

Katz et al. discovered a hydrophobic region within the MDM2 N-terminal domain that binds exclusively to the dimeric form of the p53 C-terminal domain. MDM2 exhibits superior binding to, hyperdegradation of, and increased nuclear exclusion of dimeric p53 when compared with tetrameric wild-type p53.
Keywords: MDM2, p53 dimer, p53 tetramer, mutant p53, nuclear export, 20S proteasome
Abstract
The p53 tumor suppressor protein is the most well studied as a regulator of transcription in the nucleus, where it exists primarily as a tetramer. However, there are other oligomeric states of p53 that are relevant to its regulation and activities. In unstressed cells, p53 is normally held in check by MDM2 that targets p53 for transcriptional repression, proteasomal degradation, and cytoplasmic localization. Here we discovered a hydrophobic region within the MDM2 N-terminal domain that binds exclusively to the dimeric form of the p53 C-terminal domain in vitro. In cell-based assays, MDM2 exhibits superior binding to, hyperdegradation of, and increased nuclear exclusion of dimeric p53 when compared with tetrameric wild-type p53. Correspondingly, impairing the hydrophobicity of the newly identified N-terminal MDM2 region leads to p53 stabilization. Interestingly, we found that dimeric mutant p53 is partially unfolded and is a target for ubiquitin-independent degradation by the 20S proteasome. Finally, forcing certain tumor-derived mutant forms of p53 into dimer configuration results in hyperdegradation of mutant p53 and inhibition of p53-mediated cancer cell migration. Gaining insight into different oligomeric forms of p53 may provide novel approaches to cancer therapy.
Situated at the hub of several regulatory pathways that can protect cells from malignant transformation, p53 is involved in cell cycle arrest, DNA repair, metabolism, apoptosis, senescence, and other outcomes (Vousden and Prives 2009; Bieging et al. 2014). Supporting evidence that p53 is a major tumor suppressor includes the facts that (1) the TP53 gene is mutated in about half of all sporadic cancers overall, (2) cancer-prone Li Fraumeni syndrome (LFS) patients harbor germline p53 mutations, (3) mice deleted of p53 acquire tumors with 100% frequency, and (4) DNA viruses such as oncogenic versions of human papillomavirus (HPV) target p53 (Hollstein et al. 1991; Vogelstein et al. 2000; Soussi and Beroud 2001). While p53 is well studied as a DNA sequence-specific transcription factor, cytoplasmic roles for the protein have also been described (Green and Kroemer 2009; Comel et al. 2014; Marchenko and Moll 2014). Structurally, p53 has the canonical features of a regulator of transcription, including a bipartite transcriptional activation domain (TADs I and II; residues 20–40 and 41–60, respectively), a centrally located conserved sequence-specific DNA-binding domain (DBD; residues 100–300), and an oligomerization domain (OD; residues 325–355). Following the OD at the extreme C terminus of the protein is a basic regulatory region (REG; amino acids 363–393) in which six lysine residues can be extensively modified.
The oligomeric status of p53 has been studied by various biophysical approaches, which have shown that the purified full-length protein exists primarily as a tetramer (Friedman et al. 1993; Laptenko et al. 2015). The structure of the p53 OD as documented by both nuclear magnetic resonance (NMR) and X-ray crystallography is a “dimer of dimers” (Clore et al. 1994; Lee et al. 1994; Jeffrey et al. 1995). Embedded in the OD is a leucine-rich nuclear export signal (NES; residues 340–351). Wahl and colleagues (Stommel et al. 1999) first proposed that the hydrophobic NES is buried and inaccessible in the tetrameric form of p53, while, in the monomeric or dimeric forms of the protein, the NES is fully exposed and available to make protein–protein interactions that can promote p53 shuttling from the nucleus. Their model posits that in nonstressed cells, p53 exists largely in the dimer form, and, upon stress signaling leading to its increased intracellular concentration, p53 shifts to tetramer conformation that can bind more efficiently to DNA and activate p53 target genes (Stommel et al. 1999; Weinberg et al. 2004; Kawaguchi et al. 2005). This model was supported by a more recent study with stably expressed mCerulean-tagged p53, which showed that the majority of p53 in resting cells is indeed in the dimer form (∼59% dimers and 13% tetramers), and, after DNA damage, the tagged p53 is converted almost exclusively to tetramers (∼4% dimers and 92% tetramers) (Gaglia et al. 2013). The tetramer state of p53 is important for many aspects of p53 function (for review, see Kamada et al. 2016). These include DNA binding and transcriptional regulation (Chene 2001; Kawaguchi et al. 2005); post-translational modifications, particularly ubiquitination (Sakaguchi et al. 1998; Maki 1999; Shieh et al. 2000; Warnock et al. 2008; Itahana et al. 2009); degradation (Kubbutat et al. 1998; Hjerpe et al. 2010); and interaction with numerous proteins such as ARC, RhoGAP, HERC2, CK2, PKC, HPV-16, TBP, and others (Xu et al. 2013; Cubillos-Rojas et al. 2014; Gaglia and Lahav 2014; for review, see Chene 2001). It is safe to say that the implications of the different oligomeric states of p53 are still not fully understood.
Central to our understanding of p53 is its relationship with its prime negative regulator, MDM2. It is well established that p53 and MDM2 form a negative feedback loop in which p53 activates transcription of MDM2, and MDM2 inhibits p53 transactivation of its target genes, promotes its degradation, and facilitates its cytoplasmic localization (for review, see Manfredi 2010) and p53 mRNA translation (Ofir-Rosenfeld et al. 2008; Karni-Schmidt et al. 2016). The crucial role of MDM2 as the obligate negative regulator needed to keep p53 in check has been well validated by extensive mouse models (Jones et al. 1995; Montes de Oca Luna et al. 1995; Xiong 2013). MDM2 functions as an E3 ligase RING finger protein that binds to E2 ubiquitin-conjugating enzymes and promotes direct ubiquitination of p53 (Honda et al. 1997; Fang et al. 2000). MDM2 can both polyubiquitinate and monoubiquitinate p53 (Li et al. 2003), and ubiquitination of p53 by MDM2 serves mainly to degrade p53 but can also influence p53 localization and activity, depending on the type of ubiquitination (polyubiquitination versus monoubiquitination) and the levels of MDM2 (Coutts et al. 2009). The role of tetramerization in p53 ubiquitination remains unclear; it was reported that the p53 tetramerization domain and the NES are required for efficient ubiquitination by MDM2 (Maki 1999), while another study demonstrated that ubiquitinated p53 can still form tetramers (Brooks et al. 2007). Although MDM2 is able to shuttle p53 from the nucleus to the cytoplasm, where p53 is degraded (Roth et al. 1998; Nie et al. 2007), it has also been shown that nuclear export is not required for p53 degradation, as MDM2 can promote p53 ubiquitination and degradation in either the nucleus or the cytoplasm (Yu et al. 2000; Xirodimas et al. 2001).
p53 was shown to possess different regions that can interact with MDM2. The primary and most well-studied MDM2-binding site is located within the first p53 TAD (TAD-I; residues 20–40) (Kussie et al. 1996). This region is very well characterized (Momand et al. 1992; Chen et al. 1993; Oliner et al. 1993; Kussie et al. 1996) and has been resolved structurally by both NMR and X-ray crystallography, which provided detailed interactions between a p53 peptide (residues 15–29) and a hydrophobic pocket (residues 50–100) within the N-terminal region of MDM2 (Kussie et al. 1996). A second p53-binding site involves the DBD, specifically within conserved regions IV and V (residues 234–286), which was shown to interact with the central acidic domain of MDM2 and is essential for proper p53 ubiquitination (Shimizu et al. 2002; Wallace et al. 2006; Yu et al. 2006). The third binding site spanning the p53 REG at the extreme C terminus was discovered previously by our group and shown to associate with a polypeptide spanning the N-terminal domain (NTD) of MDM2 (amino acids 10–139) (Poyurovsky et al. 2010).
Here we identified new binding interfaces between p53 and Mdm2 that are distinct from the above-mentioned interacting regions within the two proteins. Our findings also show that the oligomeric status of p53 can dramatically affect its regulation by MDM2 with respect to both degradation and cellular localization.
Results
MDM2 preferentially binds and regulates the dimer form of p53
Following our discovery of an interaction between the MDM2 NTD and the p53 REG (Poyurovsky et al. 2010), we considered the possibility that binding by the p53 C terminus is regulated by the oligomeric state of the protein. Accordingly, binding by the MDM2 NTD polypeptide (residues 10–139) to either the wild-type p53 region that spans the OD and C-terminal domain (CTD; residues 293–393 [p53 CTD293–393]) or a mutated version of this region (p53 CTD293–393/L344A) that is known to exist predominantly as a dimer (Mateu and Fersht 1998) was measured by quantitative isothermal titration calorimetry (ITC). Strikingly, by this assay, the MDM2 NTD bound only to the dimeric version of the p53 CTD293–393/L344A, with virtually no binding detected to the wild-type p53 CTD293–393 (which forms tetramers at the concentrations used) (Fig. 1A). The MDM2 NTD bound the mutant p53 CTD293–393/L344A with an affinity of Kd = 1.7 µM. The binding affinity of the REG that we reported previously (Poyurovsky et al. 2010) is at least one order of magnitude less than our measurement of the MDM2 NTD bound to the mutant p53 CTD293–393 and thus is likely below the detection level in this assay. To assess preferential binding of full-length dimeric p53 and MDM2, we carried out an enzyme-linked immunosorbent assay (ELISA). Consistent with the ITC data, MDM2 bound consistently better to dimeric mutant p53 than wild-type p53, although the differences were more modest, which is to be expected from secondary binding site interactions (Fig. 1B).
Figure 1.

MDM2 preferentially binds and degrades dimer-forming mutant versions of p53. (A) Polypeptides from wild-type or L344A p53 CTDs (residues 293–393; 0.5 mM) were titrated into a solution containing 0.02 mM an MDM2 NTD polypeptide (residues 10–139), and their interactions were measured by ITC as described in the Materials and Methods. The MDM2 NTD bound the mutant p53 CTD (L344A) with an affinity of 1.7 µM. (B) The top panel shows a binding curve of p53 to MDM2 from one representative experiment. Purified full-length wild-type or E343K mutant p53 (0.03 mM) proteins were used to coat ELISA plates followed by addition of full-length purified wild-type MDM2 protein at the indicated concentrations. Complexes were detected using anti-MDM2 antibody (N-20) and enzyme-conjugated secondary antibody as described in the Materials and Methods. The bottom panel graphically depicts the cumulative binding data from six replicative experiments. The data represent the mean ± SEM for six biological replicates with two technical replicates each. (***) P-value = 0.0002, calculated by nonparametric one-way ANOVA as described in the Materials and Methods. (C, top panel) A diagram of a p53 protein showing the TAD (residues 20–60), proline domain (Pro; residues 63–97), DBD (residues 94–312), OD (residues 324–355), and C-terminal REG (residues 363–393). The positions of the dimer-forming (D) and monomer-forming (M) mutations are indicated. The two panels below the diagram are typical experiments showing results of transfecting U2OS cells with increasing amounts of Flag-MDM2 (0, 375, and 750 ng) and a constant amount of HA-tagged wild-type or OD mutant versions of p53 (150 ng) as indicated. Twenty-four hours later, cells were harvested, and lysates were used for immunoblotting with the indicated antibodies. (D) U2OS cells were transfected with 1.2 µg of Flag-MDM2 and 150 ng of HA-tagged wild-type or OD mutant p53, as indicated. Twenty-four hours after transfection, 100 µg/mL cycloheximide (CHX) was added, and cells were harvested at the indicated times. (Top panels) Cell lysates were subjected to immunoblotting with the indicated antibodies. (Bottom panels) Quantification of the immunoblotting data was carried out using ImageJ software.
