

Abstract
Caspase-3 is well known as the “executioner” whose activation commits the cell to an apoptotic fate, but low levels of caspase-3 activity also play key roles in development. A new study explains how cells can balance these functions, using biophysical, structural, and computational approaches to demonstrate the mechanism by which phosphorylation of conserved sites on a distal surface loop reduces or abolishes catalytic activity. These results provide new insights into allosteric regulation mechanisms and offer new opportunities for development of caspase-3 modulators.
Keywords: caspase, apoptosis, allosteric regulation, phosphorylation, protein evolution
Introduction
Caspases are cysteine proteases that play a number of critical roles in immune responses, cellular development, differentiation, migration, and the initiation of apoptosis. In general, caspases can be divided functionally into proinflammatory and proapoptotic family members, and the apoptotic caspases can be further subdivided into the initiator caspases and downstream executioner caspases (1). Caspase-3 is the terminal executioner protease in apoptosis, where it initiates dismantling of cellular components via cleavage of structural proteins, disruption of the nuclear envelope, and breakdown of genomic DNA through activation of caspase-activated DNase (1, 2). Cells have intrinsic mechanisms to control caspase activity, as low levels of caspase-3 activity are necessary for critical developmental processes in a number of cell types (2). As a result, a variety of strategies have evolved to modulate caspase-3 activity to allow such developmental processes to proceed without initiating cell death. There is also therapeutic interest in modulating caspase-3 activation, which is known to contribute to the progression of Huntingon's, Parkinson's, and Alzheimer's diseases, as well as ALS (3). Although the specific role of caspase-3 varies in each disease, in all cases reduction of caspase-3 activity could disrupt disease progression or limit severity. New work from Thomas et al. (4) provides an impressive exploration of the evolutionary and mechanistic basis of one cellular regulatory mechanism, in which phosphorylation of an allosteric site tunes enzyme function.
Caspase-3 is regulated by activation and maturation from a zymogen form; the mature caspase is considered to be a dimer of protomers, although each protomer in the mature enzyme is further cleaved into large and small subunits. A number of posttranslational modifications are also known to modulate caspase-3 activity, including nitrosylation of the catalytic cysteine (5) and phosphorylation events that either control processing of the zymogen or allosterically modulate protease activity (6). In particular, both Ser150 and Thr152 on the helix-3 C-terminal loop (called H3CL)2 are known sites of phosphorylation (7), and phosphorylation of Ser150 has been shown to have functional outcomes, but the mechanism by which this modification, far from the active site, impacts activity is unknown.
To gain insight into this question, Thomas et al. (4) began with a phylogenetic analysis of the five residues in the H3CL from >1,000 caspase sequences ranging from fish to mammals. These data revealed that the Ser150 phosphorylation site is an ancient adaptation that evolved after the apoptotic caspases diverged from the inflammatory caspases, whereas the Thr152 site evolved much later, in early mammals.
To characterize the effects of phosphorylation at the Ser150 and Thr152 sites, the authors analyzed a panel of caspase-3 variants, including phosphomimetic and “phospho-null” side chains, using a combination of enzymatic, spectroscopic, and structural approaches. First, Thomas et al. (4) measured steady-state kinetic parameters and specificity constants for the cleavage of a tetrapeptide substrate by WT caspase-3 and the mutants. In parallel, they assayed cleavage of an intact protein substrate, caspase-7, whose prodomain and intersubunit linker is cleaved by caspase-3. Interestingly, they found that the phosphomimetic T152D variant lost activity in both tetrapeptide and caspase-7 cleavage assays, whereas Ser150 substitutions had no effect on the tetrapeptide cleavage kinetics but did show reduced activity against caspase-7. The authors propose that phosphorylation of Ser150 is a widely conserved switch found in many caspases that reduces, but does not abolish, caspase-3 activity. In contrast, phosphorylation of Thr152 appears to be a more recent development of a “kill switch” that can abolish proteolytic activity in mammalian caspases.
Loss of activity due to phosphorylation events in the H3CL could be due to a loss of catalytic efficiency in the intact caspase dimer or due to dissociation of the dimer, as each active site contains loops from both protomers. The authors provide evidence that both mechanisms play a role in the loss of cleavage effector function. Fluorescence experiments demonstrated active-site conformational changes in the T152D variant, and the Ser150 variants and T152D all showed decreased stability of the dimer at low pH, which is relevant since the pH of the cytoplasm decreases from 7.4 to ∼6.8 during apoptosis.
Given that the H3CL phosphorylation sites are located ∼33 Å from either active site (Fig. 1), how do these posttranslational modifications propagate their inhibitory effect to the active sites? Thomas et al. (4) addressed this question using X-ray crystallography and molecular dynamics simulations of 14 different structures solved at high resolution. The crystal structures showed that alterations in the Ser150 or Thr152 side chains caused shifts in the position of the H3CL relative to the neighboring helix-2 loop. Molecular dynamics simulations indicated that in the S150E (and T152D) phosphomimetic variants, this displacement of the H3CL could propagate through helix 2 and helix 3 to the β1–β3 sheet that forms the base of the active site and contains catalytic histidine His121; His121 can then adopt an alternate configuration, approaching within hydrogen-bonding distance of neighboring residue Glu123, presumably reducing the rate of proton transfer. Both Ser150 and Glu123 are highly conserved residues across the apoptotic caspases, providing additional support to the idea that the Ser150 phosphorylation switch is an ancient modification common to the apoptotic caspases. In addition to this intraprotomer pathway for allosteric control via Ser150, the crystal structures revealed that several Thr152 variants caused alterations in the packing of an adjacent cluster of hydrophobic residues that help to stabilize some of the active site loops across the dimer interface, suggesting a mechanism for interprotomer allosteric control.
