

Abstract
The cystic fibrosis transmembrane conductance regulator (CFTR) is a Cl− channel that apparently has evolved from an ancestral active transporter. Key to the CFTR's switch from pump to channel function may have been the appearance of one or more “lateral portals.” Such portals connect the cytoplasm to the transmembrane channel pore, allowing a continuous pathway for the electrodiffusional movement of Cl− ions. However, these portals remain the least well-characterized part of the Cl− transport pathway; even the number of functional portals is uncertain, and if multiple portals do exist, their relative functional contributions are unknown. Here, we used patch-clamp recording to identify the contributions of positively charged amino acid side chains located in CFTR's cytoplasmic transmembrane extensions to portal function. Mutagenesis-mediated neutralization of several charged side chains reduced single-channel Cl− conductance. However, these same mutations differentially affected channel blockade by cytoplasmic suramin and Pt(NO2)42− anions. We considered and tested several models by which the contribution of these positively charged side chains to one or more independent or non-independent portals to the pore could affect Cl− conductance and interactions with blockers. Overall, our results suggest the existence of a single portal that is lined by several positively charged side chains that interact electrostatically with both Cl− and blocking anions. We further propose that mutations at other sites indirectly alter the function of this single portal. Comparison of our functional results with recent structural information on CFTR completes our picture of the overall molecular architecture of the Cl− permeation pathway.
Keywords: cystic fibrosis transmembrane conductance regulator (CFTR), chloride channel, ABC transporter, ion channel, electrophysiology, cytoplasmic portals, electrostatic attraction, surface charge
Introduction
Cystic fibrosis is caused by loss-of-function mutations in the cystic fibrosis transmembrane conductance regulator (CFTR),2 an epithelial cell Cl− channel. CFTR is a member of the ATP-binding cassette family of membrane transport proteins, most members of which function as ATP-dependent pumps (1, 2). It is presumed that some evolutionary change has allowed CFTR to function not as a pump but as an ion channel (3–5). Specifically, as an ion channel, CFTR must exhibit a continuous aqueous pathway that allows free electrodiffusional movement of Cl− and other small anions between the cytoplasm and the extracellular solution in the channel open state. Such a continuous open pathway is incompatible with the function of a pump.
As with other ATP-binding cassette proteins, CFTR is made up of two membrane-spanning domains each comprising six transmembrane (TM) segments and two cytoplasmic nucleotide binding domains (NBDs). The TMs form long extensions into the cytoplasm (TMEs) to contact the NBDs. Recently the molecular structure of CFTR was observed using cryo-electron microscopy (cryo-EM), first in an inactive, closed state with separated NBDs (Fig. 1A) (6, 7) and subsequently in a “near-open” state with dimerized NBDs (Fig. 1D) (8). Both of these structures show a continuous transmembrane channel pore that is wide at its cytoplasmic end and apparently closed at its extracellular end (7, 8). These structural data are in broad agreement with longstanding functional models of the channel pore and with a large body of structure–function work identifying the molecular architecture of the pore (9). Briefly, functional models suggest that the transmembrane part of the pore is made up of a wide inner vestibule that is accessible from the cytoplasm both in open and in closed channels, a narrow region that forms the anion selectivity filter, and a shallower outer vestibule accessible from the extracellular solution (9).
Figure 1.

Structural and functional models of the cytoplasmic entrance to the CFTR channel pore. A, overall structure of human CFTR in the channel closed state (7). In this state, the TMs and TMEs are arranged in an “inward facing” conformation, such that the transmembrane permeation pathway is open to the cytoplasm and closed at its extracellular end. Those positively charged amino acid side chains that have been proposed, based on functional data, to attract cytoplasmic Cl− ions to the permeation pathway are shown as space-filling models. These positive charges contribute to the overall positive electrostatic potential of this part of the protein (8). B, close-up view of the isolated TMEs (derived from A), showing the location of these positively charged side chains as labeled in the same colors. C, the wide-open nature of the TMEs suggests that Cl− and other substances could be “funneled” toward the transmembrane pore in this closed state. D, overall structure of zebrafish CFTR in a near-open state (8). In this state, the TMEs are much more closely associated, and there is no “central” pathway from the cytoplasm to the pore (such as apparently exists in A). Positively charged amino acid side chains corresponding to those in A are shown using the same color scheme. E, close-up view of the isolated TMEs (derived from D). Note that the numbering of these positively charged amino acids is slightly different in zebrafish CFTR. F, tight closure of the inner ends of the TMEs led us to suggest that cytoplasmic Cl− accesses the open channel pore by at least one, and possibly two, cytoplasmic portals that are lined by positively charged amino acid side chains that play an important role in electrostatic attraction of Cl− to the permeation pathway (17).
Comparison of the structures of CFTR in “closed” (Fig. 1, A and B) and “open” (Fig. 1, D and E) states is consistent with NBD dimerization resulting in channel opening and drawing together of the inner ends of the TMEs. The large-scale rearrangement of the TMEs implied by Fig. 1 (A and D) suggests that cytoplasmic substances might be able to access the pore-forming TMs via different routes in closed channels and in open channels. It is known that large cytoplasmic substances can access the inner vestibule of the pore lined by the TMs not only in the open state, but also when the channel is closed (10–14). Although there appears to be a central pathway between the TMEs that allows free access to the TM pore in the closed state (Fig. 1, A–C), tight closure of the inner ends of the TMEs appears to close off this pathway in the open state (Fig. 1, D and E). Instead, access to the TM pore is thought to occur via one or more lateral portals or tunnels formed between individual TMEs (8, 9, 15–18) (Fig. 1F). The existence of up to four such portals within the TMEs was originally postulated based on atomic homology modeling (15, 16). Based on the effect of mutations and chemical modifications of different amino acids on Cl− conductance, we suggested that one or two portals may be involved in Cl− access to the pore: a functionally major portal formed between TMEs 4 and 6 and perhaps a functionally lesser portal on the opposite side of the protein, between TMEs 10 and 12 (17). In particular, we suggested that positively charged amino acid side chains located in several different TMEs (Fig. 1, B and E) attracted cytoplasmic Cl− ions to the pore electrostatically (17). Although the cryo-EM structure of Fig. 1D appears to confirm the presence of a portal between TMEs 4 and 6, only a small gap of unknown functional importance was identified between TMEs 10 and 12 (8). Furthermore, positive electrostatic potential around this single large portal supports the idea that it is involved in electrostatic attraction of cytoplasmic Cl− ions (8).
