

Abstract
BACKGROUND
In contrast to lung cancer, few precision treatments are available for colorectal cancer (CRC). One rapidly emerging treatment target in CRC is ERBB2 (human epidermal growth factor receptor 2 [HER2]). Oncogenic alterations in HER2, or its dimerization partner HER3, can underlie sensitivity to HER2‐targeted therapies.
METHODS
In this study, 8887 CRC cases were evaluated by comprehensive genomic profiling for genomic alterations in 315 cancer‐related genes, tumor mutational burden, and microsatellite instability. This cohort included both colonic (7599 cases; 85.5%) and rectal (1288 cases; 14.5%) adenocarcinomas.
RESULTS
A total of 569 mCRCs were positive for ERBB2 (429 cases; 4.8%) and/or ERBB3 (148 cases; 1.7%) and featured ERBB amplification, short variant alterations, or a combination of the 2. High tumor mutational burden (≥20 mutations/Mb) was significantly more common in ERBB‐mutated samples, and ERBB3‐mutated CRCs were significantly more likely to have high microsatellite instability (P<.002). Alterations affecting KRAS (27.3%) were significantly underrepresented in ERBB2‐amplified samples compared with wild‐type CRC samples (51.8%), and ERBB2‐ or ERBB3‐mutated samples (49.0% and 60.8%, respectively) (P<.01). Other significant differences in mutation frequency were observed for genes in the PI3K/MTOR and mismatch repair pathways.
CONCLUSIONS
Although observed less often than in breast or upper gastrointestinal carcinomas, indications for which anti‐HER2 therapies are approved, the percentage of CRC with ERBB genomic alterations is significant. Importantly, 32% of ERBB2‐positive CRCs harbor short variant alterations that are undetectable by routine immunohistochemistry or fluorescence in situ hybridization testing. The success of anti‐HER2 therapies in ongoing clinical trials is a promising development for patients with CRC. Cancer 2018;124:1358‐73. © 2018 Foundation Medicine, Inc. Cancer published by Wiley Periodicals, Inc. on behalf of American Cancer Society.
Keywords: colorectal adenocarcinoma; comprehensive genomic profiling; ERBB2, ERBB3; human epidermal growth factor receptor 2 (HER2); lapatinib; microsatellite instability; pertuzumab; trastuzumab; tumor mutational burden
Short abstract
Greater than 6% of colorectal cancer cases harbor activating alterations in ERBB2 or ERBB3, representing a significant population of patients who may benefit from therapies targeting human epidermal growth factor receptor 2 (HER2) and the ERBB pathway. The success of anti‐HER2 therapies in ongoing clinical trials is a promising development for patients with colorectal cancer.
INTRODUCTION
Altered human epidermal growth factor receptor 2 (HER2) signaling caused by genomic amplification of ERBB2 or mutations is oncogenic and has been observed in multiple cancer types.1, 2, 3 Amplification of wild‐type (nonmutated) ERBB2 is observed in 15% to 20% of breast carcinomas and a similar proportion of gastric and gastroesophageal junction adenocarcinomas.4, 5, 6 This observation led to the development of therapeutic antibodies targeting this receptor, such as trastuzumab, pertuzumab, and ado‐trasuzumab emtansine, as well as pan‐ERBB small molecule inhibitors, such as lapatinib or afatinib.7, 8, 9, 10, 11 To identify patients for whom anti‐HER2 therapy is predicted to be most beneficial, fluorescence in situ hybridization (FISH) to evaluate ERBB2 amplification and immunohistochemistry (IHC) to test for HER2 protein overexpression are routinely performed as part of the standard clinical care for breast and upper gastrointestinal tract adenocarcinomas.4, 5 In addition to copy number changes in ERBB2, genomic sequencing studies have identified missense mutations and small indels within the kinase domain of HER2 in approximately 2% of lung cancers.12, 13, 14 Activating ERBB2 mutations have also been found in approximately 2% of breast cancers and are enriched 10‐fold in invasive lobular carcinomas that harbor concurrent CDH1 mutations.15, 16, 17 Similarly, activating extracellular domain mutations have been observed in approximately 40% of micropapillary urothelial carcinomas.18 Extensive preclinical studies have demonstrated that these mutations are oncogenic and sensitive to inhibitors targeting HER2, and that targeting ERBB2 mutations affecting either the kinase or extracellular domains has shown efficacy in a wide variety of tumor types.11, 19, 20, 21, 22, 23, 24, 25 Alterations in the HER2 dimerization partner HER3, encoded by ERBB3, can also activate HER2 signaling and underlie sensitivity to targeted therapies.11, 26, 27, 28
Recent studies of ERBB2 amplification and sequence mutations in colorectal cancer (CRC) suggest that HER2 is a therapy target in this disease,29, 30, 31, 32, 33 in addition to being a mechanism of resistance to epidermal growth factor receptor (EGFR)‐targeted therapies such as cetuximab and panitumumab.34, 35, 36, 37, 38 Similarly, reports of high‐level ERBB3 amplification being a negative prognostic factor within the context of CRC suggest that HER3 may also be a target in this tumor type.27, 39, 40 These studies encourage continued research into the effects and prevalence of ERBB alterations in patients with recurrent and metastatic CRC, as well as their importance for treatment. Data from the HERACLES36 and MyPathway41 studies demonstrate objective response rates of 30% to 38% for patients with HER2‐overexpressing CRC who are treated with trastuzumab plus lapatinib or trastuzumab plus pertuzumab, respectively. In the following study of nearly 9000 clinically advanced and metastatic CRC (mCRC) cases, the relative frequencies of ERBB amplification and sequence alterations were evaluated and evidence of the clinical efficacy of anti‐HER2 targeted therapies in ERBB‐driven mCRC presented.
MATERIALS AND METHODS
Comprehensive genomic profiling was performed for 8887 consecutive cases of primarily recurrent CRC, refractory CRC, and mCRC during the course of routine clinical care. Approval for the current study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act (HIPAA) waiver of authorization, was obtained from the Western Institutional Review Board (protocol no. 20152817). The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin‐stained slides and all samples forwarded for DNA extraction contained a minimum of 20% tumor nuclei.
The sequencing methods used for comprehensive genomic profiling, including validation of copy number and variant calling affecting ERBB2, have been described in detail elsewhere.42 Sample processing and sequencing analysis was performed in a laboratory accredited under the Clinical Laboratory Improvement Amendments (CLIA) and by the College of American Pathologists (CAP). In brief, samples undergo pathologist review to ensure sufficient tumor material (minimum 20% tumor nuclei) and to resolve any conflicts with the provided histological description. From a minimum of 40 microns for each sample provided as formalin‐fixed, paraffin‐embedded tissue blocks, at least 50 ng of DNA was extracted. The samples were assayed using adaptor‐ligation and hybrid capture next‐generation sequencing (FoundationOne; Foundation Medicine, Cambridge, Massachusetts) for all coding exons from 287 (version 1) or 315 (version 2) cancer‐related genes, plus select introns from 19 (version 1) or 28 (version 2) genes frequently rearranged in cancer (see Supporting Information Tables 1 and 2). Sequencing of captured libraries was performed using Illumina HiSeq technology (Illumina, San Diego, California) to a mean exon coverage depth of >500×, and resultant sequences were analyzed using both an algorithmic pipeline and manual curation for base substitutions, small insertions or deletions, copy number alterations (amplifications and homozygous deletions), and select gene fusions, as previously described.42 Clinically relevant genomic alterations were defined as alterations that are targetable by anticancer drugs currently available on the market or in registered clinical trials. Germline variants documented in the dbSNP database (dbSNP142; http://www.ncbi.nlm.nih.gov/SNP/), with ≥ 2 counts in the ExAC database (http://exac.broadinstitute.org/), or recurrent variants of unknown significance that were predicted by an internally developed algorithm to be germline were removed, with the exception of known driver events.42 Confirmed somatic alterations deposited in the Catalog of Somatic Mutations in Cancer (COSMIC v62) were highlighted as biologically significant.43 All inactivating events (ie, truncating mutations and deletions) in known tumor suppressor genes were also called as significant. To maximize mutation‐detection accuracy (sensitivity and specificity) in impure clinical specimens, the test was optimized and validated to detect base substitutions at a ≥5% mutant allele frequency, indels with a ≥10% mutant allele frequency with ≥99% accuracy, and fusions occurring within baited introns/exons with > 99% sensitivity.42
Table 1.
