

Zhang et al. determined the crystal structures of both apo AF10OM-LZ and its complex with the coiled-coil domain of DOT1L. Disruption of the DOT1L–AF10 interface abrogates MLL-AF10-associated leukemic transformation.
Keywords: DOT1L, MLL-AF10, crystal structure, leukemogenesis
Abstract
The mixed-lineage leukemia (MLL)-AF10 fusion oncoprotein recruits DOT1L to the homeobox A (HOXA) gene cluster through its octapeptide motif leucine zipper (OM-LZ), thereby inducing and maintaining the MLL-AF10-associated leukemogenesis. However, the recognition mechanism between DOT1L and MLL-AF10 is unclear. Here, we present the crystal structures of both apo AF10OM-LZ and its complex with the coiled-coil domain of DOT1L. Disruption of the DOT1L–AF10 interface abrogates MLL-AF10-associated leukemic transformation. We further show that zinc stabilizes the DOT1L–AF10 complex and may be involved in the regulation of the HOXA gene expression. Our studies may also pave the way for the rational design of therapeutic drugs against MLL-rearranged leukemia.
Aberration of the mixed-lineage leukemia (MLL; also known as MLL1, ALL1, HRX, or HTRX) gene is widely implicated in pathogenesis of acute leukemia (Ziemin-van der Poel et al. 1991). One major MLL aberration is the MLL rearrangement, which is found in ∼10% of acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) cases with a relatively dismal prognosis (Muntean and Hess 2012). The MLL gene encodes a histone H3 Lys4 (H3K4) methyltransferase that positively regulates homeobox (HOX) gene expression (Benjamin et al. 1995). Several functional domains involved in chromatin association (CXXC, AT hooks, plant homeodomain fingers, and bromodomain) are present in the N-terminal portion (Cosgrove and Patel 2010), while within the C-terminal portion lies a SET [Su(var)3–9, enhancer of zeste, and trithorax] domain that catalyzes H3K4 methylation, an epigenetic mark of active transcription (Shilatifard 2012). Chromosomal rearrangements generate MLL fusion proteins in which the N-terminal portion of MLL is fused to one of ∼70 distinct partner proteins (Slany 2016). The N-terminal chromatin-binding domains (AT hooks and CXXC) are consistently retained in the MLL fusion proteins, whereas the SET domain is absent. Intriguingly, most frequent translocation partners implicated in leukemia are transcriptional activators, which is in agreement with the notion that the target gene activation of MLL is the fundamental mechanism in MLL-rearranged leukemia.
The MLL-AF10 fusion is widely distributed among AML subtypes (DiMartino et al. 2002; Meyer et al. 2006). AF10 together with AF4, AF6, AF9, and ENL are the most common fusion partners, found in >80% of all MLL-rearranged leukemia (Krivtsov and Armstrong 2007; Thirman 2017). The MLL-AF10 fusion protein retains the octapeptide motif (OM) leucine zipper (LZ) region of AF10 (AF10OM-LZ), which recruits the H3K79 methyltransferase DOT1L, resulting in H3K79 hypermethylation and dysregulation of HOXA and MEIS1 expression (Okada et al. 2005). Previous studies have demonstrated that MLL-rearranged leukemia depends on aberrant H3K79 methylation and up-regulation of HOXA gene clusters (Ayton and Cleary 2003; Okada et al. 2005; Chen et al. 2013). The OM-LZ domain of AF10 is essential for both MLL-AF10- and CALM-AF10-induced leukemogenesis (DiMartino et al. 2002; Okada et al. 2006). AF10OM-LZ is also known to interact with the coiled-coil domains of GAS41 and Ikaros (Okada et al. 2006; Yoshida et al. 2006), suggesting a more general scaffolding role for the OM-LZ domain of AF10. AF10 has been identified as a key regulator of progressive H3K79 methylation and HOXA gene expression in various AML subtypes (Deshpande et al. 2014), indicating that disruption of DOT1L–AF10 association would be an attractive therapeutic option for a wide spectrum of AML. However, the molecular mechanism of how AF10 recognizes DOT1L in MLL-rearranged leukemia remains unknown.
Here we identified and characterized the coiled-coil domain of DOT1L as the AF10-binding site by biophysical and structural analyses. Our in vivo results revealed that disruption of the DOT1L–AF10 binding impairs the transforming ability of MLL-AF10 and decreases the HOXA gene expression. Our crystal structures also revealed a previously uncharacterized zinc-binding site in the DOT1L–AF10 complex. Zinc stabilizes the complex formation and might contribute to the regulation of Hoxa10 gene expression. In addition to elucidating the DOT1L–AF10 recognition mechanisms, our results also shed light on the potential development of DOT1L–AF10-specific therapeutic drugs in AML.