To determine the physiological relevance of these observations, we moved to cell-based assays and performed a set of transient cotransfection experiments with full-length MDM2 and either wild-type or missense mutant forms of p53 that form dimers instead of tetramers. The reasons why we chose to use ectopically expressed mutant forms of p53 in U2OS cells that harbor wild-type p53 are outlined in the Discussion. We used Flag-tagged MDM2 and HA-tagged p53 variants to distinguish between the endogenously and exogenously expressed versions of these proteins. p53 dimers are reportedly formed cotranslationally (Nicholls et al. 2002), and we confirmed that dimer-forming mutant p53 proteins cannot oligomerize with wild-type p53 (data not shown). We tested several known mutant p53 proteins (L344A, E343A, E343K, A347T, and L348A), each of which has been shown to favor the dimeric form of p53 (Mateu and Fersht 1998; Kawaguchi et al. 2005; Kamada et al. 2011). The dimeric state of these p53 variants was confirmed using glutaraldehyde cross-linking analyses (Fig. 4, below; Supplemental Fig. S1; data not shown). We also used two p53 variants known to form monomers: L330A and F341C (Kawaguchi et al. 2005; Kamada et al. 2011). All five dimeric mutants of p53 were significantly better substrates for MDM2 degradation when compared with wild-type tetrameric p53, while, as reported previously (Kubbutat et al. 1998; Hjerpe et al. 2010), the two monomer-forming mutant versions of p53 were very poorly degraded by MDM2 (Fig. 1C). To verify that the endogenous p53 in U2OS cells was not a contributing factor, we carried out the same experiment in p53-null U2OS cells that were generated by CRISPR/Cas9 (see the Materials and Methods). Our results showed that even in the absence of endogenous p53, all five dimeric mutants of p53 were significantly better substrates for MDM2 degradation when compared with wild-type tetrameric p53 (Supplemental Fig. S2). Consistent with their increased degradation, cycloheximide chase experiments showed that dimeric p53 proteins displayed increased turnover compared with wild-type p53 in the presence of Mdm2 (Fig. 1D). While there were differences in the intrinsic turnover rate of the different mutant forms of p53 without coexpressed MDM2, each variant was degraded at a faster rate than wild-type p53 in the presence of Mdm2 (Supplemental Fig. S3). N-terminally deleted versions of p53 (Δ42) with either wild-type or dimer-forming mutant ODs were not degraded by coexpressed MDM2, confirming that the primary binding site within the p53 N terminus is required for MDM2 to degrade p53 regardless of the oligomerization state of the protein (Supplemental Fig. S4).
Figure 4.

Tetrameric but not dimeric p53 is degraded by the 26S proteasome. (A) U2OS parental (par) or U2OS CRISPR cell lines expressing mutated dimeric p53 (E343K) (het 10, hom 8, and hom 84) were harvested, and lysates were treated with increasing amounts of glutaraldehyde (0, 0.01% and 0.1%) for 20 min at room temperature and then used for immunoblotting with p53 antibodies (1801/DO1). (B) U2OS parental (par) and U2OS CRISPR cell lines expressing mutated dimeric p53 (E343K) (hom 8 and hom 84) were transfected with 30 nM nonspecific siRNAs (siC) and two different Rpn2 siRNAs. Forty-eight hours later, cells were harvested, and lysates were used for immunoblotting with antibodies versus the indicated proteins. (C) U2OS cells were either untreated (NT) or transfected with 30 nM three different Rpn2 siRNAs and 30 nM two different nonspecific siRNAs (siC and siLuc). Forty-eight hours later, cells were harvested, and lysates were subjected to glutaraldehyde cross-linking as in A (right panel) or used for immunoblotting with the antibodies versus the indicated proteins (left panel). (D) Model depicting possible degradation mechanisms of wild-type and dimeric versions of p53. As shown in many previous studies, wild-type p53 is degraded mostly by the well-characterized ubiquitin-dependent 26S proteasomal degradation pathway, while dimeric p53 with a partially unstructured conformation is preferentially degraded by the 20S proteasome in a ubiquitin-independent manner.
MDM2-dependent cytoplasmic localization of p53 dimers
The presence of MDM2 can affect p53 cellular distribution, shifting it from the nucleus to the cytoplasm (Freedman and Levine 1998; Roth et al. 1998; Stommel et al. 1999; Tao and Levine 1999; Geyer et al. 2000; Nie et al. 2007). However, it is unclear whether or how the p53 oligomeric status (tetramer/dimer/monomer) affects this shuttling process. We expressed different p53 oligomeric variants in U2OS cells that were then treated with the nuclear export inhibitor leptomycin B (LMB). This led to increased accumulation of nuclear dimeric p53 when compared with wild-type p53 specifically in the presence of MDM2 (Fig. 2A). The data suggested that under basal (unstressed) conditions, dimeric p53 localizes more to the cytoplasm than wild-type p53. Moreover, when comparing the levels of dimeric p53 with the initial amount of wild-type p53, LMB treatment was able to rescue dimeric p53 from degradation by MDM2, indicating that MDM2-mediated degradation of p53 occurs mostly in the cytoplasm under our experimental setup (Fig. 2A). To confirm the subcellular distribution of wild-type and dimer-forming mutant p53 variants, we performed immunofluorescence assays. When transfected alone, both wild-type p53 and dimeric p53 were mostly nuclear, while, upon coexpression with MDM2, p53 was partially exported to the cytoplasm, again indicating that this relocalization occurs in an MDM2-dependent manner (Fig. 2B; Supplemental Fig. S5A,B; note that the graph in Fig. 2B shows results of all cells with cytoplasmic p53, while that in Supplemental Fig. S5B shows the distribution of cells with either N > C or C > N staining, where N is nuclear, and C is cytoplasmic). While ∼50% of wild-type p53 was translocated from the nucleus in the presence of MDM2, with each dimer-forming mutant p53 variant, there was a significantly greater fraction in the cytoplasm, supporting the results above that MDM2 preferentially targets dimeric p53 for nuclear export. Both wild-type and mutant forms of p53 remained nuclear upon LMB treatment in an MDM2-independent manner (Supplemental Fig. S5C). As additional validation, subcellular fractionation assays were performed (Fig. 2C). In the absence of MDM2, both wild-type (and presumably tetrameric) p53 and most of the dimer-forming p53 variants were found in both nuclear and cytoplasmic fractions, with a slight preference for the nuclear fraction (with the exception of mutant p53 proteins A347T and L344A). Upon coexpression with ectopic MDM2, while both wild-type mutant forms of p53 were relatively more abundant in the cytoplasm, the ratio of cytoplasmic:nuclear p53 was greater with the dimer-forming mutant versions of p53. Furthermore, mutant p53 (A347T) and p53 (L344A) that were initially predominantly cytoplasmic displayed even greater localization in this cellular compartment when coexpressed with MDM2. Note that upon treatment with LMB, the trend was reversed, and the dimeric p53 mutant became more predominantly nuclear (Supplemental Fig. S5D). Together, our observations confirm the role of MDM2 in exporting p53 from the nucleus to the cytoplasm and strongly indicate that MDM2 preferentially translocates dimeric p53 over its tetrameric counterpart.
Figure 2.

MDM2-dependent cytoplasmic localization of dimer-forming p53 variants. (A, top panels) U2OS cells were transfected with 150 ng of HA-p53 (wild type or mutants) in the absence or presence of 900 ng of Flag-MDM2. Twenty-four hours after transfection, cells were treated with 5 ng/mL LMB or DMSO for 5 h. Cell lysates were subjected to immunoblotting with the indicated antibodies. (Bottom panels) Quantification of the immunoblotting data was carried out using ImageJ software. In each case, values for p53 protein variants were plotted relative to their untreated counterparts (DMSO), which were taken as 1. The bar graphs represent the average of three independent experiments. P-values were calculated by comparing the LMB-treated samples with the DMSO-treated samples within each pair (B, left) Cellular localization of 150 ng of wild-type or mutant versions of p53 was detected in U2OS cells transfected with the p53 variants indicated at the top in the absence or presence of 750 ng of Flag-MDM2. Twenty-four hours after transfection, cells were fixed with 4% paraformaldehyde and analyzed by immunofluorescence microscopy after staining with anti-HA antibodies (green). Arrows indicate the presence of p53 in the cytoplasm. For the full images, including MDM2, see Supplemental Figure S5A. (Right) Quantification of cell distribution was done for three independent experiments, as described in the Materials and Methods. (C, top) U2OS cells were transfected with 150 ng of HA-p53 (wild type or mutants) in the absence or presence of 1050 ng of Flag-MDM2. Cell extracts were fractionated into cytoplasmic (Cyto) and nuclear (Nuc) portions and then analyzed by immunoblotting with the indicated antibodies. PARP and GFP were used as cytosolic and nuclear controls, respectively. (Bottom) Quantification of the immunoblotting data was carried out using ImageJ software. The graph represents the average of three independent experiments. P-values were analyzed by comparing the cytoplasmic fraction versus the nuclear fraction within each pair.
MDM2 degradation of dimeric p53 occurs via a ubiquitination-independent mechanism
Based on these findings, we predicted that the enhanced degradation of dimer-forming mutant p53 proteins by MDM2 would be reflected in their relatively increased ubiquitination. To test this, we examined the impact of MDM2 on conjugation of either wild-type ubiquitin or a lysine-free ubiquitin (UbKO) that can be linked to the substrate but cannot form a polyubiquitin chain to the above-mentioned p53 oligomerization variants. Unexpectedly, none of the tested mutants displayed the predicted increase in polyubiquitination or monoubiquitination (Fig. 3A,B). In fact, with the exception of mutant p53 (E343K), these p53 variants were actually markedly underubiquitinated compared with wild-type p53. Furthermore, since the E343K mutation endows p53 with an extra lysine in the OD region, this may account for its relatively increased ubiquitination compared with the other dimer-forming mutant versions of p53. However, even here, ubiquitination of mutant p53 (E343K) did not exceed that of wild-type p53, as would have been predicted.
Figure 3.

Dimeric p53 is underubiquitinated by MDM2 and structurally distinct from wild-type p53. (A,B) Wild-type U2OS cells were transfected with 160 ng of HA-p53 (wild type or the indicated mutant forms) in the absence or presence of 1.2 µg of Flag-MDM2 as indicated along with 0.8 µg of Flag-ubiquitin (A) or 0.7 µg of His-UbKO monoubiquitin (B). Cells were treated with 20 µM MG132 for 4 h before harvesting, and cell lysates were immunoprecipitated with anti-HA antibody followed by immunoblotting with anti-p53 antibody (FL393-G) to detect ubiquitinated p53. (C) U2OS cells were transfected with 200 ng of constructs expressing wild type or the indicated dimer-forming mutant versions of HA-p53 or the tumor-derived core domain mutant p53 (R175H). Twenty-four hours later, cell lysates were prepared and subjected to immunoprecipitation (IP) with anti-p53 PAb 240 or Mab 1620 antibodies followed by immunoblotting with anti-HA antibody. (D) Forty nanograms of p53 proteins (wild-type p53, dimeric p53E343A, and monomeric p53L330A) that was purified from HCT116 cells grown in medium containing photo-L-methionine was premixed without or with increasing amounts of recombinant purified MDM2 (190–760 ng). After irradiation with UV as described in the Materials and Methods, samples were resolved by SDS-PAGE, and cross-linked p53 species were detected by immunoblotting using anti-p53 antibody (Fl-393).
The extreme C terminus of wild-type p53 harbors six lysine residues that have been shown to be the primary sites of ubiquitination and degradation by MDM2 (Rodriguez et al. 2000). We introduced into the dimer-forming mutants additional changes within the six extreme C-terminal lysines, changing them to arginine (6KR) to preserve charge but prevent any lysine modifications, including ubiquitination. The 6KR mutated form of p53 is known to be both resistant to MDM2-induced degradation and refractory to MDM2-mediated ubiquitination (Rodriguez et al. 2000). Dimeric 6KR p53 mutants still underwent enhanced degradation in the presence of MDM2, thereby corroborating that MDM2 degradation of dimer-forming mutant proteins is unlikely to be due to increased ubiquitination (Supplemental Fig. S6).