Figure 1.
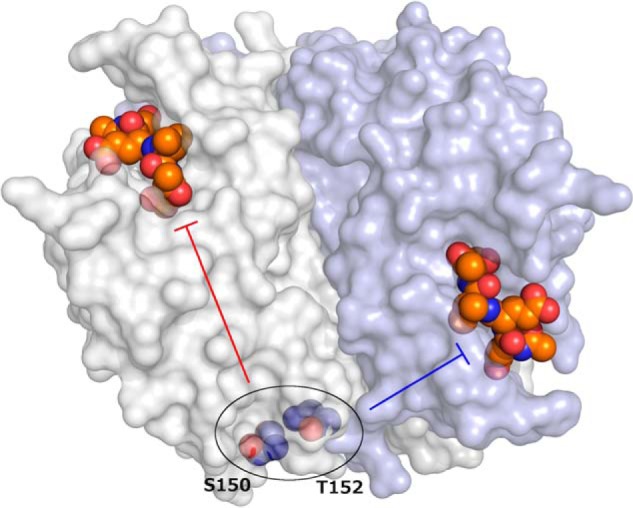
Caspase-3 catalytic activity can be allosterically modulated by phosphorylation at Ser150 or Thr152. The caspase-3 dimer (Protein Data Bank code 2J30) is illustrated with one protomer shown as a white surface and the other as a light blue surface. Active sites are shown with the Ac-DEVD-CMK inhibitor as colored spheres, with orange carbon atoms. Conformational changes that occur upon phosphorylation of Ser150 or Thr152 (colored spheres with blue carbon atoms) are propagated to the active sites through distinct intraprotomer (red bar-headed arrow) or interprotomer (blue bar-headed arrow) pathways.
This study provides interesting insights into the mechanisms by which phosphorylation of the H3CL exerts allosteric control of caspase-3 activity. From a technical standpoint, the results demonstrate that tetrapeptide substrates do not always accurately reflect the full story on caspase activity, so future studies would do well to assess cleavage of intact protein substrates in parallel. Perhaps the most exciting aspect that emerges from this work is the potential for developing new therapeutic agents to specifically target caspase-3 in a way that mimics phosphorylation at the Ser150 or Thr152 sites. For example, small-molecule inhibitors binding near the H3CL could shift its position relative to the helix-2 loop and trigger the same allosteric effect of phosphorylation at the H3CL. Therapeutic targeting of the caspases has been particularly challenging due to overlapping substrate specificity, which makes active site targeting of particular caspases difficult (8). Thus, targeting allosteric sites such as Thr152 in caspase-3 may allow specific targeting while minimizing off-target effects. Efforts to target caspase-3 in neurons, for example to treat the neurodegenerative diseases described above, face a double challenge: Any therapeutic agent would need to cross both the blood-brain barrier as well as the plasma membrane to access caspase-3 in the cytoplasm. Therefore, small-molecule allosteric inhibitors could be more tractable than protein or peptide-based therapeutics for these diseases. This study thus provides exciting new opportunities to flip the caspase-3 off switch.
Acknowledgments
I thank Dr. Joe Maciag for helpful discussions and comments on the manuscript. The Herr laboratory is supported by National Institutes of Health Grants U19-AI070235, R21-AI132822, and R21-HD090856.
The author declares that he has no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
- H3CL
- helix-3 C-terminal loop.
References
- 1. Pop C., and Salvesen G. S. (2009) Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 284, 21777–21781 10.1074/jbc.R800084200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Crawford E. D., and Wells J. A. (2011) Caspase substrates and cellular remodeling. Annu. Rev. Biochem. 80, 1055–1087 10.1146/annurev-biochem-061809-121639 [DOI] [PubMed] [Google Scholar]
- 3. D'Amelio M., Cavallucci V., and Cecconi F. (2010) Neuronal caspase-3 signaling: not only cell death. Cell Death Differ. 17, 1104–1114 10.1038/cdd.2009.180 [DOI] [PubMed] [Google Scholar]
- 4. Thomas M. E., Grinshpon R., Swartz P., and Clark A. C. (2018) Modifications to a common phosphorylation network provide individualized control in caspases. J. Biol. Chem. 293, 5447–5461 10.1074/jbc.RA117.000728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mannick J. B., Schonhoff C., Papeta N., Ghafourifar P., Szibor M., Fang K., and Gaston B. (2001) S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 154, 1111–1116 10.1083/jcb.200104008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parrish A. B., Freel C. D., and Kornbluth S. (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harb. Perspect. Biol. 5 10.1101/cshperspect.a008672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alvarado-Kristensson M., Melander F., Leandersson K., Rönnstrand L., Wernstedt C., and Andersson T. (2004) p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils. J. Exp. Med. 199, 449–458 10.1084/jem.20031771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poreba M., Strozyk A., Salvesen G. S., and Drag M. (2013) Caspase substrates and inhibitors. Cold Spring Harb. Perspect. Biol. 5, a008680 10.1101/cshperspect.a008680 [DOI] [PMC free article] [PubMed] [Google Scholar]