Results and Discussion
AF10 binds to the coiled-coil domain of DOT1L
The OM-LZ region of AF10 (AF10OM-LZ) has been reported previously to bind to DOT1L (Okada et al. 2005). As the central region of the DOT1L is predicted to fold into three coiled-coil domains (Fig. 1A; Supplemental Fig. S1A,B; Finn et al. 2011), we hypothesized that the coiled-coil interactions between the OM-LZ region of AF10 and the coiled-coil domains of DOT1L might mediate the DOT1L–AF10 binding. Our GST pull-down assays using recombinant proteins indeed revealed that the coiled-coil domains of DOT1L bound to AF10OM-LZ (Fig. 1B). To further map the AF10-binding region on DOT1L, we produced recombinant proteins for each individual coiled-coil domain of DOT1L. Our isothermal titration calorimetry (ITC) studies showed that the first (DOT1LCC1) and the second (DOT1LCC2) domains exhibit submicromolar affinities toward AF10. In addition, the third coiled-coil domain (DOT1LCC3) binds AF10 with micromolar affinity (Fig. 1C). Thus, DOT1L engages the AF10 binding at three sites. Both the coiled-coil domains of DOT1L and the OM-LZ region of AF10 are evolutionarily conserved across species (Supplemental Figs. S1A, 2A), suggesting a conserved binding mode between DOT1L and AF10. Sequence alignment shows that AF10 and AF17 share a conserved C-terminal OM-LZ domain (Supplemental Fig. S2A), suggesting that AF17 might bind to DOT1L in a manner similar to that of AF10. Indeed, both GST pull-down and ITC studies confirmed the direct binding between the coiled-coil domains of DOT1L and AF17 (Fig. 1B; Supplemental Fig. S2B).
Figure 1.
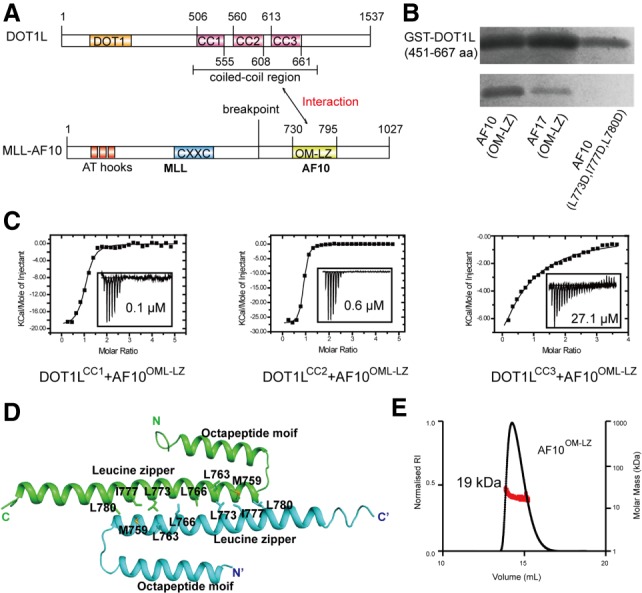
The coiled-coil region of DOT1L binds to AF10 and AF17. (A) Schematic diagrams of the domain organization of DOT1L and MLL-AF10. Interactions between DOT1L and AF10 are indicated by a two-way arrow. There are three coiled-coil regions in DOT1L. (B) The coiled-coil region of DOT1L robustly binds to AF10 and AF17 by GST pull-down assay. Triple mutation AF10L773D, I777D, L780D abolishes the DOT1L binding. (C) ITC-based measurements quantifying the binding affinities between the coiled-coil region of DOT1L and AF10OM-LZ. (NB) No binding. (D) Cartoon representation of the AF10OM-LZ structure. The two chains are colored cyan and green, respectively. The residues involved in the dimer interface are shown in stick representations. (E) Size exclusion chromatography coupled with multiangle light-scattering analysis of AF10OM-LZ.
Crystal structure of AF10OM-LZ
To elucidate the molecular basis of the DOT1L–AF10 interaction, we first determined the crystal structure of AF10OM-LZ (residues 720–795) (Supplemental Table S1). The asymmetric unit contains two all-helical AF10OM-LZ protomers in which the OMs (residues 731–750) pack against the LZs (residues 753–794) (Fig. 1D). The LZs from these two protomers dimerize and form anti-parallel coiled coils, resulting in a buried surface area of 681 Å2 (Krissinel and Henrick 2007). Static light-scattering studies revealed that AF10OM-LZ has a measured mass of ∼19 kDa, which matches the theoretical mass of dimeric AF10OM-LZ (∼18 kDa) (Fig. 1E). There are 3.5 heptad repeats in each AF10OM-LZ LZ domain, with predominantly hydrophobic residues at the a and d positions (Supplemental Fig. S3A). These hydrophobic residues are invariant across species (Supplemental Fig. S2A), suggesting that the dimerization mode is preserved among different organisms. Polar interactions are also present in the two-stranded helical coiled coils, such as S762 at the g position hydrogen-bonding with Q776 at the g′ position (Supplemental Fig. S3B). Substitutions of hydrophobic residues with glutamic acid significantly disrupted the homodimer formation (Supplemental Fig. S3C).