Dimeric mutant p53 proteins are structurally distinct from wild-type p53
The 20S proteasome degrades proteins that contain unstructured regions by a ubiquitin-independent mechanism (Ben-Nissan and Sharon 2014). To address the possibility that dimer-forming mutant versions of p53 might have unstructured regions, we first examined structure-relevant properties of dimeric p53 compared with wild-type p53. The p53 variants were immunoprecipitated with two different p53 monoclonal antibodies that have structure-dependent epitopes: PAb 1620 that was reported to be specific for the “wild-type” conformation (Cook and Milner 1990) and PAb 240 that binds to unfolded p53; i.e., p53 in the “mutant” conformation (Gannon et al. 1990; Legros et al. 1994; Vojtesek et al. 1995). Wild-type p53 was immunoprecipitated almost exclusively by PAb1620 and not PAb240, while a tumor-derived mutant form of p53 (R175H) known to be extensively unfolded (Bargonetti et al. 1993; Joerger and Fersht 2007) was virtually exclusively immunoprecipitated by PAb 240. We found that dimeric p53 proteins were relatively more efficiently brought down by PAb240 than wild-type p53, while they were relatively less immunoprecipitated by PAb 1620, indicating that dimeric p53 is at least partially unstructured (Fig. 3C).
Evidence for distinct conformational changes in dimeric p53 in the presence of MDM2 was derived from UV-induced cross-linking experiments in which recombinant purified wild-type p53, dimeric p53 (E343A), and monomeric p53 (L330A) proteins containing photo-L-methionine were premixed with increasing amounts of recombinant purified MDM2. After UV irradiation, proteins were separated by SDS-PAGE. While there were almost no differences between the cross-linking products of wild-type p53 and monomeric p53 in the presence of MDM2, dimeric p53 had a significantly different pattern of cross-linked species, with a shifted product (Fig. 3D, arrow) that was more prominent in the presence of MDM2 (Fig. 3D). Of the 12 methionine residues in p53, only one (M340) is present in the OD, which indicates that the change seen upon binding to MDM2 is the result of a conformational shift specifically within this region of the protein only when it is in dimer configuration. Even though monomeric p53 is reportedly also a target of the 20S proteasome (Hjerpe et al. 2010; Lang et al. 2014) and yet is very poorly degraded by MDM2, our data suggest that the dimer form of p53 assumes a unique conformation that allows it to be degraded rapidly by MDM2.
The dimer form of endogenous p53 dimers is not degraded by the 26S proteasome
The above-mentioned experiments showed differences between dimeric mutant and wild-type versions of p53 proteins when expressed in U2OS cells via transient transfection. Of particular note, ectopically expressed dimer-forming mutant versions of p53 were hyperdegraded, markedly underubiquitinated, and partially unfolded compared with wild-type p53. In turn, this predicts that only the tetramer and not the dimer form of p53 is a target of the 26S proteasome, which requires that its target proteins be ubiquitinated (in contrast to the 20S proteasome) (Lecker et al. 2006; Collins and Goldberg 2017). To extend these studies in a more physiological context, we aimed to confirm the above results with p53 expressed from the endogenous locus. To that end, we used CRISPR–Cas9 gene editing to change the resident wild-type p53 in U2OS cells to the dimer-forming mutant p53 (E343K). Three dimeric p53 CRISPR cell lines were generated: one E343K heterozygous clone (+/E343K; het 10) and two E343K homozygous clones (E343K/E343K; hom 8 and hom 84). We used glutaraldehyde cross-linking analyses to determine the relative oligomeric states of p53 protein in these clones (Fig. 4A). In this analysis, compared with p53 in the parental U2OS cells, there was strikingly more dimeric than tetrameric p53 in each of the cell lines expressing one or both mutant TP53 (E343K) alleles. To our knowledge, this is the first example of detection of dimer and tetramer forms of basally (i.e., non-stress-induced) p53 protein expressed from the endogenous locus. It is interesting that in either case, despite their different ratios, both dimeric and tetrameric cross-linked species were detected in either wild-type- or mutant p53-expressing U2OS cells. This suggests that these oligomeric forms exist in an equilibrium that shifts dramatically when the OD is mutated. To further test our hypothesis that dimeric p53 is preferentially degraded by the 20S proteasome, we used siRNA to down-regulate the Rpn2 subunit that is positioned in the base of the 19S proteasomal cap (Wehmer and Sakata 2016). Loss of Rpn2 would prevent the assembly of 26S proteasomes while not affecting the 20S proteasome (Tsvetkov et al. 2009). By specifically ablating the 26S proteasome, we would predict that wild-type p53 should accumulate, since we are disturbing its main degradation pathway, whereas dimeric p53 in the CRISPR cell lines should be relatively unaffected by loss of the 19S cap. Indeed, following knockdown of Rpn2 (see Supplemental Fig. S7 for siRNA depletion of Rpn2 siRNA), there was a significant increase in the levels of wild-type p53 protein in in parental U2OS cell line, while levels of dimeric p53 in the CRISPR mutated cell lines (hom 84 and hom 8) were relatively unaffected (Fig. 4B). As a control, Bim, known to be a target of the 20S proteasome (Wiggins et al. 2011; Ben-Nissan and Sharon 2014), was unaffected by Rpn2 knockdown.
We then asked whether endogenously expressed wild-type p53 tetramers and dimers differed in their requirement for degradation by the 26S proteasome. To test this, we knocked down Rpn2 in the parental U2OS cells and then subjected cell lysates to glutaraldehyde cross-linking and immunoblotting to visualize p53 oligomeric species. Strikingly, when the 26S proteasome was selectively ablated, only cross-linked p53 tetramers accumulated, while levels of dimeric p53 remained relatively unchanged (Fig. 4C, left panel). Note that in Nutlin-treated cells, the tetramer form of wild-type p53 was greatly increased, while there was, at best, only a modest increase in the dimer form. This is consistent with the hypothesis that dimers are prevalent in unstressed conditions and that tetramers are selectively increased when Mdm2 cannot associate with p53. Controls for this experiment are shown in Figure 4C (right panel), where, again, Bim was unaffected by Rpn2 knockdown, while levels of cdc25A, a known 26S target (Mailand et al. 2000), were significantly increased. We found that the ratio between p53 tetramer and dimer species in unstressed cells differed somewhat between experiments (e.g., Fig. 4, cf. “par” lanes in A and the “NT,” “siC,” and “siLuc” lanes in C). This is potentially explained by the appearance of cross-linked species being affected by the total protein concentration of cell lysate used for a given glytaraldehyde concentration. Nevertheless, each experiment was internally controlled and reproducible, so our conclusions that dimeric p53 cannot be stabilized by blocking the 26S proteasome are valid. Our results showing that only tetrameric p53 requires the 26S proteasome strongly indicate that dimeric p53 proteins are primarily targets of the 20S proteasome. In support of this, we found that endogenous p53 (E343K) protein in the hom8 and hom84 CRISPR cell lines could be well be stabilized by Mg132 that inhibits both the 20S and 26S proteasome (Supplemental Fig. S8).
Based on the above results, we propose a model in which wild-type p53 is degraded mostly (but not exclusively) by the well-characterized ubiquitin-dependent 26S proteasomal degradation pathway, while dimeric p53, with its partially unstructured conformation, is preferentially (but also not exclusively) degraded in a ubiquitin-independent manner by the 20S proteasome (Fig. 4D).
Identification of a region within the N terminus of MDM2 that binds to the dimeric p53 OD
We discovered previously that the NTD of MDM2 (residues 10–110) can interact with the p53 CTD293–393 region spanning the OD + REGs, although the precise binding region within the MDM2 NTD was not determined (Poyurovsky et al. 2010). To gain more precise information about the MDM2 sequences involved, we used a combination of peptide array screening and fluorescence anisotropy. First, an array consisting of overlapping peptides derived from the NTD of MDM2 (amino acids 10–139) was screened for binding the p53 CTD 293–393 (see the Materials and Methods for the peptide-binding assay), which revealed several candidate peptides with different signal intensities (Supplemental Fig. S9A; Supplemental Table S1). Fluorescence anisotropy was used to further measure each peptide binding to either the wild-type p53 CTD293–393 or dimeric mutant p53 CTD293–393/L344A. Of these, one peptide spanning MDM2 residues 33–43 bound to dimeric p53 CTD293–393 but not wild-type p53 CTD293–393 with an affinity of 121 µM ± 1 µM (Fig. 5A; Supplemental Fig. S9B), while none of the others bound to either version of p53 (Supplemental Fig. S9B). Since the MDM2 sequence between residues 33 and 43 is extremely hydrophobic (LLLKLLKSVGA), as is the p53 NES (340MFRELNEALELK351), the nature of the interaction is likely to be salt-insensitive, which we confirmed using fluorescence anisotropy (Supplemental Fig. S9C). Our results therefore show that the p53 C-terminal region has an additional binding site that is distinct from our previously discovered MDM2-interacting region situated within the p53 REG363–393 region. They also imply that this new site requires exposure of the NES region within the p53 OD that occurs only in dimeric p53. We did not determine whether MDM2 binds to monomeric p53, although our and others’ (Kubbutat et al. 1998) data show that MDM2 cannot degrade p53 in the monomeric state.
Figure 5.
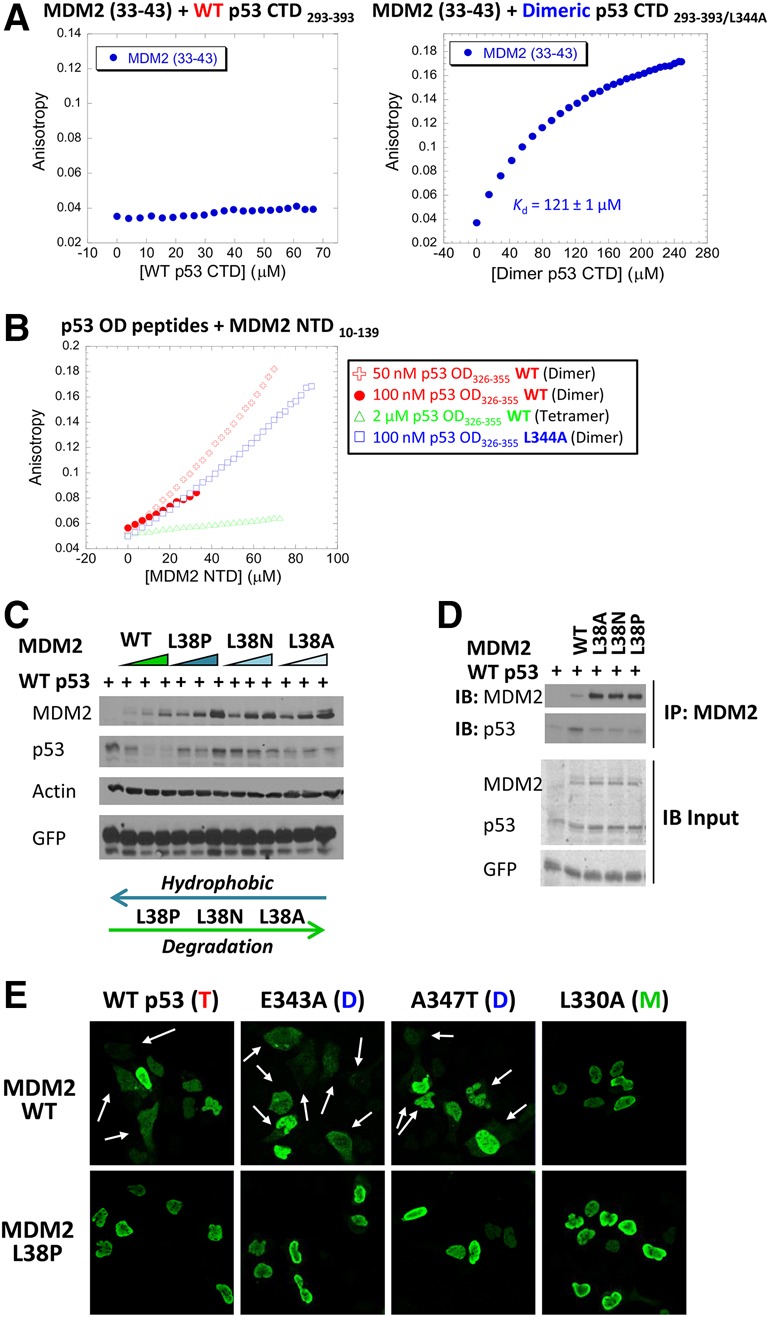
Mapping and characterizing the MDM2 N-terminal region required for binding to the dimer form of the p53 C terminus. (A) Fluorescence anisotropy binding studies were performed to measure the binding of an MDM2 peptide (residues 33–43) to wild-type (left panel) or the L344A dimer-forming mutant (right panel) versions of the p53 CTD293–393 polypeptide. L344A p53 CTD293–393 bound the MDM2 peptide with an affinity of 121 µM ± 1 µM. (B) Fluorescence anisotropy binding was performed to measure the interaction between the MDM2 NTD (amino acids 10–139) and wild-type or mutant p53 OD (residues 326–355) peptides. The wild-type p53 OD peptide was provided at concentrations of 50 nM (red +), 100 nM (red •), or 2 µM (green Δ) and were compared with dimeric mutant peptide p53 (L344A) at 100 nM (blue □). The binding curve shape is not suitable for accurate quantification because the peptide had a tendency to aggregate shortly after the analysis. (C) U2OS cells were transfected with increasing amounts of Flag-tagged version of wild-type (WT) or the indicated mutant forms of MDM2 (0, 0.5, 1, and 2 µg) along with 142 ng of HA-p53. Twenty-four hours later, cell lysates were prepared and used for immunoblotting with the indicated antibodies. (D) U2OS cells were transfected with 300 ng of wild-type or the indicated mutant forms of Flag-MDM2 along with wild-type HA-p53 (150 ng was used with wild-type MDM2, and 75 ng was used with MDM2 mutants). Cell lysates were subjected to immunoprecipitation with anti-Flag antibody followed by immunoblotting with anti-HA antibody. (E) U2OS cells were transfected with 300 ng of wild-type or mutant forms of HA-p53 as indicated in the presence of 1500 ng of wild-type or mutant (L38P) Flag-MDM2. Twenty-four hours later, cells were fixed with 4% paraformaldehyde and visualized by immunofluorescence microscopy after staining with anti-HA antibodies (green). Arrows indicate localization of p53 in the cytoplasm. For the full images, including MDM2, see Supplemental Figure S11A.