In addition to their functional importance in Cl− conduction (17, 18), the portals formed by the TMEs may also be the site of action of some CFTR inhibitors. In particular, it was shown that Arg303 in TME5 interacts electrostatically with the large channel blocking anion suramin (19). Furthermore, it has been proposed that the appearance of the portals may have been a key step in the evolution of CFTR from a pump into an ion channel (5). Despite their functional importance and mechanistic significance, the portals remain the least well-characterized part of the CFTR permeation pathway. As described above, even the number of portals utilized by Cl− ions to access the pore remains open to question, and if more than one portal exists, their relative functional contributions remain unknown. In this work we have studied the effect of mutating positively charged residues potentially contributing to portal function (Fig. 1, B and E) on both Cl− conductance and block by cytoplasmic suramin and Pt(NO2)42− ions. Our results suggest that CFTR possesses one dominant lateral portal with a cytoplasmic entrance between TMEs 4 and 6 and that a number of positively charged side chains surrounding this portal act to attract cytoplasmic anions to the pore.
Results
Role of fixed positive charges in chloride conductance
Previously we showed that mutagenesis of several positively charged arginine and lysine residues in the TMEs to cysteine (in a cysteine-less CFTR background) caused a significant reduction in unitary current amplitude (17). The putative location of these positively charged residues in CFTR is shown in Fig. 1. Fig. 2 shows the effect of mutating these residues (Lys190, Arg248, Arg303, Lys370, Lys1041, and Arg1048) to neutrally charged glutamine in a wildtype CFTR background. In each case, the neutralizing mutation caused small but significant decreases in Cl− conductance to between 51 ± 1% in K190Q (n = 5) and 91 ± 2% in K370Q (n = 5) of wildtype conductance (Fig. 2). We also found that neutralization of another nearby positively charged side chain, Lys978 (that we could not previously investigate in cysteine-less CFTR for technical reasons (17)), caused a similar, significant reduction in Cl− conductance (Fig. 2C). These results are consistent with the earlier suggestion (17, 27) that these positively charged side chains (now extended to include Lys978) electrostatically attract cytoplasmic Cl− ions to the pore.
Figure 2.

Effect of charge-neutralizing mutations on single channel conductance. A, example single channel currents carried by the named channel variants at a membrane potential of −50 mV. The closed state is indicated by the line to the far left. B, mean single channel current–voltage relationships for these channel variants. C, mean single channel conductance measured from the slope of the current–voltage relationship from individual patches. Asterisks indicated a significant difference from wildtype (p < 0.001). The means of data from 4–8 patches are shown in B and C.
Role of fixed positive charges in suramin block
Previously we showed that Arg303 in TME5 plays an apparently electrostatic role in interacting with a very large polyanionic blocker of CFTR, suramin (19). The blocking effects of intracellular suramin are compared for wildtype and charge-neutralizing mutant forms of CFTR in Fig. 3. Not only R303Q as shown previously (19) but also K190Q, R248Q, and K370Q significantly weakened the blocking effects of suramin, consistent with these positively charged side chains also normally interacting with negatively charged suramin molecules. In contrast, R1048Q had no significant effect on suramin inhibition, whereas two other mutations, K978Q and K1041Q, unexpectedly led to an apparent strengthening of channel current inhibition by intracellular suramin (Fig. 3). These results appear inconsistent with simple models of the cytoplasmic entrance to the pore (such as those shown in Fig. 1, C and F) in which all of these positively charged side chains act in a similar way to attract negatively charged Cl− and other anions (such as suramin) into the permeation pathway.
Figure 3.

Effect of charge-neutralizing mutations on suramin block. A, example macroscopic current–voltage relationships for different channel variants in inside-out patches. In each case currents were recorded before (control, black) and after (red) addition of 10 μm suramin to the intracellular solution. B, mean fraction of control current remaining following addition of this concentration of suramin as a function of voltage in these channel variants. C, mean Kd (at −100 mV) calculated from data such as that shown in B as described under “Experimental Procedures” (Equation 1). Asterisks indicate a significant difference from wildtype. *, p < 0.05; **, p < 0.0005. The means of data from 4–10 patches are shown in B and C.
