have lost various key components of the apoptotic machinery (e.g., casp3−/− mice, casp9−/− mice, and apaf-1−/− mice) show severe developmental defects, including defective neural tube closure and embryonic/prenatal lethality (Kuida et al. 1996, 1998; Cecconi et al. 1998; Yoshida et al. 1998). In contrast, mutant mice that lack key elements of the necroptosis machinery (e.g., ripk3−/− and mlkl−/− mice) or lack the kinase activity of RIPK1 (including three different kinase-dead knock-in RIPK1 mutant mouse lines carrying D138N, K45A, or ΔG26F27 mutations) show normal development and do not exhibit any obvious phenotype (Newton et al. 2004; Wu et al. 2013; Polykratis et al. 2014; Shutinoski et al. 2016; Liu et al. 2017b). Thus, necroptosis and RDA are not involved in mediating development or adult function under normal conditions. This is likely due to the fact that necroptosis and RDA are activated primarily in response to cytokines induced by pathological stimuli (e.g., TNFα and mediated by death receptor signaling) that are not present or activated under normal conditions.
The genes involved in mediating necroptosis, including RIPK1, RIPK3, and MLKL, are not found in primitive organisms. RIPK1 orthologs are present in most vertebrate species and mammals but not in more primitive organisms such as nematodes or flies (Dondelinger et al. 2016). RIPK3 and MLKL are found only in certain vertebrates and mammals and not in some specific Craniata clades and other discrete species. Within the Mammalia class, the whole Carnivora order (including domestic dogs [Canis lupus familiaris], domestic cats [Felis catus], and ferrets [Mustela putorius furo]) lacks the MLKL gene, whereas the infraclass of Marsupialia (opossums, Tasmanian devils, and wallabies) lacks both RIPK3 and MLKL genes. These observations argue against the view that necroptosis is evolved as an essential host defense mechanism.
We analyzed the breeding patterns of mammalian species with fully sequenced genomes (Stockley and Hobson 2016) and found that organisms that possess an MLKL ortholog have statistically significantly smaller litter sizes and longer interbirth intervals (Fig. 3). While the mean litter size of organisms that have an MLKL ortholog is 2.3, the mean litter size for organisms that lack an MLKL ortholog is 4.7. Similarly, while the mean interbirth interval for organisms that have an MLKL ortholog is 1053.6 d, the mean interbirth interval for organisms that lack an MLKL ortholog is 254.5 d. The exception to this rule appears to be rodents, which have MLKL but also large litter sizes and short interbirth intervals. Thus, organisms that produce fewer offspring per litter and have longer interbirth intervals may rely on necroptosis-mediated elimination of unfit offspring early in the pregnancy to allow for sooner de novo pregnancy and eliminate the burden of rearing unhealthy offspring. Such a developmental checkpoint, with a likely positive influence on breeding and population growth, could be an adaptation to compensate for the smaller litter sizes and longer interbirth intervals. Thus, necroptosis may have evolved as a mechanism to ensure the vitality of precious vertebrate offspring.
Figure 3.
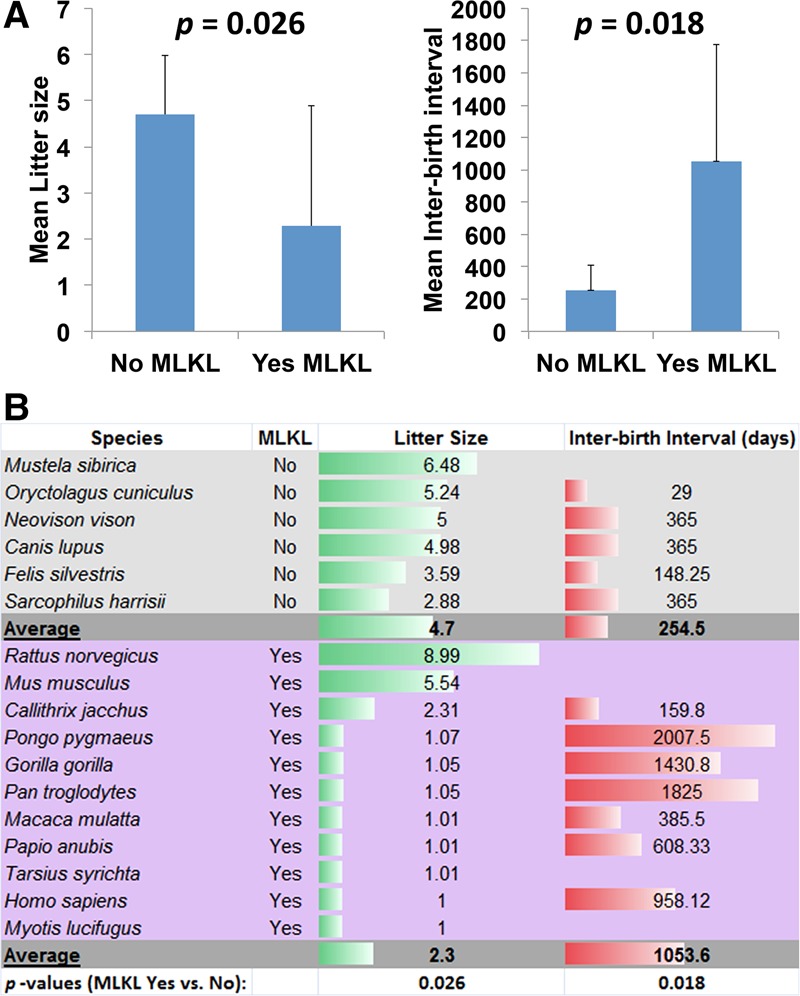
MLKL expression and necroptosis in fast- and slow-breeding mammalian species. (A) Comparison of litter sizes and interbirth intervals in mammals that express or do not express MLKL. Mean values are shown, and error bars represent standard deviation. Student's t-test was used to calculate the P-values. (B) List of species used in A and their mean litter sizes and mean interbirth intervals.