Crystal structure of the DOT1LCC2–AF10OM-LZ complex
Because the DOT1LCC2 protein behaves better than DOT1LCC1 and DOT1LCC3, we chose DOT1LCC2 for structural studies of the DOT1L–AF10 interaction. To reconstitute the complex, we mixed stoichiometric amounts of recombinant human DOT1LCC2 and AF10OM-LZ proteins and monitored complex formation using analytical size exclusion chromatography. Apparent coelution of DOT1LCC2 and AF10OM-LZ indicated formation of the DOT1LCC2-AF10OM-LZ complex (Fig. 2A).
Figure 2.
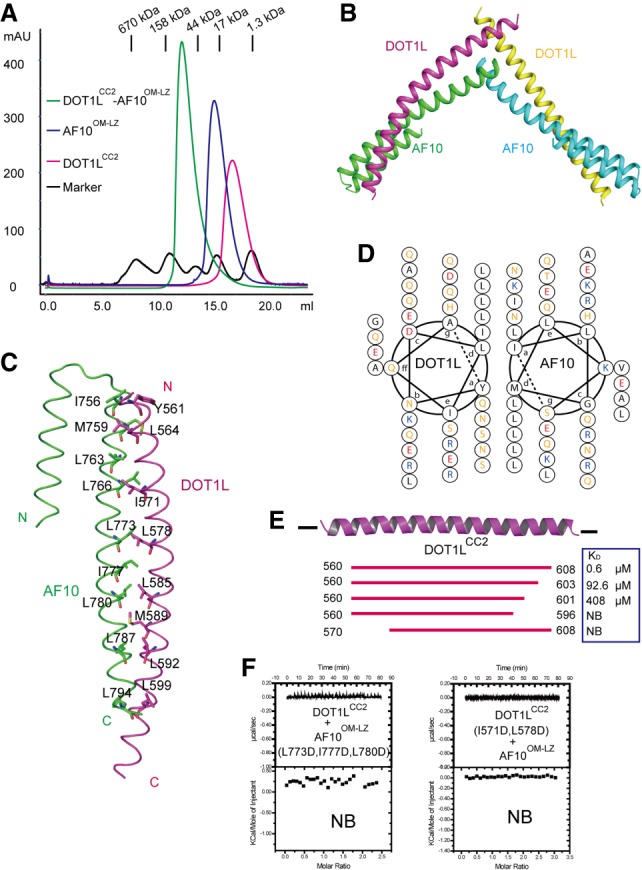
Structure of the DOT1LCC2–AF10OM-LZ complex. (A) Superimposed gel filtration profiles of DOT1LCC2, AF10OM-LZ, and the DOT1LCC2–AF10OM-LZ complex. The elution positions of standard marker proteins are indicated at the top. The results indicate that DOT1LCC2 and AF10OM-LZ exist as a dimer in solution. (B) Overall structure of DOT1LCC2–AF10OM-LZ. (C) The interaction interface between DOT1LCC2 and AF10OM-LZ. DOT1LCC2 forms the parallel coiled coils with AF10OM-LZ. Hydrophobic residues at the heterodimer interface are indicated. (D) Helical wheel presentation of the coiled-coil motifs formed by DOT1LCC2 and AF10OM-LZ. (E) ITC-based measurements of the AF10OM-LZ binding for wide-type and variant DOT1LCC2. (F) ITC results showing that the double mutant on DOT1L or triple mutant on AF10 disrupts the complex binding.
We next tried to crystallize the DOT1LCC2–AF10OM-LZ complex. However, we could obtain only needle-shaped crystals that diffracted poorly. Extensive optimization (including seeding, protein truncations, mutations, and DOT1L–AF10 fusion protein) of these crystals was not successful. Nevertheless, we were able to crystallize and determine the structure of a chimeric complex of human AF10OM-LZ and Danio rerio (zebrafish) DOT1LCC2 (Supplemental Table S1). The human DOT1L and zebrafish DOT1L have a high degree of sequence homology in the coiled-coil domains (Supplemental Fig. S1A), and our ITC results showed that the AF10OM-LZ binds to zebrafish DOT1LCC2 and human DOT1LCC2 with similar affinities (Fig. 1C; Supplemental Fig. S4A). Therefore, the interactions between DOT1LCC2 and AF10OM-LZ are highly conserved, and the structural insight from the chimeric complex could be applied to the human DOT1L–AF10 interactions as well. The DOT1LCC2–AF10OM-LZ complex was found to form a heterotetramer in solution (Supplemental Fig. S4B). In the complex structure, each asymmetric unit contains four molecules composed of two DOT1LCC2–AF10OM-LZ heterodimers (Fig. 2B). The heterotetramer complex structure adopts a V-shaped architecture. The two heterodimers aligned well with each other (Supplemental Fig. S4C) and were linked by a disulfide bridge involving C604 in DOT1LCC2 (Supplemental Fig. S4D). Of note, the disulfide bond is almost absent in the larger DOT1L fragment (Supplemental Fig. S4E), thus suggesting that the observed disulfide bond may be an artifact.