The effects of the K190Q, R248Q, R303Q, and K370Q mutations on Cl− conductance (Fig. 2) and suramin block (Fig. 3) are consistent with these positive charges attracting both Cl− and suramin to the pore. In contrast, the effects of K978Q, K1041Q, and R1048Q (Figs. 2 and 3) appear inconsistent with such a role for these residues. It is striking that Lys190, Arg248, Arg303, and Lys370 were all previously suggested to line a primary cytoplasmic portal to the pore (17) (Fig. 1E), whereas Lys1041 and Arg1048, as well as perhaps Lys978, are expected to lie closer to the opposite side of the channel (Fig. 1E) and potentially to contribute to a secondary portal (17). The distinct effect of mutations at these two groups of sites led us to consider a model in which a differential contribution of these positively charged residues to the function of two separate portals could explain the similar effects of neutralizing mutations on Cl− conductance (Fig. 2) but the seemingly opposite effects of different mutations on suramin block (Fig. 3). One model by which two portals could potentially explain these results is shown in cartoon form in Fig. 4A. In this model, both Cl− and suramin can enter the cytoplasmic part of the pore via a primary portal, whereas only Cl− can enter via a secondary portal. Although mutations affecting the function of the primary portal will then impair both Cl− conductance and suramin block, mutations affecting the secondary portal may reduce Cl− conductance but perhaps strengthen suramin block (for example, by reducing the relative contribution of a suramin-insensitive Cl− entrance pathway to the pore). This model would predict that the fraction of Cl− current that passes through the secondary portal should be resistant even to very high concentrations of suramin, potentially allowing the relative contributions of the two portals to Cl− conductance to be estimated from the suramin-insensitive fraction of the overall current. Furthermore, the suramin-insensitive component of the current might be increased in channel mutants that impaired Cl− entry through the main portal (because a larger proportion of the remaining current might then flow through the secondary portal), and by the same logic the suramin-resistant component of current might be reduced (or abolished) in mutants that impair Cl− entry through the secondary portal. We therefore compared the blocking effects of very high concentrations of suramin (1–10 mm) on example mutants that weakened (R303Q and K370Q in the putative primary portal) or strengthened (K1041Q in the putative secondary portal) block by suramin. As shown in Fig. 4 (B and C), all channel variants studied were almost completely inhibited by high concentrations of suramin, contrary to the predictions of the model of Fig. 4C and outlined above. Fitting of the suramin concentration-inhibition relationships as described under “Experimental Procedures” (Equation 2) suggested only a very small suramin-insensitive component to the current of between 3 and 6% in all channel variants studied (Fig. 4C). Furthermore, whereas each of these three charge-neutralizing mutants showed significantly different degrees of block compared with wildtype at lower suramin concentrations (1–10 μm) (p < 0.05), at high concentrations (1–10 mm), none showed significant differences from wildtype (Fig. 4C). These results suggest that suramin binds in the cytoplasmic part of the permeation pathway at a position at which it likely occludes all Cl− permeation, irrespective of the route(s) by which Cl− enters this pathway.
Figure 4.

Effect of high concentrations of suramin. A, hypothetical model (derived from Fig. 1F) in which both Cl− (green) and suramin (red) could access the open pore via one “primary” portal (from the left as shown), whereas a “secondary” portal (from the right) allows access by Cl− but not suramin (for example, because of restricted dimensions) (see “Results”). B, example macroscopic current–voltage relationships for the weakly suramin-sensitive mutant K370Q (left panel) and the strongly suramin-sensitive mutant K1041Q (right panel) in inside-out patches. In both cases, current was recorded before (control, black) and after (red) addition of 1 mm suramin to the intracellular solution. C, mean fraction of control current remaining after addition of different concentrations of suramin to the intracellular solution for the different channel variants indicated. The mean data have been fitted as described under “Experimental Procedures” (Equation 2) with the following parameters: wildtype Kd 11.6 μm, nh 0.87, rf 0.047; R303Q Kd 32.3 μm, nh 0.93, rf 0.06; K370Q Kd 39.5 μm, nh 1.10, rf 0.034; and K1041Q Kd 2.0 μm, nh 1.19, rf 0.060. The asterisk indicates concentrations at which mutations showed a significant difference from wildtype (p < 0.05). At high concentrations (1–10 mm), none of the mutants studied showed a significant difference from wildtype. The means of data from 4–10 patches are shown.
Even if suramin occludes the entire Cl− permeation pathway, reducing Cl− flow through a secondary portal (by mutagenesis) could still lead to an overall strengthening of suramin block if that Cl− were able to compete very effectively with suramin for binding inside the permeation pathway (Fig. 5A). Although intracellular Cl− and other permeant anions are known to compete with other blocking anions for sites located more deeply into the pore (26), anion competition with suramin has not previously been reported. To investigate possible competition between intracellular Cl− and suramin, intracellular NaCl was replaced by sodium formate or NaSCN (Fig. 5). In wildtype, block by intracellular suramin was significantly strengthened when current was carried by formate (p < 0.005 compared with Cl− current; Fig. 5, B and D) and greatly weakened when current was carried by SCN− (p < 0.0001; Fig. 5, C and D). This suggests that, as with other blocker binding sites in the pore (26), formate is less able to compete with suramin compared with Cl−, and SCN− more effectively competes than does Cl−. Similar competition between suramin and these permeant anions was observed in the weakly suramin-sensitive, putative primary portal mutant K370Q (Fig. 5E) using a higher concentration of suramin. This indicates that even when suramin entry through the primary portal is impaired, competition with permeant anions does not appear to be altered. In contrast, in the strongly suramin-sensitive, putative secondary portal mutant K1041Q, block by a low concentration of suramin was similar when Cl− or formate carried current (Fig. 5F), although SCN− substitution still led to a significant weakening of suramin block (p < 0.0001; Fig. 5F). Thus it appears that the K1041Q mutation, in addition to causing an overall strengthening of suramin block (Fig. 3), also subtly alters the interaction between suramin and intracellular permeant anions. However, even when current was carried by formate ions and permeant ion interactions were minimal, K1041Q showed significantly stronger block compared with wildtype (p < 0.0001; Fig. 5G). The apparent effect of K1041Q on the permeant ion dependence of suramin block therefore appears unlikely to explain the overall strengthening of block observed in this mutant.
Figure 5.