At high concentrations (>150 nM), purified wild-type p53 OD exists primarily as a tetramer, while, at lower concentrations, the OD peptide is present mostly in the dimer conformation (Brandt et al. 2009; Rajagopalan et al. 2011). Additional fluorescence anisotropy experiments were performed to examine binding between the MDM2 NTD10–139 and different concentrations of a peptide derived from the wild-type p53 OD (p53 OD326–355). Indeed, only when present in relatively dilute concentrations (50 and 100 nM) was binding to the MDM2 NTD peptide detected to the same extent as the dimeric mutant p53 OD (L344A), while, at higher concentrations (2 µM), the wild-type p53 OD showed no detectable binding to the MDM2 NTD (Fig. 5B). These data confirm and extend our findings that dimer-forming mutant versions of p53 are preferentially degraded by Mdm2. We point out that the original peptide array screen in Supplemental Figure S9A used the wild-type p53 CTD polypeptide and acknowledge a discrepancy between the results of the peptide array screening using wild-type p53 that led to the initial identification of the Mdm2 region between residues 33 and 43, while fluorescent anisotropy showed that only dimeric p53 binds to this sequence. Peptide array screening is a very robust and sensitive assay, and, speculatively, even if there was a minor component of dimers in the wild-type p53 CTD, this would provide a positive signal.
To confirm the hydrophobic nature of the interaction and the importance of the 33–43 region, we generated mutations within residue L38 (L38A, L38N, and L38P) in full-length MDM2 that reduce the hydrophobicity of the MDM2-binding region. These mutations led to the decreased ability of MDM2 to degrade (Fig. 5C) or bind (Fig. 5D) p53. Moreover, immunofluorescence experiments showed that mutated MDM2 (L38P) impairs p53 nuclear export (Fig. 5E; Supplemental Fig. S10A). When the p53 variants (wild-type or dimeric) were cotransfected along with wild-type MDM2 or mutant MDM2 (L38P) in the presence or absence of LMB, mutant MDM2 (L38P) caused p53 to accumulate to the same extent as cells treated with LMB (Supplemental Fig. S10B). These results indicate that this region of MDM2 is essential for p53 shuttling from the nucleus.
The MDM2 (L38P) mutation within the region that we identified here (residues 33–43) is located within an α helix situated behind the canonical N-terminal-binding pocket (residues 50–100) (Kussie et al. 1996) of p53, which suggested the possibility that this mutation may induce conformational changes that prevent p53 binding to the canonical pocket. In this scenario, the mutation would not be relevant to the interaction of this region with the dimeric C terminus, and levels of p53 in cells treated with Nutlin-3a that effectively separates p53 from MDM2 in vivo should not be further increased by mutation of L38 in MDM2. Since the combination of MDM2 (L38P) and Nutlin-3a stabilized p53 to a greater extent than each one alone (Supplemental Fig. S11), this supports the possibility that there are two different sites in the MDM2 NTD: one affected by Nutlin-3a (the p53 N terminus of p53) and the other affected by the MDM2 mutation (the p53 OD). Taken together, our results show that the region of the MDM2 NTD that is required for binding to the p53 OD as a dimer spans residues 33–43 and indicate that this interaction involves hydrophobic interactions with the NES.
Tumor-derived mutant p53 proteins are superior targets for full-length MDM2-mediated degradation when present as dimers
It is now well established that some of the frequently occurring tumor-derived mutant forms of p53 have pro-oncogenic properties in cell-based assays and in mice (Freed-Pastor and Prives 2012; Muller and Vousden 2014). These “hot spot” mutations within p53 are found within the central conserved DBD (Olivier et al. 2010), and thus two out of three of the regions to which MDM2 binds (the primary N-terminal region and the secondary C-terminal region) are intact. A number of reports have queried the roles of MDM2 in binding to and promoting the degradation of these tumor-derived mutants in cell-based studies (Lukashchuk and Vousden 2007; Parrales et al. 2016) as well as an elegant study in mice implicating MDM2 as one of the E3 ligases that degrades mutant p53 (Terzian et al. 2008). It was of interest to determine whether MDM2, as we found with wild-type p53, might be able to preferentially target tumor-derived mutant forms of p53 that are present as dimers. To test this, we generated three p53 variants harboring single hot spot core domain mutations along with dimer-forming mutations within the OD. MDM2 was more effective in degrading two of these doubly mutated p53 proteins (R248W and R175H) compared with tetrameric mutant p53 in both U2OS and H1299 cell lines (Fig. 6A; Supplemental Fig. S12A). We then asked whether this increased degradation by MDM2 might impact a known “gain-of-function” activity of mutant p53; namely, migration into an artificial “wound” in cell culture. We chose one of the most common hot spot mutant forms of p53 (R175H) for this assay and compared the ability of U2OS cells (Fig. 6B) or H1299 cells (Supplemental Fig. S12B) transfected with R175H p53 with either a wild-type OD or dimer-forming (E343K) second mutation in the presence or absence of cotransfected MDM2 to fill the experimentally induced cleared space on the culture dish. The experiment was done in the presence of mitomycin C to prevent growth and thereby rule out secondary effects caused by increased cell number. By 24 h, there was a marked (∼85%) decrease in the cleared area, with mutant p53 alone either with or without the dimer-forming mutation. Notably, the presence of MDM2 had a striking impact in preventing wound closure only of the dimer-forming version of R175H p53. These results not only extend our findings to different versions of p53 found in tumors but present the possibility of novel therapeutic approaches as discussed below.
Figure 6.
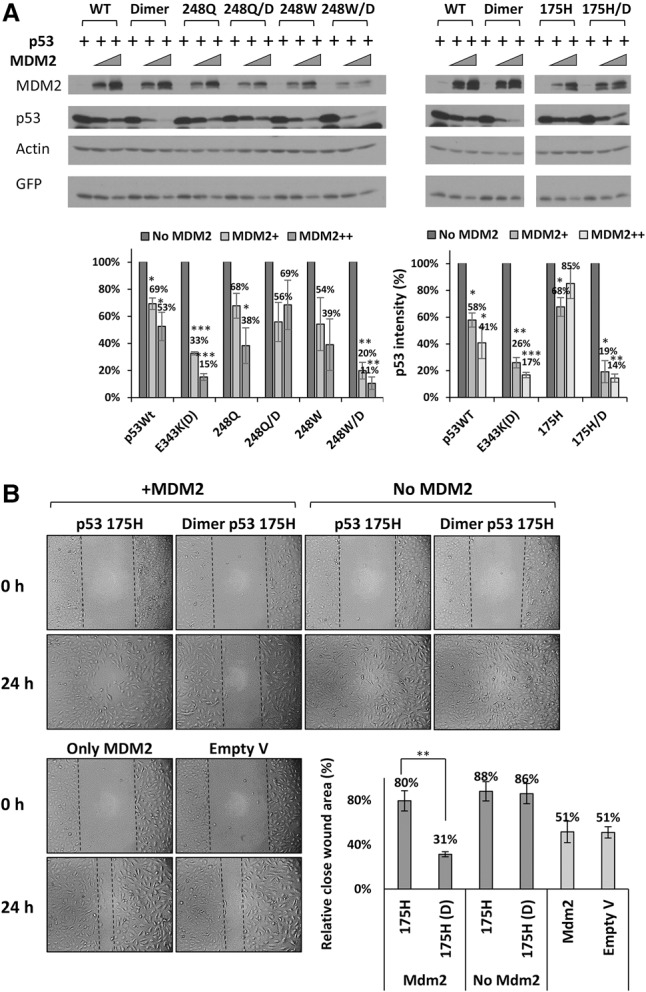
MDM2 preferentially degrades and regulates cancer-derived core domain mutant forms of p53 when dimeric. (A) U2OS cells were transfected with increasing amounts of Flag-MDM2 (0, 500, and 1000 ng) and constant amounts of wild-type or the indicated core domain mutant forms of HA-p53 (200 ng), each of which contained either a wild-type OD or dimer-forming mutation (E343K) in the OD, as indicated. Twenty-four hours after transfection, cell lysates were prepared and used for immunoblotting with the indicated antibodies. Quantification of the immunoblotting data was carried out using ImageJ software. The graph represents the average of five independent experiments. P-values were calculated by comparing p53 with MDM2 with the initial levels of p53 without MDM2. (B) U2OS cells were transfected with mutant p53 (R175H) containing either wild-type OD or E343K dimer-forming mutant OD (200 ng) in the absence or presence of 1200 ng of Flag-MDM2. Twenty-four hours after transfection, cultures were scratch-wounded with a 200-µL pipette tip and treated with 2.5 µg/mL mitomycin C to prevent cell proliferation. Images of wound closure were captured by phase-contrast microscopy at 0 h and after 24 h. The graphed quantification of migration is presented as the distance between the edges of each scratch relative to 0 h. The graph represents the average of three independent experiments. Student's t-test was used to compare different mutant p53 (175) variants with or without the presence of MDM2.
Discussion
Given the centrality of the relationship between p53 and MDM2, it is not surprising that their physical interactions are quite complex. Prior to the work described in this study, p53 was shown to possess three distinct regions that can interact with MDM2. Here, using a number of independent methods (ITC, florescence anisotropy, ELISA, and peptide array screening), we discovered a fourth region on p53 that is recognized by MDM2. We also mapped the region on MDM2 that recognizes the new p53-binding site to a sequence spanning amino acids 33 and 43 within its N terminus. What is unique to this p53–MDM2 interaction is the requirement that p53 be in dimer configuration.
To study the role of MDM2 in p53 oligomerization, previous studies used mutations such as deletion of the OD or OD-fused proteins (Maki 1999; Carter et al. 2007; Hjerpe et al. 2010; Davis et al. 2013), which in some cases might cause structural changes that could alter p53 protein folding. Furthermore, these gross mutations might not allow discrimination between monomers, dimers, or tetramers. To avoid these drawbacks, our findings were based on the transient expression of several dimer- and monomer-forming missense mutant versions of p53 that differ in charge, hydrophobicity, polarity, and length of amino acid side chain. Although the bulk of our experiments was performed with ectopically expressed proteins in U2OS cells, there are several reasons that validated this approach. First, using ectopically expressed p53 variants that exist primarily in only one oligomeric form allowed for more unambiguous results. Second, as U2OS cells harbor wild-type p53, their p53/MDM2 circuitry is likely to be relatively intact, and, in fact, these cells have been used for studying MDM2 regulation of p53 in several laboratories (Grossman et al. 1998; Geyer et al. 2000; Dai and Lu 2004; Lindstrom et al. 2007; Sasaki et al. 2007). Third, our findings were supported by results of a binding study in vitro comparing the wild-type p53 C-terminal polypeptide at low concentrations when p53 is present mostly as a dimer with a high concentration of the polypeptide that would render it likely exclusively in tetramer conformation. Finally, we were able to confirm with both wild-type and dimer-forming mutant p53 (E343K) expressed from the endogenous Tp53 locus the key finding that tetrameric but not dimeric p53 is degraded by the 26S proteasome.