Clinical and Genomic Characteristics of ERBB2‐ and ERBB3‐mutated mCRC
| All mCRC | ERBB2 Positive | ERBB2 Positive | ERBB3 Positive | Cooccurring ERBB2/3 | ||
|---|---|---|---|---|---|---|
| Amplification | Short Variants | Amp + SV | ||||
| No. of cases | ||||||
| Total | 8887 | 251 (2.8%) | 135 (1.5%) | 35 (0.4%) | 140 (1.6%) | 8 (0.1%) |
| Colonic CRC | 7599 | 215 (2.8%) | 112 (1.5%) | 28 (0.4%) | 113 (1.5%) | 7 (0.1%) |
| Rectal CRC | 1288 | 36 (2.8%) | 23 (1.8%) | 7 (0.5%) | 27 (2.1%) | 1 (<0.1%) |
| Sample site | ||||||
| Colorectal | 4660 | 124 | 79 | 21 | 64 | 4 |
| Distant | 4176 | 124 | 55 | 14 | 74 | 4 |
| Stage | ||||||
| IV | 100% | 100% | 100% | 100% | 100% | 100% |
| Patient demographics | ||||||
| Median age (range), y | 56 (8‐96) | 54 (22‐88) | 59 (31‐79) | 57 (29‐87) | 54 (14‐83) | 53 (46‐80) |
| Sex | ||||||
| Female | 45% | 43% | 41% | 46% | 39% | 50% |
| Male | 55% | 57% | 59% | 54% | 60% | 50% |
| ERBB mutation type | ||||||
| Amplification | NA | 251 | ‐ | ‐ | 2 | 0 |
| Short variant | NA | ‐ | 135 | ‐ | 138 | 8 |
| Amp + SV | NA | ‐ | ‐ | 35 | 0 | 0 |
| Global mutation metrics | ||||||
| TMB (mut/Mb) | ||||||
| Range | 0‐854.1 | 0‐230.6 | 0‐230.6 | 0‐10.1 | 0‐854.1 | 6.3‐126.1 |
| Median | 3.8 | 3.6 | 5.4 | 3.8 | 5.4 | 44.2 |
| <6 mut/Mb | 6294 (70.8%) | 179 (71.3%) | 68 (50.4%) | 27 (77.1%) | 79 (56.4%) | 0 (0.0%) |
| 6‐20 mut/Mb | 2173 (24.5%) | 72 (28.7%) | 38 (28.1%) | 8 (22.9%) | 36 (25.7%) | 2 (25.0%) |
| ≥20 mut/Mb | 420 (4.7%) | 0 (0%) | 29 (21.5%) | 0 (0%) | 25 (17.9%) | 6 (75.0%) |
| P | ‐ | <<.0005 | <.0001 | NS | <<.0001 | <<.0001 |
| MSI | ||||||
| No. of cases evaluated | 5899 | 171 | 77 | 24 | 83 | 5 |
| Stable | 5389 (91.4%) | 169 (98.8%) | 64 (83.1%) | 24 (100%) | 69 (83.1%) | 1 (20%) |
| Ambiguous | 103 (1.7%) | 2 (1.2%) | 1 (1.3%) | 0 (0%) | 1 (1.2%) | 1 (20%) |
| High | 407 (6.9%) | 0 (0%) | 12 (15.6%) | 0 (0%) | 12 (14.5%) | 3 (60%) |
| P | ‐ | <.005 | <.005 | NS | <.05 | <<.0001 |
Abbreviations: Amp, amplification; mCRC, metastatic colorectal cancer; MSI, microsatellite instability; NA, not applicable, NS, not significant; mut/Mb, mutations per megabase; TMB, tumor mutational burden; SV, short variant.
Samples in the cooccurring column had both ERBB2 and ERBB3 alterations, whereas other samples had only ERBB2 or ERBB3 alterations. Sample site was defined as colorectal for the colon or rectum and distant for all others; a subset of samples did not have the exact sample site defined. Significance values for TMB and MSI were calculated by the chi‐square test and compared the distribution of samples positive for a given alteration type with the distribution for all other samples in the data set.
Table 2.
Short Variant Alterations Observed in ERBB2‐Mutated and ERBB3‐Mutated mCRC
| ERBB2 | ||||
|---|---|---|---|---|
| Mutation Type | Alteration | Count | Cases With Multiple Alterations | |
| SV Only | Amplification | |||
| Missense | N=181 | |||
| ECD | ||||
| P122L | 1 | |||
| E265K | 1 | |||
| G292R | 1 | |||
| S310F | 19 | 6 | ||
| S310Y | 8 | 1 | 1 | |
| L313V | 1 | |||
| TM | ||||
| V659E | 1 | 1 | ||
| G660D | 2 | 1 | ||
| S653C | 1 | 1 | ||
| JM | ||||
| R678Q | 44 | 1 | 3 | |
| KD | ||||
| T733I | 1 | 1 | ||
| L755S | 12 | 3 | 1 | |
| I767M | 2 | |||
| D769H | 1 | |||
| D769N | 2 | |||
| D769Y | 6 | 3 | 1 | |
| V773M | 2 | |||
| G776S | 1 | 1 | ||
| G776V | 6 | |||
| V777L | 19 | 2 | 9 | |
| V777M | 4 | |||
| V842I | 31 | 4 | 2 | |
| T862A | 11 | 1 | 2 | |
| H878Y | 2 | 1 | ||
| R896C | 1 | |||
| R896H | 1 | |||
| Truncationa | N=5 | |||
| A1232fs*25+ | 2 | 1 | ||
| G1189fs*9 | 1 | |||
| P1170fs*88+ | 1 | |||
| Q1136fs*5 | 1 | |||
| Indel | N=1 | |||
| P780_Y781insGSP | 1 | |||
| Deletion exon 16 | N=2 | |||
| Splice site 1899‐59_1945del106 | 1 | |||
| Deletion exon 16 | 1 | |||
| ERBB3 | ||||
|---|---|---|---|---|
| Mutation Type | Alteration | Count | Cases with Multiple SV | |
| Missense | N=154 | |||
| ECD | ||||
| M60K | 7 | 1 | ||
| M91I | 1 | |||
| R103H | 2 | 1 | ||
| V104L | 17 | |||
| V104M | 35 | 2 | ||
| N126K | 1 | |||
| A232V | 17 | 2 | ||
| A245V | 2 | |||
| R258H | 2 | |||
| G284R | 26 | 1 | ||
| D297Y | 14 | |||
| K329E | 4 | |||
| E332K | 3 | |||
| T355A | 1 | |||
| T355I | 1 | 1 | ||
| Y464C | 1 | |||
| G582V | 1 | 1 | ||
| KD | N=13 | |||
| S846I | 5 | 1 | ||
| E928G | 8 | 2 | ||
| Other | N=5 | |||
| A1023T | 1 | 1 | ||
| S1049G | 1 | |||
| P1212S | 3 | |||
Abbreviations: ECD, extracellular domain; JM, juxtamembrane region; KD, kinase domain; mCRC, metastatic colorectal cancer; SV, short variant; TM, transmembrane domain.
Frameshift alterations are as follows: first amino acid and position changed, fs* to note variant type and termination codon, position of termination codon relative to first amino acid changed. A plus sign (+) indicates that no termination codon was observed in the new frame before the end of the original coding sequence.
Each tumor sample is analyzed alongside an internally validated mixture of 10 heterozygous diploid HAPMAP control samples, which custom algorithms use to normalize the sequence coverage distribution across baited targets. Normalized coverage data for exonic, intronic, and single‐nucleotide polymorphism (SNP) targets accounting for stromal admixture are plotted on a logarithmic scale and minor allele SNP frequencies are concordantly plotted across the genome. Further cluster groupings of targets and minor allele SNPs are used to define upper and lower bounds of genomic segments. Empirical Bayesian algorithms use a distribution of parameters including purity and base ploidy and probability matrices are derived using different statistical sampling methodologies to fit these data and generate copy number alteration variant calls; all computational models are reviewed by expert analysts for each sample. Given that each copy number model is dynamically generated for each individual sample, credibility and confidence intervals vary with sample data; however, copy number calling achieves high performance (sensitivity was 99% with positive predictive value >99%) within a range of 20% to 75% tumor content, as previously described.42
Previous studies have demonstrated high levels of concordance between the current method of detecting ERBB2 amplification and FISH.42 In that study, 2 cohorts of breast carcinomas were shown to have 100% (42 of 42 cases) and 97% (29 of 30 cases) concordance with FISH results.42 Amplification of ERBB2 as described here includes the detection of ≥5 copies of ERBB2 above the overall ploidy of the tumor sample.
Alteration nomenclature in general follows the recommendations of the Human Genome Variation Society.44 Briefly, frameshift alterations are described as follows: first amino acid changed, position of first amino acid change, fs to designate frame shift variant and * to designate termination codon, position of termination site relative to first amino acid changed. A plus sign is used to designate that no termination site is encountered in that frame before the end of the normally encoded protein sequence. For example, A1232fs*25 + indicates the initiation of a frame shift event at alanine 1232, with 25 amino acids of novel sequence before the normal termination codon of the protein (amino acid 1255). No termination codon is observed in this particular sequence.
Tumor mutational burden (TMB) was determined on 0.83 to 1.14 megabase (Mb) of sequenced DNA using a mutation burden estimation algorithm that, based on the genomic alterations detected, extrapolates to the genome as a whole.45 For purposes of mutation burden estimation, all coding short variant alterations (SV) (base substitutions and indels), including synonymous alterations, are counted. Subtracted from this number are functionally oncogenic or germline alterations, as defined below. Germline alterations are those listed in the dbSNP database (http://www.ncbi.nlm.nih.gov/SNP), those with ≥ 2 counts in the ExAC database (http://exac.broadinstitute.org), or those predicted by a somatic‐germline zygosity algorithm to be germline in the specimen being assessed (unpublished data). Functionally oncogenic mutations are those occurring as known somatic alterations in the COSMIC database (http://cancer.sanger.ac.uk/cosmic) or with likely functional status (disruptive alterations in tumor suppressor genes). Finally, to calculate the mutation burden per Mb (mut/Mb), the total number of relevant mutations is divided by the coding region target territory of the test (0.83 Mb for version 1 and 1.14 Mb for version 2). High TMB was defined in this study as ≥20 mutations/Mb of sequenced DNA; a subset of those cases are designated as hypermutated and have ≥50 mutations/Mb.