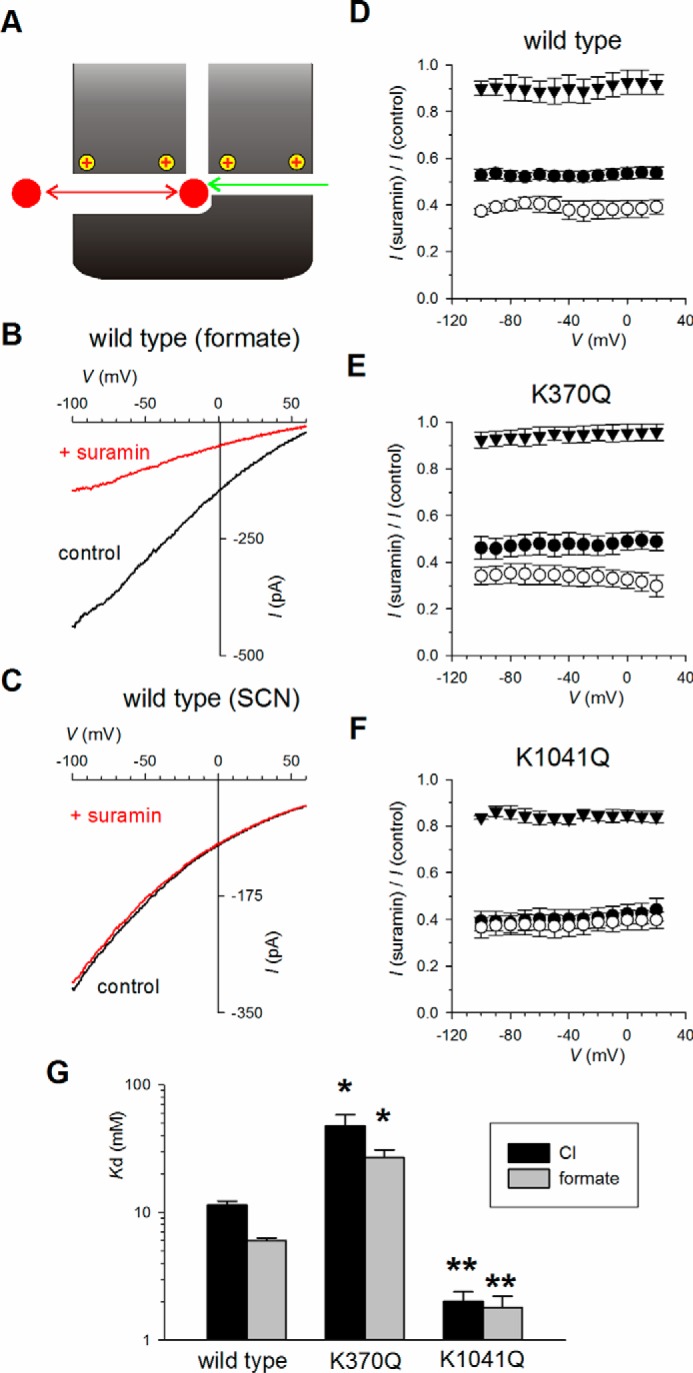
Interaction between intracellular permeant anions and suramin. A, hypothetical model (derived from Fig. 1F) in which permeant anions entering via a secondary portal are able to compete with suramin entering via a separate, primary portal and thereby indirectly weaken suramin's blocking effect (see “Results”). B and C, example macroscopic current–voltage relationships for wildtype in inside-out patches when extracellular NaCl (see Fig. 3A) had been substituted by sodium formate (B) or NaSCN (C). In each case currents were recorded before (control, black) and after (red) addition of 10 μm suramin to the intracellular solution. D–F, mean fraction of control current remaining following addition of suramin as a function of voltage in wildtype (D), K370Q (E), or K1041Q (F), when the extracellular solution contained NaCl (●), sodium formate (○), or NaSCN (▾). Note that different concentrations of suramin were used to give similar degrees of inhibition (with Cl−-containing solutions) in these differentially suramin-sensitive channel constructs: 10 μm (wildtype), 50 μm (K370Q), and 3 μm (K1041Q). G, mean Kd (at −100 mV) calculated from data such as that shown in D–F as described under “Experimental Procedures” (Equation 1) when the extracellular solution contained NaCl (black bars) or sodium formate (gray bars). Asterisks indicate a significant difference from wildtype. *, p < 0.01; **, p < 0.0001. Because block by this concentration of suramin was so weak when SCN− carried current, Kd could not be determined accurately for any variant. The means of data from 4–6 patches are shown in D–G.
Effects of additional mutations on conductance and block
The charge dependence of the roles of key residues in the primary (Arg303 and Lys370) and secondary (Lys978 and Lys1041) portals was further investigated by mutation to negatively charged glutamate (Fig. 6). As expected, R303E and K370E led to a further dramatic decrease in Cl− conductance (Fig. 6, A and C); the conductance of R303E was 19 ± 0% (n = 4) of R303Q and that of K370E was 60 ± 1% (n = 7) of K370Q. In contrast, the conductance of K1041E was only slightly decreased to 91 ± 0% (n = 5) of K1041Q (Fig. 6C). Even more strikingly, the conductance of K978E was significantly (p < 0.02) increased to 111 ± 4% (n = 6) of K978Q (Fig. 6C). Also as expected, R303E and K370E showed a further decrease in suramin sensitivity compared with R303Q and K370Q (Fig. 6, B and D). In contrast, the suramin sensitivity of K1041E was similar to or slightly less than that of K1041Q (Fig. 6D). Again the most striking result was observed with K978E; whereas K978Q significantly strengthened suramin block compared with wildtype (Figs. 3C and 6D), suramin block of K978E was significantly weaker than for wildtype (p < 0.005, n = 5) (Fig. 6, B and D). Although these results further support an electrostatic role for Arg303 and Lys370 in interactions with anions, they do not support a purely electrostatic interaction between Lys978 and Lys1041 and either Cl− (Fig. 6C) or suramin (Fig. 6D).