Quantification of the interaction between either full-length proteins or purified domains (the MDM2 NTD [residues 10–139] and the p53 C-terminal region [residues 293–393]) using both ELISA and ITC, respectively, showed these interactions to be in the low micromolar range (Kd = 1–10 µm) and therefore relatively weak. The MDM233–43 peptide alone bound the p53 C-terminal region with a still weaker binding affinity in the hundred micromolar range (Kd = 121 µm). This implies that, in addition to the region between residues 33 and 43, there are additional contributions of the MDM2 N terminus that participate in the interaction with the p53 C terminus. One such possibility is the presence of a flexible “lid” in the extreme N terminus of MDM2 (residues 16–23) whose phosphorylation was predicted to stabilize Mdm2–p53 interactions (Worrall et al. 2010). We note that this affinity is similar to that of a previously observed secondary binding site between the peptides from the MDM2 acidic region and the p53 core domain (Kd = 10–100 µM) (Shimizu et al. 2002; Wallace et al. 2006; Yu et al. 2006). Speculatively, the low affinity of the p53 C-terminal regions for MDM2 may be essential for allowing modifications of the lysines in this region of the protein, as was discussed in our earlier study (Poyurovsky et al. 2010), or allowing p53 to shift from dimer to tetramer as needed. A tighter interaction between MDM2 and the OD could potentially stabilize the p53 as a dimer and impede conversion of p53 to the tetrameric form that is needed for its nuclear functions.
The binding regions that we characterized on both p53 and Mdm2 harbor numerous hydrophobic residues. Taken together with the facts that the interaction is independent of ionic strength (Supplemental Fig. S9C) and that the MDM2 L38P mutation that reduces hydrophobicity impairs p53 degradation and binding by MDM2 (Fig. 5C,D), we conclude that the association between these two regions is mediated mainly by hydrophobic interactions. Modeling based on structural analyses predicts that both dimers and monomers have the hydrophobic NES exposed, in contrast to the tetramer conformation, where the NES would be masked. It is thus somewhat puzzling that monomeric p53 proteins are not targets of MDM2 in any of the assays that we used (degradation, turnover, or nuclear exclusion) and as others have reported (Kubbutat et al. 1998; Hjerpe et al. 2010; Lang et al. 2014). We can suggest two possible and not necessarily mutually exclusive explanations. First, having only one NES in the p53 monomer might not allow for a sufficiently strong interaction with the MDM2 NTD. Second, the dimer form of p53 may uniquely possess structural features that are required for MDM2 regulation. Favoring this second possibility, our photo-L-methionine cross-linking experiment (Fig. 3D) showed that the dimer form of p53 undergoes a conformational change in the presence of MDM2 that is distinct from that seen with either tetrameric or monomeric forms of p53. Further experiments will be needed to reveal more precisely the nature of the structural differences between different oligomeric states of p53.
Implications of findings that MDM2 preferentially degrades p53 as a dimer
Our work revealed that as a dimer, p53 is degraded mainly via the 20S rather than the 26S proteasome. At first glance, these results may be at variance with the traditional model of MDM2 ubiquitin-mediated proteasomal degradation of p53 through the 26S proteasome. However, evidence has suggested that ubiquitination of p53 may be independent of degradation (O'Keefe et al. 2003) and that ubiquitination does not impair the ability of p53 to form tetramers (Brooks et al. 2007). Our results support the idea of “degradation by default” that was established by the Shaul group (Asher et al. 2006; Asher and Shaul 2006; Tsvetkov et al. 2010), and we believe it to be even more relevant when p53 is a dimer. It is interesting that relatively more efficient degradation by MDM2 via this pathway is unique to dimeric p53 via the 20S proteasome. While seemingly at odds with the prior assumption that p53 monoubiquitination is required for its nuclear export (Li et al. 2003), it was reported that the function of monoubiquitination is to expose the NES within a p53 tetramer (Carter et al. 2007). In that scenario, monoubiquitination would be unnecessary for nuclear export in dimer-forming mutant versions of p53. Monomeric p53 was also shown to undergo ubiquitin-independent degradation by the 20S proteasome, although that process appears to be independent of MDM2 (Hjerpe et al. 2010; Lang et al. 2014).
We also examined the impact of p53 oligomeric status on cellular localization using immunofluorescence, cell fractionation, and treatment with the nuclear export inhibitor LMB. Our results support the model proposed by the Wahl group (Stommel et al. 1999) and later extended by the Lahav group (Gaglia et al. 2013), which suggested that dimeric forms of p53 are present and abundant in nonstressed conditions, and p53 as a dimer would be shuttled from the nucleus to the cytoplasm. We extended their findings in critical ways. In addition to demonstrating preferential degradation of the dimer form of p53 by MDM2, we (1) provided strong evidence for these interactions being an important component of the ability of MDM2 to shuttle p53 from the nucleus to the cytoplasm, (2) identified novel interacting regions in MDM2 and dimeric p53, and (3) provided evidence for a ubiquitin-independent role for MDM2 in degradation of p53 by the 20S proteasome.
Clinical implications of MDM2 degradation of the dimer form of tumor-derived mutant p53 proteins
p53 mutations in many human tumors are frequently of the missense variety and are expressed as full-length proteins. The common “hot spot” mutations that occur with higher frequency than other mutations in many human tumors are located in the DBD, and it is now well established that some of these mutant forms of p53 have oncogenic properties in cell-based assays and in mice (Freed-Pastor and Prives 2012; Muller and Vousden 2014). Such p53 variants are often present at far higher levels than wild-type p53, and their protein levels correlate with the extent to which mutant p53 can promote malignant behavior of cells (Rivlin et al. 2011; Freed-Pastor and Prives 2012; Hanel and Moll 2012; Muller and Vousden 2013; Kim et al. 2015). Our results showed that introducing the dimer-forming mutation into certain mutant p53 proteins causes p53 proteins to be degraded significantly better by MDM2 when compared with tetrameric mutant p53. This finding was extended in assays showing that in the presence of MDM2, the dimeric form of a cancer-derived mutant p53 migrates much less efficiently into an artificial wound in cell cultures. It is interesting that as a dimer, R248Q was not susceptible to increased degradation by MDM2. Why this mutation is not further destabilized is not understood at present. Future studies will be needed to identify those cancer-derived p53 mutations that would benefit by being forced into dimer conformation. Regardless, these results suggest the potential benefit of identifying ways to stabilize or induce the dimer state of mutant p53 as a means to lower its levels and reduce its oncogenic gain of function in cancer cells. Several studies have presented different strategies for shifting the oligomerization equilibrium of p53 toward the tetrameric conformation (for review, see Gabizon and Friedler 2014). We propose that an effort should also be made to shift the equilibrium toward dimeric p53, since some tumor-derived mutant forms of p53 might then be degraded faster by MDM2. Relatedly, a p53 peptide spanning the NES was shown to inhibit p21 expression via heterotetramerization with endogenous p53 protein (Wada et al. 2012).
We used gene-editing technology in two ways. First, we generated U2OS p53−/− cells and found that the results with ectopically expressed proteins were essentially the same as those obtained in the parental p53+/+ cells, thereby eliminating the possibility that the presence of endogenously expressed wild-type p53 was influencing our results. We also generated clones of U2OS cells where the dimer-forming mutant p53 (E343K) is expressed from the endogenous locus. While these cells provided strong evidence that the dimer form of p53 cannot be degraded by the 26S proteasome and instead is likely targeted by the 20S proteasome, they could not be used to show that Mdm2 preferentially degrades this dimeric mutant. The clones of p53 (E343K) that were obtained consistently had a lower ratio of Mdm2 to p53 than parental U2OS cells. This could reflect a partial transcriptional impairment of this dimer-forming mutant p53, and, in fact, a subset of such dimer-forming mutants is transcriptionally impaired in yeast-based assays (Kawaguchi et al. 2005; Gaglia et al. 2013; Fischer et al. 2016). In such cases, MDM2 would likely be underexpressed, leading to higher rather than lower levels of mutant p53 (and even wild-type p53 if cells are heterozygous). Our results with ectopically expressed mutant forms of p53 are therefore particularly relevant, as they allowed us to identify and follow the connection between MDM2 and dimeric p53 without the added complexity of the MDM2–p53 feedback loop.
Relevantly, patients with LFS that harbors germline mutations in p53 are highly cancer-prone (Malkin 2011), and, whereas ∼72% of their missense mutations are located in the DBD, 19% of LFS mutations occur in the p53 OD (Kamada et al. 2016). Given the relative length of the OD (∼30 residues) compared with the DBD (∼200 residues), the frequency of LFS p53 mutations relative to the length of the domain in the DBD and the OD is almost the same (Kamada et al. 2011, 2016). Indeed, the fact that LFS mutations occur within the p53 oligomerization region with considerable frequency indicates that transcriptional impairment and deregulated MDM2 circuitry are likely contributors to their pathology. We hope in the future to address such issues, as they are likely to provide another glimpse into the complexity of p53.
Materials and methods
Plasmids, drugs, cells, and antibodies
Plasmids
Flag-MDM2, HA-p53, HA-ubiquitin, His-ubiquitin, and GFP plasmids were described previously (Zhu et al. 2009). Mutations within MDM2 or p53 were introduced using a QuikChange II site-directed mutagenesis kit (Agilent Technologies) and were verified by sequencing (GeneWiz).
Drugs
Cycloheximide (100 µg/mL; Sigma-Aldrich) and 10 µM Nutlin-3 (Sigma-Aldrich) were given to cells for the times indicated. LMB (5 ng/mL; Sigma-Aldrich), 25 µM MG132 (Calbiochem), and 5 µM clasto-lactacystin β-lactone (Sigma-Aldrich) were given to cells 6 h before harvesting.
Antibodies
Antibodies used in this study were anti-Flag (Sigma, M2), anti-GFP (Santa Cruz Biotechnology, B2), anti-HA (Covance, 16B12), anti-MDM2 (a mixture of 3G5, 4B11, and 5B10 hybridomas or Santa Cruz Biotechnology, N-20), anti-p53 (a mixture of 1801 and D01 hybridoma supernatants or Santa Cruz Biotechnology, FL393-G), anti-actin (Sigma, A2066), anti- PARP (Cell Signaling Technology, 9542S), anti-Bim (Cell Signaling Technology, 2933s), anti-CDC25a (Santa Cruz Biotechnology, sc-7389), and anti RPN2 (Bethyl Laboratories, PSMD1 A303851A-T, or Santa Cruz Biotechnology, sc- 166038).
Cells
U2OS osteosarcoma and H1299 lung carcinoma cell lines were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) at 37°C.
To generate p53 knockout clones, 7 × 105 U2OS cells were transfected with 2 µg of p53 CRISPR/Cas9 knockout plasmid (Santa Cruz Biotechnologiy) using Lipofectamine 2000 (Thermo Fisher). Two days later, cells were treated with 10 µM Nutlin-3 for 14 d to inhibit proliferation of cells with wild-type p53, thereby enriching for p53 knockout cells as described previously (Malina et al. 2013). Single-cell clones were selected via limiting dilution, and p53 knockout clones were confirmed by Western blotting using FL-393 polyclonal antibody.
U2OS p53 (E343K) mutant cells were generated using a modification of CRISPR/Cas9 genome-editing technology (Lin et al. 2014). A 100-base-pair single-stranded oligodeoxynucleotide (ssODN) flanking the cut site and containing the mutation of interest was used as a donor template (CTCCTCTGTTGCTGCAGATCCGTGGGCGTGAGCGCTTCGAGATGTTCCGTAAGCTGAACGAGGCTTTGGAACTCAAGGATGCCCAGGCTGGGAAGGAGCCA). U2OS cells (2 × 106) were electroporated with 200 pmol of ssODN (IDT), 100 pmol of EnGen Cas9 NLS (New England Biolabs), and 120 pmol of single-guide RNA (Synthego) using an Amaxa Nucleofector II device. Single-cell clones were selected via limiting dilution, and mutant clones were confirmed by genomic DNA and cDNA sequencing. We obtained one heterozygous (+/E343K; het10) and two homozygous (E343K/E343K; hom 84 and hom 8) p53 CRISPR cell lines. It is important to note for future use that the hom 8 cell lines were somewhat unstable in that we were able to obtain highly reproducible results only with earlier-passaged cells (around passage 17), after which the results became more variable.