Consistent with this idea, necroptosis may be activated during embryonic development to abort fetuses with severe developmental abnormalities, as RIPK3 deficiency and MLKL deficiency can block the embryonic lethality induced by mutations in multiple genes during different stages of embryonic development. While this hypothesis should be examined further in the future, here we review four examples in which embryonic/perinatal lethality of mutant mice can be blocked by inhibition of necroptosis and sometimes apoptosis. We predict that many such examples exist, as the activation of necroptosis and apoptosis might provide a common strategy to abort defective mutant embryos.
Activation of necroptosis and apoptosis in embryonic day 10.5 (E10.5) embryos mutant for caspase-8/fadd/cflar
Genetic ablation of caspase-8, FADD, or FLIP (cflar) results in defective vascular and heart development and lethality at E10.5 during mouse development (Varfolomeev et al. 1998; Yeh et al. 1998, 2000). Embryonic lethality of casp8−/− and fadd−/− mice is fully rescued when these mice are bred in a ripk3−/− or mlkl−/− background. casp8−/− ripk3−/−, fadd−/− ripk3−/−, casp8−/− mlkl−/−, and fadd−/− mlkl−/− mice are normal, including in their vascular and cardiac systems. Furthermore, consistent with the essential role of RIPK1 in mediating necroptosis, the embryonic lethality of casp8−/− and fadd−/− mice can also be rescued by RIPK1 deficiency until birth (Zhang et al. 2011; Dillon et al. 2014; Alvarez-Diaz et al. 2016). On the other hand, the lethality of cflar−/− mice cannot be suppressed by ripk3−/− alone but is blocked by double deficiency of FADD and RIPK3 as in cflar−/− casp8−/− ripk3−/−, cflar−/− fadd−/− ripk3−/−, and cflar−/− fadd−/− mlkl−/− mice (Kaiser et al. 2011; Oberst et al. 2011; Dillon et al. 2012; Alvarez-Diaz et al. 2016). Thus, the caspase-8/FADD/FLIP complex is required for normal embryonic vascular development. Defective embryos without caspase-8 or FADD are eliminated by activation of necroptosis, whereas embryos that lack FLIP are eliminated through the activation of both necroptosis and apoptosis.
In addition, while casp8−/−ripk3−/−, fadd−/−ripk3−/−, and casp8−/−mlkl−/− double-deficient mice were viable and matured into fertile adults, they rapidly developed severe lymphadenopathy similar to FASL- or FAS-deficient mice (Watanabe-Fukunaga et al. 1992; Kaiser et al. 2011; Oberst et al. 2011; Dillon et al. 2012; Alvarez-Diaz et al. 2016). These results are consistent with the requirement for caspase-8/FADD in mediating signaling pathways downstream from Fas to control lymphadenopathy and autoimmunity. However, inactivation of RIPK1 (as in mice with inactivated RIPK1 D138N or K45A knock-in mutations), which blocks both necroptosis and RDA mediated by TNFα, does not lead to immune system dysfunction such as that of caspase-8/FADD null mutations.
Activation of necroptosis and apoptosis in E15.5 embryos mutant for p65/RelA
NF-κB, consisting of p50 and p65/RelA, is critically involved in mediating gene expression in response to infection, inflammation, and stress. p65/RelA−/− mice die at E15.5 with defective vascular development, massive hemorrhage, and cell death in multiple tissues. The embryonic lethality of RelA−/− mice is fully rescued by the combined ablation of FADD and RIPK3/MLKL or by blocking RIPK1 kinase activity by D138N or K45A knock-in mutations (Xu et al. 2018). RelA−/− MEFs are sensitized to both apoptosis and necroptosis. Since the ablation of TNFα or TNFR1 can also rescue RelA-deficient mice from embryonic lethality (Doi et al. 1999; Alcamo et al. 2001), this suggests that defective NF-κB signaling in developing embryos promotes the activation of RIPK1 to mediate both necroptosis and apoptosis through TNFR1.
Consistent with the role of RelA to suppress necroptosis during embryonic development, intestinal epithelial cell (IEC)-specific knockout of RelA also leads to increased IEC apoptosis and reduced Paneth cell numbers in ileal crypts, which can be blocked by RIPK1 D138N mutation (Vlantis et al. 2016).
Activation of necroptosis in E18.5 embryos mutant for ABIN-1
ABIN-1, encoded by the gene Tnip1, is recruited into complex I in TNFα-stimulated cells to facilitate the recruitment of A20, an important ubiquitin-editing enzyme that suppresses the activation of RIPK1 (Dziedzic et al. 2018). Abin-1 deficiency promotes the activation of RIPK1 and necroptosis. Abin-1−/− mice die at E18.5 with massive liver degeneration, which can be completely blocked by the RIPK1 D138N knock-in mutation or RIPK3 deficiency. Thus, mutant embryos that are deficient for Abin-1 are aborted through the activation of necroptosis, demonstrating that necroptosis can be activated in defective embryos without inactivation of caspase-8/FADD.