Interaction of DOT1LCC2 with AF10OM-LZ
The DOT1LCC2–AF10OM-LZ complex forms a parallel coiled-coil heterodimer (Fig. 2C), packing in a “knobs into holes” manner (Lupas 1996). Residues at the d positions of both DOT1LCC2 and AF10OM-LZ heptad repeats are all hydrophobic and highly conserved (Fig. 2D; Supplemental Figs. S1A, 2A), and hydrophobic interactions are predominantly responsible for stabilizing the heterodimer. Polar residues are prevalent at a positions of both molecules, especially for the DOT1LCC2 heptad repeats. The OM of AF10 is not directly involved in the DOT1LCC2 binding, and deletion of this OM had no obvious effect on the DOT1LCC2 binding (Supplemental Fig. S5A). However, previous studies have shown that both the OM and LZ are essential for DOT1L–AF10 interactions in vivo (DiMartino et al. 2002). Indeed, removal of the GST tag led to precipitation of virtually all of the AF10 protein without the OM, suggesting that the OM may play a role in stabilization of the LZ moiety. Similarly, deletion of the N terminus or C terminus of DOT1LCC2 also impaired the interaction between DOT1LCC2 and AF10OM-LZ in vitro (Fig. 2E). To discern the residues critical for the DOT1LCC2–AF10OM-LZ complex formation, we introduced mutations into the a and/or d positions of the heptad repeats of DOT1LCC2 or AF10OM-LZ. No detectable AF10 binding was observed for the double mutation (I571D/L578D) of DOT1LCC2 in either GST pull-down or ITC studies (Fig. 2F; Supplemental Fig. S5A). Likewise, the triple mutation (L773D/I777D/L780D) in AF10OM-LZ abolished binding of the DOT1L coiled-coil domains (Figs. 1B, 2F; Supplemental Fig. S5B,C), indicating similar AF10-binding modes for these three coiled-coil domains of DOT1L. Furthermore, the majority of these key residues for AF10-binding is conserved among DOT1LCC1, DOT1LCC2, and DOT1LCC3 (Supplemental Fig. S1B).
It was reported previously that the L736R mutation in the OM or the L787R mutation in the LZ of AF10 significantly reduces its binding to DOT1L (Okada et al. 2005). Based on our complex structure, the impaired binding may arise from electrostatic repulsion between the arginine substitutions and their neighboring basic residues from AF10 or DOT1L (Supplemental Fig. S5D).
DOT1L–AF10 interaction is essential for MLL-AF10-associated leukemogenesis
To evaluate the role of the DOT1L–AF10 interaction in MLL-AF10 leukemogenesis, we cotransduced DOT1L-floxed (DOT1Lf/f ErCre+/−) murine bone marrow cells with pMSCB-DOT1L or pMSCB-DOT1L (I571D/L578D) together with pMSCV-MLL-AF10 or pMSCV-MLL-AF10 (L773D/I777D/L780D) (Okada et al. 2005). The effects of DOT1L or AF10 mutations on proliferation of MLL-AF10 cells were analyzed by liquid culture proliferation and colony-forming assays (Zhou et al. 2016). Expression of the DOT1LI571D/L578D mutation led to a significant growth inhibition of MLL-AF10 transformed cells (Fig. 3A; Supplemental Fig. S6A). The triple AF10L773D/I777D/L780D mutation almost inhibited cell proliferation and colony formation (Fig. 3A,B). Furthermore, wild-type transformed cells formed compact and blast-like colonies in the methylcellulose medium, whereas the DOT1LI571D/L578D or AF10L773D/I777D/L780D mutant grew in small and diffuse colonies (Fig. 3C; Supplemental Fig. S6B). Consistent with these results, Wright-Giemsa staining suggested that DOT1LI571D/L578D or AF10L773D/I777D/L780D mutant cells display an increased myeloid differentiation with a larger cytoplasm and smaller nucleus compared with wild type (Fig. 3C; Supplemental Fig. S6B).
Figure 3.
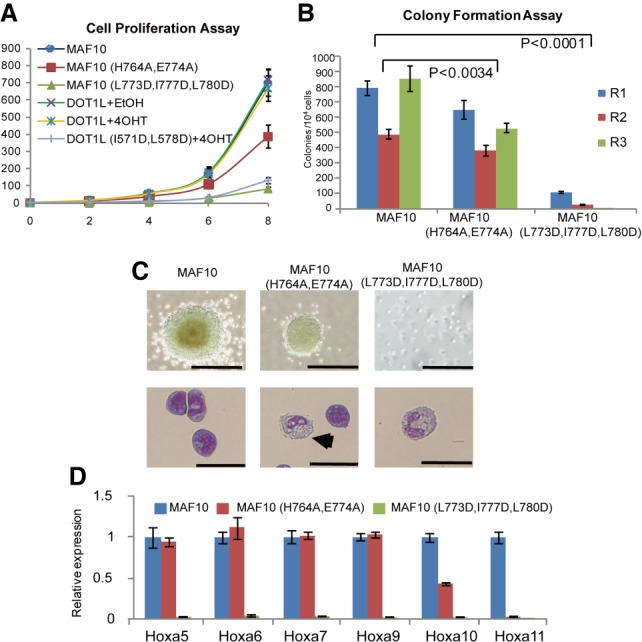
The DOT1L–AF10 binding is essential for MLL-AF10-associated leukemogenesis. (A) Cell proliferation assay for MLL-AF10 (MAF10) wild-type or mutant transduced bone marrow cells as well as MAF10 wild type transduced with or without DOT1L deletion bone marrow cells with DOT1L wild-type or mutant overexpression. Error bars indicate standard deviation (SD) from duplicates. The results were repeated at least three times. (B) Myeloid colony formation assay for transduced bone marrow cells with MAF10 wild type or mutants as indicated. Colony counts were summarized from primary, secondary, and tertiary plating on methycellulose medium in the presence of IL3, IL6, stem cell factor (SCF), and granulocyte-macrophage colony-stimulating factor (GM-CSF). Error bars indicate SD from duplicates. The results were repeated at least three times. P < 0.0001, two-way ANOVA test. (C) Representative colonies (top) and Wright-Giemas-stained cells (bottom) from the tertiary plating of MAF10 wild type or mutant transduced bone marrow cells are shown. Bars: colonies, 200 µm; Wright-Giemas-stained cells, 50 µm. (D) Real-time PCR for HOXA genes in cells as indicated. Gene expression was normalized against GAPDH and is presented as fold change against their respective levels in MAF10 wild-type cells, which was arbitrarily set at 1. Means and SDs (as error bars) from at least three independent experiments are presented.