The 114 loci used to evaluate microsatellite instability (MSI) status were selected from a total set of 1897 loci that have adequate coverage on both the version 1 and version 2 bait sets. Among the 1897 microsatellites, the 114 that maximized variability between samples were chosen. Each chosen locus was intronic and had hg19 reference repeat length of 10‐20 base pairs (bp). This range of repeat lengths was chosen such that the microsatellites are long enough to produce a high rate of DNA polymerase slippage, while short enough such that they are well within the 49‐bp read length of next‐generation sequencing to facilitate alignment to the human reference genome. Using the 114 loci, for each sample we calculated the repeat length in each read that spans the locus. We recorded the means and variances of repeat lengths across the reads, forming 228 data points per sample. In a large training set of data from clinical specimens, we then used principal components analysis to project the 228‐dimension data onto a single dimension (the first principal component) that maximizes the data separation, producing a next‐generation sequencing‐based “MSI score.” There was no need to extend beyond the first principal component, because it explained approximately 50% of the total data variance, whereas none of the other principal components explained >4% each. Ranges of the MSI score were assigned MSI‐high, MSI ambiguous, or microsatellite stable. MSI‐low calls are not made because there was no gold‐standard test set, but we presume such samples would significantly overlap with the MSI‐ambiguous category reported here. For samples with low coverage (<250 times the median), a status of MSI unknown is assigned.
RESULTS
The comprehensive genomic profiles from a series of 8887 consecutive cases of mCRC, including both colonic adenocarcinomas (7599 cases; 85.5%) and rectal adenocarcinomas (1288 cases; 14.5%), were evaluated for clinically relevant genomic alterations (Table 1, Supporting Information Table 3) (Figs. 1A‐1E). The distribution of patients with CRC harboring ERBB2/3 alterations was 45% females and 55% males. The median age was 56 years (range, 8‐96 years). Samples positive for ERBB2 amplification and ERBB3 alterations tended to be from younger patients: median age of 54 years (range, 22‐88 years) for patients with ERBB2 amplification; 54 years (range, 14‐83 years) for patients with ERBB3 alterations; 53 years (range, 46‐80 years) for patients with both ERBB2 and ERBB3 alterations; compared with 59 years (range, 31–79 years) for patients with only ERBB2 SV.
Figure 1.

Genes commonly altered in metastatic colorectal cancer (mCRC) and cooccurrence with mutations in ERBB2 or ERBB3. Statistically significant differences in mutation frequencies (P<.05) by the Fisher exact text are indicated with an asterisk; differences without an asterisk were not statistically significant. (A) The frequency of gene mutations in 8887 colonic (denoted by C) and rectal (denoted by R) adenocarcinomas. (B) Genes coaltered with ERBB2 in colonic and rectal mCRCs. (C) Genes coaltered with ERBB3 in colonic adenocarcinomas. (D) Mutation frequencies for genes in the mismatch repair pathway in all samples, ERBB2‐mutated, and ERBB3‐mutated samples for colonic and rectal mCRC. Statistically significant (P<.05) differences between ERBB2‐mutated or ERBB3‐mutated and nonmutated samples are highlighted with an asterisk. (E) Differences in mutation frequencies among samples with ERBB2 amplification (AMP) only, short variants (SV) only, or cooccurring AMP and SV (the statistical significance of observations illustrated in Figure 1E is reported in Table 3). MLH1 indicates MutL homolog 1; MSH2, mutS homolog 2; MSH6, mutS homolog 6.
A total of 569 samples (6.4%) harbored alterations affecting ERBB2 (429 cases; 4.8%), ERBB3 (148 cases; 1.7%), or both ERBB2 and ERBB3 (8 cases; 0.1%) (Fig. 2A). The ERBB2‐positive mCRC cases featured samples with ERBB2 amplification only (251 cases; 58.5%), a SV sequence alteration in ERBB2 (135 cases; 31.5%) (Fig. 2B), or cooccurring SV and amplification alterations in ERBB2 (35 cases; 8.2%). The 8 samples with cooccurring mutations in ERBB2 and ERBB3 (0.1%) harbored only ERBB SV. No activating ERBB2 genomic rearrangements were identified. A total of 189 ERBB2 SV were detected across 178 mCRC samples, represented by 33 different alterations (Table 2) (Fig. 2B). Of these, 180 of 189 (95.8%) were of known activating alterations. The vast majority of ERBB2 SV detected encode missense alterations, although 1 instance of an exon 20 insertion (P780_Y781insGSP) and 2 alterations expected to delete exon 16 were detected (Table 2). The remaining 4 alterations were frameshift mutations located at the C‐terminus of the protein that may affect regulation of HER2 (Table 2) (Fig. 2B). Of the 148 samples harboring ERBB3 alterations, 2 had amplification only (1.4%) and 138 harbored only SV in ERBB3 (93.2%) (Table 2) (Fig. 2A). Of these SV, 110 of 154 cases (71.4%) were of known activating alterations,26 with the remaining 44 instances distributed among suspected activating alterations and somatically recurrent cancer‐related mutations (Table 2) (Fig. 2B).43
Figure 2.
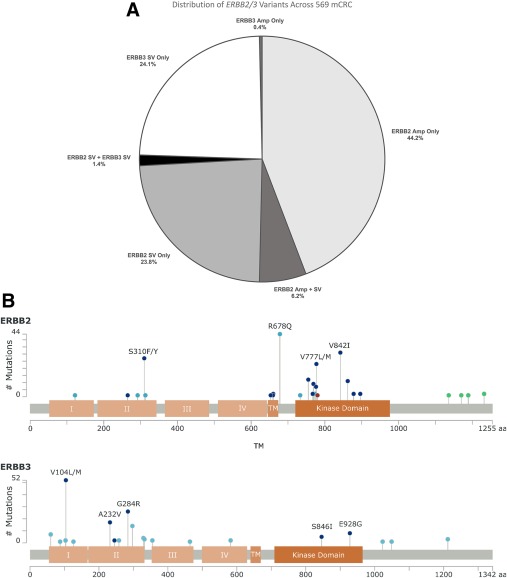
(A) Distribution of the ERBB2/3 variants in 569 metastatic colorectal cancer (mCRC) cases. (B) Alterations most commonly observed in (Top) ERBB2 and (Bottom) ERBB3. Shown here are the extracellular (I‐IV), transmembrane (TM), and kinase (KD) domains of human epidermal growth factor receptor 2 (HER2) and HER3. Dark blue dots represent known activating missense alterations, whereas light blue dots are missense mutations suspected to be activating or recurrent in cancer. Green dots represent truncating frameshift alterations that are expected to remove a regulatory phosphorylation site from the C‐terminus of HER2. Amp indicates amplification; SV, short variants.
Amplification of ERBB2 was defined as ≥ 5 copies of ERBB2 above the average ploidy of the tumor sample. Of the 286 samples with amplification of ERBB2, 284 of 286 had focal amplification (defined as ≤ 20 Mb) of the region surrounding ERBB2 and 2 of 286 samples harbored amplification of a region >20 Mb (45.6 Mb and 55.8 Mb, respectively). The size of the amplified segment containing ERBB2 for each sample is reported in Supporting Information Table 4.
Table 4.
Mutation Frequency Differences Between Samples With ERBB2 Amplification or Short Variants
| ERBB2 Alteration(s) | ||||
|---|---|---|---|---|
| AMP Only | SV Only | Cooccurring AMP and SV | Fisher Exact Test P a | |
| TP53 | 93.2% | 64.3% | 74.3% | <.00001 |
| APC | 72.5% | 72.0% | 62.9% | NS |
| TOP2A | 30.3% | 0.0% | 38.7% | <.00001 |
| KRAS | 17.1% | 49.0% | 11.4% | <.00001 |
| MYC | 12.0% | 9.1% | 8.6% | NS |
| PIK3CA | 10.0% | 23.8% | 5.7% | <.001 |
| CDK12 | 8.9% | 3.5% | 5.7% | NS |
| PTEN | 5.6% | 13.3% | 0.0% | <.02 |
| ARID1A | 5.2% | 16.8% | 8.6% | <.001 |
| RNF43 | 3.2% | 12.7% | 0.0% | <.001 |
| GNAS | 2.8% | 9.8% | 0.0% | <.01 |
| PIK3R1 | 2.4% | 10.5% | 0.0% | <.001 |
| FAM123B | 2.0% | 9.2% | 5.7% | <.01 |
| ATM | 1.6% | 7.7% | 5.7% | <.01 |
| SOX9 | 1.5% | 11.2% | 12.9% | <.001 |
| ASXL1 | 1.2% | 9.2% | 5.7% | <.001 |
| NF1 | 1.2% | 8.4% | 0.0% | <.001 |
| CIC | 0.8% | 9.2% | 2.9% | <.0001 |
| BCORL1 | 0.8% | 8.5% | 5.7% | <.001 |
| TET2 | 0.8% | 6.3% | 0.0% | <.01 |
| MLL2 | 0.4% | 13.4% | 0.0% | <.00001 |
| ERBB3 | 0.0% | 5.6% | 0.0% | <.001 |
| MSH6 | 0.4% | 6.3% | 0.0% | <.001 |
| MSH2 | 0.0% | 9.1% | 0.0% | <.00001 |
| MLH1 | 0.0% | 4.9% | 0.0% | <.001 |
| PMS2 | 0.0% | 4.2% | 0.0% | <.01 |
| Median TMB, mut/Mb | 3.6 | 6.3 | 3.8 | |
| TMB range, mut/Mb | 0‐16.2 | 0‐230.6 | 0‐10.1 | |
| High MSI | 0/172 (0.0%) | 15/88 (17.0%) | 0/24 (0.0%) | <.00001 |
AMP, amplification; APC, adenomatous polyposis coli; ARID1A, AT‐rich interaction domain 1A; ASXL1, additional sex combs‐like 1; ATM, ataxia‐telangiectasia mutated; BCORL1, BCL6 corepressor‐like 1; CDK12, cyclin‐dependent kinase 12; CIC, Capicua transcriptional repressor; MLH1, MutL homolog 1; MLL2, mixed linage leukemia gene 2; MSH2, mutS homolog 2; MSH6, mutS homolog 6; MSI, microsatellite instability; mut/Mb, mutation burden per megabase; NF1, neurofibromatosis type 1; NS, not significant; PIK3CA, phosphatidylinositol 3‐kinase; PIK3R1, phosphoinositide‐3‐kinase regulatory subunit 1; PTEN, phosphatase and tensin homolog; RNF43, ring finger protein 43; SOX9, SRY‐box 9; SV, short variants; TMB, tumor mutational burden; TET2, Tet methylcytosine dioxygenase 2 ; TOP2A, topoisomerase (DNA) II alpha; TP53, tumor protein p53.