Cell-based assays
DNA transfection and immunoblot analysis
Transfections were performed using Lipofectamine 2000 (Invitrogen). In all cases, the pcDNA3 plasmid was added at the appropriate quantities to ensure that equal amounts of total DNA were used for transfecting cells. Where indicated, a GFP construct was included as a transfection and loading control. Old medium was removed, and the transfected cells were lysed with lysis buffer A (10 mM Tris-HCl at pH 7.5, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40 with 50 nM PMSF, and inhibitor cocktail containing 100 µM benzamidine, 300 µg/µL leupeptin, 100 mg/mL bacitracin, 1 mg/mL a2-macroglobulin). Cell lysates were cleared by spinning at 4000 rpm for 10 min at 4°C. Protein concentrations were determined using the Bradford assay (Bio-Rad protein assay, Life Science Research). Equivalent amounts of each transfected and clarified cell lysate were supplemented with protein sample buffer and incubated for 10 min at 95°C, after which samples were loaded onto an 11% polyacrylamide gel and separated using constant voltage. Proteins were transferred to nitrocellulose membranes (Bio-Rad), blocked with PBS containing 0.1% Tween 20 (Sigma-Aldrich) and 5% nonfat dry milk, and probed with the indicated antibodies. Membranes were washed with PBS supplemented with 0.1% Tween 20 prior to the addition of secondary antibodies. In some cases, goat anti-mouse or anti-rabbit conjugated to horseradish-peroxidase (HRP) (Sigma) was used, and membranes were visualized using ECL (GE Healthcare). In other cases, fluorescent green goat anti-mouse (IRDye 800CW, LI-COR Biosciences) and fluorescent red donkey anti-rabbit (IRDye 680LT, LI-COR Biosciences) secondary antibodies were used in conjunction with the Odyssey Imaging System (LI-COR Biosciences).
Transfection of siRNAs
siRNA transfections were performed for 48 h using Lipofectamine RNAiMAX transfection reagent (Life Technologies). The sequences for siRNA directed against luciferase (siLuc) were published previously (Urist et al. 2004). Negative control siRNA (siC) and Rpn2 siRNAs (siRPN2 #1, #2, and #3) were purchased from Ambion Life Technologies (siRNA ID 4390843, s11386, s11387, and s11388) and used at a concentration of 30 nM. For RNAi experiments, U2OS cells were plated at 80% confluence and transfected twice: once with the siRNA and, 24 h later, again with the indicated DNA. Twenty-four hours after the second transfection, cells were harvested, and cell lysates were subjected to immunoblotting with the indicated antibodies.
Immunoprecipitation
U2OS cells were transfected with various Flag-MDM2 or HA-p53 constructs, as indicated. The cells were harvested in lysis buffer A and cleared by centrifugation at 13,000 rpm for 10 min at 4°C. The total protein concentration was measured using the Bio-Rad protein assay (Life Science Research). All of the following steps were performed at 4°C. Equivalent amounts (100–200 µg) of each clarified cell lysate were subjected to immunoprecipitation with 1 µg of the purified antibody as indicated in each experiment for 2 h. Antibodies used included anti-Flag (Sigma, M2) for Flag-MDM2 and anti-HA (Covance, 16B12) for HA-p53 or other anti-p53 antibodies (PAb 240 and Mab 1620) as indicated. Next, 35 µL of protein G beads (GE Healthcare) that were preblocked with bovine serum albumin (BSA) (New England BioLabs) was added for an additional hour. Following three washes with 1 mL of lysis buffer A (without the protease inhibitors), proteins were eluted by adding protein sample buffer and incubated for 10 min at 95°C. Immunoblotting analysis was performed as described above.
Cell fractionation
U2OS cells were transfected with Flag-MDM2 or various HA-p53 constructs as indicated. Cell fractionation was performed using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific). Briefly, transfected cells were harvested with cold PBS and then centrifuged at 13,000g for 30 sec. Cells were washed with PBS and pelleted again by centrifugation at 13,000g for 30 sec. Supernatants were discarded, and 100 µL of ice-cold CER I buffer was added to the cell pellet, which was vortexed vigorously for 15 sec to fully suspend the cell pellet. After incubation for 10 min on ice, 5.5 µL of cold CER II buffer was added to the samples, which were vortexed for 5 sec and then incubated for 1 min on ice followed by another 5 sec of vigorous vortexing. After centrifuging at 16,000g for 5 min, the supernatant was saved as cytoplasmic extract, and the pellet was resuspended in 50 µL of ice-cold NER buffer followed by vortexing vigorously for 15 sec. Samples were incubated on ice and vortexed four times for 15 sec every 10 min. Samples were centrifuged at 16,000g for 10 min, and the supernatant was used as the nuclear extract. Immunoblotting analysis was performed as described above. To determine P-values, the intensity of the p53 proteins of the different variants was analyzed by comparing the cytoplasmic fraction versus the nuclear fraction within each pair.
Protein turnover assay
U2OS cells were transfected with Flag-MDM2 and various HA-p53 constructs as indicated. Twenty-four hours after transfection, cells were treated with 100 µg/mL cycloheximide (Sigma) and harvested at the indicated time points. Cell lysates were subjected to immunoblotting with the indicated antibodies, and band intensities were quantified using Odyssey software (LI-COR Biosciences).
Ubiquitination and degradation assays
U2OS cells were transfected with constructs expressing either Flag-tagged (Flag-Ub) or histidine-tagged (His-Ub) ubiquitin, HA-tagged p53, and MDM2 (not tagged), as indicated. Eighteen hours after transfection, cells were treated with 25 µM MG132 (Calbiochem) for 6 h. Equivalent amounts of clarified cell lysates were immunoprecipitated (as described above) with 1 µg of anti-HA-coupled protein G Sepharose beads (GE Healthcare) followed by immunoblotting with anti-p53 antibody (Sigma, FL393-G) to detect ubiquitinated p53.
Immunofluorescence analysis
U2OS cells grown on coverslips were transfected with Flag-MDM2 and a wild-type or mutant version of HA-tagged p53 as indicated. DNA transfections were performed with FuGENE6 (Promega). Twenty-four hours after transfection, cells were washed twice with 1× PBS and fixed with 4% paraformaldehyde for 1 h. Cells were again washed twice with 1× PBS and permeabilized with PBS/0.5% Triton X-100 for 1.5 min followed by blocking with 0.5% BSA (Sigma) for 30 min at room temperature. Immunofluorescence staining was performed by adding 100 µL of diluted (1:1000) primary antibody solution (rabbit anti-Flag and mouse anti-Ha) to coverslips. After 1 h of incubation at room temperature, coverslips were washed with 1× PBS and then incubated for 1 h with 100 µL of diluted (1:100) secondary antibody (Alexa fluor 488 goat anti-rabbit or Alexa fluor 594 goat anti-mouse antibodies; Life Technologies). Coverslips were washed three times with 1× PBS after each of the above steps and mounted on a microscope slide with VectaShield (mounting medium with DAPI; Vector Laboratories), and images were analyzed by confocal laser-scanning microscopy (Zeiss, LSM 700). Distribution of proteins in nuclei or cytoplasm was analyzed quantitatively in 65 individual cells for each experimental variable in three independent experiments. Cellular distribution of p53 was classified into three different categories: (1) exclusively in the nucleus (N), (2) more in nucleus than in cytoplasm (N > C), and (3) equal or more in the cytoplasm (N ≤ C). To determine P-values, the number of cells in the different classes (N > C, N ≤ C, and N + C) were compared with the number of cells in which p53 was found exclusively in the nucleus (N).
In vivo cross-linking
U2OS or H1299 cells were transfected with the indicated plasmids and lysed with phosphate buffer (PBS, 10% glycerol, 10 mM EDTA, 0.5% NP-40, 0.1M KCl, protease inhibitor mixture) 24 h after transfection. Glutaraldehyde was added to the lysate to final concentrations of 0.05% and 0.1%. After incubating the lysate for 20 min at room temperature, the reactions were stopped by adding 3× loading sample buffer, and the samples were heated for 5 min at 100°C and resolved by 8% SDS-PAGE. Western blot analysis was performed with anti-p53 antibody (Sigma, DO-1/18O1 or FL393-G).
Wound migration assay
U2OS and H1299 cells (1 × 105) were seeded on six-well plates in Complete medium overnight to reach ∼80% confluency and then were transfected with constructs expressing MDM2 and/or p53 variants (core domain tetrameric and dimeric mutants) as indicated. Twenty-four hours later, cultures were washed twice with 1× PBS, and a 200-µL pipette tip was used to produce a straight cell-free “wound.” Each well was washed twice with Complete medium to remove any debris followed by addition of fresh medium supplemented with 2.5 µg/mL mitomycin C (Sigma, M4287) to prevent cell proliferation. Cells were imaged at 0 and 24 h (approximate wound closure time for the control sample). Imaging was done using a phase-contrast microscope (Nikon, Diaphot 3000) with 10× magnification. The distances between the two edges of the scratch were analyzed quantitatively relative to 0 h for three images per biological replicate. To determine P-values, the values of the wound size for cotransfected MDM2 and mutant p53 (175) were calculated with respect to the mutant p53 (175) alone.
Protein chemistry experiments
Protein purification
Insect Sf9 cells were infected with recombinant viruses encoding either Flag-MDM2, Flag-wild-type p53, or Flag-L344A p53. Cells were lysed in lysis buffer B (50 mM Tris-Cl at pH 8.0, 300 mM NaCl, 1% Triton, 10 mM βME, 0.35 mM PMSF, 0.1% aporotomimin, protease inhibitors) and sonicated. Cell debris and chromatin were precipitated by centrifugation at 15,000 rpm for 25 min at 4°C in a SS-34 rotor using a Sorvall centrifuge, and the supernatant was bound to prewashed anti-Flag M2 resin (Sigma-Aldrich, A2220) by shaking for 3 h at 4°C. The bound material was washed extensively with wash buffer A (50 mM Tris-Cl at pH 8.0, 250 mM NaCl, 10% glycerol). Proteins were eluted with wash buffer A supplemented with 0.4 mg/mL Flag peptide (Sigma-Aldrich, F4799) and 2 mM DTT. Proteins were fast-frozen in liquid nitrogen and stored at −80°C until needed. Purified proteins were analyzed by 10% SDS-PAGE along with standard amounts of BSA, and their concentration was determined by densitometry.
ITC
ITC measurements were carried out at 10°C with an isothermal titration calorimeter (MicroCal, VP–ITC 200). All samples were dissolved in 20 mM HEPES (pH 7.4), 150 mM NaCl, and 0.02% NaN3. Twenty aliquots of 2 µL of wild-type p53 CTD293–393 or mutant p53 CTD 293–393/L344A proteins were injected every 4 sec into 0.02 mM MDM2 NTD (10–139) polypeptide. A 180-sec delay between injections was allowed for the system to return to equilibration. Controls included the titration of p53 CTD293–393 proteins into buffer alone, titration of buffer into MDM2 protein, and titration of buffer into buffer solution. The ITC data were analyzed using Origin 7.0 software.