We next examined the roles of the DOT1L–AF10 interaction in the expression of MLL-AF10 target genes using quantitative RT–PCR. As expected, the HOXA gene cluster was significantly down-regulated in cells expressing the DOT1LI571D/L578D or AF10L773D/I777D/L780D mutant (Fig. 3D; Supplemental Fig. S6C), thereby suggesting that the expression of HOXA genes depends on DOT1L–AF10 binding. Consistently, chromatin immunoprecipitation studies demonstrated that the DOT1LI571D/L578D mutant shows a reduction in expression of HOXA genes in H3K79me2 (Supplemental Fig. S6D). Taken together, these data clearly demonstrated that the DOT1L–AF10 interactions are essential for MLL-AF10-induced oncogenic transformation.
The DOT1LCC2–AF10OM-LZ complex specifically binds zinc
Merging statistics for the diffraction data collected near the selenium absorption edge indicated an unexpected presence of a significant anomalous signal, which we then exploited for structure solution by means of the single-wavelength anomalous diffraction technique (Wang 1985). The sites of the most significant scatterers appeared to be coordinated by protein side chains and solvent molecules and were tentatively assigned as Zn(II) ions because a fluorescence spectrum revealed peaks that approximately corresponded to the characteristic zinc emission energies. A binding survey of various divalent cations confirmed that the DOT1LCC2–AF10OM-LZ complex specifically bound to Zn(II), with very weak or no binding detected with other divalent cations tested (Fig. 4A).
Figure 4.
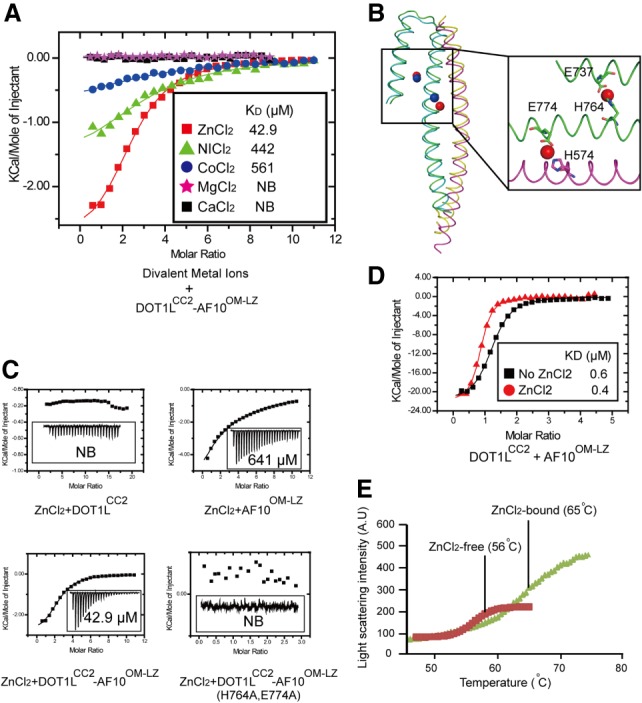
Zinc enhances the binding ability of DOT1LCC2 to AF10OM-LZ. (A) Summary of the quantitative binding constants between various divalent metal ions and the DOT1LCC2–AF10OM-LZ complex. (B) There are two conserved zinc-binding sites in the DOT1LCC2–AF10OM-LZ complex. (C) Dissociation constants of zinc with DOT1LCC2, AF10OM-LZ, and the DOT1LCC2–AF10OM-LZ complex. Mutation of zinc-coordinating residues (AF10H764A/E774A) abolishes the zinc binding. (D) ITC data show that zinc enhances the binding of DOT1LCC2 to AF10OM-LZ. (E) The thermal shift melting curves of the DOT1LCC2–AF10OM-LZ complex in the presence or absence of zinc measured by differential static light scattering.