Significance of the difference between cases harboring only amplification or only short variant alterations in ERBB2 calculated by the Fisher exact test.
The median TMB in the ERBB‐positive mCRC cases was 4.5 mut/Mb (ERBB2) overall, 3.6 mut/Mb for cases with amplification only, 6.3 mut/Mb for cases with SV only, and 3.8 mut/Mb for cases with both SV and amplification (Table 1). This is compared with 5.4 mut/Mb in ERBB3‐mutated samples and 3.8 mut/Mb for wild‐type mCRC. A TMB score ≥ 20 mut/Mb was significantly more common in the mCRC with ERBB SV, with 24.5% of ERBB2‐mutated, 20.9% of ERBB3‐mutated, and 75% (6 of 8) of ERBB2/3‐mutated mCRC having high TMB (<<.0001) (Table 1). By contrast, it is significant that none of the samples with ERBB2 amplification were high TMB (P<.0005). A significant number of ERBB2‐mutated and ERBB3‐mutated mCRC were MSI‐high, at 17.2% to 18.3%, compared with 6.9% of wild‐type mCRC (P<.05). In addition, 3 of 5 of the ERBB2/3‐mutated mCRC (60%) tested for MSI demonstrated high levels of MSI (P<.004); whereas none of the samples with ERBB2 amplification did (P<.005).
A survey of the genomic alterations observed in the colonic and rectal mCRC cases are shown in Figures 1A to 1E and Table 3. The genes most frequently coaltered with ERBB alterations are shown in Figure 1B to 1E. Similar to ERBB wild‐type samples, tumor protein p53 (TP53), adenomatous polyposis coli (APC), and KRAS were the most frequently altered genes in ERBB‐mutated samples (Figs. 1B‐1C) (Table 3). However, the frequency of KRAS alterations was significantly reduced in ERBB2 amplification samples (17.1%) compared with all mCRC (51.8%) samples, ERBB2 SV only (49.0%), or ERBB3‐mutated mCRC (60.8%) (P<.00001). KRAS alterations were much more likely to cooccur with SV in ERBB2 than amplification (P<.00001). Similar results were observed for NRAS (Table 3).
Table 3.
Significant Differences in Gene Mutation Frequencies Between ERBB2/3‐Mutated Colonic and Rectal mCRC
| Colonic | ||||||
|---|---|---|---|---|---|---|
| Gene | All | ERBB2+ | Fisher Exact Test P a | ERBB3+ | Fisher Exact Test P a | Fisher Exact Test P b |
| TP53 | 74.9% | 82.3% | <.001 | 60.0% | <.001 | <.00001 |
| APC | 75.3% | 72.7% | NS | 80.8% | NS | NS |
| KRAS | 51.6% | 28.2% | <.00001 | 62.5% | <.02 | <.00001 |
| PIK3CA | 18.5% | 14.4% | <.05 | 24.2% | NS | <.02 |
| SMAD4 | 15.4% | 15.7% | NS | 16.7% | NS | NS |
| SOX9 | 10.2% | 6.7% | <.05 | 11.8% | NS | NS |
| FBXW7 | 9.4% | 12.7% | <.05 | 18.3% | <.01 | NS |
| MYC | 9.0% | 11.3% | NS | 4.2% | NS | <.02 |
| BRAF | 8.6% | 3.6% | <.001 | 5.0% | NS | NS |
| PTEN | 8.1% | 8.3% | NS | 14.2% | <.05 | NS |
| ARID1A | 6.8% | 9.1% | NS | 14.2% | <.01 | NS |
| FAM123B | 6.4% | 4.7% | NS | 20.0% | <.00001 | <.00001 |
| BCL2L1 | 5.0% | 3.1% | NS | 0.0% | <.05 | NS |
| RNF43 | 4.4% | 6.7% | <.05 | 12.5% | <.001 | NS |
| NRAS | 4.3% | 1.7% | <.01 | 3.3% | NS | NS |
| MLL2 | 3.6% | 5.0% | NS | 8.3% | <.02 | NS |
| NF1 | 2.5% | 3.6% | NS | 5.8% | <.05 | NS |
| TOP2A | 1.2% | 23.5% | <.00001 | 2.4% | NS | <.00001 |
| CDK12 | 1.0% | 7.0% | <.00001 | 3.3% | <.05 | NS |
| PIK3R1 | 3.6% | 5.8% | <.05 | 8.3% | <.02 | NS |
| ASXL1 | 3.6% | 4.5% | NS | 12.5% | <.0001 | <.01 |
| LRP1B | 3.6% | 4.4% | NS | 9.2% | <.01 | NS |
| MAP2K4 | 2.7% | 4.4% | <.05 | 3.3% | NS | NS |
| BCORL1 | 2.2% | 3.9% | <.05 | 7.5% | <.01 | NS |
| MSH6 | 2.0% | 2.8% | NS | 10.8% | <.00001 | <.0001 |
| MLH1 | 1.1% | 1.9% | NS | 7.5% | <.00001 | <.01 |
| MSH2 | 1.1% | 3.6% | <.001 | 5.0% | <.01 | NS |
| PMS2 | 0.3% | 1.7% | <.001 | 2.5% | <.01 | NS |
| Rectal | ||||||
|---|---|---|---|---|---|---|
| Gene | All | ERBB2+ | Fisher Exact Test P a | ERBB3+ | Fisher Exact Test P a | Fisher Exact Test P b |
| TP53 | 79.6% | 80.6% | NS | 67.9% | NS | NS |
| APC | 77.7% | 65.7% | <.05 | 57.1% | <.02 | NS |
| KRAS | 53.0% | 22.4% | <.00001 | 53.6% | NS | <.01 |
| PIK3CA | 12.9% | 13.4% | NS | 25.0% | NS | NS |
| SMAD4 | 12.3% | 11.9% | NS | 28.6% | <.02 | NS |
| SOX9 | 8.9% | 0.0% | <.02 | 5.0% | NS | NS |
| ARID1A | 8.2% | 10.4% | NS | 21.4% | <.05 | NS |
| TOP2A | 0.6% | 9.8% | <.00001 | 0.0% | NS | NS |
| CDK12 | 0.6% | 6.1% | <.001 | 0.0% | NS | NS |
| PIK3R1 | 2.3% | 0.0% | NS | 10.7% | <.05 | <.05 |
| BCORL1 | 0.9% | 3.0% | NS | 7.1% | <.05 | NS |
| SMAD2 | 3.3% | 6.0% | NS | 14.3% | <.02 | NS |
| FAM123B | 5.2% | 4.5% | NS | 3.6% | NS | NS |
Abbreviations: APC, adenomatous polyposis coli; ARID1A, AT‐rich interaction domain 1A; ASXL1, additional sex combs‐like 1; BCL2L1, Bcl‐2‐like 1; BCORL1, BCL6 corepressor‐like 1; CDK12, cyclin‐dependent kinase 12; FBXW7, F‐box/WD repeat‐containing protein 7; LRP1B, low‐density lipoprotein receptor‐related protein 1B; MAP2K4, mitogen‐activated protein kinase 4; mCRC, metastatic colorectal cancer; MLH1, MutL homolog 1; MLL2, mixed linage leukemia gene 2; MSH2, mutS homolog 2; MSH6, mutS homolog 6; NF1, neurofibromatosis type 1; NS, not significant; PIK3CA, phosphatidylinositol 3‐kinase; PIK3R1, phosphoinositide‐3‐kinase regulatory subunit 1; PTEN, phosphatase and tensin homolog; RNF43, ring finger protein 43; SOX9, SRY‐box 9; TOP2A, topoisomerase (DNA) II alpha; TP53, tumor protein p53.
Significance values for the difference in frequency between all mCRC and ERBB2‐mutated or ERBB3‐mutated samples, respectively.
Significance values for the difference in frequency between ERBB2‐mutated and ERBB3‐mutated samples.