Early postnatal lethality of ripk1−/− mice
Ripk1−/− newborns appear normal at birth but fail to thrive and die at 1–3 d of age (Kelliher et al. 1998). Since the kinase activity of RIPK1 is prodeath and involved in mediating both apoptosis and necroptosis, it is intriguing why ripk1−/− mice might die. The early postnatal lethality of ripk1−/− mice can be fully blocked upon inhibition of both apoptosis and necroptosis in ripk1−/− fadd−/− ripk3−/−, ripk1−/− casp8−/− ripk3−/−, and ripk1−/− fadd−/− mlkl−/− mice, and the triple-mutant animals survive and live grossly normal lives but develop autoimmune lymphoproliferative syndrome (ALPS) later, likely due to the lack of caspase-8 activity (Dillon et al. 2014; Kaiser et al. 2014; Rickard et al. 2014b; Alvarez-Diaz et al. 2016). At the cellular level, RIPK1 has an indispensible role in the initiation of necroptosis in response to TNFα stimulation, as ripk1−/− MEFs cannot undergo necroptosis upon TNFα stimulation (Dillon et al. 2014). Therefore, RIPK1 is unlikely to be directly involved in suppressing the activation of necroptosis. On the other hand, since RIPK1 serves as a scaffold in coordinating the activation of NF-κB (Kelliher et al. 1998; Ea et al. 2006), it is possible that the early postnatal lethality of ripk1−/− mice might be due to defects in the activation of NF-κB, which mediates the expression of multiple key regulators of apoptosis and necroptosis; e.g., cFLIPL, cIAP1, and A20. Consistently, the expression of cFLIPL in ripk1−/− mice and ripk1−/− MEFs is reduced (Dillon et al. 2014). Since the embryonic lethality of RelA−/− mice is fully rescued by the combined ablation of FADD and RIPK3/MLKL (Xu et al. 2018), the defects in the activation of NF-κB might be involved in mediating the perinatal lethality of ripk1−/− mice.
The signals responsible for necroptosis in the absence of RIPK1 are still not well understood. One possible candidate is ZBP1 (DAI), as it can induce necroptosis through directly binding to RIPK3 (Lin et al. 2016; Newton et al. 2016b). Mice with epidermis-specific RIPK1 deficiency develop skin inflammation due to RIPK3/MLKL-dependent keratinocyte necroptosis. The expression of ZBP1 is elevated in ripk1−/− keratinocytes and can act to compensate for the loss of RIPK1 and directly interact with RIPK3 in ripk1−/− keratinocytes to promote necroptosis. However, since mice with epidermis-specific RIPK1 deficiency do not die perinatally, it remains to be tested whether the regulation of ZBP1 is also important in other RIPK1-deficient cells and involved in mediating the perinatal lethality of ripk1−/− mice.
Necroptosis in diseases
As discussed above, necroptosis might serve as a critical checkpoint during embryonic development to ensure the vitality of vertebrate offspring by aborting embryos with severe developmental defects. Interestingly, necroptosis can be reactivated during adult life and in aging in response to acute and chronic insults. The activation of RIPK1 and necroptosis has been implicated in many human diseases by mediating cell death and inflammation. The application of Nec-1s has shown efficacy in ameliorating tissue injuries in animal models of diseases ranging from ischemic brain, kidney, and heart injuries to multiple sclerosis, ALS, and Alzheimer's disease (AD) (Zhou and Yuan 2014; Ofengeim et al. 2015, 2017; Ito et al. 2016; Caccamo et al. 2017). Thus, the activation of RIPK1 and necroptosis may represent an important pathological mechanism involved in mediating cell death and inflammation in many human diseases. Here we focus on reviewing those human diseases with direct genetic links to the activation of RIPK1 and necroptosis.
ALS
ALS is a lethal degenerative motor neuron disease with an average survival of 3–5 yr after the diagnosis. Loss-of-function mutations in the OPTN gene have been implicated in both familial and sporadic ALS cases (Maruyama et al. 2010; Beeldman et al. 2015; Cirulli et al. 2015). Mutant mice with either total or conditional loss of OPTN in oligodendrocytes or microglia develop progressive dysmyelination and axonal degeneration (Ito et al. 2016). Inhibition of RIPK1 kinase activity by D138N knock-in or by pharmacological inhibition using Nec-1s in mice restores the myelination and axonal integrity. Furthermore, the engagement of necroptotic machinery in the CNS, including RIPK1, RIPK3, and MLKL in mediating axonal pathology, is also observed in SOD1G93A transgenic mice and pathological samples from human ALS patients. Thus, inhibiting RIPK1 kinase may provide an axonal protective strategy for the treatment of ALS and other human degenerative diseases characterized by axonal degeneration.
ABIN-1 was found to be associated with both ALS and schizophrenia in humans in genome-wide association studies (McLaughlin et al. 2017). Although the implications of ABIN-1 in ALS and schizophrenia have not yet been examined in animal models, the involvement of ABIN-1 in regulating the activation of RIPK1 and necroptosis during embryonic development suggests that, like OPTN, ABIN-1 may be another ubiquitin-binding protein involved in regulating the activation of RIPK1 in ALS and possibly also in schizophrenia. If so, this may raise the intriguing possibility for a role of RIPK1-mediated necroptosis and apoptosis in schizophrenia and other mental disorders.
A20 and Abin-1 deficiency-associated immunopathologies
The systemic inflammation and early postnatal lethality of A20−/− mice can be significantly ameliorated by either catalytically inactive RIPK1 or RIPK3 deficiency but not MLKL deficiency (Onizawa et al. 2015; Newton et al. 2016a). Since A20 deficiency promotes the activation of RIPK1 and necroptosis (Dziedzic et al. 2018), blocking RIPK1 and necroptosis might provide treatments for human diseases with reduced A20 expression and/or function. A20 has been identified as a susceptibility locus in humans with genetic predisposition to multiple immunopathologies, including Crohn's disease (CD), rheumatoid arthritis, systemic lupus erythematosus, psoriasis, and type 1 diabetes (Vereecke et al. 2009). Furthermore, mucosal biopsies from CD patients confirmed a consistent down-regulation of mucosal A20 expression (Arsenescu et al. 2008). In addition, ABIN-1 also shows strong association with psoriasis in humans (Nair et al. 2009). Since A20-deficient myeloid cells are sufficient to elicit the spontaneous inflammatory phenotype as seen in the full-body A20 knockout mice (Turer et al. 2008), the activation of RIPK1 and necroptosis in myeloid lineages may play an important role in driving proinflammatory signaling under A20-deficient conditions.