Figure 6.

Effect of additional mutations on chloride conductance and suramin block. A, mean single channel current–voltage relationships for the glutamate-substituted channel mutants listed. Wildtype data as in Fig. 2B. B, mean fraction of control current remaining following addition of 10 μm suramin to the intracellular solution as a function of voltage in the same glutamate-substituted channel mutants listed in A. For each mutant shown, block was significantly different from wildtype (black symbols; data from Fig. 3B) under these conditions (p < 0.005). C, mean single channel conductance (measured from the slope of the current–voltage relationship from individual patches) as a function of side chain charge at the different positions listed. Black symbols represent wildtype (charge +1); charge 0 represents glutamine (Q) substitution at each site, and charge −1 glutamate (E) substitution. D, mean fraction of control current remaining following addition of 10 μm suramin to the intracellular solution (at −100 mV) for the same channel variants shown in C. E, mean single channel conductance as a function of the number of positively charged side chains neutralized by mutation to glutamine, in single mutants (change in charge −1) and double mutants (change in charge −2) as listed. F, mean fraction of control current remaining following addition of 10 μm suramin to the intracellular solution (at −100 mV) for the same channel variants shown in E. The means of data from 4–8 patches are shown.
To investigate the independence of effects of mutations at different sites, we also combined charge-neutralizing mutations predicted to be near the periphery (K370Q) and core (R303Q) of the primary portal and within the secondary portal (K1041Q). Overall, the double mutant channels R303Q/K370Q, R303Q/K1041Q, and K370Q/K1041Q had effects on Cl− conductance (Fig. 6E) and suramin sensitivity (Fig. 6F) that appeared broadly additive of the effects of the individual mutations.
Role of fixed positive charges in Pt(NO2)42− block
In addition to determining the apparent affinity of suramin block (19), Arg303 has been proposed to play a role in channel block by smaller, divalent Pt(NO2)42− (22), even though these Pt(NO2)42− ions are able to penetrate much further into the pore than suramin and interact with sites within the TMs (22, 28). To test whether portal mutations would also have differential effects on Pt(NO2)42− block, we investigated suramin-resistant K370Q and suramin-sensitive K1041Q. As described previously for R303Q (22), K370Q led to a significant weakening of block by intracellular Pt(NO2)42− (p < 0.0005; Fig. 7), consistent with Pt(NO2)42− entering the pore via the primary portal. In contrast, K1041Q showed significantly strengthened block by Pt(NO2)42− (p < 0.005; Fig. 7).
Figure 7.

Differential effect of mutations on Pt(NO2)42− block. A, example macroscopic current–voltage relationships for different channel variants in inside-out patches. In each case currents were recorded before (control, black) and after (red) addition of 50 μm Pt(NO2)42− to the intracellular solution. B, mean fraction of control current remaining following addition of this concentration of Pt(NO2)42− as a function of voltage in these channel variants. The means of data from 5–7 patches are shown.
Discussion
Structural (7, 8), functional (17, 18), and molecular modeling studies (15, 16) concur that cytoplasmic Cl− ions use lateral portals, rather than a directly central route, to enter the CFTR channel protein and so begin their journey across the membrane. These portals may be a key factor in CFTR functioning as an ion channel rather than a pump (5). However, how these portals are arranged—and even how many portals exist and what their relative functional contributions to Cl− transport might be—remains to be defined. Previously we speculated, based on functional investigation, that two portals might exist (17). A functionally dominant portal was proposed between TMEs 4 and 6, and lined by Lys190 (TME3), Arg248 (TME4), Arg303 (TME5), and Lys370 (TME6) (17). We also speculated that a functionally lesser portal might be located on the opposite side of the protein, between TMEs 10 and 12 and possibly involving Lys1041 and Arg1048 in TME10 (17). This tentative model was based on the finding that mutation and chemical modification of amino acid side chains in different TMEs had qualitatively similar (although quantitatively different) effects on single channel Cl− current amplitudes (17). A possible contribution of positively charged amino acid side chains to distinct portals coming from opposite sides of the TMEs also appeared consistent with atomic homology models of that time (15, 17). However, we will argue that our present results investigating the effects of mutations of these same, functionally important positively charged side chains on both Cl− conductance and interactions with blockers are more consistent with the existence of a single portal from the cytoplasm to the pore, with its mouth located between TMEs 4 and 6. This model appears consistent with the reported cryo-EM structure of the TMEs in the near-open state (Fig. 8A), which shows a large portal between TMEs 4 and 6 but not between TMEs 10 and 12 (8).
Figure 8.

Models of cytoplasmic portal structure and function. A and B, structure of the TMEs in zebrafish CFTR in a near-open state (8) (see also Fig. 1, D and E). Positively charged amino acids mutated in the present study are shown as space-filling models, viewed from above (A) or from the side (B; looking toward the portal entrance). In A, the red arrow indicates the entrance to the portal (note that no similar sized portal was observed on the “opposite” side of the TMEs (8)). In B, the black dotted line indicates the approximate extent of the apparent portal entrance. As noted in Fig. 1, the numbering of these positively charged amino acids is slightly different in zebrafish CFTR. C–E, models of portal function. C, Arg248 and Lys370 are proposed to lie close to the mouth of a single portal, with Lys190 and Arg303 located more internally. All four of these positively charged residues attract cytoplasmic Cl− (green) and suramin (red) electrostatically. Mutation of Lys978, Lys1041, and Arg1048 affects the function of this portal at a distance, perhaps via propagated structural changes, to impact Cl− and suramin entry indirectly. D, alternative to C, Lys978, Lys1041 and/or Arg1048 could contribute to a second entry pathway for Cl−, whereas at the same time indirectly impacting suramin entry via the main portal. E, location of Lys978, Lys1041 and/or Arg1048 in a “recessed” part of the pore might allow direct interaction with small Cl− ions but not large suramin molecules. Again, an indirect impact on suramin entry would also have to be supposed.