Conversely, alterations in TP53 were far more common in ERBB2 amplification samples (86.7%‐91.7%) compared with ERBB2 SV samples (64.4%‐71.9%), ERBB2 SV plus amplification samples (74.1%‐75.0%), ERBB3‐mutated samples (60.0%‐67.9%), or the cohort in general (74.9%‐79.6%). Coamplification of topoisomerase (DNA) II alpha (TOP2A), which is colocalized with ERBB2 on chromosome 12, was found in 34.5% of samples harboring ERBB2 amplification but not in any samples with only ERBB2 SV (Table 4). An example of cooccurring amplification of ERBB2 and TOP2A is shown in Figure 3. Alterations in BRAF, phosphatidylinositol 3‐kinase (PIK3CA), or SRY‐box 9 (SOX9) were less common in ERBB2 amplification samples, whereas alterations in CDK12 and ring finger protein 43 (RNF43) were more likely (Table 3) (Table 4).
Figure 3.
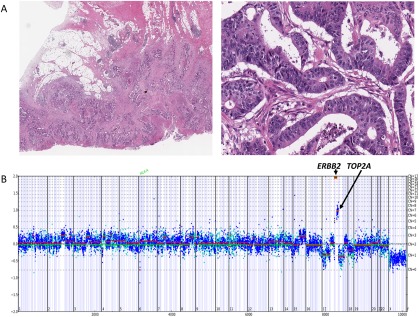
(A) Moderately differentiated adenocarcinoma of the colon in a 72‐year‐old white man. The tumor invaded through the colon wall and involved numerous pericolonic lymph nodes (pathologic classification T3N2A). The patient rapidly developed stage IV disease. (B) The copy number plot below the histologic images demonstrates extremely high‐level amplification of ERBB2 at 60 copies, associated with lower level coamplification of topoisomerase (DNA) II Alpha (TOP2A) at 7 copies. Using comprehensive genomic profiling, this metastatic colorectal cancer also harbored base substitutions in KRAS (G12D), F‐box/WD repeat‐containing protein 7 (FBXW7) (R479Q), adenomatous polyposis coli (APC) (Q1367*), SRY‐box 9 (SOX9) (D274fs*22), and tumor protein p53 (TP53) (C275W).
Significant increases in mutation frequencies in ERBB3‐mutated samples were noted for FAM123B (20.0% vs 4.7‐6.4%; P<.00001) and genes responsible for mismatch repair (mutS homolog 6 [MSH6], mutS homolog 2 [MSH2], MutL homolog 1 [MLH1], and PMS2) (Table 4) (Fig. 1D). Alterations in ASXL1, LRP1B, MLL2, BCORL1, and phosphoinositide‐3‐kinase regulatory subunit 1 (PIK3R1) were more common in ERBB3‐mutated colonic mCRC, whereas SMAD2 and SMAD4 were more often altered in ERBB3‐mutated rectal samples (Table 4).
The frequency of cooccurring mutations in other genes also differed slightly between the colonic and rectal mCRC cases, as shown in Figures 1A to 1C. In the context of ERBB2, the only significant difference in mutation frequency between colonic and rectal samples was for amplification of TOP2A, which was more common in colonic mCRC (23.5% vs 9.8%) (P<.05). For ERBB3, there were striking differences in mutation frequencies for APC (80.8% for colonic vs 57.1% for rectal; P<.02) and FAM123B (20.0% for colonic vs 3.6% for rectal; P<.05).
An example of ERBB2‐amplified mCRC responding to anti‐HER2 targeted therapy is shown in Figure 4.46 This widely disseminated rectal mCRC in a 39‐year‐old woman that was refractory to systemic chemotherapy and multiple metastasectomies responded to a trastuzumab‐based regimen. In case Colonic mCRC 220, a 72‐year‐old woman with CRC that was metastatic to the liver, ERBB2 was amplified to 163 copies (Fig. 5). Also present were the SV alterations FBXW7 M118fs*52, TP53 splice site 672G>A, APC R1450*, and FAM123B R531*. This tumor responded to a combination of trastuzumab and lapatinib for 6 months after prior failure of 4 separate lines of cytotoxic chemotherapy.
Figure 4.
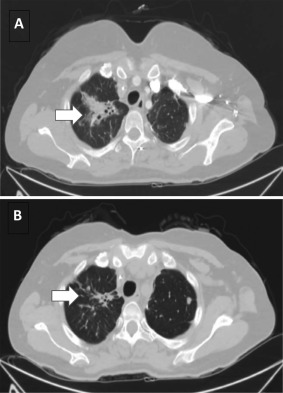
Response of an ERBB2‐amplified metastatic colorectal cancer (mCRC) to antihuman epidermal growth factor receptor 2 (HER2)‐targeted therapy. A 39‐year‐old woman with a pT3N0 rectal adenocarcinoma developed widespread metastatic disease and was treated with systemic chemotherapy and metastasectomies. The mCRC was found to be KRAS wild‐type on routine single‐gene testing and anti‐EGFR therapy with cetuximab was used until disease progression. Comprehensive genomic profiling was performed on a metastasis sample at that time and revealed ERBB2 amplification at 21 copies and a tumor protein p53 (TP53) base substitution. Combination therapy with trastuzumab with a backbone of capecitabine and oxaliplatin was initiated. Treatment with trastuzumab continued for 12 months, after which time the patient's symptoms returned with biomarkers and radiology confirming progressive disease. Representative computed tomography scan image of upper lung metastasis is shown (A) at baseline and (B) after 3 months of trastuzumab and chemotherapy. The arrow indicates significantly regressed tumor burden accounting for improved pulmonary symptoms. The targeted therapy using trastuzumab in combination with cytotoxic chemotherapy maintained a strong response in the patient over a 1‐year course of therapy, reducing tumor burden and improving quality of life.
Figure 5.
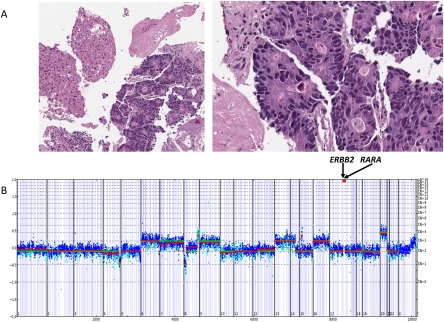
(A) Metastatic colorectal cancer to the liver in a 72‐year‐old woman whose tumor had progressed after 4 separate lines of chemotherapy. (B) Comprehensive genomic profiling revealed both ERBB2 (163 copies) and retinoic acid receptor alpha (RARA) (35 copies) amplification as well as multiple untargetable short variant genetic alterations. This tumor responded clinically for 6 months to a combination of trastuzumab and lapatinib, with a significant decrease in serum carcinoembryonic antigen levels.
DISCUSSION
In December 1998, the simultaneous approvals of the anti‐HER2 targeting antibody trastuzumab and the slide‐based IHC test to select patients for therapy ushered in the era of personalized medicine for solid tumors. The following 15 years then saw 2 major evolutions in anti‐HER2 therapies: 1) the development and approval of oral anti‐HER2 small molecule kinase inhibitors and additional anti‐HER2 antibody therapeutics47; and 2) the expanded indication from breast cancer to upper gastroesophageal carcinomas.48 The expanded use of anti‐HER2 drugs was coordinated by slide‐based tests including IHC, FISH, and chromogenic in situ hybridization (CISH).4 Additional approaches to detect increased HER2 activity were evaluated, such as measuring HER2 mRNA expression levels, but failed to achieve broad clinical usefulness, possibly due to technical limitations.49 More recently, the comprehensive genomic analysis of DNA extracted from formalin‐fixed, paraffin‐embedded samples has been used to survey mCRC, breast, and gastroesophageal cancers for genomic alterations affecting ERBB2 copy number and sequence.29, 30, 34, 50, 51, 52 When combined with the published data from The Cancer Genome Analysis and the COSMIC database, these studies have reported similar frequencies of ERBB2 amplification and short variant mutation.34, 43, 53
CRC continues to be a major cause of morbidity and mortality worldwide in both developed and, to a lesser extent, underdeveloped countries.54 In the United States, CRC is the second most prevalent cancer in males at 724,690 cases and the third most prevalent in women with 727,350 cases.55 The standard‐of‐care treatment for mCRC using multiagent chemotherapy is effective at slowing the progress of the disease, but long‐term remissions are rare and the treatment side effects are often significant.56, 57 This has prompted the development of less toxic targeted therapies for the disease, but progress has been slow.58, 59, 60, 61 Although anti‐EGFR antibody therapeutics have been approved for the treatment of mCRC for several years, the use of these agents has been personalized by determining which patients should not be treated due to predicted resistance rather than identifying individuals significantly likely to benefit from treatment.62 Thus, interest has emerged in finding targeted therapies for which biomarkers can positively predict patient benefit from therapy.58, 59, 60, 61
Based on the current and previously published studies, ERBB2 has now emerged as an important target in mCRC. Given the high incidence worldwide of mCRC, the nearly 5% frequency of ERBB2 genomic alterations makes this an attractive target for future regulatory approval of anti‐HER targeted therapies. Two basic strategies for targeting HER2 in mCRC have been taken: 1) targeting HER2 as the primary driver of the disease when appropriate; and 2) attempting to overcome the resistance to other targeted therapies mediated by ERBB2 genomic alterations.34 Initial studies targeting ERBB2 in mCRC focused exclusively on ERBB2‐amplified cases detected by either FISH or direct sequencing methods.31, 36, 63, 64 Figure 4 provides an example of an ERBB2‐amplified mCRC vigorously responding to anti‐HER2 targeted therapy.46 Various levels of success have been achieved targeting ERBB2 amplification in CRC, and use of both antibody and small molecule treatments has been described.31, 36, 46, 63, 64, 65 To our knowledge, tyrosine kinase inhibitors such as afatinib and lapatinib have yielded limited clinical efficacy as monotherapies in patients with mCRC, suggesting antibody therapeutics or combination therapies may be more beneficial in this tumor type.66, 67, 68 In one report of a widely disseminated, ERBB2‐amplified mCRC, various combinations of trastuzumab, trastuzumab‐DM1 and pertuzumab achieved prolonged patient response and disease control.69
Two effective therapeutic options have emerged in the management of ERBB2‐amplified colorectal cancers. The HERACLES trial36 investigated a combination of the ERBB2‐binding monoclonal antibody, trastuzumab, and the ERBB tyrosine kinase inhibitor lapatinib. Of 27 heavily pretreated patients with CRC with ERBB2 amplification, 30% of patients achieved an objective response and 44% achieved disease stabilization. One patient experienced a complete response. Patients with an ERBB2 copy number > 9.45 derived a significantly better outcome than those with lower levels of amplification. In the current study, 86.9% of samples with ERBB2 amplification had ≥ 10 copies of the gene.