We modeled several zinc ions in the DOT1LCC2–AF10OM-LZ heterodimer structures (Supplemental Fig. 7); two of those sites are conserved in both heterodimers (Fig. 4B). One of the Zn(II) ions is coordinated by Glu737 and His764 of AF10, and a second Zn(II) ion is located at an DOT1L–AF10 interface coordination site formed by Glu774 of AF10 and His574 of DOT1L (Fig. 4B), suggesting that the DOT1L–AF10 complex formation is crucial for Zn(II) binding. Indeed, ITC studies revealed Zn(II) dissociation constants (KD) of ∼40 µM for the DOT1LCC2–AF10OM-LZ complex, ∼600 µM for AF10OM-LZ, and no binding for DOT1LCC2 (Fig. 4C). The positioning of Zn(II) ions also suggests a structural role that they may play in strengthening the DOT1LCC2–AF10OM-LZ interaction. Indeed, the ZnCl2-containing buffer resulted in a slightly increased binding affinity between DOT1LCC2 and AF10OM-LZ (Fig. 4D). We also observed an increased melting temperature for the protein complex in the presence of the zinc ion (∼65°C) compared with a control in the absence of the zinc ion (∼56°C) (Fig. 4E), indicating a pronounced stabilization effect of the zinc ion. Notably, the residues involved in zinc coordination are conserved between AF10 and AF17 proteins (Supplemental Fig. S2A), implying that they have a conserved function in zinc binding. Indeed, the zinc ion exhibited a similar effect on the DOT1LCC2–AF17OM-LZ complex, suggesting a conserved role of Zn(II) in MLL-AF10/AF17-associated leukemogenesis (Supplemental Fig. S2C).
A role of zinc in the leukemogenesis
Zinc is involved in numerous cellular pathways and is essential for normal functions. Abnormal cellular zinc homeostasis has been associated with many diseases, including cancer, diabetes, and neurodegeneration (Maret 2013). Previous studies suggest that zinc functions as a regulator of monocyte differentiation in the acute myeloid leukemia cell line HL-60 (Dubben et al. 2010). To test the validity of the Zn(II) binding observed in the crystal structure, we mutated residues involved in zinc coordination and then assessed leukemogenesis. Double mutation (H764A/E774A) of AF10 abolished its zinc-binding ability as examined by ITC (Fig. 4C). Consistent with the modest role of zinc in mediating the DOT1L–AF10 binding, AF10H764A/E774A had a modest inhibitory effect on proliferation and differentiation of MLL-AF10 cells (Fig. 3A–C). On the other hand, although the expression of Hoxa7 and Hoxa9 was just slightly affected in cells expressing the H764A/E774A mutant, Hoxa10 and Hoxa11 were significantly down-regulated. Furthermore, a decrease of H3K79me2 at the Hoxa10 and Hoxa11 was detected (Supplemental Fig. S6E). Given that overexpression of Hoxa10 results in suppression of megakaryocyte (MK) development (Magnusson et al. 2007), zinc may play a role in MLL-AF10-mediated leukemogenesis through the regulation of postnatal hematopoietic development. Taken together, zinc ions may promote leukemogenesis by supporting cell proliferation and Hoxa10/Hoxa11 expression via enhancing the DOT1L–AF10 interactions.
In summary, we characterized the DOT1L–AF10 interaction by providing the first structural model for how AF10 recruits DOT1L and probed the effect of this interaction on MLL-AF10-mediated leukemic transformation. Apart from binding of AF10 and AF17, DOT1L is also involved in the binding of other MLL fusion partners such as AF9 and ENL (Zhang et al. 2006; Mueller et al. 2007; Kuntimaddi et al. 2015), albeit through distinct binding sites: AF10 and AF17 specifically interact with the coiled-coil domains of DOT1L based on our results, whereas AF9 and ENL bind mainly to the C-terminal intrinsically disordered region of DOT1L (Mueller et al. 2007; Kuntimaddi et al. 2015). Although AF9 and AF10 bind to DOT1L through different binding sites, both binding events involve more than one binding site on DOT1L. The three DOT1L-binding sites for AF10 are proximal to each other (Fig. 1A); in contrast, AF9 binds to three spatially separated sites of DOT1L (Kuntimaddi et al. 2015). Multiple binding motifs on DOT1L would increase the DOT1L-binding affinity and enhance HOXA gene expression, an event reminiscent of the role of oligomerization proposed for MLL. Oligomerization of MLL via the self-association motifs of fusion partners has been reported to be important and sufficient to immortalize hematopoietic cells (Martin et al. 2003; Smith et al. 2017). Our DOT1L–AF10 data together with the previous DOT1L–AF9 studies revealed another mode of oligomerization of the MLL fusion proteins; i.e., heterodimerization between DOT1L and AF9 or AF10.
Interestingly, the first DOT1L-binding site for AF9, which spans residues 628–653, partially overlaps with the DOT1L-binding site for AF10. Consistently, AF17 has been reported to compete with AF9 for interaction with DOT1L, thereby regulating the DOT1L expression (Reisenauer et al. 2009). Thus, it is conceivable that AF10 would also compete with AF9 for interaction with DOT1L.