The cytoplasmic portal(s) appear to be the site of action of the large cytoplasmic channel blocker suramin (19) (Figs. 3–6). Investigation of the concentration dependence of suramin block (Fig. 4C) is consistent with this large anionic molecule occluding the entire Cl− permeation pathway, most likely by interacting with a single binding site. However, the finding that charge-neutralizing mutations can either weaken or strengthen block by suramin (Figs. 3–6), as well as Pt(NO2)42− (Fig. 7), appears inconsistent with the simplest models in which all positively charged amino acid side chains studied electrostatically funnel Cl− and blocking anions to the pore (Fig. 1, C and D).
All results concerning residues previously suggested to contribute to the primary cytoplasmic portal, namely Lys190, Arg248, Arg303, and Lys370 (17), do appear consistent with an electrostatic effect on the attraction of Cl− (Figs. 2 and 6), suramin (Figs. 3 and 6), and Pt(NO2)42− (Fig. 7) (22) to the pore. These four positively charged residues may therefore line a bona fide entry pathway into the pore from the cytoplasm, not only for Cl− but also blocking anions such as suramin and Pt(NO2)42− (Fig. 8C). This is in fact consistent with the reported structure of the TMEs in the near-open state (Fig. 8A), which also shows a positive electrostatic potential surrounding the portal between TMEs 4 and 6 (8).
Although neutralization of other nearby positively charged residues (Lys978, Lys1041, and Arg1048) also reduces Cl− conductance (Fig. 2), other results argue against an electrostatic role of these residues in attracting anions to the pore. First, mutations at these sites either have no effect (Fig. 3) or actually strengthen block by suramin and Pt(NO2)42− (Figs. 3–7). Second, the effects of mutations at Lys978 do not appear to be related to amino acid side chain charge (Fig. 6, C and D) as would be expected of any kind of electrostatic effect. Mutations at Lys1041 also have effects that are only weakly related to side chain charge at best (Fig. 6, C and D), especially when compared with the strongly charge-dependent effects of mutations at Arg303 and Lys370 (Fig. 6, C and D). In fact, deposition of a negative charge at position Lys1041 or Arg1048 (by cysteine mutagenesis followed by modification with a negatively charged cysteine-reactive reagent) was previously shown to have only a small effect on Cl− conductance when compared with Lys190, Arg248, Arg303, or Lys370 (17), again suggesting that these groups of positively charged residues had differential effects on pore function. Thus Lys978, Lys1041, and Arg1048 do not appear to influence channel properties by the same mechanism as Lys190, Arg248, Arg303, and Lys370.
We considered different models in which these amino acids might contribute to a secondary portal and thereby indirectly affect suramin binding (Figs. 4A and 5A). Several of our findings argue against the existence of such a secondary portal. As described above, mutation of these positively charged residues have effects on anion interactions with the channel that are either weakly charge-dependent or charge-independent. Furthermore, there does not appear to be a substantial suramin-insensitive component to the current (Fig. 4C) that might be expected for two independent entrances to the pore (Fig. 4A). In addition, suramin block of K1041Q remains strong even under low [Cl−] conditions that minimize competition between suramin and permeant anions (Fig. 5G), arguing against the existence of two non-independent pore entrances (Fig. 5A). Finally, whereas mutations at Lys978, Lys1041, and Arg1048 clearly affect both Cl− conductance (Figs. 2, 6) and suramin block (Figs. 3 and 6), there is no obvious pattern linking the effects of different mutations on these two aspects of pore function. Because we are unable to find compelling evidence that Lys978, Lys1041, and Arg1048 interact directly with suramin (or, in the case of Lys1041, with Pt(NO2)42−), we speculate that the effects of mutations at these sites on channel block are indirect; in other words, that mutations at these sites somehow alter the structure of the primary portal in a way that promotes suramin entry into and/or binding within this portal (Fig. 8C). If this is the case, then it may also be that the weakly charge-dependent or charge-independent effect of mutations (Fig. 6C) or chemical modifications (17) on Cl− conductance might also be ascribed to an indirect effect on Cl− entry via the primary portal (Fig. 8C). For example, one speculative mechanism by which this might take place is if these mutations lead indirectly to a widening of the primary portal, which could in theory facilitate the entry of large blocker molecules while at the same time impair entry of Cl− ions because of a reduction in the electrostatic influence of positive charges in the primary portal. Alternatively, we cannot rule out that Lys978, Lys1041, and/or Arg1048 directly influence the entry of a small amount of Cl− from the cytoplasm via a separate pathway (Fig. 8D) or that these residues lie in some recess that can be accessed from the primary portal that allows them to directly interact with Cl− ions but not with suramin (Fig. 8E). In either of these cases, the side chains of Lys978, Lys1041, and/or Arg1048 could contribute to the overall positive electrostatic potential that influences the movement of cytoplasmic Cl− ions into the pore. However, in either of these cases we would have to assume that mutation of these residues has both direct (electrostatic) and indirect (structural) effects on portal function, an assumption for which direct experimental evidence is lacking.