Another emerging option for this population consists of the combination of trastuzumab plus pertuzumab, which allows for a more effective inhibition of ERBB2/ERBB3 signaling. In a preliminary analysis, the MyPathway trial reported a response rate of 37.5% with this combination in heavily pretreated patients with CRC with ERBB2 amplification.41 The intergroup is in the process of activating a randomized clinical trial of trastuzumab plus pertuzumab in comparison with cetuximab plus irinotecan in the second‐line/third‐line treatment of ERBB2‐amplified CRCs (SWOG S1613).
Targeting activating ERBB2 mutations in CRC is less well‐defined clinically. Based on patient‐derived tumor xenograft studies, these tumors appear more resistant to HER2‐targeting monoclonal antibodies and are more sensitive to the combinations of trastuzumab plus pan‐ERBB or HER2‐specific tyrosine kinase inhibitors.70
An additional 1.7% of mCRCs harbor alterations in the HER2 dimerization partner HER3, which may represent a second group of patients for whom HER2‐targeted treatments would be relevant. Indeed, clinical reports of activity of ERBB2 tyrosine kinase inhibitors with or without trastuzumab have already been reported in breast and urothelial cancers with ERBB3 mutations.71, 72
Mutations affecting HER2 or HER3 can drive downstream processes impacting proliferation and invasiveness and resistance to apoptosis, and have emerged as potential therapy targets.12, 13, 14, 15, 16, 17, 18, 26, 73 The distribution of kinase versus extracellular domain mutations appears to vary by tumor type. For breast cancer, HER2 kinase domain mutations are more common, whereas in urinary bladder cancer, extracellular domain mutations predominate.3 Some tumor types also feature specific types of alteration, such as the ERBB2 insertion mutations commonly identified in non‐small cell lung cancers.3 For mCRC, SV mutations in the ERBB2 sequence accounted for approximately one‐third of all ERBB2 genomic alterations, with rectal tumors having slightly more SV alterations than colonic lesions (Fig. 1B). Recent studies have further emphasized the potential of targeting ERBB2 sequence mutations in the absence of ERBB2 amplification in mCRC using combinations of kinase inhibitors and antibody therapeutics.36, 69, 70, 74, 75 The preliminary success described in these reports have generated further interest in expanding clinical trials to include ERBB2 SV mutations when there are no copy number changes in ERBB2.
In the current study, a variety of SV alterations in ERRB2 were observed (Fig. 2B), with the vast majority being characterized as activating missense alterations (eg, S310F/Y) or missense mutations alterations that are highly recurrent in cancer (eg, R678Q). Missense alterations were clustered in the extracellular domain, the transmembrane domain, and the kinase domain, with the exception of R678Q, which lies within the juxtamembrane region. In addition, we observed several frameshift alterations predicted to truncate HER2, an exon 20 insertion, and 2 alterations predicted to delete exon 16 and hyperactive HER2 signaling.76, 77, 78 A similar variety of alterations was observed for ERBB3, with the majority affecting the extracellular domain (Fig. 2B). Two recurrent alterations in the kinase domain (S846I and E928G) are located at the dimerization interface and have been shown to increase HER3‐induced signaling.79 In addition, preclinical experiments have shown that activating alterations in HER3 can underlie sensitivity to therapies targeting HER2, such as lapatinib or trastuzumab.26, 28 The wide variety of alterations observed reinforces the clinical usefulness of sequencing technologies that can comprehensively interrogate oncogenic genes in an unbiased fashion.
Preclinical and clinical data have suggested that ERBB2 amplification in mCRC is associated with a lack of response to the EGFR antibodies cetuximab and panitumumab.34, 80, 81, 82 In recent reports, a combination of therapies targeting both EGFR and HER2 was effective in treating tumors with ERBB2 amplification in preclinical experiments.81, 83, 84 Mutations in KRAS are widely accepted as predictors of resistance to anti‐EGFR antibody therapies in patients with mCRC.85 In the current study and previous reports,34, 86 ERBB2 amplification strongly correlated with a lack of RAS and BRAF alterations. In addition, although we did not observe a difference between rectal and colonic tumors in terms of frequency of ERBB2 amplifications, other studies have demonstrated a strong correlation between ERBB2 amplification and left colonic tumors.31, 87 Greater than 5% of left colonic tumors with RAS wild‐type status will harbor ERBB2 amplifications and with a resultant relative resistance to anti anti‐EGFR therapy. The early identification of these genomic alterations will impact the choice of biological therapies in the management of these patients and/or guide them toward the appropriate HER2‐targeting clinical trials.
Both TMB and MSI status were evaluated in the current study. The median TMB was higher in cases with ERBB2 or ERBB3 SV, and a greater percentage of cases harboring ERBB SV had TMB scores ≥20 mut/Mb compared with ERBB wild‐type or ERBB2 amplified mCRC (Table 1). High MSI was also found for ERBB mCRC, and was significantly enriched in ERBB3‐mutated mCRC. A significant association also was observed between ERBB3 mutation and alterations in the genes responsible for mismatch repair (MSH2, MSH6, MLH1, and PMS2). To the best of our knowledge, this association between ERBB3 and the DNA mismatch repair pathway has not been previously reported. For patients with mCRC, both MSI high status and high TMB have been associated with responsiveness to checkpoint inhibitor immunotherapies.88, 89, 90 Given the enrichment in mutational load for ERBB SV samples, combining immunotherapies with anti‐HER2 targeting agents for these patients becomes an intriguing possibility.
The use of comprehensive genomic profiling on such a large cohort of CRC cases allowed us to identify several striking differences in mutation frequencies between colonic and rectal adenocarcinomas (Fig. 1) (Table 3), predominantly in the PI3K/MTOR and WNT/β‐catenin pathways. Differences also were observed in the mutation frequencies of genes coaltered with ERBB2 amplification versus SV (Table 4). CRCs harboring ERBB2 SV had higher mutation frequencies in the PI3K (PIK3CA, PTEN, PIK3R1, and NF1), mismatch repair (MSH6, MSH2, MLH1, and PMS2), and Wnt (RNF43, SOX9, and FAM123B) pathways, among others. It remains to be determined whether the high MSI found in a significant percentage of cases with only ERBB2 SV (17%) is one mechanism underlying these associations. TOP2A amplification was frequently observed within the context of ERBB2 amplification; TOP2A is located near ERBB2 on chromosome 17.
Although more often observed in breast and upper gastrointestinal carcinomas, for which anti‐HER2 therapies currently are approved indications, the frequency of ERBB genomic alterations in mCRC (6.4%) is nonetheless significant. It is important to note that nearly one‐third of ERBB2‐altered mCRCs harbor SV alterations only, which are not detectable by routine IHC and FISH testing. Given the successful use of anti‐HER2 therapies to treat ERBB2‐driven mCRC in case studies and ongoing clinical trials, HER2‐targeted therapies may one day become approved precision treatments for patients with mCRC.
FUNDING SUPPORT
No specific funding was disclosed.
CONFLICT OF INTEREST DISCLOSURES
Jeffrey S. Ross, Siraj M. Ali, Julia A. Elvin, Alexa B. Schrock, James Suh, Jo‐Anne Vergilio, Shakti Ramkissoon, Eric Severson, Sugganth Daniel, David Fabrizio, Garrett Frampton, James Sun, Vincent A. Miller, Philip J. Stephens, and Laurie M. Gay are employees of and shareholders in Foundation Medicine Inc.
AUTHOR CONTRIBUTIONS
Conceptualization: Jeffrey S. Ross, Marwan Fakih, Siraj M. Ali, Alexa B. Schrock, and Laurie M. Gay. Data curation: Julia A. Elvin, James Suh, Jo‐Anne Vergilio, Shakti Ramkissoon, Eric Severson, Sugganth Daniel, David Fabrizio, Garrett Frampton, and James Sun. Formal analysis and investigation: Jeffrey S. Ross, and Laurie M. Gay. Methodology and software: David Fabrizio, Garrett Frampton, and James Sun. Supervision: Jeffrey S. Ross, Siraj M. Ali, Julia A. Elvin, Vincent A. Miller, and Philip J. Stephens.
Supporting information
Additional supporting information may be found in the online version of this article.