A heterozygous C243Y mutation in A20 in a Japanese family has been found to be responsible for autosomal-dominant Behçet's disease, which is characterized by the chronic inflammation of the blood vessels (Shigemura et al. 2016). A reduced effect of C243Y A20 compared with wild-type A20 in suppressing cytokine production in cultured cells suggests that it is a loss-of-function allele. Consistently, heterozygous germline mutations in A20 have been found in six unrelated families with early-onset systemic inflammation, a disorder resembling Behçet's disease (Zhou et al. 2016). These patients express mutant truncated A20 proteins, which are likely to act through haploinsufficiency. These results suggest that A20 might be a dose-dependent suppressor of RIPK1 and necroptosis in mediating the inflammatory pathology of these human diseases.
NEMO deficiency syndromes
NEMO is a ubiquitin-binding protein with a UBAN domain and can bind M1 ubiquitin chains (Hauenstein et al. 2017). While the well-established role of NEMO is to facilitate the efficient recruitment of IKKα/β into complex I and support their activation in mediating NF-κB activation (Ea et al. 2006; Wu et al. 2006), recent studies suggest that NEMO is an important suppressor of RIPK1 (Legarda-Addison et al. 2009; Kondylis et al. 2015; Pescatore et al. 2016; Vlantis et al. 2016). NEMO deficiency promotes the activation of RIPK1 and RDA independent of NF-κB (Legarda-Addison et al. 2009). IEC-specific ablation of NEMO causes apoptosis of Paneth cells and colonocytes and microbiota-driven chronic inflammation in the colon, which is not reproduced in a combined IEC-specific ablation of RelA, c-Rel, and RelB. These data suggest that NEMO suppresses colon inflammation through NF-κB-independent functions. On the other hand, inhibition of RIPK1 kinase activity or combined deficiency of FADD and RIPK3 prevented epithelial and Paneth cell death and colitis development in mice with epithelial NEMO deficiency (Vlantis et al. 2016). Inhibition of RIPK1 by D138N mutation can also block the embryonic lethality of NEMO−/− mice. Therefore, NEMO prevents intestinal inflammation and embryonic lethality by inhibiting RIPK1 kinase activity. These observations indicate that RIPK1 inhibitors could be effective in the treatment of colitis in patients with NEMO mutations and possibly in inflammatory bowel disease (IBD). Human patients with immunodeficiency (ectodermal dysplasia with immune deficiency [EDA-ID]) caused by hypomorphic NEMO mutations also suffer from colitis that is not resolved by hematopoietic stem cell transplantation (Fish et al. 2009), suggesting that NEMO deficiency in stromal cells also causes colon inflammation in humans.
NEMO has also been implicated in suppressing RIPK1 activation to inhibit the development of steatohepatitis and hepatocellular carcinoma (HCC) (Kondylis et al. 2015). The loss of NEMO immunoreactivity has been found in a significant percentage of human HCC samples and correlated with a poor 5-yr overall survival (Aigelsreiter et al. 2012). Mice with NEMO ablation specifically in liver parenchymal cells spontaneously develop steatohepatitis and HCC, which cannot be phenocopied by inhibition of NF-κB signaling. Interestingly, inactivation of RIPK1, but not deficiency of RIPK3, prevented hepatocyte apoptosis and the development of HCC. Thus, the lack of NEMO in liver parenchymal hepatocytes may promote RDA and inflammation to lead to the development of HCC.
Patients who carry hypomorphic NEMO mutations develop X-linked ectodermal dysplasia with anhidrosis and immunodeficiency, also known as NEMO syndrome. A subset of NEMO syndrome patients carries NEMO C-terminal deletion (ΔCT-NEMO) mutations and develops inflammatory skin and intestinal disease in addition to ectodermal dysplasia with anhidrosis and immunodeficiency (Zilberman-Rudenko et al. 2016). ΔCT-NEMO mutants show impaired interactions with A20, leading to prolonged accumulation of K63 ubiquitinated RIPK1 within the TNFR1 signaling complex. Given the roles of NEMO and A20 in suppressing the activation of RIPK1, it is possible that the activation of RIPK1 is involved in mediating skin inflammation and intestinal diseases in these patients.
LUBAC deficiency syndrome
Patients who have biallelic loss-of-function mutations in HOIL-1, a component of the LUBAC, develop a fatal human inherited disorder characterized by chronic autoinflammation, invasive bacterial infections, and muscular amylopectinosis (Boisson et al. 2012). These patient-derived HOIL-1-deficient fibroblasts show reduced levels of IKK kinase phosphorylation, slower IκBα degradation, and a deficit in the recruitment and ubiquitination of NEMO in response to TNFα stimulation. Given that an important function of the LUBAC is to recruit and activate NEMO by adding M1 ubiquitination, HOIL-1 deficiency also impairs the function of NEMO and sensitizes cells to apoptosis. The livers of HOIL-1−/− mice show increased activation of caspase-3 in response to TNFα (Tokunaga et al. 2009). In addition, the loss of another LUBAC component, SHARPIN, has been shown to lead to an inflammatory pathology that is at least partially driven by both caspase-8-dependent apoptosis and RIPK3-dependent necroptosis in cpdm mice (Rickard et al. 2014a). Thus, it is possible that HOIL-1 deficiency in humans also promotes RDA.
Immunodeficiency associated with defective caspase-8
Specific loss of caspase-8 in T cells leads to failure in antigen-driven expansion and promotes necroptosis. However, loss of caspase-8 does not affect the cell cycle progression or NF-κB activation (Ch'en et al. 2008). Similarly, T cells with specific loss of FADD respond poorly to TCR triggering and die by necroptosis, which can be inhibited by Nec-1 (Osborn et al. 2010). Mice with T-cell-specific loss of FADD show immunodeficiency and succumb to Toxoplasma gondii infection more readily than wild-type mice. Thus, caspase-8 and FADD function to suppress the activation of RIPK1 in T cells during the early phases of T-cell clonal expansion, which is important for mediating a host defense response.