In conclusion, our functional results suggest that a single portal is sufficient to explain entry of cytoplasmic Cl− and blocking ions into the CFTR pore. This conclusion appears consistent with the reported structure of the TMEs (Fig. 8, A and B). Positive charges at Arg248 and Lys370 appear located close to the mouth of this portal (Fig. 8A), where they strongly impact suramin block (Fig. 3). Closer to the interior of the protein (Fig. 8, A and B), Lys190 and Arg303 strongly attract Cl− ions “upwards” toward the TM pore (Figs. 2 and 6), consistent with the important role proposed previously (17, 27). Suramin molecules may be too large to penetrate into the interior of the protein via this route. Around the periphery of the portal, Lys978, Lys1041, and Arg1048 may play minor roles in interactions with Cl− ions that, as described above, may be direct or indirect.
Experimental procedures
Experiments were carried out on baby hamster kidney cells transiently transfected with human CFTR, as described in detail previously (20). Mutant forms of CFTR were generated using the QuikChange site-directed mutagenesis system (Agilent Technologies, Santa Clara, CA) and verified by DNA sequencing. Functional characterization of wildtype and mutant CFTR channels was carried out using single channel and macroscopic patch-clamp recordings from inside-out membrane patches, as described in detail previously (20). Following patch excision and recording of background currents, CFTR channels were activated by exposure to protein kinase A catalytic subunit (PKA) plus MgATP (1 mm) in the intracellular solution. As in earlier studies (19, 21, 22), single-channel currents were recorded after weak PKA stimulation (1–5 nm), whereas all macroscopic CFTR currents were recorded after maximal PKA stimulation (∼20 nm) and subsequent treatment with sodium pyrophosphate (2 mm) to stabilize the channel open state. As in previous studies of both single-channel conductance (17, 18, 23) and channel block (24–26), the intracellular (bath) solution contained 150 mm NaCl, 2 mm MgCl2, 10 mm TES, pH 7.4, whereas the extracellular (pipette) solution contained 150 mm sodium gluconate, 2 mm MgCl2, 10 mm TES, pH 7.4, resulting in an outwardly directed Cl− concentration gradient. Current traces were filtered at 100 (for macroscopic currents) or 50 Hz (for single channel currents) using an eight-pole Bessel filter, digitized at 250 Hz, and analyzed using pCLAMP10 software (Molecular Devices, Sunnyvale, CA). Measurement of single channel and macroscopic current amplitudes and construction of leak-subtracted macroscopic current–voltage relationships were carried out as described previously (20). Membrane voltages were corrected for liquid junction potentials calculated using pCLAMP software.
Block of macroscopic current by intracellular suramin and Pt(NO2)42− was investigated as described previously (13, 19, 22). Apparent blocker dissociation constant, Kd, was approximated from the following equation,
| (1) |
where [B] is blocker concentration, I is current amplitude following the addition of blocker, and I0 is the control, unblocked current amplitude. Suramin concentration–inhibition relationships (Fig. 4) were fitted by an equation that assumes a fixed fraction of the current is suramin-insensitive,
| (2) |
where nh is the slope factor or Hill coefficient, and rf is the suramin-resistant fraction of the current (0–1).
The experiments were carried out at room temperature, 21–24 °C. The values are presented as the means ± S.E. For graphical presentation of mean values, the error bars represent S.E., and where no error bars are visible, S.E. is smaller than the size of the symbol. Tests of significance were carried out using Student's two-tailed t test, with p < 0.05 being considered statistically significant. All chemicals were from Sigma–Aldrich except for K2Pt(NO2)4 (Strem Chemicals, Newburyport, MA) and PKA (Promega, Madison, WI).
Author contributions
M.-S. L. and P. L. data curation; M.-S. L. and P. L. investigation; E. A. C., Y. E. H., and P. L. supervision; E. A. C., Y. E. H., and P. L. project administration; Y. E. H. and P. L. conceptualization; Y. E. H. and P. L. writing-review and editing; P. L. formal analysis; P. L. funding acquisition; P. L. writing-original draft.
Acknowledgment
We thank Christina Irving for technical assistance.
This work was supported by Cystic Fibrosis Canada. The authors declare that they have no conflicts of interest with the contents of this article.
- CFTR
- cystic fibrosis transmembrane conductance regulator
- cryo-EM
- cryo-electron microscopy
- NBD
- nucleotide binding domain
- PKA
- protein kinase A
- TES
- N-tris[hydroxymethyl]methyl-2-aminoethanesulfonate
- TM
- transmembrane helix
- TME
- transmembrane helical extension.