Supporting Information Table T1 and T2
Supporting Information Table T3
Supporting Information Table T4
REFERENCES
- 1. Schechter AL, Stern DF, Vaidyanathan L, et al. The neu oncogene: an erb‐B‐related gene encoding a 185,000‐Mr tumour antigen. Nature. 1984;312:513‐516. [DOI] [PubMed] [Google Scholar]
- 2. Moasser MM. The oncogene HER2: its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene. 2007;26:6469‐6487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chmielecki J, Ross JS, Wang K, et al. Oncogenic alterations in ERBB2/HER2 represent potential therapeutic targets across tumors from diverse anatomic sites of origin. Oncologist. 2015;20:7‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN. The HER‐2 receptor and breast cancer: ten years of targeted anti‐HER‐2 therapy and personalized medicine. Oncologist. 2009;14:320‐368. [DOI] [PubMed] [Google Scholar]
- 5. Bang YJ, Van Cutsem E, Feyereislova A, et al; ToGA Trial Investigators . Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2‐positive advanced gastric or gastro‐oesophageal junction cancer (ToGA): a phase 3, open‐label, randomised controlled trial. Lancet. 2010;376:687‐697. [DOI] [PubMed] [Google Scholar]
- 6. Ali SM, Sanford EM, Klempner SJ, et al. Prospective comprehensive genomic profiling of advanced gastric carcinoma cases reveals frequent clinically relevant genomic alterations and new routes for targeted therapies. Oncologist. 2015;20:499‐507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slamon DJ, Leyland‐Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783‐792. [DOI] [PubMed] [Google Scholar]
- 8. Verma S, Miles D, Gianni L, et al. Trastuzumab emtansine for HER2‐positive advanced breast cancer. N Engl J Med. 2012;367:1783‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baselga J, Cortes J, Kim S‐B, et al. Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366:109‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Van Cutsem EV, Kang Y, Chung H, et al. Efficacy results from the ToGA trial: a phase III study of trastuzumab added to standard chemotherapy (CT) in first‐line human epidermal growth factor receptor 2 (HER2)‐positive advanced gastric cancer (GC) [abstract]. J Clin Oncol. 2009;27:18(suppl). Abstract LBA4509. https://doi.org/10.1200/jco.2009.27.18s.lba4509 [Google Scholar]
- 11. Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13:663‐673. [DOI] [PubMed] [Google Scholar]
- 12. Shigematsu H, Takahashi T, Nomura M, et al. Somatic mutations of the HER2 kinase domain in lung adenocarcinomas. Cancer Res. 2005;65:1642‐1646. [DOI] [PubMed] [Google Scholar]
- 13. Buttitta F, Barassi F, Fresu G, et al. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int J Cancer. 2006;119:2586‐2591. [DOI] [PubMed] [Google Scholar]
- 14. Greulich H, Kaplan B, Mertins P, et al. Functional analysis of receptor tyrosine kinase mutations in lung cancer identifies oncogenic extracellular domain mutations of ERBB2. Proc Natl Acad Sci U S A. 2012;109:14476‐14481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ross JS, Wang K, Sheehan CE, et al. Relapsed classic E‐eadherin (CDH1)‐mutated invasive lobular breast cancer shows a high frequency of HER2 (ERBB2) gene mutations. Clin Cancer Res. 2013;19:2668‐2676. [DOI] [PubMed] [Google Scholar]
- 17. Ross JS, Gay LM, Wang K, et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: an emerging opportunity for anti‐HER2 targeted therapies. Cancer. 2016;122:2654‐2662. [DOI] [PubMed] [Google Scholar]
- 18. Ross JS, Wang K, Gay LM, et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin Cancer Res. 2014;20:68‐75. [DOI] [PubMed] [Google Scholar]
- 19. Kaidar‐Person O, Billan S, Kuten A. Targeted therapy with trastuzumab for advanced salivary ductal carcinoma: case report and literature review. Med Oncol. 2012;29:704‐706. [DOI] [PubMed] [Google Scholar]
- 20. Cappuzzo F, Bemis L, Varella‐Garcia M. HER2 mutation and response to trastuzumab therapy in non‐small‐cell lung cancer. N Engl J Med. 2006;354:2619‐2621. [DOI] [PubMed] [Google Scholar]
- 21. Mazieres J, Peters S, Lepage B, et al. Lung cancer that harbors an HER2 mutation: epidemiologic characteristics and therapeutic perspectives. J Clin Oncol. 2013;31:1997‐2003. [DOI] [PubMed] [Google Scholar]
- 22. Mazieres J, Barlesi F, Filleron T, et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2‐targeted drugs: results from the European EUHER2 cohort. Ann Oncol. 2016;27:281‐286. [DOI] [PubMed] [Google Scholar]
- 23. Ou SI, Schrock AB, Bocharov EV, et al. HER2 transmembrane domain (TMD) mutations (V659/G660) that stabilize homo‐ and heterodimerization are rare oncogenic drivers in lung adenocarcinoma that respond to afatinib. J Thorac Oncol. 2017;12:446‐457. [DOI] [PubMed] [Google Scholar]
- 24. Ben‐Baruch NE, Bose R, Kavuri SM, Ma CX, Ellis MJ. HER2‐mutated breast cancer responds to treatment with single‐agent neratinib, a second‐generation HER2/EGFR tyrosine kinase inhibitor. J Natl Compr Canc Netw. 2015;13:1061‐1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ali SM, Alpaugh RK, Downing SR, et al. Response of an ERBB2‐mutated inflammatory breast carcinoma to human epidermal growth factor receptor 2‐targeted therapy. J Clin Oncol. 2014;32:e88‐e931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jaiswal BS, Kljavin NM, Stawiski EW, et al. Oncogenic ERBB3 mutations in human cancers. Cancer Cell. 2013;23:603‐617. [DOI] [PubMed] [Google Scholar]
- 27. Karachaliou N, Lazzari C, Verlicchi A, Sosa AE, Rosell R. HER3 as a therapeutic target in cancer. BioDrugs. 2017;31:63‐73. [DOI] [PubMed] [Google Scholar]
- 28. Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014;20:1410‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bai J, Gao J, Mao Z, et al. Genetic mutations in human rectal cancers detected by targeted sequencing. J Hum Genet. 2015;60:589‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El‐Deiry WS, Vijayvergia N, Xiu J, et al. Molecular profiling of 6,892 colorectal cancer samples suggests different possible treatment options specific to metastatic sites. Cancer Biol Ther. 2015;16:1726‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richman SD, Southward K, Chambers P, et al. HER2 overexpression and amplification as a potential therapeutic target in colorectal cancer: analysis of 3256 patients enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials. J Pathol. 2016;238:562‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jauhri M, Bhatnagar A, Gupta S, Shokeen Y, Minhas S, Aggarwal S. Targeted molecular profiling of rare genetic alterations in colorectal cancer using next‐generation sequencing. Med Oncol. 2016;33:106. [DOI] [PubMed] [Google Scholar]
- 33. Dienstmann R, Tabernero J. Spectrum of gene mutations in colorectal cancer: implications for treatment. Cancer J. 2016;22:149‐155. [DOI] [PubMed] [Google Scholar]
- 34. Rankin A, Klempner SJ, Erlich R, et al. Broad detection of alterations predicted to confer lack of benefit from EGFR antibodies or sensitivity to targeted therapy in advanced colorectal cancer [published online ahead of print 2016 September 28, 2016]. Oncologist. doi: 10.1634/theoncologist.2016-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barry GS, Cheang MC, Chang HL, Kennecke HF. Genomic markers of panitumumab resistance including ERBB2/ HER2 in a phase II study of KRAS wild‐type (wt) metastatic colorectal cancer (mCRC). Oncotarget. 2016;7:18953‐18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sartore‐Bianchi A, Trusolino L, Martino C, et al. Dual‐targeted therapy with trastuzumab and lapatinib in treatment‐refractory, KRAS codon 12/13 wild‐type, HER2‐positive metastatic colorectal cancer (HERACLES): a proof‐of‐concept, multicentre, open‐label, phase 2 trial. Lancet Oncol. 2016;17:738‐746. [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Castanaro C, Luan B, et al. ERBB3/HER2 signaling promotes resistance to EGFR blockade in head and neck and colorectal cancer models. Mol Cancer Ther. 2014;13:1345‐1355. [DOI] [PubMed] [Google Scholar]
- 38. Raghav KP, Overman MJ, Yu R, et al. HER2 amplification as a negative predictive biomarker for anti‐epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J Clin Oncol. 2016;34(suppl 15):3517‐3517. [DOI] [PubMed] [Google Scholar]
- 39. Bai JW, Xue HZ, Zhang C. Down‐regulation of microRNA‐143 is associated with colorectal cancer progression. Eur Rev Med Pharmacol Sci. 2016;20:4682‐4687. [PubMed] [Google Scholar]
- 40. Ledel F, Hallstrom M, Ragnhammar P, Ohrling K, Edler D. HER3 expression in patients with primary colorectal cancer and corresponding lymph node metastases related to clinical outcome. Eur J Cancer. 2014;50:656‐662. [DOI] [PubMed] [Google Scholar]
- 41. Hurwitz H, Raghav KPS, Burris HA, et al. Pertuzumab + trastuzumab for HER2‐amplified/overexpressed metastatic colorectal cancer (mCRC): interim data from MyPathway. J Clin Oncol. 2017;35(suppl 4):676‐676. [Google Scholar]
- 42. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Forbes SA, Beare D, Gunasekaran P, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805‐D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564‐569. [DOI] [PubMed] [Google Scholar]