ELISA
ELISA experiments were performed as described (Katz et al. 2008). Briefly, 200 µL of ligand (purified full-length wild-type or mutant E343A p53 proteins) diluted in carbonate buffer to a final concentration of 10 µM was incubated overnight at 4°C on Maxisorp plates (Nunc). After incubation, the supernatant solution was removed, the plates were washed three times with 1× PBS, and then 200 µL of 7% BSA (Sigma) in PBS was added for 2 h at room temperature. After rewashing with 1× PBS, the analyte (full-length MDM2 purified protein) was dissolved in 1× PBS to give the final concentrations as indicated and incubated for 1 h at room temperature. Following three washes with 1× PBS, 200 µL of primary antibody was added to diluted (1:750) anti-MDM2 (rabbit anti-N20) for 1 h at room temperature. Next, after three washes with 200 µL of 1× PBS, diluted (1:2500) secondary antibody (goat HRP-conjugated anti-rabbit) was added for an additional hour at room temperature. The enzymatic activity of HRP was estimated by monitoring the optical density of the product at 490 nm using an ELISA plate reader (Tecan Sunrise). The binding values for the different conditions (wild-type, mutant, and no p53) were plotted as a function of concentration of MDM2. To determine P-value, the area under the curve was calculated for each of the curves (three different conditions) in six different experiments, each with two technical replicates. Next, the ratio of area under the curve with respect to that of wild-type p53 was calculated for each of the curves in each experiment. The fold changes of areas under the curve from six different experiments were averaged to obtain the final mean ± SEM represented in the figures. We then used the nonparametric one-way ANOVA (Kruskal-Wallis) to compute the P-values. Prism software (version 7) was used to perform these calculations.
Peptide array screening
An array consisting of 22 partly overlapping peptides (between 10 and 30 residues) (see Supplemental Table S1) derived from the MDM2 NTD (10–139) was designed based on the secondary and tertiary structures of MDM2 (Protein Data Bank: 1Z1m; Uhrinova et al. 2005). The peptide array was synthesized by Intavis Bioanalytical Instruments AG. Peptide–cellulose conjugates were synthesized and spotted on glass slides (Intavis). The peptide array was screened as described (Katz et al. 2008). The array was immersed for 4 h in blocking solution (BS; 50 mM Tris HCl at pH 7.5, 0.15 M NaCl, 0.05% Tween 20, milk 3.5%) and prewashed three times in TBST (50 mM Tris-HCl at pH 7.5, 0.15 M NaCl, Tween 0.05%). The His-tagged p53 CTD (amino acids 293–393) polypeptide was diluted with blocking solution to a final concentration of 5 µM and incubated with the array overnight at 4°C. Washing steps included two washes for 5 min in 5% BSA and three washes for 5 min in TBST. The binding was detected with anti-His-conjugated HRP antibody (Sigma) using a chemiluminescence blotting substrate (Super Signal reagent, Beit Haemek).
Peptide synthesis and purification
Peptides were synthesized on a Liberty MAPS (microwave-assisted peptide synthesizer; CEM) using standard Fmoc [N-(9-fluorenyl)methoxycarbonyl] chemistry as described (Katz et al. 2008). For peptides without any fluorophore (Trp or Tyr), Trp was added at the peptides’ N termini for UV spectroscopy. Peptides were purified on a Gilson high-performance liquid chromatograph using a reverse-phase C8 semipreparative column (ACE) with a gradient from 5% to 60% acetonitrile in water (both containing 0.001% [v/v] trifluoroacetic acid). Peptides were analyzed using MALDI-TOF mass spectrometry on a Voyager DE-Pro instrument (Applied Biosystems). For fluorescence anisotropy experiments, the peptides were also labeled with fluorescein at their N termini as described (Katz et al. 2008).
Fluorescence anisotropy
Fluorescence anisotropy binding studies were performed in 20 mM HEPES buffer (pH 7.3), 42 mM NaCl, and 5 mM DTT. Measurements were performed at 10°C using a PerkinElmer LS-55 luminescence spectrofluorimeter equipped with a Hamilton Microlab 500 dispenser (Friedler et al. 2002). Fluorescein-labeled peptides derived from MDM2 as indicated were dissolved in the buffer and diluted to a final concentration of 100 nM. The labeled ligand solution (1000 µL) was placed in a cuvette, and the analyte (wild-type or mutant L344A unlabeled p53 CTD293–393 purified protein) was titrated into the labeled ligand in aliquots of 4–10 µL with 30 sec of mixing and 1-min intervals. The total fluorescence and anisotropy were measured after each addition using an excitation wavelength of 480 nm and an emission wavelength of 530 nm. For a simple 1:1 binding reaction, the data were fit to the following single-site model equation:
where r is the measured fluorescence anisotropy value, Δr is the amplitude of the fluorescence anisotropy change from the initial value (peptide only) to the final value (peptide in complex), r0 is the starting anisotropy value corresponding to the free peptide, [P] is the protein concentration, and Kd is the dissociation constant.
UV-induced photo-cross-linking of photo-L-methionine-modified p53 proteins
HCT116 p53-null cells were grown in DME + 10% FBS until ∼80% confluence and transfected with 5 µg of constructs expressing wild-type p53 or E343K or L330A mutant p53 proteins that had an N-terminal Flag-PKA fusion tag. Six hours after transfection, the medium was replaced with DMEM-LM (Thermo Scientific, 30030) supplemented with 10% dialyzed FBS (Thermo Scientific, 89986) and a mixture of 2 mM photo-L-methionine/0.8 mM L L-leucine (Sigma-Aldrich, 61819). Cells were then treated with 10 µM Nutlin-3a and cultured for an additional 20 h. p53 proteins were purified via one-step anti-Flag affinity chromatography according to Laptenko et al. (2015). To obtain unmodified wild-type p53 expressed in the same cell line, we used 30 mg/L natural L-methionine and 105 mg/L L-leucine. Reaction mixtures (40 µL) contained 40 ng of purified unmodified or photo-L-methionine-containing p53 with or without 190–760 ng of purified MDM2 in 1× EMSA buffer (20 mM HEPES at pH 7.8, 25 mM KCl, 10% glycerol, 2 mM MgCl2, 0.1 mM EDTA, 0.02% NP-40, 0.5 mM DTT) supplemented with 100 g/mL BSA. Mixtures were incubated for 20 min at 22°C, transferred into a 96-well plate, and irradiated for 15 min on ice at ∼1-cm distance with UV at 366 nm (Ultraviolet Products, UVSL-58). UV-irradiated samples were separated by 7% TG SDS-PAGE. Cross-linked p53 species were detected by immunoblotting using anti-p53 antibody (FL-393).
Statistical analysis
Statistical analyses were conducted in Excel and Graphoad Prism 7. Results are expressed as mean + SEM. Statistical significance for hypothesis testing was considered as a P-value of 0.05 via two-tailed Student's t-test of unknown variance. Bonferroni correction was used to correct for multiple testing during determination of P-values where appropriate. The following format was used to assign significance based on P-value: P < 0.05 (*), P < 0.01 (**), P < 0.001 (***).
Supplementary Material
Acknowledgments
We are grateful to members of the Prives laboratory for helpful suggestions and comments. We thank Ella Freulich for expert technical assistance. This work was supported by grant CA58316 from the National Cancer Institute.
Author contributions: C.P. conceived and mentored this study. C.K. designed and performed experiments, analyzed data, and wrote the manuscript. A.M.L.-C. and J.H.C. designed and performed experiments. O.L. performed protein purification and in vitro biochemical assays. D.T. generated several U2OS cell lines using CRISPR/Cas technology. Y.Z. gave technical support and conceptual advice. J.-S.N.J.-C. and F.G. performed experiments. A.F. mentored all in vitro studies that were obtained in the early stages of the study at the Hebrew University of Jerusalem. All authors discussed the results and commented on the manuscript.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.304071.117.
References
- Asher G, Shaul Y. 2006. Ubiquitin-independent degradation: lessons from the p53 model. Isr Med Assoc J 8: 229–232. [PubMed] [Google Scholar]
- Asher G, Reuven N, Shaul Y. 2006. 20S proteasomes and protein degradation ‘by default’. Bioessays 28: 844–849. [DOI] [PubMed] [Google Scholar]
- Bargonetti J, Manfredi JJ, Chen XB, Marshak DR, Prives C. 1993. A proteolytic fragment from the central region of P53 has marked sequence-specific DNA-binding activity when generated from wild-type but not from oncogenic mutant P53-protein. Genes Dev 7: 2565–2574. [DOI] [PubMed] [Google Scholar]
- Ben-Nissan G, Sharon M. 2014. Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4: 862–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieging KT, Mello SS, Attardi LD. 2014. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14: 359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T, Petrovich M, Joerger AC, Veprintsev DB. 2009. Conservation of DNA-binding specificity and oligomerisation properties within the p53 family. BMC Genomics 10: 628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks CL, Li M, Gu W. 2007. Mechanistic studies of MDM2-mediated ubiquitination in p53 regulation. J Biol Chem 282: 22804–22815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S, Bischof O, Dejean A, Vousden KH. 2007. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat Cell Biol 9: 428–435. [DOI] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ. 1993. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 13: 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chene P. 2001. The role of tetramerization in p53 function. Oncogene 20: 2611–2617. [DOI] [PubMed] [Google Scholar]
- Clore GM, Omichinski JG, Sakaguchi K, Zambrano N, Sakamoto H, Appella E, Gronenborn AM. 1994. High-resolution structure of the oligomerization domain of p53 by multidimensional NMR. Science 265: 386–391. [DOI] [PubMed] [Google Scholar]
- Collins GA, Goldberg AL. 2017. The logic of the 26S proteasome. Cell 169: 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comel A, Sorrentino G, Capaci V, Del Sal G. 2014. The cytoplasmic side of p53's oncosuppressive activities. FEBS Lett 588: 2600–2609. [DOI] [PubMed] [Google Scholar]
- Cook A, Milner J. 1990. Evidence for allosteric variants of wild-type p53, a tumour suppressor protein. Br J Cancer 61: 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutts AS, Adams CJ, La Thangue NB. 2009. p53 ubiquitination by Mdm2: a never ending tail? DNA Repair (Amst) 8: 483–490. [DOI] [PubMed] [Google Scholar]
- Cubillos-Rojas M, Amair-Pinedo F, Peiro-Jordan R, Bartrons R, Ventura F, Rosa JL. 2014. The E3 ubiquitin protein ligase HERC2 modulates the activity of tumor protein p53 by regulating its oligomerization. J Biol Chem 289: 14782–14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai MS, Lu H. 2004. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem 279: 44475–44482. [DOI] [PubMed] [Google Scholar]
- Davis JR, Mossalam M, Lim CS. 2013. Controlled access of p53 to the nucleus regulates its proteasomal degradation by MDM2. Mol Pharm 10: 1340–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Jensen JP, Ludwig RL, Vousden KH, Weissman AM. 2000. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 275: 8945–8951. [DOI] [PubMed] [Google Scholar]
- Fischer NW, Prodeus A, Malkin D, Gariepy J. 2016. p53 oligomerization status modulates cell fate decisions between growth, arrest and apoptosis. Cell Cycle 15: 3210–3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman DA, Levine AJ. 1998. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol 18: 7288–7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor WA, Prives C. 2012. Mutant p53: one name, many proteins. Genes Dev 26: 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedler A, Hansson LO, Veprintsev DB, Freund SM, Rippin TM, Nikolova PV, Proctor MR, Rudiger S, Fersht AR. 2002. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc Natl Acad Sci 99: 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman PN, Chen X, Bargonetti J, Prives C. 1993. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc Natl Acad Sci 90: 3319–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabizon R, Friedler A. 2014. Allosteric modulation of protein oligomerization: an emerging approach to drug design. Front Chem 2: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia G, Lahav G. 2014. Constant rate of p53 tetramerization in response to DNA damage controls the p53 response. Mol Syst Biol 10: 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaglia G, Guan Y, Shah JV, Lahav G. 2013. Activation and control of p53 tetramerization in individual living cells. Proc Natl Acad Sci 110: 15497–15501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon JV, Greaves R, Iggo R, Lane DP. 1990. Activating mutations in p53 produce a common conformational effect. A monoclonal antibody specific for the mutant form. EMBO J 9: 1595–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer RK, Yu ZK, Maki CG. 2000. The MDM2 RING-finger domain is required to promote p53 nuclear export. Nat Cell Biol 2: 569–573. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. 2009. Cytoplasmic functions of the tumour suppressor p53. Nature 458: 1127–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman SR, Perez M, Kung AL, Joseph M, Mansur C, Xiao ZX, Kumar S, Howley PM, Livingston DM. 1998. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol Cell 2: 405–415. [DOI] [PubMed] [Google Scholar]