References
- 1. Dean M., Rzhetsky A., and Allikmets R. (2001) The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 11, 1156–1166 10.1101/gr.GR-1649R [DOI] [PubMed] [Google Scholar]
- 2. Rees D. C., Johnson E., and Lewinson O. (2009) ABC transporters: the power to change. Nat. Rev. Mol. Cell Biol. 10, 218–227 10.1038/nrm2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gadsby D. C., Vergani P., and Csanády L. (2006) The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature 440, 477–483 10.1038/nature04712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Callebaut I., Hoffmann B., Lehn P., and Mornon J.-P. (2017) Molecular modeling and molecular dynamics of CFTR. Cell. Mol. Life Sci. 74, 3–22 10.1007/s00018-016-2385-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Linsdell P. (2017) Structural changes fundamental to gating of the cystic fibrosis transmembrane conductance regulator anion channel pore. Adv. Exp. Med. Biol. 925, 13–32 [DOI] [PubMed] [Google Scholar]
- 6. Zhang Z., and Chen J. (2016) Atomic structure of the cystic fibrosis transmembrane conductance regulator. Cell 167, 1586–1597 10.1016/j.cell.2016.11.014 [DOI] [PubMed] [Google Scholar]
- 7. Liu F., Zhang Z., Csanády L., Gadsby D. C., and Chen J. (2017) Molecular structure of the human CFTR ion channel. Cell 169, 85–95 10.1016/j.cell.2017.02.024 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Z., Liu F., and Chen J. (2017) Conformational changes of CFTR upon phosphorylation and ATP binding. Cell 170, 483–491 10.1016/j.cell.2017.06.041 [DOI] [PubMed] [Google Scholar]
- 9. Linsdell P. (2017) Architecture and functional properties of the CFTR channel pore. Cell. Mol. Life Sci. 74, 67–83 10.1007/s00018-016-2389-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. El Hiani Y., and Linsdell P. (2010) Changes in accessibility of cytoplasmic substances to the pore associated with activation of the cystic fibrosis transmembrane conductance regulator chloride channel. J. Biol. Chem. 285, 32126–32140 10.1074/jbc.M110.113332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bai Y., Li M., and Hwang T.-C. (2011) Structural basis for the channel function of a degraded ABC transporter, CFTR (ABCC7). J. Gen. Physiol. 138, 495–507 10.1085/jgp.201110705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W., El Hiani Y., and Linsdell P. (2011) Alignment of transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Gen. Physiol. 138, 165–178 10.1085/jgp.201110605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Linsdell P. (2014) State-dependent blocker interactions with the CFTR chloride channel: implications for gating the pore. Pflügers Arch. 466, 2243–2255 [DOI] [PubMed] [Google Scholar]
- 14. Zhang J., and Hwang T.-C. (2015) The fifth transmembrane segment of cystic fibrosis transmembrane conductance regulator contributes to its anion permeation pathway. Biochemistry 54, 3839–3850 10.1021/acs.biochem.5b00427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mornon J.-P., Hoffmann B., Jonic S., Lehn P., and Callebaut I. (2015) Full-open and closed CFTR channels, with lateral tunnels from the cytoplasm and an alternative position of the F508 region, as revealed by molecular dynamics. Cell. Mol. Life Sci. 72, 1377–1403 10.1007/s00018-014-1749-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Corradi V., Vergani P., and Tieleman D. P. (2015) Cystic fibrosis transmembrane conductance regulator (CFTR): closed and open state channel models. J. Biol. Chem. 290, 22891–22906 10.1074/jbc.M115.665125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. El Hiani Y., and Linsdell P. (2015) Functional architecture of the cytoplasmic entrance to the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 290, 15855–15865 10.1074/jbc.M115.656181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. El Hiani Y., Negoda A., and Linsdell P. (2016) Cytoplasmic pathway followed by chloride ions to enter the CFTR channel pore. Cell. Mol. Life Sci. 73, 1917–1925 10.1007/s00018-015-2113-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. St Aubin C. N., Zhou J.-J., and Linsdell P. (2007) Identification of a second blocker binding site at the cytoplasmic mouth of the cystic fibrosis transmembrane conductance regulator chloride channel pore. Mol. Pharmacol. 71, 1360–1368 10.1124/mol.106.031732 [DOI] [PubMed] [Google Scholar]
- 20. Zhou J.-J., Li M.-S., Qi J., and Linsdell P. (2010) Regulation of conductance by the number of fixed positive charges in the intracellular vestibule of the CFTR chloride channel pore. J. Gen. Physiol. 135, 229–245 10.1085/jgp.200910327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ge N., Muise C. N., Gong X., and Linsdell P. (2004) Direct comparison of the functional roles played by different transmembrane regions in the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 279, 55283–55289 10.1074/jbc.M411935200 [DOI] [PubMed] [Google Scholar]
- 22. Zhou J.-J., Fatehi M., and Linsdell P. (2007) Direct and indirect effects of mutations at the outer mouth of the CFTR chloride channel pore. J. Membr. Biol. 216, 129–142 10.1007/s00232-007-9056-6 [DOI] [PubMed] [Google Scholar]
- 23. Negoda A., El Hiani Y., Cowley E. A., and Linsdell P. (2017) Contribution of a leucine residue in the first transmembrane segment to the selectivity filter region in the CFTR chloride channel. Biochim. Biophys. Acta 1859, 1049–1058 10.1016/j.bbamem.2017.02.014 [DOI] [PubMed] [Google Scholar]
- 24. Linsdell P. (2005) Location of a common inhibitor binding site in the cytoplasmic vestibule of the cystic fibrosis transmembrane conductance regulator chloride channel pore. J. Biol. Chem. 280, 8945–8950 10.1074/jbc.M414354200 [DOI] [PubMed] [Google Scholar]
- 25. Linsdell P. (2015) Interactions between permeant and blocking anions inside the CFTR chloride channel pore. Biochim. Biophys. Acta 1848, 1573–1590 10.1016/j.bbamem.2015.04.004 [DOI] [PubMed] [Google Scholar]
- 26. Linsdell P. (2016) Anion conductance selectivity mechanism of the CFTR chloride channel. Biochim. Biophys. Acta 1858, 740–747 10.1016/j.bbamem.2016.01.009 [DOI] [PubMed] [Google Scholar]
- 27. St Aubin C. N., and Linsdell P. (2006) Positive charges at the intracellular mouth of the pore regulate anion conduction in the CFTR chloride channel. J. Gen. Physiol. 128, 535–545 10.1085/jgp.200609516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. El Hiani Y., and Linsdell P. (2012) Tuning of CFTR chloride channel function by location of positive charges within the pore. Biophys. J. 103, 1719–1726 10.1016/j.bpj.2012.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]