- 45. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Disel U, Germain A, Yilmazel B, et al. Durable clinical benefit to trastuzumab and chemotherapy in a patient with metastatic colon adenocarcinoma harboring ERBB2 amplification. Oncoscience. 2015;2:581‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2‐positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9:16‐32. [DOI] [PubMed] [Google Scholar]
- 48. Boku N. HER2‐positive gastric cancer. Gastric Cancer. 2014;17:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dabbs DJ, Klein ME, Mohsin SK, Tubbs RR, Shuai Y, Bhargava R. High false‐negative rate of HER2 quantitative reverse transcription polymerase chain reaction of the Oncotype DX test: an independent quality assurance study. J Clin Oncol. 2011;29:4279‐4285. [DOI] [PubMed] [Google Scholar]
- 50. D'Haene N, Le Mercier M, De Neve N, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PloS One. 2015;10:e0138245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shanmugam V, Ramanathan RK, Lavender NA, et al. Whole genome sequencing reveals potential targets for therapy in patients with refractory KRAS mutated metastatic colorectal cancer. BMC Med Genomics. 2014;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang L, Chen L, Sah S, et al. Profiling cancer gene mutations in clinical formalin‐fixed, paraffin‐mbedded colorectal tumor specimens using targeted next‐generation sequencing. Oncologist. 2014;19:336‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cancer Genome Atlas Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66:683‐691. [DOI] [PubMed] [Google Scholar]
- 55. Miller KD, Siegel RL, Lin CC, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271‐289. [DOI] [PubMed] [Google Scholar]
- 56. Cho M, Gong J, Fakih M. The state of regional therapy in the management of metastatic colorectal cancer to the liver. Expert Rev Anticancer Ther. 2016;16:229‐245. [DOI] [PubMed] [Google Scholar]
- 57. Jawed I, Wilkerson J, Prasad V, Duffy AG, Fojo T. Colorectal cancer survival gains and novel treatment regimens: a systematic review and analysis. JAMA Oncol. 2015;1:787‐795. [DOI] [PubMed] [Google Scholar]
- 58. Seeber A, Gastl G. Targeted therapy of colorectal cancer. Oncol Res Treat. 2016;39:796‐802. [DOI] [PubMed] [Google Scholar]
- 59. Ohhara Y, Fukuda N, Takeuchi S, et al. Role of targeted therapy in metastatic colorectal cancer. World J Gastrointest Oncol. 2016;8:642‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Seow HF, Yip WK, Fifis T. Advances in targeted and immunobased therapies for colorectal cancer in the genomic era. OncoTargets Ther. 2016;9:1899‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Palma S, Zwenger AO, Croce MV, Abba MC, Lacunza E. From molecular biology to clinical trials: toward personalized colorectal cancer therapy. Clin Colorectal Cancer. 2016;15:104‐115. [DOI] [PubMed] [Google Scholar]
- 62. Kassouf E, Tabchi S, Tehfe M. Anti‐EGFR therapy for metastatic colorectal cancer in the era of extended RAS gene mutational analysis. BioDrugs. 2016;30:95‐104. [DOI] [PubMed] [Google Scholar]
- 63. Frank D, Jumonville A, Loconte NK, et al. A phase II study of capecitabine and lapatinib in advanced refractory colorectal adenocarcinoma: a Wisconsin Oncology Network study. J Gastrointest Oncol. 2012;3:90‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rubinson DA, Hochster HS, Ryan DP, et al. Multi‐drug inhibition of the HER pathway in metastatic colorectal cancer: results of a phase I study of pertuzumab plus cetuximab in cetuximab‐refractory patients. Invest New Drugs. 2014;32:113‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cremolini C, Pietrantonio F. How the lab is changing our view of colorectal cancer. Tumori. 2016;102:541‐547. [DOI] [PubMed] [Google Scholar]
- 66. Johnsson A, Hagman H, Frodin J‐E, et al. A randomized phase III trial on maintenance treatment with bevacizumab alone or in combination with erlotinib after chemotherapy and bevacizumab in metastatic colorectal cancer: the Nordic ACT Trial. Ann Oncol. 2013;24:2335‐2341. [DOI] [PubMed] [Google Scholar]
- 67. Ma BB, Chan SL, Ho WM, et al. Intermittent versus continuous erlotinib with concomitant modified “XELOX” (q3W) in first‐line treatment of metastatic colorectal cancer: correlation with serum amphiregulin and transforming growth factor alpha. Cancer. 2013;119:4145‐4153. [DOI] [PubMed] [Google Scholar]
- 68. Bouche O, Maindrault‐Goebel F, Ducreux M, et al. Phase II trial of weekly alternating sequential BIBF 1120 and afatinib for advanced colorectal cancer. Anticancer Res. 2011;31:2271‐2281. [PubMed] [Google Scholar]
- 69. Kloth M, Ruesseler V, Engel C, et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan‐HER inhibitors in Lynch and Lynch‐like colorectal cancer. Gut. 2016;65:1296‐1305. [DOI] [PubMed] [Google Scholar]
- 70. Kavuri SM, Jain N, Galimi F, et al. HER2 activating mutations are targets for colorectal cancer treatment. Cancer Discov. 2015;5:832‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bidard F‐C, Ng CKY, Cottu P, et al. Response to dual HER2 blockade in a patient with HER3‐mutant metastatic breast cancer. Ann Oncol. 2015;26:1704‐1709. [DOI] [PubMed] [Google Scholar]
- 72. Choudhury NJ, Campanile A, Antic T, et al. Afatinib activity in platinum‐refractory metastatic urothelial carcinoma in patients with ERBB alterations. J Clin Oncol. 2016;34:2165‐2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shimamura T, Ji H, Minami Y, et al. Non‐small‐cell lung cancer and Ba/F3 transformed cells harboring the ERBB2 G776insV_G/C mutation are sensitive to the dual‐specific epidermal growth factor receptor and ERBB2 inhibitor HKI‐272. Cancer Res. 2006;66:6487‐6491. [DOI] [PubMed] [Google Scholar]
- 74. Parikh A, Atreya C, Korn WM, Venook AP. Prolonged response to HER2‐directed therapy in a patient with her2‐amplified, rapidly progressive metastatic colorectal cancer. J Natl Compr Cancer Netw. 2017;15:3‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Aung KL, Stockley TL, Serra S, Kamel‐Reid S, Bedard PL, Siu LL. Testing ERBB2 p.L755S kinase domain mutation as a druggable target in a patient with advanced colorectal cancer. Cold Spring Harb Mol Case Stud. 2016;2:a001016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Akiyama T, Matsuda S, Namba Y, Saito T, Toyoshima K, Yamamoto T. The transforming potential of the c‐erbB‐2 protein is regulated by its autophosphorylation at the carboxyl‐terminal domain. Mol Cell Biol. 1991;11:833‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Marchini C, Gabrielli F, Iezzi M, et al. The human splice variant Δ16HER2 induces rapid tumor onset in a reporter transgenic mouse. PloS One. 2011;6:e18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8:2152‐2162. [DOI] [PubMed] [Google Scholar]
- 79. Littlefield P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal. 2014;7:ra114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 80. Siravegna G, Mussolin B, Buscarino M, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:827. [DOI] [PubMed] [Google Scholar]
- 81. Bertotti A, Migliardi G, Galimi F, et al. A molecularly annotated platform of patient‐derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab‐resistant colorectal cancer. Cancer Discov. 2011;1:508‐523. [DOI] [PubMed] [Google Scholar]
- 82. Takegawa N, Yonesaka K, Sakai K, et al. HER2 genomic amplification in circulating tumor DNA from patients with cetuximab‐resistant colorectal cancer. Oncotarget. 2016;7:3453‐3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yonesaka K, Zejnullahu K, Okamoto I, et al. Activation of ERBB2 signaling causes resistance to the EGFR‐directed therapeutic antibody cetuximab. Sci Transl Med. 2011;3:99ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Guan SS, Chang J, Cheng C‐C, et al. Afatinib and its encapsulated polymeric micelles inhibits HER2‐overexpressed colorectal tumor cell growth in vitro and in vivo. Oncotarget. 2014;5:4868‐4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Knickelbein K, Zhang L. Mutant KRAS as a critical determinant of the therapeutic response of colorectal cancer. Genes Dis. 2015;2:4‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nam SK, Yun S, Koh J, et al. BRAF, PIK3CA, and HER2 oncogenic alterations according to KRAS mutation status in advanced colorectal cancers with distant metastasis. PloS One. 2016;11:e0151865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Seo AN, Kwak Y, Kim DW, et al. HER2 status in colorectal cancer: its clinical significance and the relationship between HER2 gene amplification and expression. PloS One. 2014;9:e98528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bupathi M, Wu C. Biomarkers for immune therapy in colorectal cancer: mismatch‐repair deficiency and others. J Gastrointest Oncol. 2016;7:713‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. George TJ, Frampton GM, Sun J, et al. Tumor mutational burden as a potential biomarker for PD1/PD‐L1 therapy in colorectal cancer [abstract]. J Clin Oncol. 2016;34(suppl). Abstract 3587. http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.3587 [Google Scholar]
- 90. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: a review. Cancer Treat Rev. 2016;51:19‐26. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article.
Supporting Information Table T1 and T2
Supporting Information Table T3
Supporting Information Table T4