- Hanel W, Moll UM. 2012. Links between mutant p53 and genomic instability. J Cell Biochem 113: 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, Torres-Ramos M, Farras R, Hay RT, Rodriguez MS. 2010. Oligomerization conditions Mdm2-mediated efficient p53 polyubiquitylation but not its proteasomal degradation. Int J Biochem Cell Biol 42: 725–735. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. 1991. p53 mutations in human cancers. Science 253: 49–53. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. 1997. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420: 25–27. [DOI] [PubMed] [Google Scholar]
- Itahana Y, Ke H, Zhang Y. 2009. p53 Oligomerization is essential for its C-terminal lysine acetylation. J Biol Chem 284: 5158–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey PD, Gorina S, Pavletich NP. 1995. Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267: 1498–1502. [DOI] [PubMed] [Google Scholar]
- Joerger AC, Fersht AR. 2007. Structure–function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 26: 2226–2242. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. 1995. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378: 206–208. [DOI] [PubMed] [Google Scholar]
- Kamada R, Nomura T, Anderson CW, Sakaguchi K. 2011. Cancer-associated p53 tetramerization domain mutants: quantitative analysis reveals a low threshold for tumor suppressor inactivation. J Biol Chem 286: 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada R, Toguchi Y, Nomura T, Imagawa T, Sakaguchi K. 2016. Tetramer formation of tumor suppressor protein p53: structure, function, and applications. Biopolymers 106: 598–612. [DOI] [PubMed] [Google Scholar]
- Karni-Schmidt O, Lokshin M, Prives C. 2016. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol 11: 617–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz C, Benyamini H, Rotem S, Lebendiker M, Danieli T, Iosub A, Refaely H, Dines M, Bronner V, Bravman T, et al. 2008. Molecular basis of the interaction between the antiapoptotic Bcl-2 family proteins and the proapoptotic protein ASPP2. Proc Natl Acad Sci 105: 12277–12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi T, Kato S, Otsuka K, Watanabe G, Kumabe T, Tominaga T, Yoshimoto T, Ishioka C. 2005. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 24: 6976–6981. [DOI] [PubMed] [Google Scholar]
- Kim MP, Zhang Y, Lozano G. 2015. Mutant p53: multiple mechanisms define biologic activity in cancer. Front Oncol 5: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubbutat MH, Ludwig RL, Ashcroft M, Vousden KH. 1998. Regulation of Mdm2-directed degradation by the C terminus of p53. Mol Cell Biol 18: 5690–5698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. 1996. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274: 948–953. [DOI] [PubMed] [Google Scholar]
- Lang V, Pallara C, Zabala A, Lobato-Gil S, Lopitz-Otsoa F, Farras R, Hjerpe R, Torres-Ramos M, Zabaleta L, Blattner C, et al. 2014. Tetramerization-defects of p53 result in aberrant ubiquitylation and transcriptional activity. Mol Oncol 8: 1026–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laptenko O, Shiff I, Freed-Pastor W, Zupnick A, Mattia M, Freulich E, Shamir I, Kadouri N, Kahan T, Manfredi J, et al. 2015. The p53 C terminus controls site-specific DNA binding and promotes structural changes within the central DNA binding domain. Mol Cell 57: 1034–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Goldberg AL, Mitch WE. 2006. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 17: 1807–1819. [DOI] [PubMed] [Google Scholar]
- Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH. 1994. Solution structure of the tetrameric minimum transforming domain of p53. Nat Struct Biol 1: 877–890. [DOI] [PubMed] [Google Scholar]
- Legros Y, Meyer A, Ory K, Soussi T. 1994. Mutations in p53 produce a common conformational effect that can be detected with a panel of monoclonal antibodies directed toward the central part of the p53 protein. Oncogene 9: 3689–3694. [PubMed] [Google Scholar]
- Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. 2003. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science 302: 1972–1975. [DOI] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. 2014. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3: e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. 2007. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol Cell Biol 27: 1056–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashchuk N, Vousden KH. 2007. Ubiquitination and degradation of mutant p53. Mol Cell Biol 27: 8284–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuasen RG, Welcker M, Bartek J, Lukas J. 2000. Rapid destruction of human Cdc25A in response to DNA damage. Science 288: 1425–1429. [DOI] [PubMed] [Google Scholar]
- Maki CG. 1999. Oligomerization is required for p53 to be efficiently ubiquitinated by MDM2. J Biol Chem 274: 16531–16535. [DOI] [PubMed] [Google Scholar]
- Malina A, Mills JR, Cencic R, Yan Y, Fraser J, Schippers LM, Paquet M, Dostie J, Pelletier J. 2013. Repurposing CRISPR/Cas9 for in situ functional assays. Genes Dev 27: 2602–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D. 2011. Li-fraumeni syndrome. Genes Cancer 2: 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi JJ. 2010. The Mdm2–p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 24: 1580–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko ND, Moll UM. 2014. Mitochondrial death functions of p53. Mol Cell Oncol 1: e955995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateu MG, Fersht AR. 1998. Nine hydrophobic side chains are key determinants of the thermodynamic stability and oligomerization status of tumour suppressor p53 tetramerization domain. EMBO J 17: 2748–2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. 1992. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69: 1237–1245. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. 1995. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378: 203–206. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. 2013. p53 mutations in cancer. Nat Cell Biol 15: 2–8. [DOI] [PubMed] [Google Scholar]
- Muller PA, Vousden KH. 2014. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25: 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls CD, McLure KG, Shields MA, Lee PW. 2002. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J Biol Chem 277: 12937–12945. [DOI] [PubMed] [Google Scholar]
- Nie L, Sasaki M, Maki CG. 2007. Regulation of p53 nuclear export through sequential changes in conformation and ubiquitination. J Biol Chem 282: 14616–14625. [DOI] [PubMed] [Google Scholar]
- Ofir-Rosenfeld Y, Boggs K, Michael D, Kastan MB, Oren M. 2008. Mdm2 regulates p53 mRNA translation through inhibitory interactions with ribosomal protein L26. Mol Cell 32: 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Keefe K, Li H, Zhang Y. 2003. Nucleocytoplasmic shuttling of p53 is essential for MDM2-mediated cytoplasmic degradation but not ubiquitination. Mol Cell Biol 23: 6396–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. 1993. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 362: 857–860. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol 2: a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrales A, Ranjan A, Iyer SV, Padhye S, Weir SJ, Roy A, Iwakuma T. 2016. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat Cell Biol 18: 1233–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky MV, Katz C, Laptenko O, Beckerman R, Lokshin M, Ahn J, Byeon IJ, Gabizon R, Mattia M, Zupnick A, et al. 2010. The C terminus of p53 binds the N-terminal domain of MDM2. Nat Struct Mol Biol 17: 982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan S, Huang F, Fersht AR. 2011. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res 39: 2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivlin N, Brosh R, Oren M, Rotter V. 2011. Mutations in the p53 tumor suppressor gene: important milestones at the various steps of tumorigenesis. Genes Cancer 2: 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Desterro JM, Lain S, Lane DP, Hay RT. 2000. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol 20: 8458–8467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth J, Dobbelstein M, Freedman DA, Shenk T, Levine AJ. 1998. Nucleo-cytoplasmic shuttling of the hdm2 oncoprotein regulates the levels of the p53 protein via a pathway used by the human immunodeficiency virus rev protein. EMBO J 17: 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. 1998. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 12: 2831–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Nie L, Maki CG. 2007. MDM2 binding induces a conformational change in p53 that is opposed by heat-shock protein 90 and precedes p53 proteasomal degradation. J Biol Chem 282: 14626–14634. [DOI] [PubMed] [Google Scholar]
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. 2000. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 14: 289–300. [PMC free article] [PubMed] [Google Scholar]
- Shimizu H, Burch LR, Smith AJ, Dornan D, Wallace M, Ball KL, Hupp TR. 2002. The conformationally flexible S9-S10 linker region in the core domain of p53 contains a novel MDM2 binding site whose mutation increases ubiquitination of p53 in vivo. J Biol Chem 277: 28446–28458. [DOI] [PubMed] [Google Scholar]
- Soussi T, Beroud C. 2001. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat Rev Cancer 1: 233–240. [DOI] [PubMed] [Google Scholar]
- Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM. 1999. A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking. EMBO J 18: 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao W, Levine AJ. 1999. Nucleocytoplasmic shuttling of oncoprotein Hdm2 is required for Hdm2-mediated degradation of p53. Proc Natl Acad Sci 96: 3077–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, Van Pelt CS, Lozano G. 2008. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22: 1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P, Reuven N, Prives C, Shaul Y. 2009. Susceptibility of p53 unstructured N terminus to 20 S proteasomal degradation programs the stress response. J Biol Chem 284: 26234–26242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov P, Reuven N, Shaul Y. 2010. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ 17: 103–108. [DOI] [PubMed] [Google Scholar]
- Uhrinova S, Uhrin D, Powers H, Watt K, Zheleva D, Fischer P, McInnes C, Barlow PN. 2005. Structure of free MDM2 N-terminal domain reveals conformational adjustments that accompany p53-binding. J Mol Biol 350: 587–598. [DOI] [PubMed] [Google Scholar]
- Urist M, Tanaka T, Poyurovsky MV, Prives C. 2004. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev 18: 3041–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408: 307–310. [DOI] [PubMed] [Google Scholar]
- Vojtesek B, Dolezalova H, Lauerova L, Svitakova M, Havlis P, Kovarik J, Midgley CA, Lane DP. 1995. Conformational changes in p53 analysed using new antibodies to the core DNA binding domain of the protein. Oncogene 10: 389–393. [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]
- Wada J, Kamada R, Imagawa T, Chuman Y, Sakaguchi K. 2012. Inhibition of tumor suppressor protein p53-dependent transcription by a tetramerization domain peptide via hetero-oligomerization. Bioorg Med Chem Lett 22: 2780–2783. [DOI] [PubMed] [Google Scholar]
- Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. 2006. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell 23: 251–263. [DOI] [PubMed] [Google Scholar]
- Warnock LJ, Knox A, Mee TR, Raines SA, Milner J. 2008. Influence of tetramerisation on site-specific post-translational modifications of p53: comparison of human and murine p53 tumor suppressor protein. Cancer Biol Ther 7: 1481–1489. [DOI] [PubMed] [Google Scholar]
- Wehmer M, Sakata E. 2016. Recent advances in the structural biology of the 26S proteasome. Int J Biochem Cell Biol 79: 437–442. [DOI] [PubMed] [Google Scholar]
- Weinberg RL, Veprintsev DB, Fersht AR. 2004. Cooperative binding of tetrameric p53 to DNA. J Mol Biol 341: 1145–1159. [DOI] [PubMed] [Google Scholar]
- Wiggins CM, Tsvetkov P, Johnson M, Joyce CL, Lamb CA, Bryant NJ, Komander D, Shaul Y, Cook SJ. 2011. BIMEL, an intrinsically disordered protein, is degraded by 20S proteasomes in the absence of poly-ubiquitylation. J Cell Sci 124: 969–977. [DOI] [PubMed] [Google Scholar]
- Worrall EG, Worrall L, Blackburn E, Walkinshaw M, Hupp TR. 2010. The effects of phosphomimetic lid mutation on the thermostability of the N-terminal domain of MDM2. J Mol Biol 398: 414–428. [DOI] [PubMed] [Google Scholar]
- Xiong S. 2013. Mouse models of Mdm2 and Mdm4 and their clinical implications. Chin J Cancer 32: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas DP, Stephen CW, Lane DP. 2001. Cocompartmentalization of p53 and Mdm2 is a major determinant for Mdm2-mediated degradation of p53. Exp Cell Res 270: 66–77. [DOI] [PubMed] [Google Scholar]
- Xu J, Zhou X, Wang J, Li Z, Kong X, Qian J, Hu Y, Fang JY. 2013. RhoGAPs attenuate cell proliferation by direct interaction with p53 tetramerization domain. Cell Rep 3: 1526–1538. [DOI] [PubMed] [Google Scholar]
- Yu ZK, Geyer RK, Maki CG. 2000. MDM2-dependent ubiquitination of nuclear and cytoplasmic P53. Oncogene 19: 5892–5897. [DOI] [PubMed] [Google Scholar]
- Yu GW, Rudiger S, Veprintsev D, Freund S, Fernandez-Fernandez MR, Fersht AR. 2006. The central region of HDM2 provides a second binding site for p53. Proc Natl Acad Sci 103: 1227–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Poyurovsky MV, Li Y, Biderman L, Stahl J, Jacq X, Prives C. 2009. Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol Cell 35: 316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.