The DOT1L–AF10 interaction has been implicated in regulating the HOXA gene cluster expression in various AML subtypes (Deshpande et al. 2014), unveiling that the DOT1L–AF10-binding interface would be an excellent therapeutic target for AML. In addition, blocking the interaction of MLL-AF10 with DOT1L is likely to have some advantages over inhibiting the DOT1L enzymatic activity, as the MLL-AF10 targeted therapy would be of high efficacy and low toxicity. Recently, it was shown that disruption of GAS41–AF10OM-LZ binding by the inhibitory peptides results in a significant reduction in HOXA gene cluster expression, further suggesting that blocking the DOT1L–AF10 interaction may be a feasible therapeutic approach in treating leukemias (Hagen et al. 2014). Similarly, the peptide mimics and small molecules that target the coiled-coil domain of HIV-1 gp41 have been successfully developed and applied to the HIV therapy (Liu et al. 2007; Welch et al. 2007; Sofiyev et al. 2017). Thus, our structures presented here could serve as a foundation for the development of specific DOT1L–AF10OM-LZ targeted drugs in the future.
Materials and methods
Protein crystallization
Crystallization experiments were performed using sitting drop vapor diffusion methods at 18°C. Crystals of AF10OM-LZ were grown by mixing 1 µL of protein solution with 1 µL of reservoir solution consisting of 0.2 M magnesium pernitrate (pH 5.5) and 20% (w/v) PEG3350. The DOT1LCC2–AF10OM-LZ crystals were obtained in the mixture solution of 0.1 M sodium acetate (pH 4.6), 0.2 M calcium chloride, 0.01 M zinc chloride, and 30% (w/v) MPD. Crystals appeared in 3 d and grew to full size in 3 wk. Before data collection, the crystals were soaked in the reservoir solution supplemented with 10% glycerol before being flash-frozen.
Additional experimental details are in the Supplemental Material.
Supplementary Material
Acknowledgments
We thank Dr. Akihiko Yokoyama for providing the pMSCV-MLL-AF10 plasmids, Dr. Guillermo Senisterra for help with the differential static light scattering, and the Ontario Institute for Cancer Research for access to multiangle light-scattering equipment. We also thank Dr. Rui-ming Xu for communicating their data prior to publication. Some X-ray diffraction experiments were performed at Argonne National Laboratory Structural Biology Center (SBC-CAT) Beamline 19-ID at the Advanced Photon Source. SBC-CAT is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research, under contract DE-AC02-06CH11357. Beamline 08ID-1 at the Canadian Light Source is supported by the Canada Foundation for Innovation, Natural Sciences, and Engineering Research Council of Canada; the University of Saskatchewan; the Government of Saskatchewan; Western Economic Diversification Canada; the National Research Council Canada; and the Canadian Institutes of Health Research. The Structural Genomics Consortium is a registered charity (no. 1097737) that receives funds from AbbVie; Bayer Pharma AG; Boehringer Ingelheim; Canada Foundation for Innovation; Eshelman Institute for Innovation; Genome Canada through Ontario Genomics Institute (OGI-055); Innovative Medicines Initiative (European Union/European Federation of Pharmaceutical Industries and Associations [EU/EFPIA]; Unrestricted Leveraging of Targets for Research Advancement and Drug Discovery [ULTRA-DD] grant no. 115766); Janssen; Merck KGaA; Merck Sharp and Dohme; Novartis Pharma AG; Ontario Ministry of Research, Innovation, and Science (MRIS); Pfizer; São Paulo Research Foundation (FAPESP); Takeda; and Wellcome. This work is also supported by the Natural Sciences and Engineering Research Council of Canada (NSERC; funding reference no. RGPIN-2016-06300).
Author contributions: H.Z., Y.D., and J.M. designed the research; H.Z., B.Z., S.Q., J.X., R.H., W.T., V.N., Y.L., and P.L. performed the research; H.Z., Y.D., and J.M. analyzed the data; and H.Z. and J.M. wrote the paper.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.311639.118.