Homozygous human individuals with an inherited genetic deficiency of caspase-8 manifest ALPS-related clinical features and also exhibit defects in their activation of T lymphocytes, B lymphocytes, and natural killer cells, which leads to immunodeficiency characterized by recurrent sinopulmonary and herpes simplex virus infections and poor responses to immunization (Chun et al. 2002). Postnatal survival of caspase-8-deficient humans may be due to the redundant function of caspase-10, the closest paralog of caspase-8 in humans. Thus, caspase-8 has a postnatal role in immune activation of naive lymphocytes by suppressing the activation of RIPK1.
Conclusion
Discovery of the necroptosis pathway represents a major breakthrough in the cell death field in the past decade. While caspase-mediated apoptosis is involved in regulating cell numbers and morphogenesis during normal embryonic development, we propose that necroptosis may be involved primarily in aborting defective embryos during development. Since the number of offspring that can be produced by a vertebrate mother is very limited compared with that of primitive organisms such as nematode and flies, the quality control mechanisms that ensure the vitality of newborn mammals could be critical for survival of the species and be mediated at least in part by necroptosis.
Necroptosis can be reactivated under pathological conditions in many human inflammatory and degenerative diseases. However, it has become increasingly clear that necroptosis is often not activated alone. The activation of necroptosis and RDA may be involved in mediating the deleterious effects of TNFα, although the contributions of each pathway and their interactions may vary under different pathological conditions. While the kinase activity of RIPK1 mediates both necroptosis and RDA, its inactivation seems to have no significant consequence on development and adult life. This is illustrated by a lack of any gross phenotype in the RIPK1 kinase-dead D138N, K45A, and ΔG26F27 knock-in mice, suggesting that RIPK1 may provide a rare therapeutic target that can be safely modulated for the treatment of chronic diseases.
In addition, the kinase domain of RIPK1 provides a structural pocket suitable for the development of specific small-molecule inhibitors (Degterev et al. 2008; Xie et al. 2013). RIPK1 inhibitors have already entered human clinical studies for the treatment of multiple inflammatory and degenerative diseases, including IBD, CD, rheumatoid arthritis, ALS, and AD (Ofengeim and Yuan 2013; Harris et al. 2017; Ofengeim et al. 2017). However, since studies on RIPK3 inhibitors and RIPK3 kinase-dead knock-in mice have revealed that RIPK3 inhibition can result in apoptosis (Mandal et al. 2014; Newton et al. 2014), this kinase is not a suitable target for pharmacological inhibition of cell death. Finally, MLKL deficiency has been shown to offer less protection in multiple animal models, such as the kidney ischemia reperfusion injury model, and no benefit in blocking the inflammation and lethality of A20−/− mice (Newton et al. 2016a), suggesting that MLKL might not be an ideal target for developing drugs for human diseases characterized by inflammation and degeneration. Thus, targeting RIPK1 may provide an important therapeutic strategy to block both cell death and inflammation for the treatment of human inflammatory and degenerative diseases.
Acknowledgments
We thank Dr. Alexei Degterev, Dr. Dimitry Ofengeim, and Dr. Palak Amin for comments on the manuscript. This work was supported in part by grants from the National Key R&D Program of China (2016YFA0501900), the China National Natural Science Foundation (31530041), and the Chinese Academy of Sciences; grants from the National Institute of Neurological Disorders and Stroke (1R01NS082257) and the National Institute on Aging (1R01AG047231 and RF1AG055521) to J.Y.; and a grant from the Natural Science Foundation of Shanghai (16ZR1443900) to B.S.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.312561.118.
References
- Aggarwal BB. 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3: 745–756. [DOI] [PubMed] [Google Scholar]
- Aigelsreiter A, Haybaeck J, Schauer S, Kiesslich T, Bettermann K, Griessbacher A, Stojakovic T, Bauernhofer T, Samonigg H, Kornprat P, et al. 2012. NEMO expression in human hepatocellular carcinoma and its association with clinical outcome. Hum Pathol 43: 1012–1019. [DOI] [PubMed] [Google Scholar]
- Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, Doerschuk CM, Hynes RO, Baltimore D. 2001. Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J Immunol 167: 1592–1600. [DOI] [PubMed] [Google Scholar]
- Alvarez-Diaz S, Dillon CP, Lalaoui N, Tanzer MC, Rodriguez DA, Lin A, Lebois M, Hakem R, Josefsson EC, O'Reilly LA, et al. 2016. The pseudokinase MLKL and the kinase RIPK3 have distinct roles in autoimmune disease caused by loss of death-receptor-induced apoptosis. Immunity 45: 513–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenescu R, Bruno ME, Rogier EW, Stefka AT, McMahan AE, Wright TB, Nasser MS, de Villiers WJ, Kaetzel CS. 2008. Signature biomarkers in Crohn's disease: toward a molecular classification. Mucosal Immunol 1: 399–411. [DOI] [PubMed] [Google Scholar]
- Beeldman E, van der Kooi AJ, de Visser M, van Maarle MC, van Ruissen F, Baas F. 2015. A Dutch family with autosomal recessively inherited lower motor neuron predominant motor neuron disease due to optineurin mutations. Amyotroph Lateral Scler Frontotemporal Degener 16: 410–411. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30: 689–700. [DOI] [PubMed] [Google Scholar]
- Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israel L, Trevejo-Nunez G, Bogunovic D, et al. 2012. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat Immunol 13: 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, Phu L, Phung Q, Maurer B, Arnott D, et al. 2010. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-κB signaling. Mol Cell 40: 548–557. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, et al. 2017. Necroptosis activation in Alzheimer's disease. Nat Neurosci 20: 1236–1246. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. 2014. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 16: 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. 1998. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737. [DOI] [PubMed] [Google Scholar]
- Chang M, Jin W, Sun SC. 2009. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol 10: 1089–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'en IL, Beisner DR, Degterev A, Lynch C, Yuan J, Hoffmann A, Hedrick SM. 2008. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci 105: 17463–17468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li W, Ren J, Huang D, He WT, Song Y, Yang C, Li W, Zheng X, Chen P, et al. 2014. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24: 105–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. 2009. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Yuan J. 2010. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22: 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Li Y, Yuan J. 2014. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol 76: 129–150. [DOI] [PubMed] [Google Scholar]
- Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, et al. 2002. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature 419: 395–399. [DOI] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ, et al. 2015. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347: 1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Boyce M, Yuan J. 2003. A decade of caspases. Oncogene 22: 8543–8567. [DOI] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. 2005. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112–119. [DOI] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. 2008. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Oberst A, Weinlich R, Janke LJ, Kang TB, Ben-Moshe T, Mak TW, Wallach D, Green DR. 2012. Survival function of the FADD–CASPASE-8–cFLIPL complex. Cell Rep 1: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, et al. 2014. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell 157: 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ, Obata Y. 1999. Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc Natl Acad Sci 96: 2994–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, Hulpiau P, Weber K, Sehon CA, Marquis RW, et al. 2014. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971–981. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Jouan-Lanhouet S, Divert T, Theatre E, Bertin J, Gough PJ, Giansanti P, Heck AJ, Dejardin E, Vandenabeele P, et al. 2015. NF-κB-independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol Cell 60: 63–76. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P. 2016. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol 26: 721–732. [DOI] [PubMed] [Google Scholar]
- Dondelinger Y, Delanghe T, Rojas-Rivera D, Priem D, Delvaeye T, Bruggeman I, Van Herreweghe F, Vandenabeele P, Bertrand MJM. 2017. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol 19: 1237–1247. [DOI] [PubMed] [Google Scholar]
- Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, Spilgies L, Surinova S, Taraborrelli L, Hartwig T, et al. 2015. LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep 13: 2258–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. 2011. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 35: 908–918. [DOI] [PubMed] [Google Scholar]
- Dziedzic SA, Su Z, Jean Barrett V, Najafov A, Mookhtiar AK, Amin P, Pan H, Sun L, Zhu H, Ma A, et al. 2018. ABIN-1 regulates RIPK1 activation by linking Met1 ubiquitylation with Lys63 deubiquitylation in TNF-RSC. Nat Cell Biol 20: 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. 2006. Activation of IKK by TNFα requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22: 245–257. [DOI] [PubMed] [Google Scholar]
- Elliott PR, Leske D, Hrdinka M, Bagola K, Fiil BK, McLaughlin SH, Wagstaff J, Volkmar N, Christianson JC, Kessler BM, et al. 2016. SPATA2 links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol Cell 63: 990–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, MacFarlane M, Hacker G, Leverkus M. 2011. cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell 43: 449–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish JD, Duerst RE, Gelfand EW, Orange JS, Bunin N. 2009. Challenges in the use of allogeneic hematopoietic SCT for ectodermal dysplasia with immune deficiency. Bone Marrow Transplant 43: 217–221. [DOI] [PubMed] [Google Scholar]
- Geng J, Ito Y, Shi L, Amin P, Chu J, Ouchida AT, Mookhtiar AK, Zhao H, Xu D, Shan B, et al. 2017. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun 8: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, Green DR. 2017. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169: 286–300 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, et al. 2009. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell 36: 831–844. [DOI] [PubMed] [Google Scholar]
- Hadian K, Griesbach RA, Dornauer S, Wanger TM, Nagel D, Metlitzky M, Beisker W, Schmidt-Supprian M, Krappmann D. 2011. NF-κB essential modulator (NEMO) interaction with linear and lys-63 ubiquitin chains contributes to NF-κB activation. J Biol Chem 286: 26107–26117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harhaj EW, Dixit VM. 2012. Regulation of NF-κB by deubiquitinases. Immunol Rev 246: 107–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris PA, Berger SB, Jeong JU, Nagilla R, Bandyopadhyay D, Campobasso N, Capriotti CA, Cox JA, Dare L, Dong X, et al. 2017. Discovery of a first-in-class receptor interacting protein 1 (RIP1) kinase specific clinical candidate (GSK2982772) for the treatment of inflammatory diseases. J Med Chem 60: 1247–1261. [DOI] [PubMed] [Google Scholar]
- Hauenstein AV, Xu G, Kabaleeswaran V, Wu H. 2017. Evidence for M1-linked polyubiquitin-mediated conformational change in NEMO. J Mol Biol 429: 3793–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. 2009. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-α. Cell 137: 1100–1111. [DOI] [PubMed] [Google Scholar]
- Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, Pierotti C, Garnier JM, Dobson RC, Webb AI, et al. 2014. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci 111: 15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, Yuan J. 2008. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135: 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Ofengeim D, Najafov A, Das S, Saberi S, Li Y, Hitomi J, Zhu H, Chen H, Mayo L, et al. 2016. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science 353: 603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaco I, Annibaldi A, Lalaoui N, Wilson R, Tenev T, Laurien L, Kim C, Jamal K, Wicky John S, Liccardi G, et al. 2017. MK2 phosphorylates RIPK1 to prevent TNF-induced cell death. Mol Cell 66: 698–710 e695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z, Johnson HJ, Nie H, Qin J, Bird TA, Li X. 2003. Pellino 1 is required for interleukin-1 (IL-1)-mediated signaling through its interaction with the IL-1 receptor-associated kinase 4 (IRAK4)–IRAK–tumor necrosis factor receptor-associated factor 6 (TRAF6) complex. J Biol Chem 278: 10952–10956. [DOI] [PubMed] [Google Scholar]
- Kaiser WJ, Upton JW, Long AB, Livingston-Rosanoff D, Daley-Bauer LP, Hakem R, Caspary T, Mocarski ES. 2011. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471: 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, Sundararajan A, Guo H, Roback L, Speck SH, et al. 2014. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci 111: 7753–7758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, Chen ZJ. 2004. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 15: 535–548. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. 1998. The death domain kinase RIP mediates the TNF-induced NF-κB signal. Immunity 8: 297–303. [DOI] [PubMed] [Google Scholar]