References
- Ayton PM, Cleary ML. 2003. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 17: 2298–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin DY, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. 1995. Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378: 505–508. [DOI] [PubMed] [Google Scholar]
- Chen L, Deshpande AJ, Banka D, Bernt KM, Dias S, Buske C, Olhava EJ, Daigle SR, Richon VM, Pollock RM. 2013. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 27: 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove MS, Patel A. 2010. Mixed lineage leukemia: a structure–function perspective of the MLL1 protein. FEBS J 277: 1832–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, Fazio M, Chen C-w, Zhu N, Koche R. 2014. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell 26: 896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMartino JF, Ayton PM, Chen EH, Naftzger CC, Young BD, Cleary ML. 2002. The AF10 leucine zipper is required for leukemic transformation of myeloid progenitors by MLL-AF10. Blood 99: 3780–3785. [DOI] [PubMed] [Google Scholar]
- Dubben S, Hönscheid A, Winkler K, Rink L, Haase H. 2010. Cellular zinc homeostasis is a regulator in monocyte differentiation of HL-60 cells by 1α, 25-dihydroxyvitamin D3. J Leukoc Biol 87: 833–844. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR. 2011. HMMER Web server: interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen S, Mattay D, Räuber C, Müller KM, Arndt KM. 2014. Characterization and inhibition of AF10-mediated interaction. J Pept Sci 20: 385–397. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. 2007. Inference of macromolecular assemblies from crystalline state. J Mol Biol 372: 774–797. [DOI] [PubMed] [Google Scholar]
- Krivtsov AV, Armstrong SA. 2007. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7: 823–833. [DOI] [PubMed] [Google Scholar]
- Kuntimaddi A, Achille NJ, Thorpe J, Lokken AA, Singh R, Hemenway CS, Adli M, Zeleznik-Le NJ, Bushweller JH. 2015. Degree of recruitment of DOT1L to MLL-AF9 defines level of H3K79 di- and tri-methylation on target genes and transformation potential. Cell Rep 11: 808–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wu S, Jiang S. 2007. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr Pharm Des 13: 143–162. [DOI] [PubMed] [Google Scholar]
- Lupas A. 1996. Coiled coils: new structures and new functions. Trends Biochem Sci 21: 375–382. [PubMed] [Google Scholar]
- Magnusson M, Brun AC, Miyake N, Larsson J, Ehinger M, Bjornsson JM, Wutz A, Sigvardsson M, Karlsson S. 2007. HOXA10 is a critical regulator for hematopoietic stem cells and erythroid/megakaryocyte development. Blood 109: 3687–3696. [DOI] [PubMed] [Google Scholar]
- Maret W. 2013. Zinc and human disease. Met Ions Life Sci 13: 389–414. [DOI] [PubMed] [Google Scholar]
- Martin ME, Milne TA, Bloyer S, Galoian K, Shen W, Gibbs D, Brock HW, Slany R, Hess JL. 2003. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell 4: 197–207. [DOI] [PubMed] [Google Scholar]
- Meyer C, Schneider B, Jakob S, Strehl S, Attarbaschi A, Schnittger S, Schoch C, Jansen M, van Dongen J, Den Boer M. 2006. The MLL recombinome of acute leukemias. Leukemia 20: 777–784. [DOI] [PubMed] [Google Scholar]
- Mueller D, Bach C, Zeisig D, Garcia-Cuellar M-P, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL. 2007. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110: 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean AG, Hess JL. 2012. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol 7: 283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Feng Q, Lin Y, Jiang Q, Li Y, Coffield VM, Su L, Xu G, Zhang Y. 2005. hDOT1L links histone methylation to leukemogenesis. Cell 121: 167–178. [DOI] [PubMed] [Google Scholar]
- Okada Y, Jiang Q, Lemieux M, Jeannotte L, Su L, Zhang Y. 2006. Leukaemic transformation by CALM–AF10 involves upregulation of Hoxa5 by hDOT1L. Nat Cell Biol 8: 1017–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisenauer MR, Anderson M, Huang L, Zhang Z, Zhou Q, Kone BC, Morris AP, LeSage GD, Dryer SE, Zhang W. 2009. AF17 competes with AF9 for binding to Dot1a to up-regulate transcription of epithelial Na+ channel α. J Biol Chem 284: 35659–35669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slany R. 2016. The molecular mechanics of mixed lineage leukemia. Oncogene 35: 5215–5223. [DOI] [PubMed] [Google Scholar]
- Smith MJ, Ottoni E, Ishiyama N, Goudreault M, Haman A, Meyer C, Tucholska M, Gasmi-Seabrook G, Menezes S, Laister RC. 2017. Evolution of AF6–RAS association and its implications in mixed-lineage leukemia. Nat Commun 8: 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofiyev V, Kaur H, Snyder BA, Hogan PA, Ptak RG, Hwang P, Gochin M. 2017. Enhanced potency of bivalent small molecule gp41 inhibitors. Bioorg Med Chem 25: 408–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thirman MJ. 2017. Paradoxical effects of MLL paralogs in MLL-rearranged leukemia. Cancer Cell 31: 729–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B-C. 1985. Resolution of phase ambiguity in macromolecular crystallography. Methods Enzymol 115: 90–112. [DOI] [PubMed] [Google Scholar]
- Welch BD, VanDemark AP, Heroux A, Hill CP, Kay MS. 2007. Potent D-peptide inhibitors of HIV-1 entry. Proc Natl Acad Sci 104: 16828–16833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Ng SY-M, Zuniga-Pflucker JC, Georgopoulos K. 2006. Early hematopoietic lineage restrictions directed by Ikaros. Nat Immunol 7: 382–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. 2006. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. J Biol Chem 281: 18059–18068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Wang J, Lee SY, Xiong J, Bhanu N, Guo Q, Ma P, Sun Y, Rao RC, Garcia BA. 2016. PRDM16 suppresses MLL1r leukemia via intrinsic histone methyltransferase activity. Mol Cell 62: 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemin-van der Poel S, McCabe NR, Gill HJ, Espinosa R, Patel Y, Harden A, Rubinelli P, Smith SD, LeBeau MM, Rowley JD. 1991. Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci 88: 10735–10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.