- Kondylis V, Polykratis A, Ehlken H, Ochoa-Callejero L, Straub BK, Krishna-Subramanian S, Van TM, Curth HM, Heise N, Weih F, et al. 2015. NEMO prevents steatohepatitis and hepatocellular carcinoma by inhibiting RIPK1 kinase activity-mediated hepatocyte apoptosis. Cancer Cell 28: 582–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppe C, Verheugd P, Gautheron J, Reisinger F, Kreggenwinkel K, Roderburg C, Quagliata L, Terracciano L, Gassler N, Tolba RH, et al. 2016. IκB kinaseα/β control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology 64: 1217–1231. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, Courtois G. 2003. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 424: 801–805. [DOI] [PubMed] [Google Scholar]
- Kreuz S, Siegmund D, Scheurich P, Wajant H. 2001. NF-κB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol 21: 3964–3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. 1996. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature 384: 368–372. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. 1998. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94: 325–337. [DOI] [PubMed] [Google Scholar]
- Kupka S, De Miguel D, Draber P, Martino L, Surinova S, Rittinger K, Walczak H. 2016. SPATA2-mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep 16: 2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Kanneganti TD. 2018. ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol 39: 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legarda-Addison D, Hase H, O'Donnell MA, Ting AT. 2009. NEMO/IKKγ regulates an early NF-κB-independent cell-death checkpoint during TNF signaling. Cell Death Differ 16: 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, McQuade T, Siemer AB, Napetschnig J, Moriwaki K, Hsiao YS, Damko E, Moquin D, Walz T, McDermott A, et al. 2012. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Devin A, Rodriguez Y, Liu ZG. 1999. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13: 2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M. 2016. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540: 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, Wang Z. 2017a. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci 114: E7450–E7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fan C, Zhang Y, Yu X, Wu X, Zhang X, Zhao Q, Zhang H, Xie Q, Li M, et al. 2017b. RIP1 kinase activity-dependent roles in embryonic development of Fadd-deficient mice. Cell Death Differ 24: 1459–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. 2014. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 56: 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, et al. 2010. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226. [DOI] [PubMed] [Google Scholar]
- McLaughlin RL, Schijven D, van Rheenen W, van Eijk KR, O'Brien M, Kahn RS, Ophoff RA, Goris A, Bradley DG, Al-Chalabi A, et al. 2017. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun 8: 14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Liu Z, Li X, Wang H, Jin T, Wu G, Shan B, Christofferson DE, Qi C, Yu Q, et al. 2018. Death-domain dimerization-mediated activation of RIPK1 controls necroptosis and RIPK1-dependent apoptosis. Proc Natl Acad Sci 115: E2001–E2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon MB, Gropengiesser J, Fischer J, Novikova L, Deuretzbacher A, Lafera J, Schimmeck H, Czymmeck N, Ronkina N, Kotlyarov A, et al. 2017. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol 19: 1248–1259. [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. 2003. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190. [DOI] [PubMed] [Google Scholar]
- Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. 2001. NF-κB signals induce the expression of c-FLIP. Mol Cell Biol 21: 5299–5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohideen F, Paulo JA, Ordureau A, Gygi SP, Harper JW. 2017. Quantitative phospho-proteomic analysis of TNFα/NFκB signaling reveals a role for RIPK1 phosphorylation in suppressing necrotic cell death. Mol Cell Proteomics 16: 1200–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moquin DM, McQuade T, Chan FK. 2013. CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8: e76841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. 2013. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39: 443–453. [DOI] [PubMed] [Google Scholar]
- Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et al. 2009. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat Genet 41: 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, Ishitani R, Kamei K, Takeyoshi I, Kawakami H, et al. 2016. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun 7: 12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda SK, Venigalla RK, Ordureau A, Patterson-Kane JC, Powell DW, Toth R, Arthur JS, Cohen P. 2011. Polyubiquitin binding to ABIN1 is required to prevent autoimmunity. J Exp Med 208: 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM. 2004. Kinase RIP3 is dispensable for normal NF-κBs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24: 1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, et al. 2014. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343: 1357–1360. [DOI] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Maltzman A, Greve JM, Hedehus M, Martin-McNulty B, Carano RA, Cao TC, van Bruggen N, Bernstein L, et al. 2016a. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ 23: 1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, et al. 2016b. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540: 129–133. [DOI] [PubMed] [Google Scholar]
- Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, Hakem R, Salvesen GS, Green DR. 2011. Catalytic activity of the caspase-8-FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471: 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Yuan J. 2013. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol 14: 727–736. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Ito Y, Najafov A, Zhang Y, Shan B, DeWitt JP, Ye J, Zhang X, Chang A, Vakifahmetoglu-Norberg H, et al. 2015. Activation of necroptosis in multiple sclerosis. Cell Rep 10: 1836–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C, Das S, Adiconis X, Chen H, Zhu H, et al. 2017. RIPK1 mediates a disease-associated microglial response in Alzheimer's disease. Proc Natl Acad Sci 114: E8788–E8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, Prodhomme T, Duong B, Whang MI, Advincula R, et al. 2015. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol 16: 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn SL, Diehl G, Han SJ, Xue L, Kurd N, Hsieh K, Cado D, Robey EA, Winoto A. 2010. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci 107: 13034–13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HH, Lo YC, Lin SC, Wang L, Yang JK, Wu H. 2007. The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu Rev Immunol 25: 561–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer N, Darding M, Walczak H. 2016. Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol 26: 445–461. [DOI] [PubMed] [Google Scholar]
- Pescatore A, Esposito E, Draber P, Walczak H, Ursini MV. 2016. NEMO regulates a cell death switch in TNF signaling by inhibiting recruitment of RIPK3 to the cell death-inducing complex II. Cell Death Dis 7: e2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polykratis A, Hermance N, Zelic M, Roderick J, Kim C, Van TM, Lee TH, Chan FK, Pasparakis M, Kelliher MA. 2014. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol 193: 1539–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. 2009. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 136: 1098–1109. [DOI] [PubMed] [Google Scholar]
- Reynoso E, Liu H, Li L, Yuan AL, Chen S, Wang Z. 2017. Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J Biol Chem 292: 17514–17524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JA, Anderton H, Etemadi N, Nachbur U, Darding M, Peltzer N, Lalaoui N, Lawlor KE, Vanyai H, Hall C, et al. 2014a. TNFR1-dependent cell death drives inflammation in Sharpin-deficient mice. eLife 3: e03464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard JA, O'Donnell JA, Evans JM, Lalaoui N, Poh AR, Rogers T, Vince JE, Lawlor KE, Ninnis RL, Anderton H, et al. 2014b. RIPK1 regulates RIPK3–MLKL-driven systemic inflammation and emergency hematopoiesis. Cell 157: 1175–1188. [DOI] [PubMed] [Google Scholar]
- Schlicher L, Wissler M, Preiss F, Brauns-Schubert P, Jakob C, Dumit V, Borner C, Dengjel J, Maurer U. 2016. SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. EMBO Rep 17: 1485–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Ma A, Harhaj EW. 2010. Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327: 1135–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemura T, Kaneko N, Kobayashi N, Kobayashi K, Takeuchi Y, Nakano N, Masumoto J, Agematsu K. 2016. Novel heterozygous C243Y A20/TNFAIP3 gene mutation is responsible for chronic inflammation in autosomal-dominant Behcet's disease. RMD Open 2: e000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, Sad S. 2016. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ 23: 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley P, Hobson L. 2016. Paternal care and litter size coevolution in mammals. Proc Biol Sci 283: 20160140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. 2002. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem 277: 9505–9511. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213–227. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et al. 2009. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 11: 123–132. [DOI] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. 2003. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature 424: 793–796. [DOI] [PubMed] [Google Scholar]
- Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, Shifrin N, Malynn BA, Ma A. 2008. Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20. J Exp Med 205: 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varfolomeev E, Vucic D. 2018. Intracellular regulation of TNF activity in health and disease. Cytokine 101: 26–32. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276. [DOI] [PubMed] [Google Scholar]
- Vassalli P. 1992. The pathophysiology of tumor necrosis factors. Annu Rev Immunol 10: 411–452. [DOI] [PubMed] [Google Scholar]
- Vereecke L, Beyaert R, van Loo G. 2009. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30: 383–391. [DOI] [PubMed] [Google Scholar]
- Vlantis K, Wullaert A, Polykratis A, Kondylis V, Dannappel M, Schwarzer R, Welz P, Corona T, Walczak H, Weih F, et al. 2016. NEMO prevents RIP kinase 1-mediated epithelial cell death and chronic intestinal inflammation by NF-κB-dependent and -independent functions. Immunity 44: 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vucic D, Dixit VM, Wertz IE. 2011. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol 12: 439–452. [DOI] [PubMed] [Google Scholar]
- Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. 2001. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412: 346–351. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. 2014. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133–146. [DOI] [PubMed] [Google Scholar]
- Wang H, Meng H, Li X, Zhu K, Dong K, Mookhtiar AK, Wei H, Li Y, Sun SC, Yuan J. 2017. PELI1 functions as a dual modulator of necroptosis and apoptosis by regulating ubiquitination of RIPK1 and mRNA levels of c-FLIP. Proc Natl Acad Sci 114: 11944–11949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. 1992. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356: 314–317. [DOI] [PubMed] [Google Scholar]
- Wei R, Xu LW, Liu J, Li Y, Zhang P, Shan B, Lu X, Qian L, Wu Z, Dong K, et al. 2017. SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes Dev 31: 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Oberst A, Beere HM, Green DR. 2017. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18: 127–136. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. 2004. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430: 694–699. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, Popovych N, Helgason E, Schoeffler A, Jeet S, et al. 2015. Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528: 370–375. [DOI] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. 2006. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-κB activation [corrected]. Nat Cell Biol 8: 398–406. [DOI] [PubMed] [Google Scholar]
- Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, Ma J, Chen W, Zhang Y, Zhou X, et al. 2013. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23: 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T, Peng W, Liu Y, Yan C, Maki J, Degterev A, Yuan J, Shi Y. 2013. Structural bsasis of RIP1 inhibition by necrostatins. Structure 21: 493–499. [DOI] [PubMed] [Google Scholar]
- Xu C, Wu X, Zhang X, Xie Q, Fan C, Zhang H. 2018. Embryonic lethality and host immunity of RelA-deficient mice are mediated by both apoptosis and necroptosis. J Immunol 200: 271–285. [DOI] [PubMed] [Google Scholar]
- Yeh WC, de la Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, et al. 1998. FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis. Science 279: 1954–1958. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, et al. 2000. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity 12: 633–642. [DOI] [PubMed] [Google Scholar]
- Yoon S, Kovalenko A, Bogdanov K, Wallach D. 2017. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47: 51–65 e57. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. 1998. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. 2009. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332–336. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. 2011. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature 471: 373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. 2012. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci 109: 5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, Yuan J. 2014. Necroptosis in health and diseases. Semin Cell Dev Biol 35: 14–23. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Wang H, Schwartz DM, Stoffels M, Park YH, Zhang Y, Yang D, Demirkaya E, Takeuchi M, Tsai WL, et al. 2016. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat Genet 48: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberman-Rudenko J, Shawver LM, Wessel AW, Luo Y, Pelletier M, Tsai WL, Lee Y, Vonortas S, Cheng L, Ashwell JD, et al. 2016. Recruitment of A20 by the C-terminal domain of NEMO suppresses NF-κB activation and autoinflammatory disease. Proc Natl Acad Sci 113: 1612–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]